

Abstract
Traumatic nerve injury or motor neuron disease leads to denervation and severe muscle atrophy. Recent evidence indicates that loss of mitochondria and the related reduction in oxidative capacity could be key mediators of skeletal muscle atrophy. As our previous study showed that heat stress increased the numbers of mitochondria in skeletal muscle, we evaluated whether heat stress treatment could have a beneficial impact on denervation-induced loss of mitochondria and subsequent muscle atrophy. Here, we report that daily heat stress treatment (mice placed in a chamber with a hot environment; 40°C, 30 min day−1, for 7 days) rescues the following parameters: (i) muscle atrophy (decreased gastrocnemius muscle mass); (ii) loss of mitochondrial content (decreased levels of ubiquinol–cytochrome c reductase core protein II, cytochrome c oxidase subunits I and IV and voltage-dependent anion channel protein); and (iii) reduction in oxidative capacity (reduced maximal activities of citrate synthase and 3-hydroxyacyl-CoA dehydrogenase) in denervated muscle (produced by unilateral sciatic nerve transection). In order to gain a better understanding of the above mitochondrial adaptations, we also examined the effects of heat stress on autophagy-dependent mitochondrial clearance (mitophagy). Daily heat stress normalized denervation-activated induction of mitophagy (increased mitochondrial microtubule-associated protein 1A/1B-light chain3-II (LC3-II) with and without blocker of autophagosome clearance). The molecular basis of this observation was explained by the results that heat stress attenuated the denervation-induced increase in key proteins that regulate the following steps: (i) the tagging step of mitochondrial clearance (increased mitochondrial Parkin, ubiquitin-conjugated, P62/sequestosome 1 (P62/SQSTM1)); and (ii) the elongation step of autophagosome formation (increased Atg5–Atg12 conjugate and Atg16L). Overall, our results contribute to the better understanding of mitochondrial quality control and the mechanisms behind the attenuation of muscle wasting by heat stress in denervated skeletal muscle.
Key points
Traumatic nerve injury or nerve disease leads to denervation and severe muscle atrophy. Recent evidence shows that mitochondrial loss could be a key mediator of skeletal muscle atrophy.
Here, we show that daily heat stress treatment rescues denervation-induced loss of mitochondria and concomitant muscle atrophy.
We also found that denervation-activated autophagy-dependent mitochondrial clearance (mitophagy) was suppressed by daily heat stress treatment. The molecular basis of this observation is explained by our results showing that heat stress treatment attenuates the increase of key proteins that regulate the tagging step for mitochondrial clearance and the intermediate step of autophagosome formation in denervated muscle.
These findings contribute to the better understanding of mitochondrial quality control in denervated muscle from a translational perspective and provide a mechanism behind the attenuation of muscle wasting by heat stress.
Introduction
Skeletal muscle constitutes 40% of total body mass, representing the largest tissue in the body. Skeletal muscle plays important roles not only in locomotion, but also in metabolic and endocrine organs, contributing to daily movement and improvement of whole-body metabolism. Skeletal muscle mass and function are regulated by motor innervation. Patients with denervation caused by conditions such as traumatic nerve injury, diabetic neuropathy, degenerative disc disease, amyotrophic lateral sclerosis and spinal muscular atrophy are forced into a vicious cycle of chronic muscle disuse myopathy, which includes muscle wasting, weakness and pain (Bongers et al. 2013). To establish effective therapies, researchers are actively exploring the cellular and molecular mechanisms involved in denervation-initiated myopathy using animal models.
Together with muscle wasting, denervation also induces mitochondrial toxicity, such as excessive accumulation of oxidative stress (Leary et al. 2012), loss of mitochondrial content (Lokireddy et al. 2012; Furuya et al. 2014) and a reduction of oxidative capacity (Wicks et al. 1991; Furuya et al. 2014) in skeletal muscle. Recent studies show that these mitochondrial toxicities could be key mediators of muscle wasting (Powers et al. 2012; Tryon et al. 2014); for example, skeletal muscle atrophy is suppressed by pharmacological scavenging of mitochondrial oxidative stress (Min et al. 2011; Talbert et al. 2013) or maintenance of mitochondrial content via genetic deletion of key proteins regulating mitochondrial degradation (mitophagy; Lokireddy et al. 2012; Furuya et al. 2014). Importantly, these studies suggest that conservation of healthy mitochondria can be an effective therapeutic target.
Recently, we have reported that heat stress treatment activates cellular signalling cascades associated with mitochondrial gene transcription and induces mitochondrial adaptations that include increased mitochondrial content and oxidative capacity in the skeletal muscle of mice (Tamura et al. 2014). Others recently reported that overexpression of heat shock protein (HSP) 72, a heat stress-inducible molecular chaperone, increases the number of mitochondria in mouse skeletal muscle (Henstridge et al. 2014). Furthermore, Naito et al. (2000) demonstrated that heat stress attenuates muscle atrophy in ‘hindlimb-suspended’ rats. These studies strongly suggest the possibility that heat stress treatment attenuates denervation-induced loss of mitochondrial content, the reduction of oxidative capacity and subsequent muscle atrophy. However, the following two important issues remain: (i) it is unknown whether heat stress treatment also rescues ‘denervation-induced’ muscle atrophy, because denervation leads to inimitable cellular responses/adaptations (Hornberger et al. 2001; Machida et al. 2012; Tang et al. 2014) and different efficacy with pharmacological intervention compared with other models of muscle disuse (MacDonald et al. 2014); and (ii) the impact of heat stress treatment on mitochondrial toxicity during muscle disuse including denervation is completely unknown.
The purpose of this study, therefore, was to examine whether daily heat stress treatment rescues denervation-induced mitochondrial loss and subsequent muscle atrophy. In order to gain a better understanding of mitochondrial adaptations, we also investigated the effects of daily heat stress treatment on cellular machinery regulating mitochondrial biogenesis and degradation in the denervated muscle.
Methods
Ethical approval
All experiments were approved by the Animal Experimental Committee of The University of Tokyo (no. 24-18). The authors have read, and all experiments complied with, the policies and regulations of Fundamental Guidelines for Proper Conduct of Animal Experiment and Related Activities in Academic Research Institutions published by the Ministry of Education, Culture, Sports, Science and Technology, Japan (no. 71, 2006) and The Journal of Physiology written by Drummond (2009).
Experimental animals
Eight-week-old male ICR mice (CLEA Japan, Tokyo, Japan; n = 110, including preliminary experiments) were used throughout this study. Mice were individually housed (cage size 16.5 cm × 9.5 cm × 10.0 cm) on a 12 h–12 h light–dark cycle (dark 07.00–19.00 h) in an air-conditioned room (22°C). All mice had access to standard chow (MF; Oriental Yeast, Tokyo, Japan) and water ad libitum. Mice were divided into a non-heat stress group (n = 6) or heat stress group (n = 6–8). All mice underwent unilateral sciatic nerve transection surgery. We performed experiments independently as follows: acute core temperature measurements, whole muscle analysis, mitochondrial isolation and/or colchicine treatment. Twenty-four hours after the final heat stress and/or colchicine treatment, mice were anaesthetized using isoflurane [4% (v/v) induction, 3% (v/v) maintenance, flow rate 0.5 l min−1] and killed by cervical dislocation. Given that some biochemical analyses require a large mass of muscle, the gastrocnemius muscles were collected and prepared for further analysis.
