

Abstract
Traditional pharmacology is defined as the science that deals with drugs and their actions. While small molecule drugs have clear advantages, there are many cases where they have proved to be ineffective, prone to unacceptable side effects, or where due to a particular disease aetiology they cannot possibly be effective. A dominant feature of the small molecule drugs is their single mindedness: they provide either continuous inhibition or continuous activation of the target. Because of that, these drugs tend to engage compensatory mechanisms leading to drug tolerance, drug resistance or, in some cases, sensitization and consequent loss of therapeutic efficacy over time and/or unwanted side effects. Here we discuss new and emerging therapeutic tools and approaches that have potential for treating the majority of disorders for which small molecules are either failing or cannot be developed. These new tools include biologics, such as recombinant hormones and antibodies, as well as approaches involving gene transfer (gene therapy and genome editing) and the introduction of specially designed self-replicating cells. It is clear that no single method is going to be a ‘silver bullet’, but collectively, these novel approaches hold promise for curing practically every disorder.
Tables of Links
| Targets | |
|---|---|
| GPCRsa | Enzymesb |
| CCR5 | GRK6 |
| Dopamine receptors |
| Ligands | |
|---|---|
| Dopamine | Insulin |
| GDP | L-DOPA |
| GTP |
These Tables list key protein targets and ligands in this article which are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Pawson et al., 2014) and are permanently archived in the Concise Guide to PHARMACOLOGY 2013/14 (a,bAlexander et al., 2013a,b).
Introduction
Most (<90%) currently used therapeutic agents are small molecules (Hopkins and Groom, 2002). The most common drug targets are GPCRs and enzymes (Rask-Andersen et al., 2011). Kinases have become major drug discovery targets, particularly for cancer therapy (Eglen and Reisine, 2011; Rask-Andersen et al., 2011; Cohen and Alessi, 2013). Recent estimates suggest that out of <20 000 human proteins, only ∼3000 are ‘druggable’ (can be regulated by small molecules) (Hopkins and Groom, 2002). At least 3000 disease-modifying genes have been identified, out of which 600–1500 were estimated to be druggable (Hopkins and Groom, 2002). Target ‘druggability’, or the probability of successful modulation of the target with small molecule drugs, is usually estimated based on previous experience, that is, on other genes from the same family successfully targeted by drugs. Conventional wisdom considers the protein targets designed to bind small molecules such as a ligand (i.e. receptors) or substrate (i.e. enzymes) as druggable, since synthetic mimics of such molecules with the desired properties can be engineered. Existing drugs target only 435 human proteins, and the rate of introduction of drugs engaging previously unexploited human targets has remained stable at approximately 4 per year for ∼30 years, despite massive investment in drug discovery (Rask-Andersen et al., 2011). Does this mean that nothing can be done about the majority of disease-associated proteins and that most diseases cannot be treated? Luckily, this is not the case. Here we discuss new developments that expand the horizons of medical research and have the potential to correct disease-associated molecular errors that cannot be addressed by small molecules (Figure 1).
Figure 1.
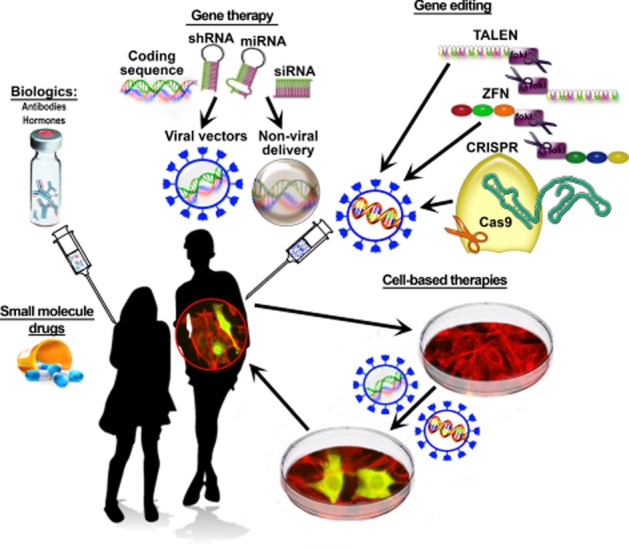
Therapeutic tools and approaches. Schematic illustration of currently used (small molecule drugs and biologics) and emerging novel therapeutic tools by category. Gene therapy includes the delivery of WT and modified genes, as well as constructs designed to reduce the expression of a particular gene (DNA sequences coding for shRNA and miRNA, as well as siRNA that are delivered directly). All constructs can be delivered using the most common virus-based, as well as non-viral, methods. Genome editing changes the actual DNA sequence in the genome. Three types of molecular tools were developed to achieve genome editing: zinc finger nucleases, TALENs, and the CRISPR-Cas system. Cell-based therapies involve the use of living cells for therapeutic purposes. The objective can be either to replace the cells that have degenerated or are missing, such as dopaminergic substantia nigra neurons in Parkinson's disease, or insulin-producing pancreatic beta cells in type I diabetes, or to introduce mammalian or bacterial cells performing a therapeutic task (e.g. producing a necessary compound).
