

Abstract
The KCNQ1 channel (also called Kv7.1 or KvLQT1) belongs to the superfamily of voltage-gated K+ (Kv) channels. KCNQ1 shares several general features with other Kv channels but also displays a fascinating flexibility in terms of the mechanism of channel gating, which allows KCNQ1 to play different physiological roles in different tissues. This flexibility allows KCNQ1 channels to function as voltage-independent channels in epithelial tissues, whereas KCNQ1 function as voltage-activated channels with very slow kinetics in cardiac tissues. This flexibility is in part provided by the association of KCNQ1 with different accessory KCNE β-subunits and different modulators, but also seems like an integral part of KCNQ1 itself. The aim of this review is to describe the main mechanisms underlying KCNQ1 flexibility.
The physiological role of KCNQ1 channel
The KCNQ1 channel is expressed in various tissues (reviewed in Jespersen et al. 2005; Abbott, 2014) where it mainly contributes to the regulation of electric activity or to maintain K+ homeostasis needed for electrolyte and hormone transport (Fig.1A). These two functional roles of KCNQ1 are perhaps best understood in the heart and inner ear, respectively. KCNQ1 in complex with the KCNE1 (also called MinK) β-subunit form the cardiac IKs channel which plays an important role in regulating cardiac action potential duration (Barhanin et al. 1996; Sanguinetti et al. 1996; Wang et al. 1996a). The outward K+ current through the IKs channel is one of the main repolarizing K+ currents in the human heart and thereby contributes to the termination of the cardiac action potential (Fig.1B). Dysfunctional cardiac IKs channels cause pathological alterations in cardiac action potential duration which can cause severe cardiac arrhythmias and sudden cardiac death (Nerbonne & Kass, 2005). In general, loss-of-function of the cardiac IKs channel tends to prolong the cardiac action potential duration (Fig.1B, grey trace) which can cause a prolonged QT interval in the ECG (long QT syndrome). KCNQ1 is also co-expressed with KCNE1 in the inner ear (Neyroud et al. 1997; Hibino et al. 2010). K+ flux through the inner ear IKs channel is important to maintain the endolymph K+ homeostasis and the endocochlear potential (Hibino et al. 2010). Dysfunctional IKs channels in the inner ear can result in decreased hearing ability or congenital deafness (Anantharam et al. 2003; Jespersen et al. 2005). KCNQ1 is also suggested to associate with KCNE1 or other KCNE subunits in other epithelial tissues such as pancreas (Hayashi & Novak, 2013), kidney (Warth & Barhanin, 2002; Vallon et al. 2005), colon and intestine (Barrett & Keely, 2000; Schroeder et al. 2000), stomach (Heitzmann & Warth, 2007), thyroid gland (Roepke et al. 2006) and airways (Grahammer et al. 2001; Preston et al. 2010) where these KCNQ1–KCNE complexes contribute to K+ homeostasis and/or maintain the proper membrane potential for transepithelial transport of, for instance, Cl− and gastric acid (Fig.1C). KCNQ1 was recently also suggested to be expressed in different brain regions and thereby contribute to the regulation of neuronal excitability (Goldman et al. 2009). The molecular composition and physiological relevance of these KCNQ1–KCNE complexes expressed throughout the body need to be further explored in some tissues. The possibility of various KCNQ1–KCNE complex combinations with different functional properties allows widely different physiological roles of KCNQ1 in different tissues.
Figure 1.
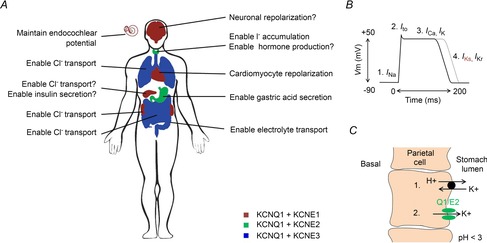
Suggested role of KCNQ1–KCNE complexes in different tissues
A, KCNQ1–KCNE1 is suggested to be expressed in the heart, inner ear, pancreas, kidney and brain and to regulate repolarization of excitable cells, enable transport, and maintain the membrane potential. KCNQ1–KCNE2 is suggested to be expressed in the thyroid gland and stomach and to regulate transport, hormone production, and gastric secretion. KCNQ1–KCNE3 is suggested to be expressed in the intestine, colon and airways and to enable electrolyte transport. B, the action potential in human ventricular cardiomyocytes has a duration of several hundred milliseconds. Cardiomyocyte depolarization (1) is caused by influx of Na+. The transient outward K+ current (Ito) causes the repolarization notch (2) and the plateau is a caused by simultaneous influx of Ca2+ and outflux of K+ (3). Cardiomyocyte repolarization is mainly caused by K+ outflux though the IKs channel and the IKr channel (4). Loss-of-function of the IKs channel tends to impair cardiomyocyte repolarization and prolong the cardiomyocyte action potential duration (grey curve). C, the parietal cells in the gastric glands of stomach epithelium secrete H+ into the stomach lumen. The H+–K+-ATPase that causes stomach acidification (1) requires K+ outflux through KCNQ1–KCNE2 (2) to recycle the K+ transported though the H+–K+-ATPase.
Architecture of KCNQ1 channel
The main part of the KCNQ1 gene was originally cloned in 1996 (Wang et al. 1996b) and the full length KCNQ1 gene was cloned shortly thereafter (Lee et al. 1997; Splawski et al. 1998). Each subunit of the tetrameric KCNQ1 channel is, in similarity to most Kv channels, composed of six transmembrane segments named S1–S6 (Fig.2A; Börjesson & Elinder, 2008; Swartz, 2008; Catterall, 2010). An assembly domain in the C-terminus is responsible for assembling four KCNQ1 subunits into a functional Kv channel (Fig.2B; Schmitt et al. 2000). Most studies on structure–function in Kv channels have been performed on Kv channels other than the KCNQ1 channel. The KCNQ1 channel is, however, believed to share most structural properties with other Kv channels. In Kv channels in general, transmembrane helices S5 and S6 from all four subunits together form the pore domain, with the central ion-conducting pore containing the K+ channel pore signature sequence GYGD (Börjesson & Elinder, 2008). An intracellular bundle crossing of S6 helices from all four subunits functions as the activation gate, shutting off the intracellular access to the pore for K+ ions in the closed state of the channel. A highly conserved PXP motif in the S6 helix of Kv channels is assumed to induce a kink in S6 of most Kv channels to allow pore opening (del Camino et al. 2000). This motif is not conserved in KCNQ1 channels, which instead possess a PXG motif. The gate movement to open the KCNQ1 channel pore may therefore look different from other Kv channels that have a conserved PXP motif. The glycine in the PXG motif may allow for more flexibility of S6 in KCNQ1 than in the canonical PXP motif in other Kv channels. Both proline and glycine are known to introduce breaks in alpha helices, but proline would introduce a more rigid bend than the glycine. The side-chain at this third position has been shown to greatly affect the gating in related Kv channels. For instance, substitution of the proline at the third position in the Shaker Kv channel (P475) with an alanine results in a non-conducting channel, while substitution with an aspartate results in an apparent constitutively open channel (Hackos et al. 2002). A proline-to-glycine substitution at this third position in Kv1.5 (P511) results in a channel with substantially shifted voltage dependence towards more positive voltages (Labro et al. 2005). In Kv channels in general, transmembrane helices S1 to S4 of each subunit form a voltage-sensor domain. The S4 segment has several positively charged residues that enable the channel to ‘sense’ the transmembrane voltage by moving across the membrane electric field in response to changes in the transmembrane voltage (Börjesson & Elinder, 2008). In Kv channels, it is thought that the movement of S4 is transferred to the pore domain, partly via the S4–S5 linker to S6, thereby coupling gate opening/closing to S4 movement. In this electro-mechanical model (Fig.2C), the outward movement of S4 would pull on the S4–S5 linker, which forms bonds with the lower end of S6. The movement of the S4–S5 linker would thereby move the lower end of S6 and open the S6 gate (Long et al. 2007; Blunck & Batulan, 2012; Vardanyan & Pongs, 2012). In, for instance, the Shaker channel, mainly interactions between hydrophobic residues in the S4–S5 linker and S6 are proposed to be important for the physical coupling between the voltage-sensor domain and the gate (reviewed in Blunck & Batulan, 2012; Fig.2D, upper panel). In the KCNQ1 channel, this region contains several basic residues that are conserved among KCNQ channels but not conserved in other Kv channels (Zaydman & Cui, 2014; Fig.2D, lower panel). This altered interface between the voltage-sensor domain and the pore domain, caused by electrostatic repulsion of basic residues, could affect the coupling between S4 and the gate. Furthermore, the S4 of KCNQ1 lacks two of the positively charged residues that have been shown in other Kv channels to be important for voltage sensing (Fig.2A). This lack of important gating charge residues has been suggested to be important for the ability of KCNQ1 to convert into a leak channel when associated with specific KCNE β-subunits (Panaghie & Abbott, 2007).