Sciatic nerve transection
We employed 10 days of unilateral sciatic nerve transection as surgical denervation, because previous studies showed that this model induced sufficient muscle atrophy and decreased the mitochondrial content in skeletal muscle (Brault et al. 2010; Kitaoka et al. 2014). Briefly, mice were anaesthetized using isoflurane [4% (v/v) induction, 3% (v/v) maintenance, flow rate 0.5 l min−1]. A small incision was made in the posterior aspect of the left hindlimb to expose the sciatic nerve at the level of the femoral trochanter. A length of at least 5.0 mm of sciatic nerve was excised using small operating scissors. The skin was closed with surgical glue. The right hindlimb served as the sham-operated control.
Daily heat stress protocol
A previous study has shown that anaesthesia during heat stress impairs the thermoregulation of normal mice (Leon et al. 2005) and induces a non-physiological situation. Therefore, we employed whole-body heat stress without anaesthesia, as previously described (Tamura et al. 2014). Briefly, mice were exposed to a hot environment chamber (40°C, 30 min day−1, from day 3 to 9 during the 10 day denervation period) without anaesthesia. We have previously reported that this heat stress method does not affect the spontaneous physical activity levels of mice during exposure and does not increase plasma corticosterone concentrations, a biomarker for systemic stress, beyond the levels of those induced by low-to-moderate intensity and volume treadmill running (Tamura et al. 2014). Mice in the non-heat stress group were exposed to a normal rearing environment chamber (22°C). Mice had no access to chow and water during exposure.
Mitophagic and autophagic flux
To evaluate mitophagic and autophagic flux, we used colchicine (C9754; Sigma-Aldrich, St Louis, MO, USA), which is a microtubule depolarizing agent, according to a previously reported protocol with minor modifications (Ju et al. 2010; Mofarrahi et al. 2013). Mice received i.p. injections of saline (without colchicine group) or colchicine (0.4 mg kg−1 day−1; with colchicine group) after the final heat stress treatment (i.e. monitoring mitophagic and autophagic flux from day 9 to 10).
Rectal temperature measurement
Rectal temperature was measured after a single bout of heat stress. Immediately after a single heat stress treatment, mice were anaesthetized using isoflurane [4% (v/v) induction, 3% (v/v) maintenance, flow rate 0.5 l min−1]. Rectal temperature was measured using a thermocouple (E52-CA20AY D = 3.2; Omron, Tokyo, Japan) connected to a data logger (GL200; Graphtec, Yokohama, Japan).
Mitochondrial enzyme activity
Maximal activities of citrate synthase (CS) and 3-hydroxyacyl-CoA dehydrogenase (3-HAD) were determined in whole muscle homogenates. Frozen and crushed gastrocnemius muscle specimens were homogenized in 100 (v/w) of 100 mm potassium phosphate buffer. Maximal CS and 3-HAD activities were measured spectrophotometrically using the methods of Srere (1969) and Bass et al. (1969), respectively.
Mitochondrial isolation
Mitochondria-enriched/associated fractions were isolated from gastrocnemius muscles using a previously described differential centrifugation method with minor modifications (Safdar et al. 2011). Briefly, gastrocnemius muscles were mildly homogenized using a polytron and Potter glass homogenizer in mitochondrial isolation buffer (67 mm sucrose, 50 mm Tris, 50 mm KCl, 10 mm EDTA and 0.2% (w/v) fatty acid-free bovine serum albumin, pH 7.4) containing protease inhibitors (complete protease inhibitor cocktail; Roche Diagnostics, Tokyo, Japan). The homogenate was centrifuged at 700g for 15 min at 4°C. The supernatant was collected and centrifuged again at 12,000g for 20 min at 4°C. The mitochondrial pellet was resuspended in mitochondrial isolation buffer and centrifuged at 12,000g for 20 min at 4°C. The supernatant was removed and the mitochondrial pellet resuspended in lysis buffer for Western blot analysis. The protein concentration was measured using the Bradford method. We confirmed the purity of the mitochondria-enriched fraction by Western blotting using antibodies against glyceraldehyde 3-phosphate dehydrogenase (cytosolic marker) and cytochrome c oxidase subunit (COX) IV (mitochondrial marker; data not shown).
The protein expressions of Parkin, ubiquitin-conjugated protein, p62/sequestosome 1 (SQSTM1) and microtubule-associated protein 1A/1B-light chain 3-II (LC3-II) in mitochondria-enriched/associated fractions were measured using Western blotting to evaluate mitophagy in accordance with previous studies (O’Leary et al. 2013; Webster et al. 2013).
Western blot analysis
Muscle samples were homogenized as previously described (Hoshino et al. 2012) using lysis buffer [1% (v/v) Triton X-100, 50 mm Tris–HCl, 1 mm EDTA, 1 mm EGTA, 50 mm sodium fluoride, 10 mm sodium 3-glycerol phosphate, 5 mm sodium pyrophosphate and 2 mm DTT; pH 7.5] containing 10 μg ml of pepstatin A, aprotinin and leupeptin, 1 mm sodium orthovanadate and 0.177 mg ml−1 phenylmethylsulfonyl fluoride. Sample proteins were measured using the Bradford method. Equal amounts (5–15 μg) of protein were separated using standard SDS-PAGE [7.5–17% (w/v) polyacrylamide gels] and transferred to a polyvinylidene difluoride membrane (Hybond-P; GE Healthcare Japan, Tokyo, Japan). Transfer of protein was confirmed by staining with Ponceau S (P7170-1L; Sigma-Aldrich). Membranes were blocked with 3–7.5% (w/v) bovine serum albumin in Tris-buffered saline containing 0.05% (v/v) Tween 20 (TBST) for 1 h and incubated overnight with the primary antibodies. After incubation, membranes were washed in TBST, incubated for 1 h at room temperature with secondary antibodies (A106PU or A102PT; American Qualex, San Clemente, CA, USA) and washed again in TBST. Chemiluminescent reagents (SuperSignal West Pico Chemiluminescent Substrate; Thermo Fisher Scientific, Waltham, MA, USA) were used to facilitate blot detection. Blots were scanned and quantified using ChemiDoc XRS (170-8071; Bio-Rad, Hercules, CA, USA) and Quantity One (170-9600, version 4.5.2, Windows; Bio-Rad). Blots of 4-hydroxy-2-nonenal (4-HNE) and ubiquitin conjugated were quantified the range between 25–150 kDa.
Primary antibodies for Western blotting
The following primary antibodies were purchased: HSP60 (ADI-SPA-806; Enzo Life Sciences, Tokyo, Japan), HSP72 (ADI-SPA-810-D; Enzo Life Sciences), COX IV (ab14744; Abcam, Cambridge, UK), MitoProfile Total OXPHOS Rodent WB Antibody Cocktail [ubiquinol–cytochrome c reductase core protein II (UQCRC2), COX I, ab110413; Abcam], voltage-dependent anion channel [VDAC, 4661S; Cell Signaling Technology (CST) Japan, Tokyo, Japan], peroxisome proliferator-activated receptor γ coactivator 1-α (PGC-1α, 516557; Millipore, CA, USA), glucose transporter 4 (GLUT4, ab33780; Abcam), fatty acid translocase/cluster of differentiation 36 (FAT/CD36, ab137320; Abcam), Parkin (ab77924; Abcam), ubiquitin (3933; CST), p62/SQSTM1 [PM045; Medical & Biological Laboratories (MBL), Nagoya, Japan], LC3 (M152-3; MBL), mitofusin-2 (Mfn2, ab124773; Abcam), Optic atrophy 1 (Opa1, 612606; BD Transduction Laboratories, Tokyo, Japan), mitochondrial fission 1 (Fis1, ab96764; Abcam), dynamin-related protein 1 (Drp1, ab56788; Abcam), 4-HNE (ab48506; Abcam), ubiquitin-specific-processing protease 30 (USP30, SAB4503385; Sigma-Aldrich), p-AMPK T172 (2535; CST), AMPK (5832; CST), phosphorylated acetyl-CoA carboxylase (p-ACC S79, 3661; CST), ACC (3662; CST), p-Akt S473 (9271; CST), Akt (9272; CST), p-P70S6K T389 (9205; CST), P70S6K (9202; CST), p-ULK1 S555 (5869; CST), p-ULK1 S757 (6888; CST), ULK1 (8054; CST), Atg13 (M183-3MS; MBL), Beclin-1 (PD017MS; MBL), Atg14L (PD026MS; MBL), Atg5–Atg12 conjugate (M153-3MS; MBL) and Atg16L (M150-3MS; MBL).