Small molecules will probably remain the best agents against bacteria, fungi and viruses. Small molecule inhibitors targeting enzymes performing functions that mammals do not have, such as building cell walls, or something bacteria-specific, like prokaryotic ribosomes, are good antimicrobials. Inhibitors of reverse transcriptases successfully work against RNA viruses; telomerase, the only eukaryotic reverse transcriptase, is quite different.
The use of small molecule drugs to treat disorders that involve signalling deregulation has one serious drawback: small molecules continuously activate or inhibit signalling proteins, but lack sensitivity to fine regulatory influences. As a result, they are prone to ‘on-target’ side effects (unwanted effects produced by the drug acting on the intended therapeutic target) and compensatory changes, such as drug tolerance, resistance and, in some cases, sensitization. These changes are caused by drug-induced alterations in the signalling pathways that modify the response of the system to the drug. Good examples are tolerance to opioid antinociceptive drugs (Christie, 2008; Dacher and Nugent, 2011), drug-resistant epilepsy (Remy and Beck, 2006) and L-DOPA-induced motor fluctuations in Parkinson's disease (Gurevich and Gurevich, 2010). To circumvent these problems, the drug discovery field is moving away from the ‘small molecule’ mentality of searching for small molecules with broader capabilities. There is a shift to drugs that ‘sense’ the status of the system, providing fine condition-dependent regulation rather than a crude on-off switch. This trend is reflected in the use of partial agonists instead of antagonists and in the effort to develop allosteric modulators for GPCRs (Nickols and Conn, 2014) and enzymes such as kinases (Eglen and Reisine, 2011; Fang et al., 2013). Partial agonists inhibit receptor activity in the presence of a high concentration of the endogenous agonist, but increase the activity when the endogenous agonist is absent or low. Similarly, the action of positive or negative allosteric modulators of GPCRs depends on the presence of the endogenous agonist, thus providing context-dependent regulation. The nascent trend is to develop pathway-biased agonists for GPCRs that engage selective pathways activated by agonist binding (Kenakin and Christopoulos, 2013). Specifically, biased GPCR agonists are being actively developed that activate either G protein-dependent or arrestin-dependent downstream signalling (DeWire and Violin, 2011).
Big molecules – biologics
One limitation of small molecules is inherent in their size: they have few chemical moieties, and therefore can bind with high affinity only to targets with deep pockets, which envelop the small molecule, maximizing interaction energy (Gurevich and Gurevich, 2014). Small molecules cannot effectively target relatively flat protein surfaces and elements lacking a rigid structure. Evolution created antibodies to bind this type of target, so monoclonal antibodies became the next therapeutic agents (Imming et al., 2006; Figure 1). These tools appeared in the late 1980s, with the development of advanced methods of protein engineering and purification (Fischbach et al., 2013). Since the Food and Drug Administration approved the first monoclonal antibody in 1986, the number of antibody drugs and their sales has outpaced the growth of the global drug market (Rask-Andersen et al., 2011). Two issues limit the therapeutic use of antibodies. First, they are proteins, which excludes oral delivery. Second, by their nature, antibodies are extracellular agents, suitable for targeting circulating antibodies, hormones and growth factors, or cell surface receptors, integrins or antigens (Imming et al., 2006). Antibodies are inherently unsuitable for targeting the great majority of disease-associated proteins, which are intracellular.
Another class of biologics, injected hormones and soluble proteins, are not amenable to fine regulation, just like small molecules. In some diseases, they are doing a good job of substituting for a lost hormone (insulin in diabetes type I). Since these are proteins, there is considerable room for improvement. Mutagenesis has been extensively used to increase the stability and pharmacokinetic properties of these biologics, change the duration of action, enhance affinity and selectivity, or reduce toxicity and other unwanted properties (Schmitz et al., 2004; Grunberger, 2013; Parkes et al., 2013).