Figure 2.
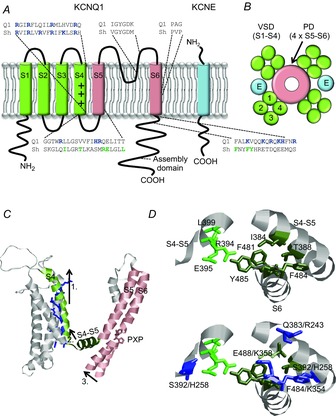
Topology of Kv7.1 and KCNE and model for electro-mechanical coupling
A, schematic topology of one KCNQ1 subunit and one KCNE subunit. S1–S4 form the voltage-sensor domain (VSD) and S5–S6 the pore domain (PD). The plus symbols in S4 denote the positive gating charges. Basic residues are shown in blue. Shaker residues suggested to make close contact between voltage-sensor domain and gate are shown in green. B, schematic top view of a tetrameric KCNQ1 channel. Blue spheres marked ‘E’ denote the deduced location of KCNE subunits in the channel complex. C, illustration of model for electro-mechanical coupling in Kv channels. S4 moves up during membrane depolarization (1). This movement pulls the S4–S5 linker (2), which forms bonds with the lower S6. The pulling of S4–S5 moves the lower end of S6 (3) which causes a kink at the PXP motif and opens the gate. Shaker model from Henrion et al. (2012). D, upper panel, molecular details of voltage-sensor domain coupling to the gate via hydrophobic residues in the S4–S5 linker and in the lower part of S6/C-terminus in the Shaker channel. Model from Henrion et al. (2012). Numbering indicates Shaker numbering. Residues in the same subunit are shown in dark green and residues in the neighbouring subunit are shown in light green. D, lower panel, corresponding position of basic KCNQ1 residues. Numbering indicates Shaker numbering/KCNQ1 numbering.
The KCNQ1 α-subunit expressed alone forms functional tetrameric voltage-gated K+ channels that open at negative voltages. Compared to most Kv channels, KCNQ1 channels activate and deactivate slowly with a time constant in the order of hundreds of milliseconds (Pusch et al. 1998; Fig.3A). The KCNE subunits (KCNE1–KCNE5) are one-transmembrane proteins (Fig.2A) that form complexes with KCNQ1 (Jespersen et al. 2005). Experiments in heterologous expression systems show that up to four KCNE subunits can associate with each tetrameric KCNQ1 channel (Wang et al. 1998; Nakajo et al. 2010). There is, however, evidence suggesting that the native cardiac IKs current is generated by KCNQ1–KCNE1 complexes with a 4:2 stoichiometry of KCNQ1–KCNE1 subunits (Plant et al. 2014). Cross-linking studies have shown that KCNE1 interacts with several regions of the KCNQ1 channel, including S1, S4, and S6 (Nakajo & Kubo, 2007; Shamgar et al. 2008; Xu et al. 2008; Chung et al. 2009), suggesting that KCNE1 probably inserts into the lipid-filled space between two adjacent voltage-sensor domains (Fig.2B; Chung et al. 2009). Co-expression of KCNQ1 with KCNE subunits in heterologous expression systems dramatically alters KCNQ1 channel properties, including the kinetics of the macroscopic current (Barhanin et al. 1996; Sanguinetti et al. 1996; Schroeder et al. 2000; Tinel et al. 2000; Angelo et al. 2002; Grunnet et al. 2002). The most pronounced kinetic effects induced by KCNEs are that KCNE1 induces a slow and sigmoidal activation time course and that KCNE3 induces a large apparent voltage-independent current component (Fig.3A). KCNE2 induces currents fairly similar to those induced by KCNE3, but with smaller current amplitude (Jespersen et al. 2005). KCNE4 and KCNE5 mainly reduce KCNQ1 current amplitude in the physiological voltage range (Jespersen et al. 2005).
Figure 3.
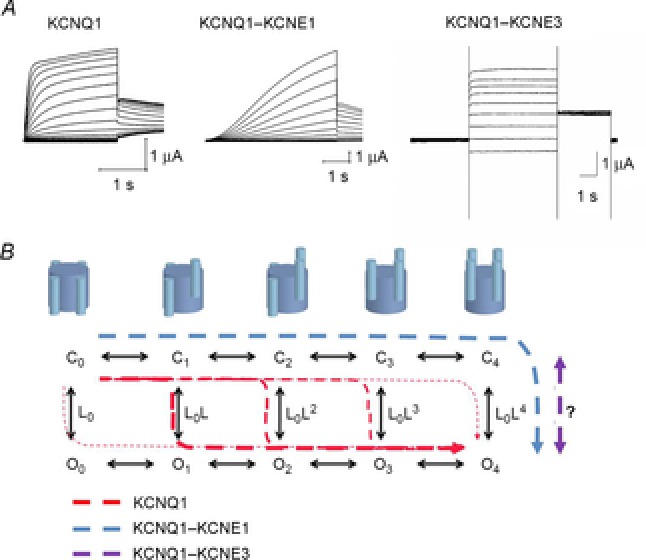
Model for KCNQ1 gating
A, representative K+ current families for KCNQ1 (stepping from a holding voltage of –80 mV to test voltages from –80 to +60 mV in 10 mV increments. Tail voltage is –30 mV), KCNQ1–KCNE1 (stepping from a holding voltage of –80 mV to test voltages from –110 to +60 mV in 10 mV increments. Tail voltage is –30 mV), and KCNQ1–KCNE3 (stepping from a holding voltage of –100 mV to test voltages from –100 to +60 mV in 20 mV increments. Tail voltage is –40 mV). B, ten-state model with five closed states (C0–C4) and five open states (O0–O4), where subscript depicts the number of activated S4s. Horizontal transitions are independent activation of S4s and vertical transition is the opening of the gate (with a potential extra concerted movement of all four S4s; Barro-Soria et al. 2014). Suggested transitions for KCNQ1 alone (red dashed lines), KCNQ1–KCNE1 (blue dashed line), and KCNQ1–KCNE3 (magenta dashed line) in response to a depolarization. Even at very negative voltages (all S4s down), KCNQ1 expressed alone displays a significant open probability that increases with more activated S4s (Osteen et al. 2012). In response to a depolarization, most KCNQ1 channels open after one or two S4s have activated (thicker red dashed lines). In contrast, KCNQ1–KCNE1 only opens after all four S4s have activated (all four S4s up; Barro-Soria et al. 2014), whereas in KCNQ1–KCNE3 the S4s are always activated independently of voltage and channel opening is relatively voltage independent (Nakajo & Kubo, 2007; Rocheleau & Kobertz, 2008).
