

Abstract
Potassium ion channels encoded by the human ether-a-go-go related gene (hERG) form the ion-conducting subunit of the rapid delayed rectifier potassium current (IKr). Although hERG channels exhibit a widespread tissue distribution they play a particularly important role in the heart. There has been considerable interest in hERG K+ channels for three main reasons. First, they have very unusual gating kinetics, most notably rapid and voltage-dependent inactivation coupled to slow deactivation, which has led to the suggestion that they may play a specific role in the suppression of arrhythmias. Second, mutations in hERG are the cause of 30–40% of cases of congenital long QT syndrome (LQTS), the commonest inherited primary arrhythmia syndrome. Third, hERG is the molecular target for the vast majority of drugs that cause drug-induced LQTS, the commonest cause of drug-induced arrhythmias and cardiac death. Drug-induced LQTS has now been reported for a large range of both cardiac and non-cardiac drugs, in which this side effect is entirely undesired. In recent years there have been comprehensive reviews published on hERG K+ channels (Vandenberg et al. 2012) and we will not re-cover this ground. Rather, we focus on more recent work on the structural basis and dynamics of hERG gating with an emphasis on how the latest developments may facilitate translational research in the area of stratifying risk of arrhythmias.
hERG K+ channels have unusual gating kinetics
Voltage-gated K+ (VGK) channels can exist in one of three main conformational states: closed, open and in-activated (Fig.1A). Differences in the kinetics and voltage dependence of the transitions between the closed and open states and between the open and inactivated states gives rise to considerable phenotypic diversity amongst the 38 different VGK channel subtypes. The Kv11.x subfamily is unique within the VGK family in that the transitions between the closed and open states are considerably slower than the transitions between the open and inactivated states. In addition the transitions between the open and inactivated states are voltage dependent (Vandenberg et al., 2012). This unusual combination of kinetics gives rise to an apparent inward rectification that is crucial for maintaining a prolonged plateau phase of the cardiac action potential (Fig.1B). Just as importantly, the rapid and voltage-dependent recovery from inactivation during the terminal phase of cardiac repolarization coupled to slow deactivation during the early diastolic period confers upon these channels an important role in the suppression of ectopic beats during the late repolarization phase (Fig.1C). There has been considerable interest in elucidating the molecular basis of the unusual gating kinetics of hERG ever since their discovery (Sanguinetti et al. 1995; Trudeau et al. 1995). In recent years, discoveries involving structural analysis of the cytoplasmic domains of hERG and application of rate equilibrium free energy relationship (REFER) analysis have started to give us a much more detailed understanding of the structural basis of the unusual behaviour of these important channels.
Figure 1.
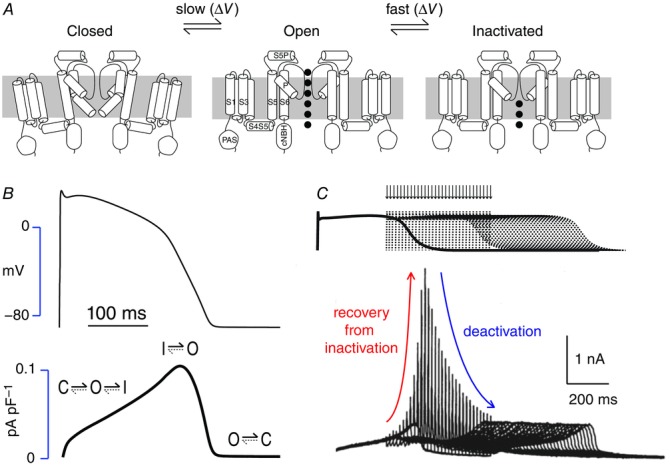
HERG K+ channel gating A, Gating scheme for hERG K+ channels showing the three main conformational states, closed, open and inactivated. Transitions between the closed and open states are slow whereas transitions between the open and inactivated states are rapid. Transmembrane segments (S1–S6 and the pore helix, P) as well as the main extracellular (S5P) and intracellular (S4S5, PAS and cNBH) domains are labelled in the open state conformation. B, Current profile for IKr (lower trace) during a ventricular action potential (upper trace). As a consequence of the unusual gating kinetics there is relatively little current flow during the first portion of the action potential plateau but as the plateau voltage slowly decreases IKr increases, due to the rapid and voltage-dependent recovery from the inactivated state back to the open state. C, Response of hERG current to premature action potential stimuli. As hERG channels recover from the inactivated stated into the open state, during repolarization an ‘ectopic beat’ will result in a large increase in outward current followed by rapid re-inactivation and hence the transient current spike. The envelope of the peak current spike reflects the recovery from inactivation followed by deactivation. Modified from Lu et al. (2001).
hERG K+ channel inactivation
Most VGK channels exhibit one, or both, of two major types of inactivation, termed N-type and C-type. N-type inactivation is caused by an occlusion of the ion-conducting pore by the intracellular N-terminal domain, and is also known as ball-and-chain inactivation. Unlike Shaker K+ channels, hERG K+ channels completely lack this N-type inactivation mechanism. Instead, the inactivation observed in hERG K+ channels more closely resembles C-type inactivation of Shaker K+ channels (Smith et al. 1996). C-type inactivation is not as well understood as N-type inactivation but is thought to involve conformational rearrangements of the selectivity filter, which directly prevent the conduction of ions (Hoshi & Armstrong, 2013). C-type inactivation in hERG K+ channels exhibits some critical differences from that of Shaker k+ channels, such as an intrinsic voltage dependence, i.e. not directly coupled to the voltage dependence of activation (Perry et al. 2013b) and much more rapid kinetics of both the forward (open-to-inactivated) and reverse (inactivated-to-open) transitions (Smith et al. 1996). Much of our understanding of the structural and molecular components that underlie C-type inactivation of hERG K+ channels has been discussed in detail elsewhere (Vandenberg et al. 2012), so this review will focus on the most recent advances in our understanding of this crucial gating mechanism.
Capturing a snapshot of the inactivated state
There is no doubt that obtaining an atomic resolution structure of the hERG K+ channel in the open-inactivated conformation would constitute a dramatic leap forward in our understanding of the C-type inactivation process, but to do this requires a high level of expression and purification of functional hERG channel protein. Despite considerable efforts this remains an elusive goal. Two recent studies utilized chimeric approaches with bacterial KcsA (Hausammann & Grütter, 2013) or rat Kv1.2 (Dhillon et al. 2014) potassium channels to help stabilize the hERG protein, with the latter reporting a degree of expression and purification of functional chimeric hERG-Kv1.2 channels in Pichia pastoris. Until we are able to obtain suitable levels of purified protein for X-ray crystallography or cryo-electron microscopy experiments, homology modelling and molecular dynamics (MD) simulations remain valuable tools for examining the molecular basis of hERG channel inactivation (Stansfeld et al. 2008; Durdagi et al. 2012; Köpfer et al. 2012; Dempsey et al. 2014). These MD simulation studies suggest that the selectivity filter of hERG channels is stabilized by an H-bonding complex involving key residues Ser620 (pore helix) and Asn629 (post-selectivity filter), in addition to water molecules and other pore helix residues, and that breakdown of this network may trigger the conformational rearrangements of the selectivity filter that underlie inactivation. In particular, ‘flipping’ of selectivity filter residues Val625, Gly626 and Phe627 has been suggested to underlie the final transition to the inactivated state (Stansfeld et al. 2008; Durdagi et al. 2012; Köpfer et al. 2012). These MD simulations are largely supported by a wealth of mutagenesis data in hERG channels.