A special issue with biologics is assessing their pharmacokinetic parameters. Many biologics have been optimized by mutagenesis to achieve better pharmacokinetics, such as a longer half-life. Yet many issues remain unresolved simply because of our limited knowledge of the chemical modifications, disposition and metabolites of biologics in the body (Ezan, 2013; Ezan et al., 2014). The continuous application of novel methods to the pharmacokinetic studies of biologics, such as the use of mass spectrometry to detect the biologic drug, its modified forms, metabolites and targets, will undoubtedly improve our understanding of the pharmacokinetic properties of biologics. One specific issue relates to the use of biologics to treat brain diseases. Since the brain is protected by the blood–brain barrier, most peripherally administered proteins do not reach it. The blood–brain barrier presents an obstacle for small molecule drugs, but some small molecules cross it passively or utilize various transporters on the luminal side, as do some larger molecules, such as hormones, whereas it is essentially impenetrable to other proteins. To introduce biologics into the brain, the ‘Trojan horse’ technology utilizing the receptor-mediated transfer across the blood–brain barrier is employed. This technology relies on the conjugation of the protein drug to monoclonal antibodies that bind transferrin or insulin receptors to cross the barrier (Boado et al., 2010a,b; Pardridge and Boado, 2012).
Gene therapy and genome editing
Many diseases are caused by mutations. Some inactivate encoded proteins, resulting in loss of function. In such cases, the natural approach is to replace the missing protein, restoring the function and curing the disease. Other mutations yield hyperactive proteins: mutations in GPCRs producing constitutively active receptors underlie many disorders including several forms of cancer (Schöneberg et al., 2004). Mutant proteins may become toxic due to misfolding or aggregation. Such mutations are considered gain-of-function, although it is often unclear which function is actually gained. Examples of genetic diseases caused by gain of poorly understood functions include α-synuclein-dependent familial Parkinson's disease and other synucleopathies (Lee and Trojanowski, 2006; Marques and Outeiro, 2012) or leucine-rich repeat kinase 2-dependent familial Parkinson's disease (Esteves et al., 2014). In these cases, providing the good protein without removal of the offending gene or correction of the mutation cannot cure the disease. Historically, the first approach aimed at supplying a functional protein to diseased cells was gene transfer with viral vectors, or gene therapy. Recently, new techniques have been developed to change the genomic sequence, collectively referred to as genome editing (Figure 2).
Figure 2.

Principles and applications of genome-editing techniques. (A) Genome-editing techniques utilize nucleases guided to specific sites by DNA-binding proteins that recognize specific DNA sequences or by RNA to cleave genomic DNA. ZFNs utilize nucleases such as Fok I coupled to zinc finger DNA-binding protein domains, which are used to create ZFNs. The TALEN-based approaches uses TALE domains linked to nucleases to create TALENs. Both systems are effective but require the design of new DNA-binding proteins for each DNA sequence to be targeted. Predicting the structure of zinc finger-binding protein domains specific for target genomic sequences remains challenging, and constructing TALENs is also demanding due to multiple repeat sequences. The newer system based on CRISPR utilizes the prokaryotic immune defense mechanisms. The main advantage of this technique is that the Cas9 nuclease is guided to the DNA locus by RNA, which is much easier to design and produce than a protein. (B) Nucleases introduce a double-stranded break in the target genomic sequence, which is repaired by the cell's DNA repair mechanism. The repair could be done via non-homologous end joining (NHEJ) creating a mismatch in the gene and thus inactivating it. Alternatively, the DNA break could be repaired using an endogenous or exogenous DNA template. In this case, a portion of a defective gene could be replaced via homology-directed repair (HDR) or a new gene could be inserted. (C) The CRISPR system could be employed to regulate the expression of endogenous genes. Deactivated Cas9, lacking nuclease activity, conjugated with a transcription activator could bring the activator to a specific gene locus. Alternatively, a transcription inhibitor could be delivered to a gene via nuclease-dead Cas9 that would block transcription initiation or elongation. For experimental purposes, deactivated Cas9 conjugated with fluorescent proteins such as GFP could be employed to visualize specific gene loci.
Gene therapy
Gene therapy relies on the expression of the protein encoded by the delivered DNA. In clinical trials conducted so far, adenoviral and retroviral vectors have been used most often (Sheridan, 2011). In the experimental work, the most popular vectors are lentiviruses (based on human HIV-1), adenoviruses, adeno-associated viruses (AAVs) or retroviruses (infect only dividing cells). All viruses were rendered self-inactivating and replication-deficient by extensive engineering, reducing the probability of viral infection to virtually zero. Since commercial helper-free systems for AAV production were developed, AAVs became the vectors of choice for gene therapy due to safety: AAVs are not associated with any human disease. Another advantage is that AAVs have many varieties (serotypes) with the ability to preferentially infect different tissues or cell types while bypassing others (tissue tropism). Mutant serotypes with desired tropisms were created (Kwon and Schaffer, 2008; Bartel et al., 2012a), so that even systemic AAV administration can achieve tissue-specific infection.