To summarize, although the overall architecture of KCNQ1 looks similar to other Kv channels, it deviates in three main aspects; KCNQ1 lacks the conserved PXP motif and two important positively charged residues in S4 and contains several basic residues in the interface between S4 and the gate. These structural differences may contribute to the intrinsic gating flexibility of KCNQ1 that makes KCNQ1 unusually sensitive to modulators (such as β-subunits) that modulate the gating of KCNQ1 and allows the KCNQ1 channel to function very differently – from a voltage-independent K+ channel to a highly voltage-dependent K+ channel – depending on its molecular composition. Among the KCNQ family (KCNQ1–KCNQ5), only KCNQ1 has been reported to associate with all five KCNE β-subunits. The ability of KCNQ1 to interact with and be modulated by all five KCNE subunits adds to its gating versatility.
Gating mechanism of KCNQ1 channel
The mechanism by which KCNQ1 channels open and close has been the subject of an active debate for more than a decade. In particular, the molecular details of how the S4 movement couples to the opening of the gate are not completely understood. For Kv channels in general, it is assumed that upon changes in voltage across the membrane, the S4 helices in the four KCNQ1 subunits move relatively independently to the activated (‘up’) state and once all four S4s are in the active state the channel can open (Börjesson & Elinder, 2008). In contrast to most Kv channels, in KCNQ1 channels the gate opens before all S4s have moved. This was demonstrated by constructing linked concatemers of KCNQ1 subunits with different numbers of active S4s (Meisel et al. 2012; Osteen et al. 2012). It was shown that KCNQ1 channels open with a substantial open probability with less than four S4s activated. Using voltage clamp fluorometry, which simultaneously tracks S4 movement (fluorescence) and channel opening (ionic current) (Mannuzzu et al. 1996), and coexpression of KCNQ1 subunits with different voltage dependences, it was shown that the four S4s move relatively independently in KCNQ1 channels (Osteen et al. 2012). Based on these experiments, a ten-state allosteric gating scheme was proposed for KCNQ1 gating (Fig.3B). In this model, the more S4s that have moved to their activated state, the higher the probability of transitioning from closed to open channel (Osteen et al. 2012). In contrast to the Shaker K+ channel, where the S4 movement from the resting to the active state alters the open probability more than 107-fold (Islas & Sigworth, 1999) and therefore the coupling of S4 movement and the gate must be very strong, in KCNQ1 the S4 movement alters the open probability less than 50-fold (Osteen et al. 2012) and therefore this coupling is relatively much looser. A 50-fold increase in open probability is less than 1 kT of energy per subunit transferred from each voltage sensor to the pore to stabilize the open state. This weak coupling might therefore account for the high gating flexibility of the KCNQ1 channel by allowing KCNE β-subunits and modulators to more easily affect KCNQ1 channel gating. The molecular mechanism underlying this weak coupling is not clear, but the PXG motif in S6 might allow for different ways to open the S6 gate or the S4–S5 linker might be differently coupled to the S6 gate compared to other Kv channels. The ten-state model offers a framework for explaining, at least in part, the gating flexibility (i.e. different gating behaviour) of the KCNQ1 channel in the context of mutations, regulatory β-subunits, and modulators.
Modulation of KCNQ1 channel by KCNE β-subunits
Different KCNE β-subunits associate with KCNQ1 and alter the properties of KCNQ1, most likely by utilizing the intrinsic gating flexibility of KCNQ1. During the last decade, different groups have revealed further insights into how different KCNE subunits alter KCNQ1 gating and permeation (Tai & Goldstein, 1998; Tapper & George, 2001; Gage & Kobertz, 2004; Melman et al. 2004; Panaghie et al. 2006; Nakajo & Kubo, 2007; Rocheleau & Kobertz, 2008; Osteen et al. 2010; Ruscic et al. 2013). The gating mechanism of KCNQ1 in complex with KCNE β-subunits is, however, still less well understood than that of KCNQ1 alone.
The association of KCNQ1 with KCNE1 (also called Mink) slows the activation kinetics, shifts the voltage dependence of KCNQ1 to positive voltages (Barhanin et al. 1996; Sanguinetti et al. 1996), increases the macroscopic current, and increases the single-channel conductance compared to KCNQ1 alone (Sesti & Goldstein, 1998; Yang & Sigworth, 1998). The slow kinetics induced by the KCNE1 subunit is critical for the role of KCNQ1–KCNE1 channels in generating the slowly activating IKs current in the heart (Nerbonne & Kass, 2005). In contrast to KCNE1, the presence of the β-subunit KCNE3 (also called MiRP2) ensures the KCNQ1 channel is always open in the voltage range of –80 to +80 mV. Association of KCNQ1 with KCNE2 results in small K+ currents with a large constitutive current (Tinel et al. 2000). This ability of KCNQ1–KCNE2 and KCNQ1–KCNE3 to remain open over a wide voltage range seems to be important for their function in non-excitable epithelial cells. In contrast, both KCNE4 and KCNE5 have inhibitory effects on KCNQ1 currents by dramatically shifting the G–V curve of KCNQ1 towards more positive voltages (Angelo et al. 2002; Grunnet et al. 2002; Jespersen et al. 2005). Because of this inhibitory effect, KCNE4 and KCNE5 have been speculated to act as KCNQ1–KCNE1 channel dampeners in the heart when forming a tripartite complex together with KCNQ1–KCNE1. KCNE4 has minor effects on KCNQ1 channel activation kinetics, while KCNE5 slows down KCNQ1 channel activation kinetics. Little is known about the mechanism by which KCNE2, KCNE4 and KCNE5 affects KCNQ1 channel activity. It has been speculated that KCNE4 might work in part by interacting with the KCNQ1 modulator calmodulin (Ciampa et al. 2011). In the next two sections, we describe the proposed molecular mechanism of action of KCNE1 and KCNE3, the two most-studied KCNEs.