Obtaining a molecular ‘movie’ of inactivation gating using REFER/Φ-value analyses
The hERG K+ channel, like all other ion channels, is a highly dynamic protein that undergoes a complex sequence of molecular and structural rearrangements in order to transition from one stable state to another (i.e. from an open state to an inactivated state). While X-ray crystallography remains the gold standard for resolving atomic resolution protein structures, it lacks a temporal dimension and so cannot be used to elucidate the molecular gymnastics that underlie the gating transitions of ion channels. REFER analysis is a powerful protein engineering technique that has been extensively applied to understand the conformational changes that occur during protein folding and unfolding (Fersht & Daggett, 2002) and to study the gating transitions of nicotinic acetylcholine receptors (Auerbach, 2003). Theoretically, REFER analysis can be applied to any transition pathway between two stable end states, but is not applicable to study processes that involve multiple successive transitions, such as the activation of voltage-gated ion channels, which requires the transition through several closed states before reaching the open conformation (Wang et al. 1997). The voltage-dependent inactivation of hERG K+ channels, however, can be well described by a two-state mechanism (Wang et al. 2011) and so is amenable to analysis using the REFER technique.
In a two-state process the kinetics of the transitions between the two states are proportional to the height of the energy barrier separating the two end states (ΔΔG‡), whereas the equilibrium of the reaction is proportional to the energy difference between the two end states (ΔΔG0) (Fig.2A). Engineered point mutations can be used to systematically probe the relative timing of individual residue or domain motions during the transition between two stable end states by comparing perturbations to the free energy changes in kinetic (ΔΔG‡) and thermodynamic (ΔΔG0) relationships. In a REFER plot, the slope of ΔΔG‡ versus ΔΔG0 is denoted as the Φ-value, and hence the technique is often referred to as Φ-value analysis. REFER analysis has shown that inactivation of hERG K+ channels does not merely involve a conformational rearrangement within the conducting pore of the channel as previously thought, but requires sequential motions of multiple interconnected domains throughout the protein (Wang et al. 2011). For instance, manipulating the external K+ concentration produced a Φ-value close to 1, indicating that a loss of K+ from the selectivity filter occurs very early in the transition from the open to the inactivated state. Diminishing Φ-values were then observed for families of mutations in the pore helix (Φ ∼ 0.85), S5 helix (Φ ∼ 0.75), S5P linker (Φ ∼ 0.6), S4 helix (Φ ∼ 0.50), S4–S5 linker (Φ ∼ 0.45), S6 helix (Φ ∼ 0.3) and pore helix again (Φ ∼ 0.25) (Wang et al. 2011; Perry et al. 2013a,b) indicating that these domains in the hERG protein undergo sequential conformational rearrangements during inactivation, similar to the sequential motion of interconnected domains that are required to open a Japanese puzzle box or in the conversion of a transformer robot.
Figure 2.
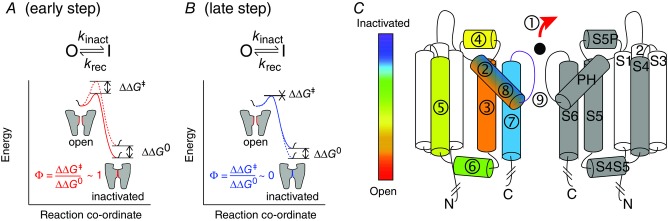
Reaction co-ordinate diagrams for hERG inactivation A, Illustration of how REFER analysis can be applied to hERG K+ channel inactivation. A perturbation to a very early step in the transition from the open state to the inactivated state (red solid line shows WT and red dotted line shows mutant) will give rise to a Φ-value close to 1; B, Illustrates how a perturbation to a very late step in the transition from the open state to the inactivated state (blue solid line shows WT and dotted line shows mutant) will give rise to a Φ-value close to 0. C, ‘Japanese puzzle box’ model of hERG inactivation. The Φ-value for removal of external K+ (1) was close to one. Mutations in the different domains of the channel resulted in a progressive decrease in Φ-value corresponding to progressively later steps in the inactivation process starting with the pore helix (2, Φ-value ∼ 0.85), S5 helix (3, Φ-value ∼ 0.7), S5P linker (4, Φ-value ∼ 0.6), S4 helix (5, Φ-value ∼0.5), S4S5 linker (6, Φ-value ∼0.45), S6 (7, Φ-value ∼ 0.3) and the pore helix again (8, Φ-value ∼0.15). The final step is presumed to involve a conformational rearrangement of the SF (9).
The importance of intra- and inter-subunit domain–domain interactions
The sequential motion of multiple interconnected domains during C-type inactivation suggests a level of domain–domain coordination that is probably achieved by an extensive network of intra- and inter-subunit interactions between different domains of the hERG channel protein. Although REFER analysis provides invaluable temporal information about the role of residues/domains during the inactivation transition pathway, it does not provide information about changes in environment or specific molecular-level interactions of that residue/domain. However, a combination of homology modelling, molecular dynamics simulations and double mutant cycle analysis data is beginning to build a picture of these molecular networks. While much of the early focus in this area surrounded interactions between the pore helix and selectivity filter (Stansfeld et al. 2008), it is becoming increasingly clear that a much more complicated network of interactions exists. Recent studies have suggested that the pore helix alone forms molecular-level interactions with the S1 helix (Colenso et al. 2013), S5 helix (Lees-Miller et al. 2009; Ferrer et al. 2011; Durdagi et al. 2012; Perry et al. 2013a) and S6 helix (Durdagi et al. 2012; Perry et al. 2013a). Crucially, REFER analysis suggests that the pore helix plays a dual role during the inter-conversion between the open and inactivated states of hERG channels. At an early stage of the inactivation transition pathway the loss of K+ ions from the selectivity filter is linked to S5 domain motion through multiple pore helix residues, including Thr618 (Perry et al. 2013a), which double mutant cycle analysis suggests is energetically coupling with His562 (Lees-Miller et al. 2009), Trp568 (Ferrer et al. 2011) and/or Ile567 (Perry et al. 2013a) residues in the S5 helix of the same subunit. In the later stages of the inactivation transition pathway, the pore helix couples S6 helix motion to a putative final rearrangement of the selectivity filter via a putative inter-subunit interaction between Phe619 of the pore helix and Ile642 of the neighbouring S6 helix (Perry et al. 2013a). Molecular dynamics simulations indicate that distal S6 motion is an important step preceding entry into the inactivation state and may, in part, underlie enhanced affinity of many drugs for the inactivated state of the channel (Chen et al. 2002; Stansfeld et al. 2007). In addition to the pore helix interactions, combined REFER analysis, molecular homology modelling and double mutant cycle analysis suggests an inter-subunit energetic coupling between hydrophobic residues on the S4 helix (Leu529, Leu530, Leu532, Val535) and S5 helix (Ile560, Ile567) of the neighbouring subunit during inactivation gating (Perry et al. 2013b).