Treatment of Leber's congenital amaurosis is the best-known gene therapy success story. This blinding disorder is caused by loss-of-function mutations in retinal pigment epithelium-specific 65 kDa protein (RPE65), which lead to a deficit of the 11-cis-retinal necessary for photopigment regeneration. Recently, three clinical trials involving delivery of the functional RPE65 gene to the patient's pigment epithelium were successful, demonstrating the power of gene replacement and the suitability of existing gene delivery methods for clinical use (Bainbridge et al., 2008; Cideciyan et al., 2008; Hauswirth et al., 2008; Maguire et al., 2008). Retinal disorders are the perfect testing ground for gene therapy because the eye is accessible and many ophthalmologists can perform subretinal injections (Colella and Auricchio, 2012; Day et al., 2014; Lopes et al., 2014; Petrs-Silva and Linden, 2014). Attempts are made to use gene therapy to treat various disorders in tissues accessible to circulating viral vectors or injections (Sheridan, 2011; Asokan and Samulski, 2013; Wang et al., 2014). The appeal of gene therapy is so strong that its application is considered for disorders afflicting the most inaccessible organ – the brain (Ramaswamy and Kordower, 2012; Bartus et al., 2013; Gelfand and Kaplitt, 2013; Gray, 2013; Bourdenx et al., 2014).
Recently, the gene therapy toolbox was enriched by virally delivered shRNAs and microRNAs to knockdown disease-associated proteins (Deng et al., 2014; Gurevich et al., 2014). Non-viral methods of delivery of therapeutic siRNAs and microRNAs are actively being developed (Yuan et al., 2011; Gherardini et al., 2014). Despite multiple challenges (off-target effects, immune response, delivery problems), siRNAs are considered very attractive for treatment of such deadly diseases as cancer (Petrocca and Lieberman, 2011) and HIV/AIDS (Zeller and Kumar, 2011), with several clinical trials already underway (Castanotto and Rossi, 2009; Burnett et al., 2011; Petrocca and Lieberman, 2011).
Gene therapy can also increase the expression of normal protein when its function needs to be augmented to compensate for pathological changes elsewhere. Even for enzymes that can potentially be regulated by small molecules, the tools to enhance their activity are often nonexistent or limited. For example, degeneration of neurons in the substantia nigra and the resulting loss of dopamine input to the striatum causes Parkinson's disease (Guigoni et al., 2005). Super-sensitivity of striatal dopamine receptors is believed to underlie dyskinesia, a devastating side effect of the most effective symptomatic therapy, dopamine replacement with its precursor L-DOPA (Guigoni et al., 2005). Since all dopamine receptors are GPCRs, their signalling is regulated via GPCR kinase (GRK)-arrestin-mediated desensitization and via facilitation of signal shutoff at the G protein level by RGS proteins. The ability of wild-type (WT) GRK6 and RGS9-2 to reduce dyskinesia was tested in animals by virus-mediated over-expression. Although GRKs are potentially druggable, few selective inhibitors have been developed so far (Thal et al., 2011), and these target only two isoforms, GRK2 and GRK3, out of the seven we have (Gurevich et al., 2011). The only feasible way to increase the activity of GRKs or RGS proteins is over-expression. Increased expression of both RGS9-2 and GRK6 in the striatum alleviated L-DOPA-induced dyskinesia (Gold et al., 2007; Ahmed et al., 2010). GRK6 was particularly interesting, because it surprisingly preserved and even prolonged the ‘good’ therapeutic effect of L-DOPA in parkinsonian monkeys, while reducing the ‘bad’ dyskinetic effect (Ahmed et al., 2010). This ‘dual’ effect of GRK6 can be explained by its mechanism of action (Carman and Benovic, 1998; Gurevich and Gurevich, 2006). At the beginning, the agonist-activated receptor is not phosphorylated, so that signalling proceeds unimpeded because G protein has an advantage over arrestins. This apparently allows the amount of signalling necessary for the therapeutic effect to go through before the extra GRK6 comes into play. With higher GRK6, receptors are phosphorylated and desensitized faster, cutting off G protein-mediated signalling just when it could become excessive. In contrast, RGS9-2, facilitating self-inactivation of GTP-liganded G protein α-subunit, probably acts from the very beginning, indiscriminately reducing both ‘good’ and ‘bad’ signalling.