KCNE1
Several models, sometimes conflicting, have been proposed for the mechanism by which KCNE1 slows the activation kinetics of KCNQ1–KCNE1 channels (Nakajo & Kubo, 2007; Rocheleau & Kobertz, 2008; Osteen et al. 2010; Ruscic et al. 2013; Barro-Soria et al. 2014). Overall, two possibilities seem plausible to explain the slow activation of KCNQ1–KCNE1 channels: (1) KCNE1 slows the outward movement of S4, or (2) KCNE1 directly slows the opening of the gate. Using different methods, two independent groups provided data in support of the idea that KCNE1 slows the movement of S4. (a) By mutating key residues in the S4 of KCNQ1 to cysteines and exposing mutated KCNQ1 to cysteine-specific methanethiosulfonate (MTS) reagents from the external solution (i.e. cysteine accessibility studies), Nakajo and Kubo showed that KCNE1 slows the modification rate of cysteines in S4 (Nakajo & Kubo, 2007). This suggests that S4 movement is slowed down by KCNE1. (b) Using voltage clamp fluorometry, Ruscic et al. found that the time course of the fluorescence had similar kinetics to the ionic current in KCNQ1 channels both with and without KCNE1. In addition, they could measure gating currents from KCNQ1 channels alone, but not from KCNQ1–KCNE1 channels, suggesting that the gating charge movement is very slow in the presence of KCNE1 (Ruscic et al. 2013). In support of the second hypothesis that KCNE1 slows the opening of the gate, a cysteine accessibility study performed by Rocheleau and Kobertz showed that the modification rate in KCNQ1–KCNE1 channels was independent of the depolarization pulse duration (≥100 ms), if the total time spent depolarized was constant (Rocheleau & Kobertz, 2008). These data suggest that S4 moves out in less than 100 ms. Because the kinetic of current activation of KCNQ1–KCNE1 channel takes more than 100 ms, the authors concluded that KCNE1 slows the opening of the gate (Rocheleau & Kobertz, 2008). In another study using voltage clamp fluorometry, Osteen et al. found that, in contrast to KCNQ1 channels alone, in KCNQ1–KCNE1 channels the time course of the fluorescence is faster than the activation of the current, indicating that KCNE1 mainly slows the gate in KCNQ1–KCNE1 channels (Osteen et al. 2010). More recently, using a combination of voltage clamp fluorometry and cysteine accessibility, our group showed that KCNE1 splits the voltage sensor movement of the KCNQ1 channel into two components (Barro-Soria et al. 2014). The first component of S4 movement occurs at negative voltages and develops faster relative to the second component of S4 movement, which develops at positive voltages simultaneously with channel opening. Gating currents in KCNQ1–KCNE1 channels were shown to occur with a similar time and voltage dependence as the first fluorescence component, suggesting that the first component of S4 movement reports on the main S4 charge movement. Although not directly detected, the second fluorescence component must involve a small gating charge movement that contributes to the opening of the pore. Our data suggest that KCNE1 affects KCNQ1 in several ways: it stabilizes an intermediate position of S4, it prevents the gate from opening unless all four S4s have reached the fully activated state, and it slows down the gate opening (and a second proposed additional movement of S4). We proposed a model for KCNQ1–KCNE1 channel gating in which the main S4 movement in the four subunits occurs quickly and independently at negative voltages and a slow, concerted further S4 movement in all four subunits opens the channel at positive voltages (Fig.3B). Our data, however, did not allow us to answer the question of whether the second fluorescence change developed during channel opening is due to an additional S4 movement or if the opening of the gate induces a conformational change that causes a second fluorescence change around S4.
More recently, a very interesting new hypothesis was put forward by Zaydman et al. in which KCNE1 neither directly affects the activation of the voltage-sensing domains nor the opening of the pore (Zaydman et al. 2014). Instead, they proposed that KCNE1 affects the state-dependent interactions between the voltage-sensing domain and the pore of KCNQ1 channels.
KCNE3
How does KCNE3 convert the voltage-dependent KCNQ1 channel into a voltage-independent channel in the physiological voltage range? Two alternatives have been suggested to explain the voltage independence of the KCNQ1–KCNE3 channel within this voltage range: (1) S4 is locked in an active state and is still physically connected to the gate through the S4–S5 linker, therefore keeping the gate always open, or (2) S4 moves upon voltage changes but the movement is uncoupled from the gate, which is always locked open by KCNE3. In support of the idea that KCNE3 locks S4 in the active state, an alanine scanning mutagenesis study from Panaghie and Abbott showed that substituting positively charged residues in the C-terminus of S4 for alanine changed the proportion of constitutively active KCNQ1–KCNE3 channels (Panaghie & Abbott, 2007). They reasoned that this variation in constitutive open channels by S4 charge neutralization would not occur if S4 were not coupled to the gate. It was therefore concluded that KCNE3 probably locks the S4s in the active state (Panaghie & Abbott, 2007). In two cysteine accessibility studies, it was shown that the time course of reaction of thiol reagents to cysteines introduced in S4 in KCNQ1–KCNE3 channels was independent of voltage (Nakajo & Kubo, 2007; Rocheleau & Kobertz, 2008). These data suggested that KCNE3 causes a stabilization of S4 in a configuration that allows accessibility to thiol reagents at all membrane voltages, implying that the S4s in KCNQ1–KCNE3 channels are in the active state independent of voltage (Nakajo & Kubo, 2007; Rocheleau & Kobertz, 2008) (Fig.3B).
How KCNE1 and KCNE3, which are quite homologous, mediate such different effects on KCNQ1 channel gating is still a matter of active study. A number of different studies have shown interactions between KCNQ1 and the N-terminal region, the transmembrane segment, and the C-terminal region for both KCNE1 and KCNE3 (see review Wrobel et al. 2012). For example, some studies have shown that the pore region or the voltage-sensing domain of KCNQ1 channel could be close enough to directly interact with the transmembrane domain of KCNEs (Melman et al. 2004; Panaghie et al. 2006; Nakajo & Kubo, 2007; Shamgar et al. 2008; Xu et al. 2008; Chung et al. 2009), while other studies have shown that the C-terminus of KCNQ1 interacts with the C-terminus of KCNEs (Gage & Kobertz, 2004; Shamgar et al. 2008). However, it is not completely clear which of all these interactions mediate the functional effects that underlie the differences between KCNE1- and KCNE3-modulation of KCNQ1. Most likely it is a combination of different interactions from different regions of the KCNEs and KCNQ1 that generates the different KCNE effects on KCNQ1 gating. We propose that the weak coupling between S4 and the gate in KCNQ1 makes it very easy to alter the gating in KCNQ1, so the differences in the KCNQ1–KCNE1 interactions and KCNQ1–KCNE3 interactions might not have to be very large for these KCNEs to have very distinct effects on KCNQ1 gating.
Regulation of KCNQ1 channel by cytosolic molecules
Due to the relevance of the KCNQ1 channel in the heart and epithelial tissues during both physiological and pathophysiological conditions, it is not surprising to find that this channel is regulated by several meta-bolic/endogenous factors. These modulators seem to use the intrinsic flexibility of KCNQ1 channels to modify the behaviour of KCNQ1–KCNEx channels. Below we will exemplify this type of regulation by discussing the role of ATP, PIP2 and PKA for KCNQ1 channel function and the potential mechanism of action of these modulators.