Statistical analysis
All data are expressed as means ± SEM. Student’s unpaired t test, two-way ANOVA (denervation × heat stress), or three-way ANOVA (denervation × heat stress × colchicine) were performed. If an interaction was observed, the Tukey–Kramer multiple-comparison test was performed. Statistical significance was defined as P < 0.05. A trend was defined as P < 0.10. Statistical analysis was performed using JMP (Version 9.0.1, Macintosh; SAS Institute, Cary, NC, USA).
Results
Validation and profile of experimental model
A single bout of heat stress treatment in this study increased rectal temperature (Fig.1A). In addition, the protein content of HSP60 and HSP72, sensitive biomarkers for mitochondrial and cellular heat shock responses (Naito et al. 2000; Oishi et al. 2002), in the gastrocnemius muscle were dramatically increased after 7 days heat stress treatment (Fig.1B–D). These observations indicate that, in this study, heat stress sufficiently induced physiological and biochemical responses. We also confirmed that daily heat stress treatment did not affect body weight and total energy intake, which potentially initiate cellular adaptations in skeletal muscle (Fig.1E and F). Denervation decreased gastrocnemius muscle mass by 38.2%. In contrast, daily heat stress treatment significantly increased sham-operated and denervated gastrocnemius muscle mass by 6.0 and 15.5%, respectively (Fig.1G). Crucially, this result demonstrates that even if denervation results in inimitable cellular adaptations compared with other muscle disuse models, daily heat stress treatment is also effective on denervation-induced muscle atrophy. Moreover, these data indicate that further investigations on mitochondrial adaptations are appropriate.
Figure 1.
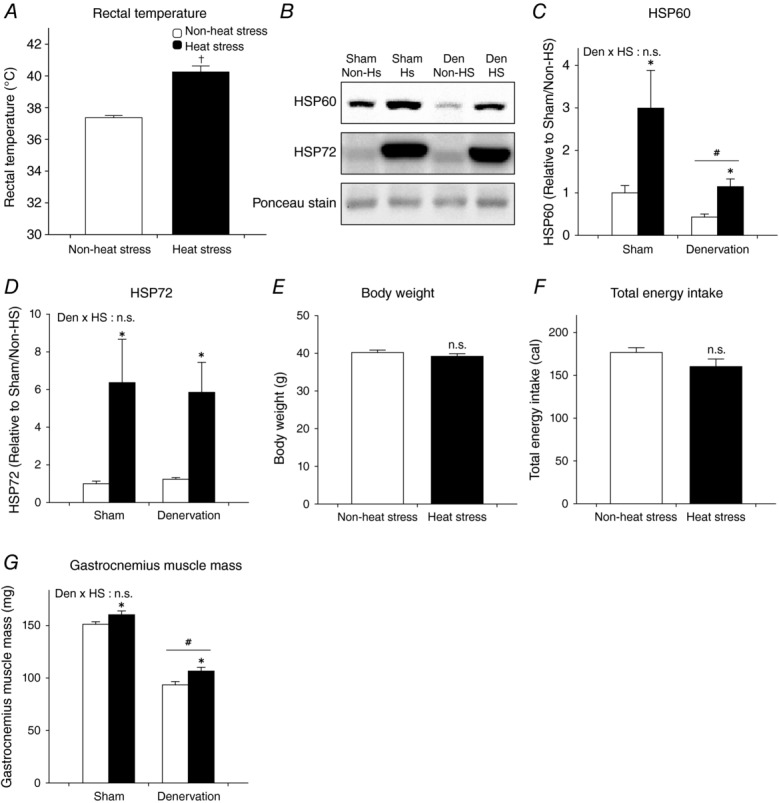
Physiological and biochemical responses/adaptations to heat stress
A, acute response to rectal temperature with a single bout of heat stress treatment. B–G, adaptation of daily heat stress treatment. Open columns, non-heat stress; filled columns, heat stress. Student’s unpaired t test (A, F and G) or two-way ANOVA was performed (C, D and E). †P < 0.05 vs. non-heat stress. #P < 0.05, main effect of denervation. *P < 0.05, main effect of heat stress. Abbreviation: HSP, heat shock protein, n.s., not significant. In this and all subsequent figures, data are shown as the means + SEM.
Daily heat stress treatment rescues loss and oxidative stress of mitochondria
In order to examine mitochondrial adaptations, we first examined the protein expression of mitochondrial enzymes and channels (UQCRC2, COX I, COX IV and VDAC) as biomarkers for mitochondrial content in the gastrocnemius muscle. Denervation dramatically decreased the protein content of these biomarker proteins (Fig.2A–E). However, daily heat stress treatment rescued the loss of these mitochondrial proteins in denervated muscle (Fig.2A–E). We also examined the maximal activity of mitochondrial enzymes (CS and 3-HAD) as biomarkers for maximal oxidative capacity in the gastrocnemius muscle. Like mitochondrial content, mitochondrial enzyme activities were reduced by denervation, but were maintained by heat stress (Fig.2F and G). Dysfunctional mitochondria excessively produce reactive oxygen species (ROS), which accumulate and cause oxidative stress in mitochondria and the cell (Min et al. 2011; Muller et al. 2007). It is therefore regarded that the accumulation of ROS is an index of mitochondrial toxicity (Wei et al. 2009; Chaiswing et al. 2005). Denervation upregulated cellular and mitochondrial 4-HNE-conjugated protein, which is one biomarker of oxidative stress (Fig.2H–J). In contrast, heat stress normalized the upregulation of 4-HNE-conjugated protein in both whole-cell and mitochondrial fractions of denervated muscle (Fig.2H–J).
Figure 2.

Daily heat stress treatment rescued denervation-induced loss of mitochondria
Open columns, non-heat stress; filled columns, heat stress. Two-way ANOVA was performed. #P < 0.05, main effect of denervation. *P < 0.05, main effect of heat stress. †P < 0.05 vs. non-heat stress/sham. Abbreviation: UQCRC2,ubiquinolcytochrome c reductase core protein II; COX, cytochrome c oxidase subunit; VDAC, voltage-dependent anion channel; CS, citrate synthase; 3-HAD, 3-hydroxyacyl-CoA dehydrogenase; 4-hydroxy-2-nonenal; n.s., not significant.
Taken together, these results indicate that daily heat stress treatment rescued denervation-induced loss of mitochondrial content and oxidative capacity in the gastrocnemius muscle, supporting our hypothesis. Importantly, heat stress-rescued mitochondria did not show an unhealthy phenotype.