Assessing subunit co-operativity during hERG inactivation gating
Techniques such as REFER analysis, coupled with double mutant cycle analysis and molecular modelling, can provide invaluable information on the molecular rearrangements that underlie inactivation in hERG channels. So far, however, these studies have only been performed on homotetrameric mutant channels and have not addressed whether there is co-operativity amongst subunits during gating. Understanding co-operativity is likely to be of physiological relevance given that a proportion of native IKr channels will be heterotetramers, comprising a mixture of hERG1a and hERG1b subunits (Jones et al. 2014).
Wu et al. (2014) recently addressed the question of whether hERG channel inactivation requires subunit co-operativity by developing a concatenated hERG channel (i.e. a channel in which subunits are joined by an intracellular linker), which makes it possible to introduce mutations separately into each of the four subunits (Wu et al. 2014). They showed that concatenated hERG channels containing an S620T mutation in the pore helix, or a double mutant (G628C + S631C) in the selectivity filter, of a single subunit can almost completely abolish inactivation. This result suggests that there is a high degree of concerted motion between all four subunits during inactivation. In stark contrast, successive T618A mutations of the pore helix produced additive perturbations to inactivation that are consistent with a sequential model of co-operative interactions, while successive M645C mutations in the pore-lining S6 domain suggested a complete lack of subunit co-operativity. While at first glance these results appear contradictory, it becomes clearer when we consider them together with the sequential ‘Japanese puzzle box’ model of inactivation gating. The S620T mutation may improve the stability of an H-bonding network between the pore helix and selectivity filter (containing Gly628) and so stabilize a conducting conformation of the selectivity filter which attenuates transition into the inactivated state (Stansfeld et al. 2008). The study of Wu and colleagues then suggests that the improved stability of the selectivity filter resulting from an S620T mutation in just a single subunit is enough to prevent the late stage transition into the inactivated state in an all-or-none fashion and promotes the idea that the final transition step requires a concerted motion of all four subunits. In contrast to Ser620, Thr618 of the pore helix is involved at a much earlier stage in the inactivation transition pathway (Perry et al. 2013a) such that conformational changes involving this residue may simply play a role in ‘priming’ each subunit to inactivate, a process which would not require a high degree of co-operativity between subunits. Thus, at least for inactivation gating in hERG channels, the degree of co-operativity between subunits may alter between different stages of the transition pathway. Whether this is also true for channel opening, or in other types of VGK channels, is not known but certainly worthy of investigation.
Role of cytoplasmic domains in regulating hERG K+ channel deactivation
Shortly after the discovery of hERG K+ channels, it was shown that deletion of the entire N-terminus, up to residue 354, resulted in a dramatic acceleration of the rate of channel closure (Schönherr & Heinemann, 1996). Later, Morais Cabral et al. (1998) showed that a similar acceleration of channel closure could be achieved by deletion of just the first 25 residues, which is often referred to as the N-Cap domain. Consistent with this, addition of a soluble peptide corresponding to the first 16 N-Cap residues was enough to partially restore the slow deactivation phenotype of hERG channels in which the entire N-terminus had been removed (Wang et al. 2000). However, X-ray crystallography carried out by Morais Cabral et al. (1998) suggested that the initial N-Cap domain did not form a stable structure, whereas residues 26–135 formed a stable PAS domain, which is a common protein–protein interaction domain in many proteins. In hERG, the PAS domain appears to be a ‘hotspot’ for long QT-causing mutations, particularly within a hydrophobic patch where mutations (e.g. F29L, I31S, I42N, Y43C, M124R) result in reduced thermal stability of the PAS and retention of the channel protein in the endoplasmic reticulum rather than expression at the membrane (Gianulis & Trudeau, 2011; Harley et al. 2012; Ke et al. 2013). More recent NMR structures of the N-Cap/PAS domains have shown that the N-Cap domain comprises a dynamic flexible tail (residues 1–12) followed by an amphipathic α-helix (residues 13–23) (Ng et al. 2011; Muskett et al. 2011; Li et al. 2010). Mutagenesis studies of the N-Cap reveal that two positively charged residues in the flexible tail (Arg4, Arg5) play an important role in slowing the deactivation rate (Ng et al. 2011; Muskett et al. 2011) while the amphipathic helix may interact with, and shield, the hydrophobic patch on the PAS domain to help stabilize the protein (Adaixo et al. 2013). From these studies a picture of the N-terminus of hERG channels is beginning to emerge, in which the PAS domain forms a stable protein–protein interaction with one or more other regions of the channel protein, while the amphipathic helix of the N-Cap may position the dynamic tail in the correct orientation to form a functional interaction elsewhere on the channel protein that slows the rate of channel closure.
In addition to the large N-terminus, the hERG protein also contains a large C-terminus, which can be roughly split into three domains: a distal C-terminal tail (∼ residues 873–1159), a cyclic nucleotide binding homology (cNBH) domain (∼ residues 749–872) and a C-linker domain (∼ residues 666–748) which connects to the distal end of the S6 helix. Removal of the distal C-terminus has little effect on channel gating (Akhavan et al. 2003; Gustina & Trudeau, 2011) whereas removal of the cNBH domain accelerates the rate of channel deactivation in a manner similar to the deletion of the N-terminus (Akhavan et al. 2003; Gustina & Trudeau, 2011). Indeed point mutations of several residues within the C-linker or cNBH domains are enough to dramatically accelerate hERG channel deactivation (e.g. Al-Owais et al. 2009). Several recent X-ray crystallography and NMR studies have obtained molecular-level structures of C-linker/cNBH domains of mosquito ERG1 (Brelidze et al. 2013; Li et al. 2014) as well as other EAG potassium channel family members such as zebrafish ELK (Brelidze et al. 2012) and mouse EAG1 (Haitin et al. 2013). Despite sharing significant structural homology with other cyclic nucleotide binding domains the Kv11.1 cNBH domain does not bind cyclic nucleotides (Brelidze et al. 2009). Rather, an additional helix immediately C-terminal to the cNBH domain folds back to occupy the cavity that normally binds cyclic nucleotides, which has been described as a self-liganded structure.