ATP
In cardiomyocytes, ATP plays a major role not only as an energy source necessary to fuel the high demands of cardiomyocyte metabolic rates, but also as a signalling molecule that regulates the cardiac action potential duration by directly modulating, for example, KCNQ1–KCNE1 (IKs) channel activity (Li et al. 2013). During a transient ischaemic event, both ATP and pH levels drop. Low pH inhibits L-type calcium channels and reduces intracellular calcium and the cardiac contraction (Lu et al. 2012). It has been speculated that the concurrent modest drop in ATP (by 50%) during a transient ischaemic event would decrease IKs activity and lead to a prolongation of the cardiac action potential, which would allow for a compensatory increase in the duration of calcium influx through calcium channels and a restoration of the cardiac contraction (Li et al. 2013). In contrast, a more drastic drop in ATP levels (to 10% of normal levels) as during prolonged ischaemia would activate KATP channels and thereby reduce cardiac activity to prevent cell damage to the cardiac tissue. Li et al. proposed that ATP promotes KCNQ1–KCNE1 channel opening by binding directly to a cluster of basic and aromatic residues in the C-terminus of KCNQ1 channels close to the S6 gate (Li et al. 2013). This proposed ATP binding site differs from classical ATP binding sites where conserved ATP binding motifs Walker A and B (e.g. CFTR) or large hydrophilic residues cassette (e.g. P2X receptors) might be located in structurally different protein domains. Loss-of-function mutations in this region of KCNQ1, which leads to long QT syndrome and could cause life-threatening cardiac arrhythmias, reduce IKs current by reducing ATP binding (Li et al. 2013). The fact that ATP removal from KCNQ1 channels completely shuts down the IKs current could be due to the loose coupling in these channels between S4 and the gate, so that the energy from S4 movement transferred to the gate does not overcome the loss of energy by the removal of ATP to stabilize the gate in the open state. Thus, the high sensitivity of KCNQ1–KCNE1 to ATP might be due to the weak S4-to-gate coupling and intrinsic flexibility of KCNQ1. The cell uses this ATP sensitivity to adjust KCNQ1–KCNE1 channel activity, and therefore the cardiac action potential duration to meet physiological needs.
PIP2
Another important regulator of the KCNQ1 channel is the acidic phospholipid PIP2 which promotes KCNQ1 channel opening (reviewed in Abbott, 2014; Zaydman & Cui, 2014). As a consequence of this, KCNQ1 current rundown is seen upon PIP2 depletion, as happens during patch excision or activation of lipid phosphatases (such as the voltage-sensing lipid phosphatase VSP). KCNE1 increases KCNQ1 sensitivity to PIP2 about 100-fold, which might be one explanation for the KCNE1-induced augmentation in KCNQ1 current amplitude, since physiological levels of PIP2 might not fully occupy the PIP2 binding sites in KCNQ1 channels (Li et al. 2011). PIP2 modulation of KCNQ1 is coupled to cardiac physiology via α1-adrenergic receptors. Stimulation of α1-adrenergic receptors in the heart activates signalling pathways that result in modulation of KCNQ1–KCNE1 activity (Jensen et al. 2011), in part via PIP2 hydrolysis. By reducing the intracellular PIP2 concentration, α1-adrenergic stimulation therefore reduces IKs channel activity and prolongs the cardiac action potential during rest. An early study proposed a model in which PIP2 stabilizes the open pore conformation by affecting the gate (Loussouarn et al. 2003). More recently, PIP2 was suggested to bind to a cluster of basic residues in the intracellular S2–S3 and S4–S5 loops and the C-terminus of KCNQ1 (reviewed in Zaydman & Cui, 2014). Mutations in this basic cluster impair the ability of PIP2 to augment KCNQ1 current amplitude, which may be part of the disease aetiology of long QT mutations in this region (Park et al. 2005; Li et al. 2011; Zaydman et al. 2013). From mutagenesis studies and voltage clamp fluorometry experiments, PIP2 has been shown to be crucial for KCNQ1 channel opening by being a requirement for coupling S4 movement to gate opening (Zaydman et al. 2013). The putative PIP2 binding site bridging the voltage-sensor domain and the pore domain is proposed to enable the PIP2 molecule to communicate conformational changes in the voltage-sensor domain to the pore domain, and vice versa. When PIP2 is depleted, the S4 and the opening of the channel are decoupled: S4 still moves in response to voltage changes, but the channel gate stays closed (Li et al. 2011; Zaydman et al. 2013). The relatively loose coupling between S4 and the gate in KCNQ1 channels might be reason for the big effect PIP2 removal has on the current. Without PIP2 there is probably not enough connection between S4 and the gate for S4 movement to affect the gate.
PKA
The KCNQ1–KCNE1 channel current in the heart is augmented by β-adrenergic stimulation, particularly in stress-mediated situations wherein the heart rate goes up and the length of the cardiac action potential shortens, to allow for enough time to fill the heart with blood in between cardiac contractions (Terrenoire et al. 2005). β1-adrenergic receptor activation by adrenaline/noradrenaline increases the intracellular level of cAMP, which leads to PKA activation. PKA, in turn, phosphorylates the N-terminus of KCNQ1 channels in the presence of the anchoring protein yotiao, which binds to the C-terminus of KCNQ1 (Marx et al. 2002). Phosphorylated KCNQ1–KCNE1 channels show not only higher current amplitude but also faster activation, which both induce a shortening of the cardiac action potential. Understanding the mechanism for β-adrenergic-induced enhancement of KCNQ1–KCNE1 activity is particularly important for patients carrying specific long QT mutations that disrupt the formation of the KCNQ1–PKA/yotiao macromolecular complex (Marx et al. 2002). The molecular mechanism for how PKA bound to yotiao in the C-terminus and phosphorylation of a residue in the N-terminus affects the gating of KCNQ1–KCNE1 channels is not understood, but it could affect the S4 movement, the gate, or the coupling between S4 and the gate.
Conclusion
The KCNQ1 channel seems to have an inherently flexible gating, largely explained by its loose coupling between S4 movement and gate opening. This loose coupling may structurally, at least in part, come from the unusual S6 PXG motif in KCNQ1, differences in S4–S5 linker/S6 interaction, and the fewer positively charged S4 residues compared to other Kv channels. The lack of important gating charge residues in S4 also reduces the amount of energy gained from S4 movement to be transferred to the gate to promote opening of the gate. S4 of KCNQ1 is therefore more easily influenced by extrinsic modulators such as KCNE β-subunits than S4 of Kv channels that have additional gating charge residues. The five different KCNE subunits take advantage of the flexible intrinsic gating of KCNQ1 to further diversify KCNQ1 channel functions. This provides the functional versatility needed from the KCNQ1 channel to be able to play different physiological roles in different tissues, from voltage-independent KCNQ1–KCNE3 channels in epithelial cells to slowly activating KCNQ1–KCNE1 channels in cardiac cells. To add a third dimension of flexibility, the KCNQ1 channel is regulated by physiological modulators that adjust KCNQ1 activity in accordance with physiological need, thereby allowing a dynamic modulation of KCNQ1 currents.
Acknowledgments
We thank Dr Daniel Wilhelms, Linköping University, Sweden, for help with the illustration in Fig.1A.
Glossary
- Kv
voltage-gated K+ channel
- MTS
methanethiosulfonate
- PIP2
phosphatidylinositol 4,5-bispho-sphate
- PKA
protein kinase A
Biographies
 Sara I. Liin completed her PhD in Neurobiology at Linköping University in the laboratory of Fredrik Elinder. She is currently a postdoctoral associate in the laboratory of H. Peter Larsson, where she is investigating the anti-arrhythmic effect of novel KCNQ1 channel modulators.
Sara I. Liin completed her PhD in Neurobiology at Linköping University in the laboratory of Fredrik Elinder. She is currently a postdoctoral associate in the laboratory of H. Peter Larsson, where she is investigating the anti-arrhythmic effect of novel KCNQ1 channel modulators.