Daily heat stress treatment does not attenuate loss of PGC-1α
Next, we attempted to clarify the mechanisms underlying the maintenance of mitochondrial content and oxidative capacity by heat stress. Loss of mitochondria due to muscle disuse is mediated by a decrease in PGC-1α (Brault et al. 2010; Cannavino et al. 2014), a regulator of mitochondrial biogenesis and oxidative metabolism (Lin et al. 2002), and can be reversed by genetic and pharmacological PGC-1α overexpression (Sandri et al. 2006; Brault et al. 2010). Therefore, we examined our hypothesis that heat stress could rescue denervation-induced loss of PGC-1α protein. Contrary to our hypothesis, loss of PGC-1α due to denervation was not inhibited by heat stress (Fig.3A and B). In order to validate this observation, we also evaluated the protein content of substrate transporters (glucose, GLUT4 and fatty acid, FAT/CD36) that are other targets of PGC-1α (Benton et al. 2008). Concomitant with the loss of mitochondria and PGC-1α, GLUT4 and FAT/CD36 were also decreased by denervation (Fig.3C and D). However, heat stress did not attenuate the loss of GLUT4 and FAT/CD36 (Fig.3C and E).
Figure 3.

Daily heat stress treatment did not rescue peroxisome proliferator-activated receptor γ coactivator 1-α (PGC-1α) levels
Open columns, non-heat stress; filled columns, heat stress. Two-way ANOVA was performed. #P < 0.05, main effect of denervation. Abbreviation: PGC-1α, peroxisome proliferator-activated receptor γ coactivator 1-α; glucose trans- porter 4; FAT/CD36, fatty acid translocase/cluster of differentiation 36; n.s., not significant.
Importantly, these observations suggest that the countering impacts of heat stress on denervation-induced mitochondrial loss are most likely to be mediated by mitochondrial selective machinery rather than a PGC-1α-associated pathway.
Daily heat stress treatment normalizes mitophagy
Mitochondrial content and oxidative capacity are determined not only by biogenesis, but also by degradation. To control mitochondrial quality, damaged and dysfunctional mitochondria are degraded by mitochondrial selective autophagy (mitophagy). The process of Parkin-mediated mitophagy, the most actively studied mitophagy, involves the following steps.
Mitochondria are tagged for clearance when Parkin, a mitochondrial E3 ubiquitin ligase, is recruited from the cytosol to the membrane of dysfunctional mitochondria, which then ubiquitinates various mitochondrial outer-membrane proteins.
Ubiquitin-conjugated mitochondria are recognized by P62/SQSTM1.
Mitochondria adapted with poly-ubiquitin and P62/SQSTM1 are engulfed by double-membrane vesicles (autophagosomes) consisting of the biomarker protein LC3-II, and are finally degraded by lysosomes.
Recent studies show that denervation continuously activates mitophagy in skeletal muscle (Lokireddy et al. 2012; O’Leary et al. 2013). Therefore, to clarify the mechanisms involving the beneficial effects of heat stress on mitochondria in denervated muscle, we examined whether daily heat stress treatment normalizes denervation-activated mitophagy. As a result, denervation increased mitophagy-related proteins, including Parkin, ubiquitin-conjugated, P62/SQSTM1 and LC3-II, in the mitochondrial fraction (Fig.4A, B, D, E and F), supporting previous reports. However, heat stress attenuated the denervation-induced upregulation of these mitophagy-related proteins without any change in USP30, which was recently identified as mitochondrial de-ubiquitinase in neurons (Bingol et al. 2014; Fig.4A and C).
Figure 4.

Daily heat stress treatment normalized continuously activated mitophagy induction
Open columns, non-heat stress; filled columns, heat stress. Two- or three-way ANOVA was performed. #P < 0.05, main effect of denervation. *P < 0.05, main effect of heat stress. †P < 0.05 vs. non-heat stress/sham. (†)P = 0.05 vs. non-heat stress/sham. §P < 0.05 vs. heat stress/denervation/colchicine. Abbreviation: USP, ubiquitin-specific-processing protease; SQSTM1, sequestosome 1; LC3, microtubule-associated protein 1A/1B-light chain 3; Mfn2, mitofusin-2; Opa1, Optic atrophy 1; Fis1, mitochondrial fission 1; Drp1, dynamin-related protein 1; n.s. not significant.
Although the presence of mitochondrial LC3-II is closely correlated with autophagosome-engulfed mitochondria and is widely used as a biomarker (O’Leary et al. 2013; Webster et al. 2013), the presence of mitochondrial LC3-II does not represent ‘mitophagic flux’. Hence, it remains an important question whether heat stress suppresses induction of mitophagy or accelerates the final degradation of damaged mitochondria. To elucidate this issue, we used colchicine, which is a pharmacological inhibitor of autophagosome degradation (Ju et al. 2010; Mofarrahi et al. 2013). In conditions where autophagosome degradation was inhibited, heat stress treatment attenuated the upregulation of mitochondrial LC3-II by denervation (Fig.4G and H). This observation indicates that heat stress attenuates the induction of mitophagy but does not accelerate the final clearance of damaged mitochondria.
Heat stress does not affect proteins associated with mitochondrial dynamics in denervated muscle
Mitochondria are highly dynamic organelles that undergo fusion and fission dependent on cellular conditions. Mitochondrial quality is tightly controlled by mitophagy and mitochondrial dynamics (Ashrafi et al. 2013; Iqbal et al. 2014). Previous studies show that mitochondrial fission events predate mitophagy (Ashrafi et al. 2013; Iqbal et al. 2014). In order to gain further understanding of the effects of heat stress on mitochondrial quality control, we next evaluated the expression of proteins that regulate mitochondrial dynamics. As a result, denervation decreased mitochondrial fusion-related proteins, such as Mfn2 and Opa1 (Fig.4I–K), but increased mitochondrial fission proteins, such as Fis1 with no change in Drp1 (Fig.4I, L and M). These results demonstrate that denervation shifts mitochondria towards fission.
In sham-operated muscle, heat stress significantly increased Mfn2 and tended to increase Opa1, but did not affect proteins involved in the regulation of mitochondrial fission (Fig.4I–K). Contrary to sham-operated muscle, in denervated muscle there was no significant effect of heat stress on proteins regulating mitochondrial dynamics. Interestingly, these results indicate that heat stress attenuates the induction of mitophagy and that the mitochondrial fission phenotype is sustained.
Heat stress suppresses denervation-activated induction of autophagy
Autophagy is excessively activated in catabolic situations, including denervation, contributing to progressive activation of mitophagy and muscle atrophy (Grumati & Bonald, 2012; Neel et al. 2013; Tyron et al. 2014). In order to gain a better understanding of the normalization of mitophagy induction and attenuation of muscle atrophy by daily heat stress, we examined whether heat stress also attenuates the activation of autophagy induction in denervated muscle. As for mitophagy, heat stress suppressed the increase in LC3-II, both with and without colchicine treatment (Fig.5A–D). These results show that heat stress normalizes denervation-activated induction of autophagy. To identify the molecular mechanism, we also examined the effects of daily heat stress on the expression of key proteins that regulate each step of autophagy induction in denervated muscle. The regulating steps of autophagy induction are often categorized as follows: (i) the initiation step; (ii) the nucleation step; and (iii) the elongation step of autophagosome formation. Denervation significantly upregulated the active (phosphorylated) form and/or total expression of proteins controlling the initiation step (AMPK, AMPK functional readout ACC, ULK1 and Atg13), nucleation step (Atg14L and Beclin 1) and elongation step (Atg5-Atg12 conjugate and Atg16L) of autophagosome formation (Fig.6A–S). In contrast, heat stress significantly attenuated the denervation-induced increase in protein expression regulating the elongation step (Atg5–Atg12 conjugate and Atg16L), but did not change the expression of proteins associated with the initiation step [including the suppressing pathway, Akt/mechanistic (mammalian) target of rapamycin complex 1 (mTORC1) and p-ULK1 Ser757] and the nucleation step of autophagosome formation (Fig.6Q–S). These observations indicate that daily heat stress normalizes autophagy induction via selective suppression of the elongation step of autophagosome formation.
Figure 5.