The shared functional consequences of N-Cap/PAS domain and C-linker/cNBH domain deletions (i.e. faster deactivation kinetics), together with their common location in the cytoplasmic region of the channel protein, make them obvious candidates for a functional interaction that mediates the slow rate of channel closure. An initial fluorescence resonance energy transfer (FRET) spectroscopy study confirmed that the N- and C-terminal regions are in close proximity in the full length channels (Miranda et al. 2008). Since then, the Trudeau group has shown that recombinant N-Cap/PAS domains (residues 1–135) can restore the slow gating phenotype of N-truncated hERG channels (Gustina & Trudeau, 2009), although not in hERG channels where the cNBH domain is removed (Gustina & Trudeau, 2011; Gianulis et al. 2013). Similarly, FRET can be observed between recombinant CFP-tagged N-Cap/PAS (donor) and either N-terminal deleted hERG channels or isolated cNBH domain protein labelled with citrine (acceptor) at the C-terminus (Gianulis et al. 2013), suggesting a direct functional interaction between the N-Cap/PAS domain and the cNBH domain. Interestingly, the co-expression of N-Cap/PAS deleted channels with cNBH domain deleted channels can also partially restore the slow deactivation gating, suggesting inter-subunit interaction between these domains (Gustina & Trudeau, 2011). These already convincing data are now supported by an X-ray crystallography study in which isolated N-Cap/PAS and cNBH domain proteins (i.e. without the transmembrane region of the channel) from the closely related EAG family member, mouse EAG1, form multiple interactions (Haitin et al. 2013). An important finding in this study was that the ‘flexible’ N-tail region was interleaved between the PAS and cNBHD and contributed to stabilization of the interaction. Furthermore, although this structure did not contain the pore domain or the first 15 residues of the N-Cap, the orientation of the tail segment was such that it was predicted not to be close to the activation gate at the cytoplasmic entrance to the pore. This does not preclude the possibility that the N-tail may still adopt different conformations in the open and closed states of the channel, but it does suggest that the N-tail may have an indirect effect on deactivation gating as its location is probably too far away from the activation gate to bind directly. The one caveat from this study is that the cNBH domain was isolated from the C-linker domain, which in the homologous HCN channels is located between the cNBH domain and the pore domain (Zagotta et al. 2003). Whether the flexible N-tail forms the same interactions with the PAS and cNBH domains when the C-linker is present one can presume is likely but not yet demonstrated.
Thermodynamic mutant cycle analysis experiments have confirmed that at least one key interaction between the PAS and cNBH domains of mEAG1 is also present in hERG channels. Specifically, Ng et al. (2014) have shown that Arg56 in the PAS domain is energetically coupled to the Asp803 residue in the cNBH domain of hERG channels and loss of this interaction may underlie the fast deactivation gating phenotype of the LQTS mutation R56Q. Furthermore, they found that positively charged arginine residues (Arg4, Arg5) within the disordered region of the N-Cap interact with negatively charged residues (Glu698, Glu699) of the C-linker domain. While they suggested that this later interaction was more transient than the PAS–cNBH domain interaction, it is strong enough to stabilize the open conformation of the channel and thus slow deactivation, possibly via an allosteric transmission to the S6 gate.
Recently, Thomson et al. (2014) used a concatenated hERG channel to address whether hERG channel deactivation requires subunit co-operativity by introducing N-Cap mutations R4A/R5A, PAS domain mutation R56Q or S6 helix mutation F656I into each of the four subunits one at a time. In all cases just one subunit mutation was enough to fully accelerate the rate of deactivation, suggesting that deactivation is a concerted fully cooperative process. The one caveat with interpreting the results from this study, however, is that the concatenation of the four subunits removes the free end of three of the N-terminal cap-domains, which are important for deactivation and so it is possible that deactivation in the concatenated channels is not precisely the same as in homotetrameric channels.
Role of the S4S5 domain in hERG K+ channels
The S4S5 linker plays an important role in electromechanical coupling of voltage sensor motion to opening and closing of the ion conduction pathway. When studied in isolation, the hERG S4S5 linker forms an amphipathic helix (Ng et al. 2012), which suggests that it may lie parallel to the membrane, at least in one conformational state of the channel. Numerous mutations in the S4S5 linker affect the voltage dependence and or kinetics of the open–closed state transitions in hERG (e.g. Alonsoron et al. 2008; Van Slyke et al. 2010). Furthermore, its location at the interface between the cytoplasmic domains and the transmembrane domains makes it ideally placed to serve as a platform for cytoplasmic domains to influence activation/deactivation gating. de la Peña et al. (2011) have shown that cysteines introduced into the flexible tail of the N-Cap domain (V3C) and in the S4S5 linker (Y542C) can form disulfide bonds. They also showed that a cysteine introduced at position 3 can interact with an endogenous cysteine residue at position 723 in the C-terminal C-linker (de la Peña et al. 2013). One must always be cautious with the interpretation of disulfide experiments, as disulfide bonds once formed are stable and two residues need only be in close proximity briefly for a bond to form. Nevertheless they were able to show that the rate of bond formation was reversible as well as being ‘voltage-dependent’, suggesting that there is state dependence for the interaction. These data appear to be at odds with the likely location of the N-Cap/flexible tail and the cNBH/C-linker domains based on the crystal structure of the mosquito PAS + cNBH domains (Haitin et al. 2013). One possibility could be that in the context of the full length channel including the pore domains and entire C-linker domain, the binding of the N-tail to the PAS domain/cNBH domain groove may not be very strong and so it may still be free to unbind and interact with the S4S5 linker – in a state-dependent manner.
The N-cap domain of hERG channels is clearly very important for the slow deactivation phenotype of these channels. One unusual feature of hERG channels with the N-Cap deleted is that whilst they have much faster deactivation kinetics, activation is minimally affected (Tan et al. 2012). That deactivation is not simply the reverse of activation in voltage-gated ion channels is a well-described phenomenon that has been reported for a wide range of voltage-gated channels and is best exemplified by the measurement of on- and off-gating currents, which clearly show quite distinct voltage dependences including for hERG (Piper et al. 2003). This phenomenon is commonly referred to as voltage sensor mode shift. In a recent study using voltage-clamp fluorimetry, Tan et al. (2012) showed that deletion of the N-tail does not affect the mode shift of the voltage sensor but uncouples the mode shift from closure of the cytoplasmic gate. The simplest explanation for these data would be that the flexible tail binds directly to the S4S5 linker and thereby modulates the coupling of voltage sensor movement with activation gate movement, consistent with the data from de la Pena and colleagues (see above). However, this remains to be definitively determined.
Congenital LQTS type 2: translational implications of understanding hERG gating kinetics
Congenital LQTS is an inherited disorder associated with a significantly increased risk of cardiac arrhythmias and sudden death. As the name suggests, this condition is defined by prolongation of the QT interval on the surface ECG, which is a result of delayed repolarization of ventricular action potentials. In theory, mutations in any of the ion channels that contribute to the cardiac action potential could result in LQTS. However, the three main subtypes (LQTS1, caused by mutations in KCNQ1, LQTS2 caused by mutations in KCNH2, which encodes the hERG K+ channel, and LQTS3 caused by mutations in SCN5a) account for >95% of genotype-confirmed cases (40–45, 35–40 and 5–10%, respectively; Splawski et al. 2000).