 Rene Barro-Soria completed his PhD in the Department of Physiology at the University of Regensburg, Germany. He is currently a postdoctoral associate in the laboratory ofH. Peter Larsson, where he is investigating the mechanism of KCNE subunit modulation of KCNQ1 channel function.
Rene Barro-Soria completed his PhD in the Department of Physiology at the University of Regensburg, Germany. He is currently a postdoctoral associate in the laboratory ofH. Peter Larsson, where he is investigating the mechanism of KCNE subunit modulation of KCNQ1 channel function.
 H. Peter Larsson is a Professor in the Department of Physiology and Biophysics at the University ofMiami. He completed his PhD in Biophysics and postdoctoral training in Neuroscience at the University of California Berkeley. His research is in the area of ion channel and neurotransmitter transporter biophysics, where he studies the molecular mechanisms of several voltage-gated ion channels and glutamate transporters.
H. Peter Larsson is a Professor in the Department of Physiology and Biophysics at the University ofMiami. He completed his PhD in Biophysics and postdoctoral training in Neuroscience at the University of California Berkeley. His research is in the area of ion channel and neurotransmitter transporter biophysics, where he studies the molecular mechanisms of several voltage-gated ion channels and glutamate transporters.
Additional information
Competing interests
None declared.
Funding
H.P.L. is funded by grants from NHLBI (R01-HL095920), NIHGM (R01-GM109762), and American Heart Association (14GRNT20380041), R.B.S. by an AHA post-doctoral fellowship (13POST17000057), and S.I.L. by a post-doctoral fellowship from the Swedish Research Council.
References
- Abbott GW. Biology of the KCNQ1 potassium channel. New J Sci. 2014;2014:237431. [Google Scholar]
- Anantharam A, Markowitz SM. Abbott GW. Pharmacogenetic considerations in diseases of cardiac ion channels. J Pharmacol Exp Ther. 2003;307:831–838. doi: 10.1124/jpet.103.054569. [DOI] [PubMed] [Google Scholar]
- Angelo K, Jespersen T, Grunnet M, Nielsen MS, Klaerke DA. Olesen SP. KCNE5 induces time- and voltage-dependent modulation of the KCNQ1 current. Biophys J. 2002;83:1997–2006. doi: 10.1016/S0006-3495(02)73961-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barhanin J, Lesage F, Guillemare E, Fink M, Lazdunski M. Romey G. KVLQT1 and IsK (minK) proteins associate to form the IKs cardiac potassium current. Nature. 1996;384:78–80. doi: 10.1038/384078a0. [DOI] [PubMed] [Google Scholar]
- Barrett KE. Keely SJ. Chloride secretion by the intestinal epithelium: molecular basis and regulatory aspects. Annu Rev Physiol. 2000;62:535–572. doi: 10.1146/annurev.physiol.62.1.535. [DOI] [PubMed] [Google Scholar]
- Barro-Soria R, Rebolledo S, Liin SI, Perez ME, Sampson KJ, Kass RS. Larsson HP. KCNE1 divides the voltage sensor movement in KCNQ1–KCNE1 channels into two steps. Nat Commun. 2014;5:3750. doi: 10.1038/ncomms4750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blunck R. Batulan Z. Mechanism of electromechanical coupling in voltage-gated potassium channels. Front Pharmacol. 2012;3:166. doi: 10.3389/fphar.2012.00166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Börjesson SI. Elinder F. Structure, function, and modification of the voltage sensor in voltage-gated ion channels. Cell Biochem Biophys. 2008;52:149–174. doi: 10.1007/s12013-008-9032-5. [DOI] [PubMed] [Google Scholar]
- Catterall WA. Ion channel voltage sensors: structure, function, and pathophysiology. Neuron. 2010;67:915–928. doi: 10.1016/j.neuron.2010.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung DY, Chan PJ, Bankston JR, Yang L, Liu G, Marx SO, Karlin A. Kass RS. Location of KCNE1 relative to KCNQ1 in the IKS potassium channel by disulfide cross-linking of substituted cysteines. Proc Natl Acad Sci USA. 2009;106:743–748. doi: 10.1073/pnas.0811897106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciampa EJ, Welch RC, Vanoye CG. George AL., Jr KCNE4 juxtamembrane region is required for interaction with calmodulin and for functional suppression of KCNQ1. J Biol Chem. 2011;286:4141–4149. doi: 10.1074/jbc.M110.158865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- del Camino D, Holmgren M, Liu Y. Yellen G. Blocker protection in the pore of a voltage-gated K+ channel and its structural implications. Nature. 2000;403:321–325. doi: 10.1038/35002099. [DOI] [PubMed] [Google Scholar]
- Gage SD. Kobertz WR. KCNE3 truncation mutants reveal a bipartite modulation of KCNQ1 K+ channels. J Gen Physiol. 2004;124:759–771. doi: 10.1085/jgp.200409114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldman AM, Glasscock E, Yoo J, Chen TT, Klassen TL. Noebels JL. Arrhythmia in heart and brain: KCNQ1 mutations link epilepsy and sudden unexplained death. Sci Transl Med. 2009;1:2ra6. doi: 10.1126/scitranslmed.3000289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grahammer F, Warth R, Barhanin J, Bleich M. Hug MJ. The small conductance K+ channel, KCNQ1: expression, function, and subunit composition in murine trachea. J Biol Chem. 2001;276:42268–42275. doi: 10.1074/jbc.M105014200. [DOI] [PubMed] [Google Scholar]
- Grunnet M, Jespersen T, Rasmussen HB, Ljungstrom T, Jorgensen NK, Olesen SP. Klaerke DA. KCNE4 is an inhibitory subunit to the KCNQ1 channel. J Physiol. 2002;542:119–130. doi: 10.1113/jphysiol.2002.017301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hackos DH, Chang TH. Swartz KJ. Scanning the intracellular S6 activation gate in the shaker K+ channel. J Gen Physiol. 2002;119:521–532. doi: 10.1085/jgp.20028569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi M. Novak I. Molecular basis of potassium channels in pancreatic duct epithelial cells. Channels (Austin) 2013;7:432–441. doi: 10.4161/chan.26100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heitzmann D. Warth R. No potassium, no acid: K+ channels and gastric acid secretion. Physiology (Bethesda) 2007;22:335–341. doi: 10.1152/physiol.00016.2007. [DOI] [PubMed] [Google Scholar]
- Henrion U, Renhorn J, Borjesson SI, Nelson EM, Schwaiger CS, Bjelkmar P, Wallner B, Lindahl E. Elinder F. Tracking a complete voltage-sensor cycle with metal-ion bridges. Proc Natl Acad Sci USA. 2012;109:8552–8557. doi: 10.1073/pnas.1116938109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hibino H, Nin F, Tsuzuki C. Kurachi Y. How is the highly positive endocochlear potential formed? The specific architecture of the stria vascularis and the roles of the ion-transport apparatus. Pflugers Arch. 2010;459:521–533. doi: 10.1007/s00424-009-0754-z. [DOI] [PubMed] [Google Scholar]