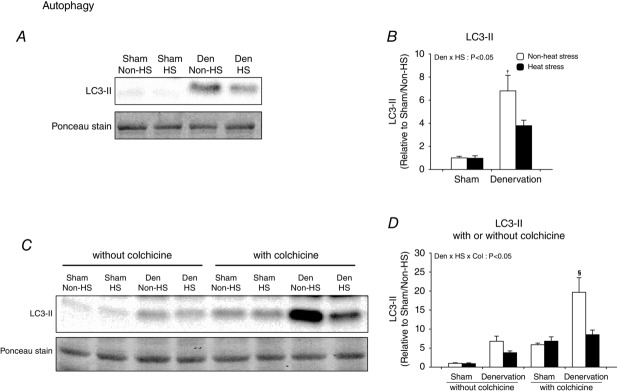
Daily heat stress treatment normalized continuously activated autophagy induction
Open columns, non-heat stress; filled columns, heat stress. Two- or three-way ANOVA was performed. #P < 0.05, main effect of denervation. *P < 0.05, main effect of heat stress. †P < 0.05 vs. non-heat stress/sham. §P < 0.05 vs. heat stress/denervation/colchicine. Abbreviation: LC3, microtubule-associated protein 1A/1B-light chain 3; n.s. not significant.
Figure 6.

Daily heat stress treatment suppressed the increase in proteins regulating the elongation step of autophagosome formation
Open columns, non-heat stress; filled columns, heat stress. #P < 0.05, main effect of denervation. *P < 0.05, main effect of heat stress. (*)P = 0.06 of main effect of heat stress. †P < 0.05 vs. non-heat stress/sham. §P < 0.05 vs. heat stress/denervation/colchicine. Abbreviation: AMPK, AMP-activated protein kinase; ACC, acetyl-CoA carboxylase; ULK1, unc-51-like kinase 1; Atg, autophagy-related gene; n.s. not significant.
Discussion
Key findings and translational significance
Traumatic nerve injury or motor neuron disease leads to denervation and subsequent severe myopathy, representing muscle atrophy (Tang et al. 2014). Unfortunately, to date, no effective therapy is established (Kanning et al. 2010). Here, we focused on heat stress treatment as a potential therapy, because it is a classic, simple and widely applicable therapy. However, the cellular and molecular responses and adaptations in skeletal muscle induced by heat stress treatment are poorly understood.
This study provides evidence that daily heat stress treatment rescues denervation-induced mitochondrial toxicity, such as loss of content, reduction of oxidative capacity, accumulation of oxidative stress and concomitant muscle atrophy. Furthermore, we also found that denervation-activated autophagy-dependent mitochondrial clearance (mitophagy) was suppressed by daily heat stress treatment. The molecular basis of this observation is explained by results showing that heat stress treatment attenuated the increase in key proteins regulating the tagging step for mitochondrial clearance and the elongation step of autophagosome formation in denervated muscle. These observations will contribute to our understanding of mitochondrial quality control in wasting skeletal muscle from a translational perspective and can explain the mechanisms behind the attenuation of muscle wasting by heat stress.
Unique effects of heat stress on mitochondrial quality control machinery
Mitochondrial quality control by mitophagy in skeletal muscle has been actively studied in recent years, because impairment to mitochondrial quality and its machinery can contribute to muscle disorders (Powers et al. 2012; Tryon et al. 2014). One of the major findings in this study is that denervation-activated mitophagic pathways were normalized by heat stress.
A recent study has shown that prolonged oxidative stress initiates further mitochondrial dysfunction, such as decreased membrane potential and impaired respiratory activity, which is followed by induction of mitophagy in skeletal muscle cells (Iqbal et al. 2014). In the present study, heat stress attenuated the denervation-induced accumulation of mitochondrial oxidative stress. Therefore, our results suggest that daily heat stress primarily conserves mitochondrial health, which consequently represses the necessity for mitochondrial clearance. The reason why muscle disuse increases mitochondrial ROS production remains unknown. Dysregulated mitochondrial protein import has been suggested to be one possible cause (Powers et al. 2012). Mitochondrial protein and transcriptional factors encoded by nuclear DNA are translated in the cytosol as precursor proteins. Precursor proteins are transported into mitochondria and refolded by HSP60 and HSP72 (Ljubicic et al. 2010). In the present study, denervation decreased HSP60, but heat stress normalized loss of HSP60 and increased HSP72 in denervated muscle. One could speculate that upregulation of HSP60 and HSP72 by heat stress may attenuate the dysfunction of mitochondrial protein import, folding and subsequent mitochondrial ROS production. This is further supported by a previous study in the hypothalamus demonstrating that decreased HSP60 protein by short hairpin RNA impairs mitochondrial respiration and induces oxidative stress (Kleinridders et al. 2013). Furthermore, a recent study demonstrated that HSP72 works interactively with Parkin as a mitochondrial stress sensor and most probably plays important roles in mitochondrial homeostasis in skeletal muscle (Drew et al. 2014). In contrast, previous studies show that HSP25 and TRAP-1 (a mitochondrial homologue of HSP90) can work against oxidative stress and mitochondrial cell death (Altieri et al. 2012; Lee et al. 2005). However, HSP25 and TRAP-1 were not changed by the heat stress treatment in this study (data not shown), suggesting that some but not all HSPs may be key players for the counter effects of heat stress on denervation-induced mitochondrial dysfunction in skeletal muscle. Therefore, our future research will be focused on the relationship between HSPs and mitochondrial health in muscle atrophying conditions.
A recent study identified that USP30, a mitochondrial de-ubiquitinase, inhibits Parkin-mediated mitophagy in neurons (Bingol et al. 2014). Therefore, USP30 may prevent mitochondrial ubiquitination. This is the first study to investigate USP30 expression with respect to induction of mitophagy in skeletal muscle. We expected that denervation would decrease USP30 expression in contrast to Parkin, because USP30 counteracts the action of Parkin. However, interestingly, denervation increased both Parkin and USP30. The physiological significance of these observations may be to prevent excessive mitochondrial ubiquitination. In contrast, heat stress suppressed the upregulation of Parkin only, suggesting that heat stress favours de-ubiquitination, thereby preventing mitochondrial clearance.
Dysfunctional mitochondria are removed by mitophagy and mitochondrial fission. A previous study demonstrated that continuous mitochondrial fission itself triggers muscle atrophy (Romanello et al. 2010). In agreement with this idea, in the present study, denervation also upregulated proteins related to mitochondrial fission and downregulated proteins related to mitochondrial fusion, concomitantly with induction of mitophagy and muscle atrophy. Interestingly, heat stress normalized mitophagy and attenuated muscle atrophy, but did not affect the protein content associated with the regulation of mitochondrial dynamics in denervated muscle. This is a unique case, in which mitochondrial fission did not always coincide with the status of mitophagy and muscle atrophy.