The clinical presentation of LQTS can be highly variable, ranging from death in utero to some patients living a full life span without ever having any symptoms. Some of this variability is related to subtype; for example, patients with LQTS3 have fewer arrhythmic events but when they do occur they are more likely to be lethal (Zareba et al. 1998). But even within one subtype there can be significant variability both between families with different mutations as well as within families, i.e. there is incomplete penetrance. Given that most families with any given mutation are relatively small (typically <10 affected individuals) and the incidence of events is still relatively rare (with severe mutations it is usually <5% per annum), it is difficult to obtain sufficient statistical power to determine which mutations are associated with a severe or a mild phenotype. Being able to determine, in advance, whether a given mutation is likely to be associated with a high likelihood of sudden death is of great clinical importance as patients with severe disease warrant management with an implantable cardiac defibrillator whereas patients with mild disease can be perfectly well managed with β-blockers (Priori et al. 2013). The dilemma of how to assess risk and decide what is the most appropriate treatment in any given individual has been described as the Achilles heel of management of patients with LQTS (Vincent, 2005). Is it possible that assessing the severity of an hERG K+ channel mutation in vitro could assist with the assessment of clinical risk?
There are many different types of clinically occurring mutations in hERG with the two most common groups being missense mutations followed by mutations introducing premature stop codons. Mutations resulting in premature stop codons will usually result in nonsense mediated RNA decay (Gong et al. 2007) and then result in a haplo-insufficiency phenotype. Missense mutations, however, are likely to have a much more varied phenotype. An estimated 80% of missense mutations result in reduced trafficking efficiency (Kuzmicki et al. 2014). However, reduced trafficking is not an all-or-none phenomenon but is graded. Furthermore some mutant channels that have reduced (but not absent) trafficking also have a gating defect (Gianulis & Trudeau, 2011; Ke et al. 2013).
If we are going to use in vitro studies to characterize the severity of LQTS2-associated mutations it is likely that we will need to combine information on how mutants affect trafficking, subunit–subunit interactions as well as gating and these data will then need to be integrated to estimate how much action potential prolongation is caused by each mutant. The easiest way to integrate such information is through computer modelling. Indeed, many groups have already shown that the basic features of LQTS2 can be reproduced in computer models of isolated cells (Clancy & Rudy, 2001), cables of cells and simulated whole hearts (Sadrieh et al. 2014). This approach, however, will not necessarily overcome the problems associated with lack of statistical power given the relatively small numbers of patients affected with each mutation. One approach to overcome this limitation is to group mutations according to their in vitro phenotype. Analyses based just on mutation location (C-terminal, N-terminal or pore-domain) have shown some promise in aiding risk stratification (Moss et al., 2002). We suggest that grouping mutations according to phenotype (e.g. severe dominant negative trafficking defects compared to mild trafficking defects with or without altered gating and haplo-insufficiency caused by premature stop codons) should be able to permit more fine-tuned stratification of risk than that based on knowledge just of the mutation location.
An additional advantage of using in silico approaches to integrate in vitro data and derive quantitative estimates of the effects on QT interval, is that one can look at how a fixed monogenic defect will be modulated by small changes in other modifying genes. This approach is sometimes referred to studying the effect of a single defect in a population of models (Sadrieh et al. 2013; Britton et al. 2013). For example, Sadrieh et al. (2014) showed that the very different phenotypes (in terms of extent of QT interval prolongation and T-wave notching) seen in two unrelated patients with the same D501N KCNH2 genotype could be reproduced by including <15% variations in the expression levels of other repolarizing currents. Whilst this does not prove that such variations are the cause of the observed variation, it establishes it as a plausible hypothesis. In this study, Sadrieh and colleagues also showed that simulation of a reduced activation-gating defect or enhanced inactivation gating defect could produce different ECG phenotypes even when the extent of QT prolongation was the same (Fig.3). Whether such differences in T-wave morphology are observed clinically in patients with different mutations remains to be determined.
Figure 3.
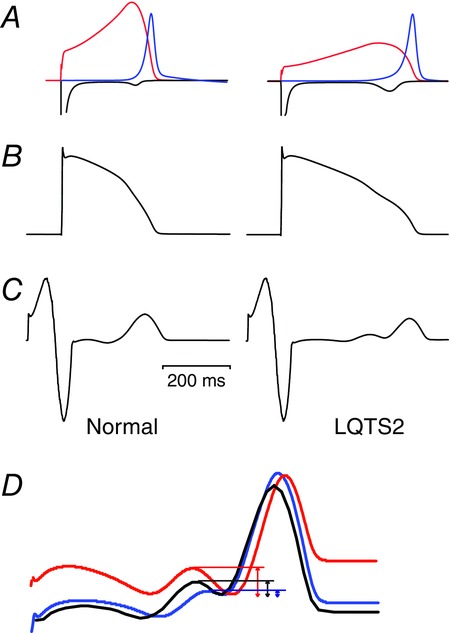
Whole heart simulations of LQTS type 2 based on the O’Hara–Rudy human ventricular cell model incorporated into the Cancer-Heart-Soft Tissue-Environment (CHASTE) that incorporates the anatomical structure as well as the epicardial, mid-myocardial and endocardial differences in ion channel conductances
LQTS2 was simulated by reducing the IKr conductance by 50%. A, IKr; B, AP waveform; and C, ECGs from normal and LQTS2 simulations. Note the bifid T-wave in the LQTS2 simulations. D, differences in the T-wave morphology observed when LQTS2 was simulated by reducing activation (blue), enhancing inactivation (red) or reducing conductance (black) such that the extent of QT prolongation was the same. The three T-waves are aligned such that the bottom of the trough is the same for each, to highlight the differences in the extent of notching: least for the inactivation mutant and most for the activation mutant. Figure modified from Sadrieh et al. (2014).
Future directions
Detailed studies of the hERG K+ channel gating are enabling the generation of biophysically accurate models of hERG gating kinetics. Incorporation of these molecular-level models into cells and ultimately whole hearts that will permit more informed predictions of how mutants affect the emergent properties of the surface ECG in patients with LQTS. Beyond the intrinsic value of understanding how things work, the real value of using computer modelling will be that it will allow for quantitative hypothesis testing of how altered hERG function, whether due to mutations that alter levels of expression and/or alter gating phenotypes, drug block or altered patterns of expression affect cardiac electrical activity in response to a range of physiological and pathophysiological stimuli. Furthermore, the explosion in big data from next generation sequencing studies and cardiac imaging, combined with the phenomenal increase in computational speeds, will contribute to the development of even better whole heart-level models and our ability to predict individual responses to specific insults. This is likely to be first applied in the area of assessing the pro-arrhythmic risk of drugs that block hERG K+ channels but in the long run this approach should also help in all inherited heart rhythm disorders and ultimately acquired arrhythmia syndromes.
Acknowledgments
J.I.V. is supported by a Senior Research Fellowship from the National Health and Medical Research Council (NHMRC). A.P.H. is supported by a Future Fellowship from the Australian Research Council. Work in the Vandenberg laboratory is supported by a Program Grant from the NHMRC (App1074386).
Biography
 Jamie Vandenberg is head of the Mark Cowley Lidwill Research Program in Cardiac Electrophysiology and co-Deputy Director of the Victor Chang Cardiac Research Institute in Sydney. He is a conjoint Professor at the University of New South Wales and an NHMRC Senior Research Fellow. He received his Bachelor of Medical Science and Medical degrees from the University of Sydney and his Doctorate, in Biochemistry, from the University of Cambridge. His research focuses on the molecular basis of disorders of heart rhythm and sudden cardiac death. His team uses a range of molecular, biophysical and computer modeling techniques to analyse arrhythmia substrates and has a particular interest in inherited arrhythmia syndromes and drug-induced arrhythmias.