- Islas LD. Sigworth FJ. Voltage sensitivity and gating charge in Shaker and Shab family potassium channels. J Gen Physiol. 1999;114:723–742. doi: 10.1085/jgp.114.5.723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen BC, O’Connell TD. Simpson PC. Alpha-1-adrenergic receptors: targets for agonist drugs to treat heart failure. J Mol Cell Cardiol. 2011;51:518–528. doi: 10.1016/j.yjmcc.2010.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jespersen T, Grunnet M. Olesen SP. The KCNQ1 potassium channel: from gene to physiological function. Physiology (Bethesda) 2005;20:408–416. doi: 10.1152/physiol.00031.2005. [DOI] [PubMed] [Google Scholar]
- Labro AJ, Raes AL. Snyders DJ. Coupling of voltage sensing to channel opening reflects intrasubunit interactions in Kv channels. J Gen Physiol. 2005;125:71–80. doi: 10.1085/jgp.200409194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee MP, Hu RJ, Johnson LA. Feinberg AP. Human KVLQT1 gene shows tissue-specific imprinting and encompasses Beckwith-Wiedemann syndrome chromosomal rearrangements. Nat Genet. 1997;15:181–185. doi: 10.1038/ng0297-181. [DOI] [PubMed] [Google Scholar]
- Li Y, Gao J, Lu Z, McFarland K, Shi J, Bock K, Cohen IS. Cui J. Intracellular ATP binding is required to activate the slowly activating K+ channel IKs. Proc Natl Acad Sci USA. 2013;110:18922–18927. doi: 10.1073/pnas.1315649110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Zaydman MA, Wu D, Shi J, Guan M, Virgin-Downey B. Cui J. KCNE1 enhances phosphatidylinositol 4,5-bisphosphate (PIP2) sensitivity of IKs to modulate channel activity. Proc Natl Acad Sci USA. 2011;108:9095–9100. doi: 10.1073/pnas.1100872108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long SB, Tao X, Campbell EB. MacKinnon R. Atomic structure of a voltage-dependent K+ channel in a lipid membrane-like environment. Nature. 2007;450:376–382. doi: 10.1038/nature06265. [DOI] [PubMed] [Google Scholar]
- Loussouarn G, Park KH, Bellocq C, Baro I, Charpentier F. Escande D. Phosphatidylinositol-4,5-bisphosphate, PIP2, controls KCNQ1–KCNE1 voltage-gated potassium channels: a functional homology between voltage-gated and inward rectifier K+ channels. EMBO J. 2003;22:5412–5421. doi: 10.1093/emboj/cdg526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Z, Wu CY, Jiang YP, Ballou LM, Clausen C, Cohen IS. Lin RZ. Suppression of phosphoinositide 3-kinase signalling and alteration of multiple ion currents in drug-induced long QT syndrome. Sci Transl Med. 2012;4:131ra150. doi: 10.1126/scitranslmed.3003623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mannuzzu LM, Moronne MM. Isacoff EY. Direct physical measure of conformational rearrangement underlying potassium channel gating. Science. 1996;271:213–216. doi: 10.1126/science.271.5246.213. [DOI] [PubMed] [Google Scholar]
- Marx SO, Kurokawa J, Reiken S, Motoike H, D’Armiento J, Marks AR. Kass RS. Requirement of a macromolecular signalling complex for beta adrenergic receptor modulation of the KCNQ1–KCNE1 potassium channel. Science. 2002;295:496–499. doi: 10.1126/science.1066843. [DOI] [PubMed] [Google Scholar]
- Meisel E, Dvir M, Haitin Y, Giladi M, Peretz A. Attali B. KCNQ1 channels do not undergo concerted but sequential gating transitions in both the absence and the presence of KCNE1 protein. J Biol Chem. 2012;287:34212–34224. doi: 10.1074/jbc.M112.364901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melman YF, Um SY, Krumerman A, Kagan A. McDonald TV. KCNE1 binds to the KCNQ1 pore to regulate potassium channel activity. Neuron. 2004;42:927–937. doi: 10.1016/j.neuron.2004.06.001. [DOI] [PubMed] [Google Scholar]
- Nakajo K. Kubo Y. KCNE1 and KCNE3 stabilize and/or slow voltage sensing S4segment of KCNQ1 channel. J Gen Physiol. 2007;130:269–281. doi: 10.1085/jgp.200709805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakajo K, Ulbrich MH, Kubo Y. Isacoff EY. Stoichiometry of the KCNQ1–KCNE1 ion channel complex. Proc Natl Acad Sci USA. 2010;107:18862–18867. doi: 10.1073/pnas.1010354107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nerbonne JM. Kass RS. Molecular physiology of cardiac repolarization. Physiol Rev. 2005;85:1205–1253. doi: 10.1152/physrev.00002.2005. [DOI] [PubMed] [Google Scholar]
- Neyroud N, Tesson F, Denjoy I, Leibovici M, Donger C, Barhanin J, Faure S, Gary F, Coumel P, Petit C, Schwartz K. Guicheney P. A novel mutation in the potassium channel gene KVLQT1 causes the Jervell and Lange-Nielsen cardioauditory syndrome. Nat Gen. 1997;15:186–189. doi: 10.1038/ng0297-186. [DOI] [PubMed] [Google Scholar]
- Osteen JD, Barro-Soria R, Robey S, Sampson KJ, Kass RS. Larsson HP. Allosteric gating mechanism underlies the flexible gating of KCNQ1 potassium channels. Proc Natl Acad Sci USA. 2012;109:7103–7108. doi: 10.1073/pnas.1201582109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osteen JD, Gonzalez C, Sampson KJ, Iyer V, Rebolledo S, Larsson HP. Kass RS. KCNE1 alters the voltage sensor movements necessary to open the KCNQ1 channel gate. Proc Natl Acad Sci USA. 2010;107:22710–22715. doi: 10.1073/pnas.1016300108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panaghie G. Abbott GW. The Role of S4 Charges in voltage-dependent and voltage-independent KCNQ1 potassium channel complexes. J Gen Physiol. 2007;129:121–133. doi: 10.1085/jgp.200609612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panaghie G, Tai KK. Abbott GW. Interaction of KCNE subunits with the KCNQ1 K+ channel pore. J Physiol. 2006;570:455–467. doi: 10.1113/jphysiol.2005.100644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park KH, Piron J, Dahimene S, Merot J, Baro I, Escande D. Loussouarn G. Impaired KCNQ1–KCNE1 and phosphatidylinositol-4,5-bisphosphate interaction underlies the long QT syndrome. Circ Res. 2005;96:730–739. doi: 10.1161/01.RES.0000161451.04649.a8. [DOI] [PubMed] [Google Scholar]