Daily heat stress also normalizes continuously activated autophagy
Skeletal muscle mass is determined by the net balance between protein synthesis and breakdown. Recently, we and others showed that heat stress treatment acutely activates mTORC1, a cellular signalling cascade associated with protein synthesis, in skeletal muscle (Yoshihara et al. 2013; Tamura et al. 2014). In contrast, the effects of heat stress on pathways related to protein breakdown remain unknown. Recently, researchers have been focusing on the autophagic signalling of proteolysis machineries. Autophagy is sometimes described as a ‘double-edged sword’, because both inhibition of requisite autophagy (Masiero & Sandri, 2014) and overstimulation of autophagy (Risson et al. 2009) can trigger mitochondrial toxicity and muscle wasting (Neel et al. 2013). Excessive autophagy is sometimes induced following physical inactivity, such as denervation (Ju et al. 2010; Lokireddy et al. 2012). Induction of autophagy is critically regulated by over 30 autophagy-related genes. In fact, denervation-activated autophagy increased various proteins associated with autophagy in this study. Interestingly, daily heat stress treatment suppressed denervation-activated induction of autophagy and the upregulation of proteins regulating the elongation step of autophagosome formation (Atg5–Atg12 conjugate and Atg16L). Atg5, Atg12 and Atg16L play a critical role in LC3 lipidation, the conversion of LC3-I to LC3-II and autophagosome formation. Recent studies demonstrate that inhibiting the increase in Atg5, Atg12 or Atg16L by small interfering RNA or short hairpin RNA suppresses the activation of autophagy in various cell types (Han et al. 2013; Wang et al. 2013; Nowag et al. 2014). Therefore, inhibiting the increase in Atg5, Atg12 and Atg16L by daily heat stress could contribute to less autophagy activation and less mitochondrial clearance and muscle atrophy. However, further studies on why daily heat stress attenuates only proteins regulating the elongation step, but not the initiation and nucleation steps of autophagosome formation are required.
Perspectives for translation
To evolve and translate our present understanding, further research is required. In the present study, we were preferentially interested in the mitochondrial content and its regulating machinery; therefore, we have limited functional information on mitochondria and muscle to report. Mitochondria play critical roles in various cellular processes, such as respiration, substrate oxidation, calcium handling, ion transport, ROS production and apoptosis, which determine muscle health and function. In future studies, we consider that multiple measurements and integrative interpretation of mitochondrial function will be very important subjects for improving our understanding the health and function of muscle as a locomotor, metabolic and endocrine organ.
Conclusion
This study provides evidence that daily heat stress treatment rescues denervation-induced mitochondrial toxicity, such as loss of content, reduction of oxidative capacity, accumulation of oxidative stress and concomitant muscle atrophy. Furthermore, we also found that denervation-activated autophagy-dependent mitochondrial clearance (mitophagy) was suppressed by daily heat stress treatment. The molecular basis of this observation is explained by results showing that heat stress treatment attenuated the increase in key proteins regulating the tagging step for mitochondrial clearance and the elongation step of autophagosome formation in denervated muscle.
Glossary
- ACC
acetyl-CoA carboxylase
- AMPK
AMP-activated protein kinase
- Atg
autophagy-related gene
- COX
cytochrome c oxidase subunit
- CS
citrate synthase
- Drp1
dynamin-related protein 1
- FAT/CD36
fatty acid translocase/cluster of differentiation 36
- Fis1
mitochondrial fission 1
- GLUT4
glucose transporter 4
- 3-HAD
3-hydroxyacyl-CoA dehydrogenase
- 4-HNE
4-hydroxy-2-nonenal
- HSP
heat shock protein
- LC3
microtubule-associated protein 1A/1B-light chain 3
- Mfn2
mitofusin-2
- mTORC1
mechanistic (mammalian) target of rapamycin complex 1
- Opa1
Optic atrophy 1
- PGC-1α
peroxisome proliferator-activated receptor γ coactivator 1-α
- ROS
reactive oxygen species
- SQSTM1
sequestosome
- TRAP-1
tumour necrosis factor receptor-associated protein-1
- ULK1
unc-51-like kinase 1
- UQCRC2
ubiquinol–cytochrome c reductase core protein II
- USP30
ubiquitin-specific-processing protease 30
- VDAC
voltage-dependent anion channel
Additional information
Competing interests
None declared.
Author contributions
Y.T. designed the study, performed all experiments, interpreted results and wrote the first draft of the manuscript. Y.K. assisted with mitochondrial isolation experiments and interpreting the results and edited the manuscript. Y.M. assisted with animal experiments and edited the manuscript. D.H. assisted with designing experiments and interpreting results and edited the manuscript. H.H. assisted with designing the study and interpreting results and edited the manuscript. All authors read and approved the final manuscript. All experiments were performed at The University of Tokyo.
Funding
This study was supported by the Grant-in-aid for Japan Society for Promotion of Science (JSPS) Fellows (no. 9688, to Y.T.).
References
- Altieri DC, Stein GS, Lian JB. Languino LR. TRAP-1, the mitochondrial Hsp90. Biochim Biophys Acta. 2012;1823:767–773. doi: 10.1016/j.bbamcr.2011.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashrafi G. Schwarz TL. The pathways of mitophagy for quality control and clearance of mitochondria. Cell Death Differ. 2013;20:31–42. doi: 10.1038/cdd.2012.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bass A, Brdiczka D, Eyer P, Hofer S. Pette D. Metabolic differentiation of distinct muscle types at the level of enzymatic organization. Eur J Biochem. 1969;10:198–206. doi: 10.1111/j.1432-1033.1969.tb00674.x. [DOI] [PubMed] [Google Scholar]
- Benton CR, Nickerson JG, Lally J, Han XX, Holloway GP, Glatz JFC, Luiken JJFP, Graham TE, Heikkila JJ. Bonen A. Modest PGC-1α overexpression in muscle in vivo is sufficient to increase insulin sensitivity and palmitate oxidation in subsarcolemmal, not intermyofibrillar, mitochondria. J Biol Chem. 2008;283:4228–4240. doi: 10.1074/jbc.M704332200. [DOI] [PubMed] [Google Scholar]
- Bingol B, Tea JS, Phu L, Reichelt M, Bakalarski CE, Song Q, Foreman O, Kirkpatrick DS. Sheng M. The mitochondrial deubiquitinase USP30 opposes parkin-mediated mitophagy. Nature. 2014;510:370–375. doi: 10.1038/nature13418. [DOI] [PubMed] [Google Scholar]