Jamie Vandenberg is head of the Mark Cowley Lidwill Research Program in Cardiac Electrophysiology and co-Deputy Director of the Victor Chang Cardiac Research Institute in Sydney. He is a conjoint Professor at the University of New South Wales and an NHMRC Senior Research Fellow. He received his Bachelor of Medical Science and Medical degrees from the University of Sydney and his Doctorate, in Biochemistry, from the University of Cambridge. His research focuses on the molecular basis of disorders of heart rhythm and sudden cardiac death. His team uses a range of molecular, biophysical and computer modeling techniques to analyse arrhythmia substrates and has a particular interest in inherited arrhythmia syndromes and drug-induced arrhythmias.
Additional information
Competing interests
There are no competing interests
References
- Adaixo R, Harley CA, Castro-Rodrigues AF. Morais-Cabral JH. Structural properties of PAS domains from the KCNH potassium channels. PLoS ONE. 2013;8:e59265. doi: 10.1371/journal.pone.0059265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akhavan A, Atanasiu R. Shrier A. Identification of a COOH-terminal segment involved in maturation and stability of human ether-a-go-go-related gene potassium channels. J Biol Chem. 2003;278:40105–40112. doi: 10.1074/jbc.M307837200. [DOI] [PubMed] [Google Scholar]
- Al-Owais M, Bracey K. Wray D. Role of intracellular domains in the function of the herg potassium channel. Eur Biophys J. 2009;38:569–576. doi: 10.1007/s00249-009-0408-2. [DOI] [PubMed] [Google Scholar]
- Alonsoron C, Delapena P, Miranda P, Dominguez P. Barros F. Thermodynamic and kinetic properties of amino-terminal and S4-S5 loop HERG channel mutants under steady-state conditions. Biophys J. 2008;94:3893–3911. doi: 10.1529/biophysj.107.116731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auerbach A. Life at the top: the transition state of AChR gating. Science Signaling. 2003;188:1–11. doi: 10.1126/stke.2003.188.re11. [DOI] [PubMed] [Google Scholar]
- Brelidze TI, Carlson AE. Zagotta WN. Absence of direct cyclic nucleotide modulation of mEAG1 and hERG1 channels revealed with fluorescence and electrophysiological methods. J Biol Chem. 2009;284:27989–27997. doi: 10.1074/jbc.M109.016337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brelidze TI, Carlson AE, Sankaran B. Zagotta WN. Structure of the carboxy-terminal region of a KCNH channel. Nature. 2012;481:530–533. doi: 10.1038/nature10735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brelidze TI, Gianulis EC, Dimaio F, Trudeau MC. Zagotta WN. Structure of the C-terminal region of an ERG channel and functional implications. Proc Natl Acad Sci USA. 2013;110:11648–11653. doi: 10.1073/pnas.1306887110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Britton OJ, Bueno-Orovio A, Van Ammel K, Lu HR, Towart R, Gallacher DJ. Rodriguez B. Experimentally calibrated population of models predicts and explains intersubject variability in cardiac cellular electrophysiology. Proc Natl Acad Sci USA. 2013;110:E2098–E2105. doi: 10.1073/pnas.1304382110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Seebohm G. Sanguinetti MC. Position of aromatic residues in the S6 domain, not inactivation, dictates cisapride sensitivity of HERG and eag potassium channels. Proc Natl Acad Sci USA. 2002;99:12461–12466. doi: 10.1073/pnas.192367299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clancy CE. Rudy Y. Cellular consequences of HERG mutations in the long QT syndrome: precursors to sudden cardiac death. Cardiovasc Res. 2001;50:301–313. doi: 10.1016/s0008-6363(00)00293-5. [DOI] [PubMed] [Google Scholar]
- Colenso CK, Sessions RB, Zhang YH, Hancox JC. Dempsey CE. Interactions between voltage sensor and pore domains in a hERG K+ channel model from molecular simulations and the effects of a voltage sensor mutation. J Chem Informat Model. 2013;53:1358–1370. doi: 10.1021/ci4000739. [DOI] [PubMed] [Google Scholar]
- Dempsey CE, Wright D, Colenso CK, Sessions RB. Hancox JC. Assessing hERG pore models as templates for drug docking using published experimental constraints: the inactivated state in the context of drug block. J Chem Informat Model. 2014;54:601–612. doi: 10.1021/ci400707h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhillon MS, Cockcroft CJ, Munsey T, Smith KJ, Powell AJ, Carter P, Wrighton DC, Rong H-L, Yusaf SP. Sivaprasadarao A. A functional Kv1.2-hERG chimaeric channel expressed in Pichia pastoris. Sci Rep. 2014;4:4201. doi: 10.1038/srep04201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durdagi S, Deshpande S, Duff HJ. Noskov SY. Modeling of open, closed, and open-inactivated states of the hERG1 channel: structural mechanisms of the state-dependent drug binding. J Chem Informat Model. 2012;52:2760–2774. doi: 10.1021/ci300353u. [DOI] [PubMed] [Google Scholar]
- Ferrer T, Cordero-Morales JF, Arias M, Ficker E, Medovoy D, Perozo E. Tristani-Firouzi M. Molecular coupling in the human ether-a-go-go-related gene-1 (hERG1) K+ channel inactivation pathway. J Biol Chem. 2011;286:39091–39099. doi: 10.1074/jbc.M111.292060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fersht AR. Daggett V. Protein folding and unfolding at atomic resolution. Cell. 2002;108:573–582. doi: 10.1016/s0092-8674(02)00620-7. [DOI] [PubMed] [Google Scholar]
- Gianulis EC. Trudeau MC. Rescue of aberrant gating by a genetically encoded PAS (Per-Arnt-Sim) domain in several long QT syndrome mutant human ether-á-go-go-related gene potassium channels. J Biol Chem. 2011;286:22160–22169. doi: 10.1074/jbc.M110.205948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gianulis EC, Liu Q. Trudeau MC. Direct interaction of eag domains and cyclic nucleotide-binding homology domains regulate deactivation gating in hERG channels. J Gen Physiol. 2013;142:351–366. doi: 10.1085/jgp.201310995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong Q, Zhang L, Vincent GM, Horne BD. Zhou Z. Nonsense mutations in hERG cause a decrease in mutant mRNA transcripts by nonsense-mediated mRNA decay in human long-QT syndrome. Circulation. 