- Plant LD, Xiong D, Dai H. Goldstein SA. Individual IKs channels at the surface of mammalian cells contain two KCNE1 accessory subunits. Proc Natl Acad Sci USA. 2014;111:E1438–1446. doi: 10.1073/pnas.1323548111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Preston P, Wartosch L, Gunzel D, Fromm M, Kongsuphol P, Ousingsawat J, Kunzelmann K, Barhanin J, Warth R. Jentsch TJ. Disruption of the K+ channel beta-subunit KCNE3 reveals an important role in intestinal and tracheal Cl– transport. J Biol Chem. 2010;285:7165–7175. doi: 10.1074/jbc.M109.047829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pusch M, Magrassi R, Wollnik B. Conti F. Activation and inactivation of homomeric KvLQT1 potassium channels. Biophys J. 1998;75:785–792. doi: 10.1016/S0006-3495(98)77568-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rocheleau JM. Kobertz WR. KCNE peptides differently affect voltage sensor equilibrium and equilibration rates in KCNQ1 K+ channels. J Gen Physiol. 2008;131:59–68. doi: 10.1085/jgp.200709816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roepke TK, Anantharam A, Kirchhoff P, Busque SM, Young JB, Geibel JP, Lerner DJ. Abbott GW. The KCNE2 potassium channel ancillary subunit is essential for gastric acid secretion. J Biol Chem. 2006;281:23740–23747. doi: 10.1074/jbc.M604155200. [DOI] [PubMed] [Google Scholar]
- Ruscic KJ, Miceli F, Villalba-Galea CA, Dai H, Mishina Y, Bezanilla F. Goldstein SA. IKs channels open slowly because KCNE1 accessory subunits slow the movement of S4 voltage sensors in KCNQ1 pore-forming subunits. Proc Natl Acad Sci USA. 2013;110:E559–E566. doi: 10.1073/pnas.1222616110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanguinetti MC, Curran ME, Zou A, Shen J, Spector PS, Atkinson DL. Keating MT. Coassembly of KVLQT1 and minK (IsK) proteins to form cardiac IKs potassium channel. Nature. 1996;384:80–83. doi: 10.1038/384080a0. [DOI] [PubMed] [Google Scholar]
- Schmitt N, Schwarz M, Peretz A, Abitbol I, Attali B. Pongs O. A recessive C-terminal Jervell and Lange-Nielsen mutation of the KCNQ1 channel impairs subunit assembly. EMBO J. 2000;19:332–340. doi: 10.1093/emboj/19.3.332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroeder BC, Waldegger S, Fehr S, Bleich M, Warth R, Greger R. Jentsch TJ. A constitutively open potassium channel formed by KCNQ1 and KCNE3. Nature. 2000;403:196–199. doi: 10.1038/35003200. [DOI] [PubMed] [Google Scholar]
- Sesti F. Goldstein SA. Single-channel characteristics of wild-type IKs channels and channels formed with two minK mutants that cause long QT syndrome. J Gen Physiol. 1998;112:651–663. doi: 10.1085/jgp.112.6.651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shamgar L, Haitin Y, Yisharel I, Malka E, Schottelndreier H, Peretz A, Paas Y. Attali B. KCNE1 constrains the voltage sensor of Kv7.1 K+ channels. PLoS One. 2008;3:e1943. doi: 10.1371/journal.pone.0001943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Splawski I, Shen J, Timothy KW, Vincent GM, Lehmann MH. Keating MT. Genomic structure of three long QT syndrome genes: KVLQT1, HERG, and KCNE1. Genomics. 1998;51:86–97. doi: 10.1006/geno.1998.5361. [DOI] [PubMed] [Google Scholar]
- Swartz KJ. Sensing voltage across lipid membranes. Nature. 2008;456:891–897. doi: 10.1038/nature07620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tai KK. Goldstein SA. The conduction pore of a cardiac potassium channel. Nature. 1998;391:605–608. doi: 10.1038/35416. [DOI] [PubMed] [Google Scholar]
- Tapper AR. George AL., Jr Location and orientation of minK within the IKs potassium channel complex. J Biol Chem. 2001;276:38249–38254. doi: 10.1074/jbc.M103956200. [DOI] [PubMed] [Google Scholar]
- Terrenoire C, Clancy CE, Cormier JW, Sampson KJ. Kass RS. Autonomic control of cardiac action potentials: role of potassium channel kinetics in response to sympathetic stimulation. Circ Res. 2005;96:e25–e34. doi: 10.1161/01.RES.0000160555.58046.9a. [DOI] [PubMed] [Google Scholar]
- Tinel N, Diochot S, Borsotto M, Lazdunski M. Barhanin J. KCNE2 confers background current characteristics to the cardiac KCNQ1 potassium channel. 2000. pp. 6326–6330. [DOI] [PMC free article] [PubMed]
- Vallon V, Grahammer F, Volkl H, Sandu CD, Richter K, Rexhepaj R, Gerlach U, Rong Q, Pfeifer K. Lang F. KCNQ1-dependent transport in renal and gastrointestinal epithelia. Proc Natl Acad Sci USA. 2005;102:17864–17869. doi: 10.1073/pnas.0505860102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang KW, Tai KK. Goldstein SA. a MinK residues line a potassium channel pore. Neuron. 1996;16:571–577. doi: 10.1016/s0896-6273(00)80076-8. [DOI] [PubMed] [Google Scholar]
- Wang Q, Curran ME, Splawski I, Burn TC, Millholland JM, VanRaay TJ, Shen J, Timothy KW, Vincent GM, de Jager T, et al. Positional cloning of a novel potassium channel gene: KVLQT1 mutations cause cardiac arrhythmias. Nat Genet. 1996;12:17–23. doi: 10.1038/ng0196-17. [DOI] [PubMed] [Google Scholar]
- Wang W, Xia J. Kass RS. MinK–KvLQT1 fusion proteins, evidence for multiple stoichiometries of the assembled IsK channel. J Biol Chem. 1998;273:34069–34074. doi: 10.1074/jbc.273.51.34069. [DOI] [PubMed] [Google Scholar]
- Vardanyan V. Pongs O. Coupling of voltage-sensors to the channel pore: a comparative view. Front Pharmacol. 2012;3:145. doi: 10.3389/fphar.2012.00145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warth R. Barhanin J. The multifaceted phenotype of the knockout mouse for the KCNE1 potassium channel gene. Am J Physiol Regul Integr Comp Physiol. 2002;282:R639–R648. doi: 10.1152/ajpregu.00649.2001. [DOI] [PubMed] [Google Scholar]
- Wrobel E, Tapken D. Seebohm G. The KCNE tango – how KCNE1 interacts with Kv7.1. Front Pharmacol. 2012;3:142. doi: 10.3389/fphar.2012.00142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu X, Jiang M, Hsu KL, Zhang M. Tseng GN. KCNQ1 and KCNE1 in the IKs channel complex make state-dependent contacts in their extracellular domains. J Gen Physiol. 2008;131:589–603. doi: 10.1085/jgp.200809976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y. Sigworth FJ. Single-channel properties of IKs potassium channels. J Gen Physiol. 1998;112:665–678. doi: 10.1085/jgp.112.6.665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaydman MA. Cui J. PIP2 regulation of KCNQ cha-nnels: biophysical and molecular mechanisms for lipid mod-ulation of voltage-dependent gating. Front Physiol. 2014;5:195. doi: 10.3389/fphys.2014.00195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaydman MA, Kasimova MA, McFarland K, Beller Z, Hou P, Kinser HE, Liang H, Zhang G, Shi J, Tarek M. Cui J. Domain–domain interactions determine the gating, permeation, pharmacology, and subunit modulation of the IKs ion channel. Elife. 2014;3 doi: 10.7554/eLife.03606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaydman MA, Silva JR, Delaloye K, Li Y, Liang H, Larsson HP, Shi J. Cui J. Kv7.1 ion channels require a lipid to couple voltage sensing to pore opening. Proc Natl Acad Sci USA. 2013;110:13180–13185. doi: 10.1073/pnas.1305167110. [DOI] [PMC free article] [PubMed] [Google Scholar]