- Bongers KS, Fox DK, Ebert SM, Kunkel SD, Dyle MC, Bullard SA, Dierdorff JM. Adams CM. Skeletal muscle denervation causes skeletal muscle atrophy through a pathway that involves both Gadd45a and HDAC4. Am J Physiol Endocrinol Metab. 2013;305:E907–E915. doi: 10.1152/ajpendo.00380.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brault JJ, Jespersen JG. Goldberg AL. Peroxisome proliferator-activated receptor γ coactivator 1α or 1β overexpression inhibits muscle protein degradation, induction of ubiquitin ligases, and disuse atrophy. J Biol Chem. 2010;285:19460–19471. doi: 10.1074/jbc.M110.113092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cannavino J, Brocca L, Sandri M, Bottinelli R. Pellegrino MA. PGC1-α over-expression prevents metabolic alterations and soleus muscle atrophy in hindlimb unloaded mice. J Physiol. 2014;592:4575–4589. doi: 10.1113/jphysiol.2014.275545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castets P, Lin S, Rion N, Di Fulvio S, Romanino K, Guridi M, Frank S, Tintignac LA, Sinnreich M. Rüegg MA. Sustained activation of mTORC1 in skeletal muscle inhibits constitutive and starvation-induced autophagy and causes a severe, late-onset myopathy. Cell Metab. 2013;17:731–744. doi: 10.1016/j.cmet.2013.03.015. [DOI] [PubMed] [Google Scholar]
- Chaiswing L, Cole MP, Ittarat W, Szweda LI, St Clair DK. Oberley TD. Manganese superoxide dismutase and inducible nitric oxide synthase modify early oxidative events in acute adriamycin-induced mitochondrial toxicity. Mol Cancer Ther. 2005;4:1056–1064. doi: 10.1158/1535-7163.MCT-04-0322. [DOI] [PubMed] [Google Scholar]
- Drummond GB. Reporting ethical matters in The Journal of Physiology: standards and advice. J Physiol. 2009;587:713–719. doi: 10.1113/jphysiol.2008.167387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drew BG, Ribas V, Le JA, Henstridge DC, Phun J, Zhou Z, Soleymani T, Daraei P, Sitz D, Vergnes L, Wanagat J, Reue K, Febbraio MA. Hevener AL. HSP72 is a mitochondrial stress sensor critical for Parkin action, oxidative metabolism, and insulin sensitivity in skeletal muscle. Diabetes. 2014;63:1488–1505. doi: 10.2337/db13-0665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furuya N, Ikeda S-I, Sato S, Soma S, Ezaki J, Trejo JAO, Takeda-Ezaki M, Fujimura T, Arikawa-Hirasawa E, Tada N, Komatsu M, Tanaka K, Kominami E, Hattori N. Ueno T. PARK2/Parkin-mediated mitochondrial clearance contributes to proteasome activation during slow-twitch muscle atrophy via NFE2L1 nuclear translocation. Autophagy. 2014;10:50–60. doi: 10.4161/auto.27785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grumati P. Bonaldo P. Autophagy in skeletal muscle homeostasis and in muscular dystrophies. Cells. 2012;1:325–345. doi: 10.3390/cells1030325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han H-E, Kim T-K, Son H-J, Park WJ. Han P-L. Activation of autophagy pathway suppresses the expression of iNOS, IL6 and cell death of LPS-stimulated microglia cells. Biomol Ther (Seoul) 2013;21:21–28. doi: 10.4062/biomolther.2012.089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henstridge DC, Bruce CR, Drew BG, Tory K, Kolonics A, Estevez E, Chung J, Watson N, Gardner T, Lee-Young RS, Connor T, Watt MJ, Carpenter K, Hargreaves M, McGee SL, Hevener AL. Febbraio MA. Activating HSP72 in rodent skeletal muscle increases mitochondrial number and oxidative capacity and decreases insulin resistance. Diabetes. 2014;63:1881–1894. doi: 10.2337/db13-0967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hornberger TA, Hunter RB, Kandarian SC. Esser KA. Regulation of translation factors during hindlimb unloading and denervation of skeletal muscle in rats. Am J Physiol Cell Physiol. 2001;281:C179–C187. doi: 10.1152/ajpcell.2001.281.1.C179. [DOI] [PubMed] [Google Scholar]
- Hoshino D, Yoshida Y, Holloway GP, Lally J, Hatta H. Bonen A. Clenbuterol, a β2-adrenergic agonist, reciprocally alters PGC-1 alpha and RIP140 and reduces fatty acid and pyruvate oxidation in rat skeletal muscle. Am J Physiol Regul Integr Comp Physiol. 2012;302:R373–R384. doi: 10.1152/ajpregu.00183.2011. [DOI] [PubMed] [Google Scholar]
- Iqbal S. Hood DA. Oxidative stress-induced mitochondrial fragmentation and movement in skeletal muscle myoblasts. Am J Physiol Cell Physiol. 2014;306:C1176–C1183. doi: 10.1152/ajpcell.00017.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ju J-S, Varadhachary AS, Miller SE. Weihl CC. Quantitation of “autophagic flux” in mature skeletal muscle. Autophagy. 2010;6:929–935. doi: 10.4161/auto.6.7.12785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanning KC, Kaplan A. Henderson CE. Motor neuron diversity in development and disease. Annu Rev Neurosci. 2010;33:409–440. doi: 10.1146/annurev.neuro.051508.135722. [DOI] [PubMed] [Google Scholar]
- Kitaoka Y, Takahashi Y, Machida M, Takeda K, Takemasa T. Hatta H. Effect of AMPK activation on monocarboxylate transporter (MCT)1 and MCT4 in denervated muscle. J Physiol Sci. 2014;64:59–64. doi: 10.1007/s12576-013-0290-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleinridders A, Lauritzen HPMM, Ussar S, Christensen JH, Mori MA, Bross P. Kahn CR. Leptin regulation of Hsp60 impacts hypothalamic insulin signaling. J Clin Invest. 2013;123:4667–4680. doi: 10.1172/JCI67615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee YJ, Lee DH, Cho CK, Chung HY, Bae S, Jhon GJ, Soh JW, Jeoung DI, Lee SJ. Lee YS. HSP25 inhibits radiation-induced apoptosis through reduction of PKCδ-mediated ROS production. Oncogene. 2005;24:3715–3725. doi: 10.1038/sj.onc.1208440. [DOI] [PubMed] [Google Scholar]
- Leon LR, DuBose DA. Mason CW. Heat stress induces a biphasic thermoregulatory response in mice. Am J Physiol Regul Integr Comp Physiol. 2005;288:R197–R204. doi: 10.1152/ajpregu.00046.2004. [DOI] [PubMed] [Google Scholar]
- Lin J, Wu H, Tarr PT, Zhang C-Y, Wu Z, Boss O, Michael LF, Puigserver P, Isotani E, Olson EN, Lowell BB, Bassel-Duby R. Spiegelman BM. Transcriptional co-activator PGC-1 alpha drives the formation of slow-twitch muscle fibres. Nature. 2002;418:797–801. doi: 10.1038/nature00904. [DOI] [PubMed] [Google Scholar]
- Ljubicic V, Joseph A-M, Saleem A, Uguccioni G, Collu-Marchese M, Lai RYJ, Nguyen LMD. Hood DA. Transcriptional and post-transcriptional regulation of mitochondrial biogenesis in skeletal muscle: Effects of exercise and aging. Biochimica et Biophysica Acta (BBA) - General Subjects. 2010;1800:223–234. doi: 10.1016/j.bbagen.2009.07.031. [DOI] [PubMed] [Google Scholar]
- Lokireddy S, Wijesoma IW, Teng S, Bonala S, Gluckman PD, McFarlane C, Sharma M. Kambadur R. The ubiquitin ligase Mul1 induces mitophagy in skeletal muscle in response to muscle-wasting stimuli. Cell Metab. 2012;16:613–624. doi: 10.1016/j.cmet.2012.10.005. [DOI] [PubMed] [Google Scholar]
- MacDonald EM, Andres-Mateos E, Mejias R, Simmers JL, Mi R, Park J-S, Ying S, Hoke A, Lee S-J. Cohn RD. Denervation atrophy is independent from Akt and mTOR activation and is not rescued by myostatin inhibition. Dis Model Mech. 2014;7:471–481. doi: 10.1242/dmm.014126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machida M, Takeda K, Yokono H, Ikemune S, Taniguchi Y, Kiyosawa H. Takemasa T. Reduction of ribosome biogenesis with activation of the mTOR pathway in denervated atrophic muscle. J Cell Physiol. 2012;227:1569–1576. doi: 10.1002/jcp.22871. [DOI] [PubMed] [Google Scholar]
- Masiero E. Sandri M. Autophagy inhibition induces atrophy and myopathy in adult skeletal muscles. Autophagy. 2014;6:307–309. doi: 10.4161/auto.6.2.11137. [DOI] [PubMed] [Google Scholar]