2007;116:17–24. doi: 10.1161/CIRCULATIONAHA.107.708818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gustina AS. Trudeau MC. A recombinant N-terminal domain fully restores deactivation gating in N-truncated and long QT syndrome mutant hERG potassium channels. Proc Natl Acad Sci USA. 2009;106:13082–13087. doi: 10.1073/pnas.0900180106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gustina AS. Trudeau MC. hERG potassium channel gating is mediated by N- and C-terminal region interactions. J Gen Physiol. 2011;137:315–325. doi: 10.1085/jgp.201010582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haitin Y, Carlson AE. Zagotta WN. The structural mechanism of KCNH-channel regulation by the eag domain. Nature. 2013;501:444–448. doi: 10.1038/nature12487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harley CA, Jesus CSH, Carvalho R, Brito RMM. Morais-Cabral JH. Changes in channel trafficking and protein stability caused by LQT2 mutations in the PAS domain of the HERG channel. PLoS ONE. 2012;7:e32654. doi: 10.1371/journal.pone.0032654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hausammann GJ. Grütter MG. Chimeric hERG channels containing a tetramerization domain are functional and stable. Biochemistry. 2013;52:9237–9245. doi: 10.1021/bi401100a. [DOI] [PubMed] [Google Scholar]
- Hoshi T. Armstrong CM. C-type inactivation of voltage-gated K+ channels: pore constriction or dilation? J Gen Physiol. 2013;141:151–160. doi: 10.1085/jgp.201210888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones DK, Liu F, Vaidyanathan R, Eckhardt LL, Trudeau MC. Robertson GA. hERG 1b is critical for human cardiac repolarization. Proc Natl Acad Sci USA. 2014;111:18073–18077. doi: 10.1073/pnas.1414945111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ke Y, Ng CA, Hunter MJ, Mann SA, Heide J, Hill AP. Vandenberg JI. Trafficking defects in PAS domain mutant Kv11.1 channels: roles of reduced domain stability and altered domain-domain interactions. Biochem J. 2013;454:69–77. doi: 10.1042/BJ20130328. [DOI] [PubMed] [Google Scholar]
- Köpfer DA, Hahn U, Ohmert I, Vriend G, Pongs O, de Groot BL. Zachariae U. A molecular switch driving inactivation in the cardiac K-channel hERG. PLoS ONE. 2012;7:e41023. doi: 10.1371/journal.pone.0041023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuzmicki CE, Childs RR, Hintz CJ, Delisle BP, Anderson CL. January CT. Large-scale mutational analysis of Kv11.1 reveals molecular insights into type 2 long QT syndrome. Nat Commun. 2014;5:1–13. doi: 10.1038/ncomms6535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de la Peña P, Alonso-Ron C, Machín A, Fernández-Trillo J, Carretero L, Domínguez P. Barros F. Demonstration of physical proximity between the N-terminus and the S4–S5 linker of the human ether-a-go-go-related gene (hERG) potassium channel. J Biol Chem. 2011;286:19065–19075. doi: 10.1074/jbc.M111.238899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de la Peña P, Machín A, Fernández-Trillo J, Domínguez P. Barros F. Mapping of interactions between the N- and C-termini and the channel core in HERG K+ channels. Biochem J. 2013;451:463–474. doi: 10.1042/BJ20121717. [DOI] [PubMed] [Google Scholar]
- Lees-Miller JP, Subbotina JO, Guo J, Yarov-Yarovoy V, Noskov SY. Duff HJ. Interactions of H562 in the S5 helix with T618 and S621 in the pore helix are important determinants of hERG1 potassium channel structure and function. Biophys J. 2009;96:3600–3610. doi: 10.1016/j.bpj.2009.01.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Q, Gayen S, Chen AS, Huang Q, Raida M. Kang C. NMR solution structure of the N-terminal domain of hERG and its interaction with the S4–S5 linker. Biochem Biophys Res Commun. 2010;403:126–132. doi: 10.1016/j.bbrc.2010.10.132. [DOI] [PubMed] [Google Scholar]
- Li Q, Ng HQ, Yoon HS. Kang C. Solution structure of the cyclic-nucleotide binding homology domain of a KCNH channel. J Struct Biol. 2014;186:68–74. doi: 10.1016/j.jsb.2014.03.008. [DOI] [PubMed] [Google Scholar]
- Lu Y, Mahaut-Smith MP, Varghese A, Huang CL, Kemp PR. Vandenberg JI. Effects of premature stimulation on HERG K+ channels. J Physiol. 2001;537:843–851. doi: 10.1111/j.1469-7793.2001.00843.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miranda P, Manso DG, Barros F, Carretero L, Hughes TE, Alonso-Ron C, Domínguez P. de la Peña P. FRET with multiply labeled HERG K+ channels as a reporter of the in vivo coarse architecture of the cytoplasmic domains. Biochim Biophys Acta. 2008;1783:1681–1699. doi: 10.1016/j.bbamcr.2008.06.009. [DOI] [PubMed] [Google Scholar]
- Morais Cabral JH, Lee A, Cohen SL, Chait BT, Li M. MacKinnon R. Crystal structure and functional analysis of the HERG potassium channel N-terminus: a eukaryotic PAS domain. Cell. 1998;95:649–655. doi: 10.1016/s0092-8674(00)81635-9. [DOI] [PubMed] [Google Scholar]
- Moss AJ, Zareba W, Kaufman ES, Gartman E, Peterson DR, Benhorin J, Towbin JA, Keating MT, Priori SG, Schwartz PJ, Vincent GM, Robinson JL, Andrews ML, Feng C, Hall WJ, Medina A, Zhang L, Wang Z. Increased risk of arrhythmic events in long-QT syndrome with mutations in the pore region of the human ether-a-go-go-related gene potassium channel. Circulation. 2002;105:794–799. doi: 10.1161/hc0702.105124. [DOI] [PubMed] [Google Scholar]
- Muskett FW, Thouta S, Thomson SJ, Bowen A, Stansfeld PJ. Mitcheson JS. Mechanistic insight into human ether-à-go-go-related gene (hERG) K+ channel deactivation gating from the solution structure of the EAG domain. J Biol Chem. 2011;286:6184–6191. doi: 10.1074/jbc.M110.199364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng CA, Hunter MJ, Perry MD, Mobli M, Ke Y, Kuchel PW, King GF, Stock D. Vandenberg JI. The N-terminal tail of hERG contains an amphipathic α-helix that regulates channel deactivation. PLoS ONE. 2011;6:e16191. doi: 10.1371/journal.pone.0016191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng CA, Perry MD, Tan PS, Hill AP, Kuchel PW. Vandenberg JI. The S4–S5 linker acts as a signal integrator for HERG K+ channel activation and deactivation gating. PLoS ONE. 2012;7:e31640. doi: 10.1371/journal.pone.0031640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng CA, Phan K, Hill AP, Vandenberg JI. Perry MD. Multiple interactions between cytoplasmic domains regulate slow deactivation of Kv11.1 channels. J Biol Chem. 2014;289:25822–25832. doi: 10.1074/jbc.M114.558379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry MD, Ng CA. Vandenberg JI. a Pore helices play a dynamic role as integrators of domain motion during Kv11.1 channel inactivation gating. J Biol Chem. 2013;288:11482–11491. doi: 10.1074/jbc.M113.461442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry MD, Wong S, Ng CA. Vandenberg JI. b Hydrophobic interactions between the voltage sensor and pore mediate inactivation in Kv11.1 channels. J Gen Physiol. 