- Min K, Smuder AJ, Kwon O-S, Kavazis AN, Szeto HH. Powers SK. Mitochondrial-targeted antioxidants protect skeletal muscle against immobilization-induced muscle atrophy. J Appl Physiol. 2011;111:1459–1466. doi: 10.1152/japplphysiol.00591.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mofarrahi M, Guo Y, Haspel JA, Choi AMK, Davis EC, Gouspillou G, Hepple RT, Godin R, Burelle Y. Hussain SNA. Autophagic flux and oxidative capacity of skeletal muscles during acute starvation. Autophagy. 2013;9:1604–1620. doi: 10.4161/auto.25955. [DOI] [PubMed] [Google Scholar]
- Muller FL, Song W, Jang YC, Liu Y, Sabia M, Richardson A. Van Remmen H. Denervation-induced skeletal muscle atrophy is associated with increased mitochondrial ROS production. Am J Physiol Regul Integr Comp Physiol. 2007;293:R1159–R1168. doi: 10.1152/ajpregu.00767.2006. [DOI] [PubMed] [Google Scholar]
- Naito H, Powers SK, Demirel HA, Sugiura T, Dodd SL. Aoki J. Heat stress attenuates skeletal muscle atrophy in hindlimb-unweighted rats. J Appl Physiol. 2000;88:359–363. doi: 10.1152/jappl.2000.88.1.359. [DOI] [PubMed] [Google Scholar]
- Neel BA, Lin Y. Pessin JE. Skeletal muscle autophagy: a new metabolic regulator. Trends Endocrinol Metab. 2013;24:635–643. doi: 10.1016/j.tem.2013.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nowag H, Guhl B, Thriene K, Romao S. Ziegler U. Macroautophagy Proteins Assist Epstein Barr Virus Production and Get Incorporated Into the Virus Particles. EBioMedicine. 2014;1:116–125. doi: 10.1016/j.ebiom.2014.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Leary MF, Vainshtein A, Iqbal S, Ostojic O. Hood DA. Adaptive plasticity of autophagic proteins to denervation in aging skeletal muscle. Am J Physiol Cell Physiol. 2013;304:C422–C430. doi: 10.1152/ajpcell.00240.2012. [DOI] [PubMed] [Google Scholar]
- O’Leary MFN, Vainshtein A, Carter HN, Zhang Y. Hood DA. Denervation-induced mitochondrial dysfunction and autophagy in skeletal muscle of apoptosis-deficient animals. Am J Physiol Cell Physiol. 2012;303:C447–C454. doi: 10.1152/ajpcell.00451.2011. [DOI] [PubMed] [Google Scholar]
- Oishi Y, Taniguchi K, Matsumoto H, Ishihara A, Ohira Y. Roy RR. Muscle type-specific response of HSP60, HSP72, and HSC73 during recovery after elevation of muscle temperature. J Appl Physiol. 2002;92:1097–1103. doi: 10.1152/japplphysiol.00739.2001. [DOI] [PubMed] [Google Scholar]
- Powers SK, Wiggs MP, Duarte JA, Zergeroglu AM. Demirel HA. Mitochondrial signaling contributes to disuse muscle atrophy. Am J Physiol Endocrinol Metab. 2012;303:E31–E39. doi: 10.1152/ajpendo.00609.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Risson V, Mazelin L, Roceri M, Sanchez H, Moncollin V, Corneloup C, Richard-Bulteau H, Vignaud A, Baas D, Defour A, Freyssenet D, Tanti JF, Le-Marchand-Brustel Y, Ferrier B, Conjard-Duplany A, Romanino K, Bauché S, Hantaï D, Mueller M, Kozma SC, Thomas G, Rüegg MA, Ferry A, Pende M, Bigard X, Koulmann N, Schaeffer L. Gangloff YG. Muscle inactivation of mTOR causes metabolic and dystrophin defects leading to severe myopathy. J Cell Biol. 2009;187:859–874. doi: 10.1083/jcb.200903131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romanello V, Guadagnin E, Gomes L, Roder I, Sandri C, Petersen Y, Milan G, Masiero E, Del Piccolo P, Foretz M, Scorrano L, Rudolf R. Sandri M. Mitochondrial fission and remodelling contributes to muscle atrophy. EMBO J. 2010;29:1774–1785. doi: 10.1038/emboj.2010.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Safdar A, Little JP, Stokl AJ, Hettinga BP, Akhtar M. Tarnopolsky MA. Exercise increases mitochondrial PGC-1α content and promotes nuclear-mitochondrial cross-talk to coordinate mitochondrial biogenesis. J Biol Chem. 2011;286:10605–10617. doi: 10.1074/jbc.M110.211466. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Sandri M, Lin J, Handschin C, Yang W, Arany ZP, Lecker SH, Goldberg AL. Spiegelman BM. PGC-1α protects skeletal muscle from atrophy by suppressing FoxO3 action and atrophy-specific gene transcription. Proc Natl Acad Sci USA. 2006;103:16260–16265. doi: 10.1073/pnas.0607795103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srere PA. Methods in Enzymology. 1969. Citrate synthase; pp. 3–5. . In:, ed. Arun KS. Elsevier, Amsterdam. [Google Scholar]
- Talbert EE, Smuder AJ, Min K, Kwon O-S, Szeto HH. Powers SK. Immobilization-induced activation of key proteolytic systems in skeletal muscles is prevented by a mitochondria-targeted antioxidant. J Appl Physiol. 2013;115:529–538. doi: 10.1152/japplphysiol.00471.2013. [DOI] [PubMed] [Google Scholar]
- Tamura Y, Matsunaga Y, Masuda H, Takahashi Y, Takahashi Y, Terada S, Hoshino D. Hatta H. Postexercise whole body heat stress additively enhances endurance training-induced mitochondrial adaptations in mouse skeletal muscle. Am J Physiol Regul Integr Comp Physiol. 2014;307:R931–R943. doi: 10.1152/ajpregu.00525.2013. [DOI] [PubMed] [Google Scholar]
- Tang H, Inoki K, Lee M, Wright E, Khuong A, Khuong A, Sugiarto S, Garner M, Paik J, DePinho RA, Goldman D, Guan K-L. Shrager JB. mTORC1 promotes denervation-induced muscle atrophy through a mechanism involving the activation of FoxO and E3 ubiquitin ligases. Sci Signal. 2014;7:ra18. doi: 10.1126/scisignal.2004809. [DOI] [PubMed] [Google Scholar]
- Tryon L, Vainshtein A, Memme JM. Crilly M. Recent advances in mitochondrial turnover during chronic muscle disuse. Integrative Medicine. 2014;3:161–171. doi: 10.1016/j.imr.2014.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang P, Zhang J, Zhang L, Zhu Z, Fan J, Chen L, Zhuang L, Luo J, Chen H, Liu L, Chen Z. Meng Z. MicroRNA 23b regulates autophagy associated with radioresistance of pancreatic cancer cells. Gastroenterology. 2013;145:1133–1143. doi: 10.1053/j.gastro.2013.07.048. [DOI] [PubMed] [Google Scholar]
- Webster BR, Scott I, Han K, Li JH, Lu Z, Stevens MV, Malide D, Chen Y, Samsel L, Connelly PS, Daniels MP, McCoy JP, Combs CA, Gucek M. Sack MN. Restricted mitochondrial protein acetylation initiates mitochondrial autophagy. J Cell Sci. 2013;126:4843–4849. doi: 10.1242/jcs.131300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wicks KL. Hood DA. Mitochondrial adaptations in denervated muscle: relationship to muscle performance. Am J Physiol. 1991;260:C841–50. doi: 10.1152/ajpcell.1991.260.4.C841. [DOI] [PubMed] [Google Scholar]
- Wei Y, Clark SE, Thyfault JP, Uptergrove GME, Li W, Whaley-Connell AT, Ferrario CM, Sowers JR. Ibdah JA. Oxidative stress-mediated mitochondrial dysfunction contributes to angiotensin II-induced nonalcoholic fatty liver disease in transgenic Ren2 rats. Am J Pathol. 2009;174:1329–1337. doi: 10.2353/ajpath.2009.080697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshihara T, Naito H, Kakigi R, Ichinoseki-Sekine N, Ogura Y, Sugiura T. Katamoto S. Heat stress activates the Akt/mTOR signalling pathway in rat skeletal muscle. Acta Physiol. 2013;207:416–426. doi: 10.1111/apha.12040. [DOI] [PubMed] [Google Scholar]