2013;142:275–288. doi: 10.1085/jgp.201310975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piper DR, Varghese A, Sanguinetti MC. Tristani-Firouzi M. Gating currents associated with intramembrane charge displacement in HERG potassium channels. Proc Natl Acad Sci USA. 2003;100:10534–10539. doi: 10.1073/pnas.1832721100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Priori SG, Wilde AA, Horie M, Cho Y, Behr ER, Berul C, Blom N, Brugada J, Chiang C-E, Huikuri H, Kannankeril P, Krahn A, Leenhardt A, Moss A, Schwartz PJ, Shimizu W, Tomaselli G. Tracy C. HRS/EHRA/APHRS expert consensus statement on the diagnosis and management of patients with inherited primary arrhythmia syndromes. Heart Rhythm. 2013;10:1932–1963. doi: 10.1016/j.hrthm.2013.07.021. [DOI] [PubMed] [Google Scholar]
- Sadrieh A, Domanski L, Pitt-Francis J, Mann SA, Hodkinson EC, Ng CA, Perry MD, Taylor JA, Gavaghan D, Subbiah RN, Vandenberg JI. Hill AP. Multiscale cardiac modelling reveals the origins of notched T waves in long QT syndrome type 2. Nat Commun. 2014;5:5069. doi: 10.1038/ncomms6069. [DOI] [PubMed] [Google Scholar]
- Sadrieh A, Mann SA, Subbiah RN, Domanski L, Taylor JA, Vandenberg JI. Hill A. Quantifying the origins of population variability in cardiac electrical activity through sensitivity analysis of the electrocardiogram. J Physiol. 2013;591:4207–4222. doi: 10.1113/jphysiol.2013.251710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanguinetti MC, Jiang C, Curran ME. Keating MT. A mechanistic link between an inherited and an acquired cardiac arrhythmia: HERG encodes the IKr potassium channel. Cell. 1995;81:299–307. doi: 10.1016/0092-8674(95)90340-2. [DOI] [PubMed] [Google Scholar]
- Schönherr R. Heinemann SH. Molecular determinants for activation and inactivation of HERG, a human inward rectifier potassium channel. J Physiol. 1996;493:635–642. doi: 10.1113/jphysiol.1996.sp021410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith PL, Baukrowitz T. Yellen G. The inward rectification mechanism of the HERG cardiac potassium channel. Nature. 1996;379:833–836. doi: 10.1038/379833a0. [DOI] [PubMed] [Google Scholar]
- Splawski I, Shen J, Timothy KW, Lehmann MH, Priori S, Robinson JL, Moss AJ, Schwartz PJ, Towbin JA, Vincent GM. Keating MT. Spectrum of mutations in long-QT syndrome genes. KVLQT1, HERG, SCN5A, KCNE1, and KCNE2. Circulation. 2000;102:1178–1185. doi: 10.1161/01.cir.102.10.1178. [DOI] [PubMed] [Google Scholar]
- Stansfeld PJ, Gedeck P, Gosling M, Cox B, Mitcheson JS. Sutcliffe MJ. Drug block of the hERG potassium channel: insight from modeling. Proteins. 2007;68:568–580. doi: 10.1002/prot.21400. [DOI] [PubMed] [Google Scholar]
- Stansfeld PJ, Grottesi A, Sands ZA, Sansom MSP, Gedeck P, Gosling M, Cox B, Stanfield PR, Mitcheson JS. Sutcliffe MJ. Insight into the mechanism of inactivation and pH sensitivity in potassium channels from molecular dynamics simulations. Biochemistry. 2008;47:7414–7422. doi: 10.1021/bi800475j. [DOI] [PubMed] [Google Scholar]
- Tan PS, Perry MD, Ng CA, Vandenberg JI. Hill AP. Voltage-sensing domain mode shift is coupled to the activation gate by the N-terminal tail of hERG channels. J Gen Physiol. 2012;140:293–306. doi: 10.1085/jgp.201110761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomson SJ, Hansen A. Sanguinetti MC. Concerted all-or-none subunit interactions mediate slow deactivation of human ether-à-go-go-related gene K+ channels. J Biol Chem. 2014;289:23428–23436. doi: 10.1074/jbc.M114.582437. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Trudeau MC, Warmke JW, Ganetzky B. Robertson GA. HERG, a human inward rectifier in the voltage-gated potassium channel family. Science. 1995;269:92–95. doi: 10.1126/science.7604285. [DOI] [PubMed] [Google Scholar]
- Van Slyke AC, Rezazadeh S, Snopkowski M, Shi P, Allard CR. Claydon TW. Mutations within the S4–S5 linker alter voltage sensor constraints in hERG K+ channels. Biophys J. 2010;99:2841–2852. doi: 10.1016/j.bpj.2010.08.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandenberg JI, Perry MD, Perrin MJ, Mann SA, Ke Y. Hill AP. hERG K+ channels: structure, function, and clinical significance. Physiol Rev. 2012;92:1393–1478. doi: 10.1152/physrev.00036.2011. [DOI] [PubMed] [Google Scholar]
- Vincent GM. Risk assessment in long QT syndrome: the Achilles heel of appropriate treatment. Heart Rhythm. 2005;2:505–506. doi: 10.1016/j.hrthm.2005.03.002. [DOI] [PubMed] [Google Scholar]
- Wang DT, Hill AP, Mann SA, Tan PS. Vandenberg JI. Mapping the sequence of conformational changes underlying selectivity filter gating in the Kv11.1 potassium channel. Nat Struct Mol Biol. 2011;18:35–41. doi: 10.1038/nsmb.1966. [DOI] [PubMed] [Google Scholar]
- Wang J, Myers CD. Robertson GA. Dynamic control of deactivation gating by a soluble amino-terminal domain in HERG K+ channels. J Gen Physiol. 2000;115:749–758. doi: 10.1085/jgp.115.6.749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S, Liu S, Morales MJ, Strauss HC. Rasmusson RL. A quantitative analysis of the activation and inactivation kinetics of HERG expressed in Xenopus oocytes. J Physiol. 1997;502:45–60. doi: 10.1111/j.1469-7793.1997.045bl.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu W, Gardner A. Sanguinetti MC. Cooperative subunit interactions mediate fast C-type inactivation of hERG1 K+ channels. J Physiol. 2014;592:4465–4480. doi: 10.1113/jphysiol.2014.277483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zagotta WN, Olivier NB, Black KD, Young EC, Olson R. Gouaux E. Structural basis for modulation and agonist specificity of HCN pacemaker channels. Nature. 2003;425:200–205. doi: 10.1038/nature01922. [DOI] [PubMed] [Google Scholar]
- Zareba W, Moss AJ, Schwartz PJ, Vincent GM, Robinson JL, Priori SG, Benhorin J, Locati EH, Towbin JA, Keating MT, Lehmann MH. Hall WJ. Influence of genotype on the clinical course of the long-QT syndrome. International long-QT syndrome registry research group. N Engl J Med. 1998;339:960–965. doi: 10.1056/NEJM199810013391404. [DOI] [PubMed] [Google Scholar]