This example highlights the advantage of using a protein with its natural fine regulations intact, as opposed to a small molecule activator that would have simply stimulated GRK6 activity. The GRK6 protein used as a therapeutic agent was able to simultaneously combat both dyskinesia caused by sensitization and the loss of L-DOPA therapeutic activity (tolerance) that unite to render the L-DOPA therapy ineffective in late Parkinson's disease (Gurevich and Gurevich, 2010). The danger of using WT proteins for therapy is that virtually every protein is multifunctional. According to the mass action law, increased concentration enhances all interactions the protein is involved in, so over-expression augments all of its functions. For example, GRK6 is not specific for dopamine receptors and can phosphorylate multiple GPCRs and regulate signalling via direct protein–protein interactions (Gurevich et al., 2011). Therefore, if the enhancement of a specific function is needed, the best choice might be a mutant with selected functions disabled or enhanced, rather than WT protein. One example, where an arrestin-1 mutant that binds activated rhodopsin regardless of its phosphorylation was used, is discussed below. The principles of this approach (and its limitations due to our insufficient knowledge) are illustrated in Figure 3.
Figure 3.
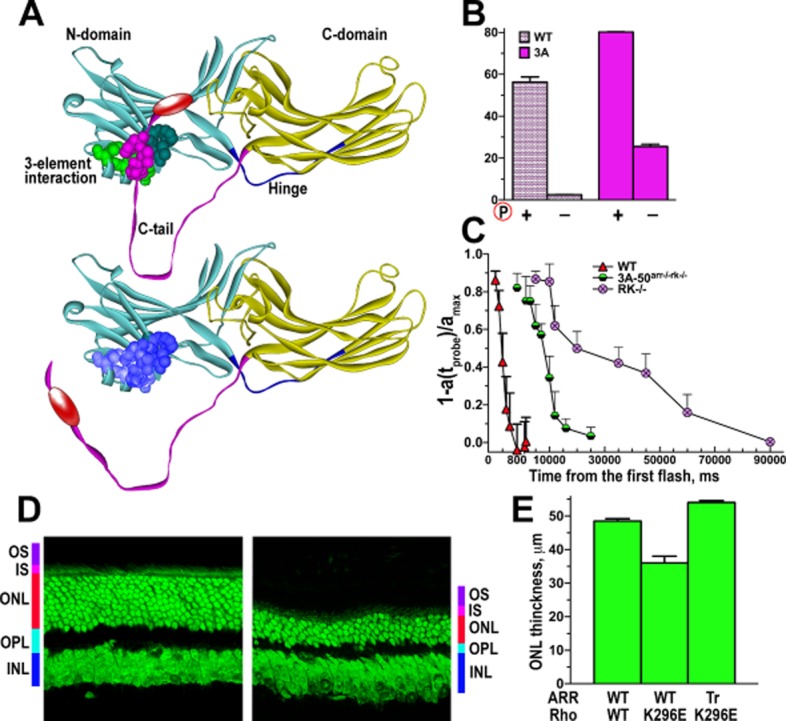
Phosphorylation-independent arrestin-1: expected compensation and unexpected side effects. (A) Mouse arrestin-1 was ‘pre-activated’ by triple alanine substitution of the hydrophobic residues in the C-tail (3A mutation; highlighted in magenta). (B) This mutation detaches the C-tail and dramatically enhances arrestin-1 binding to light-activated unphosphorylated rhodopsin (Rh*). (C) WT arrestin-1 cannot shut off rhodopsin signalling in the absence of rhodopsin kinase (RK−/−), so the time of half-recovery changes from ∼0.4 to ∼19 s. The expression of the arrestin-1-3A mutant instead of WT arrestin-1 in the absence of rhodopsin kinase (3A-50arr−/−rk−/−) accelerates the recovery, reducing the time of half-recovery to ∼6 s (data from Song et al., 2009). (D) However, the 3A mutation also reduced the ability of arrestin-1 to oligomerize, greatly increasing the concentration of free monomer in the animals expressing the arrestin-1-3A mutant. This caused rapid photoreceptor degeneration, which is shown as a reduction in the outer nuclear layer (ONL) containing photoreceptor nuclei (data from Song et al., 2013). OS, outer segments, where rhodopsin and all components of the signalling cascade are localized; IS, inner segments, where photoreceptors rich in mitochondria are localized; ONL, outer nuclear layer, where the nuclei of photoreceptors reside; OPL, outer plexiform layer, where photoreceptor synapses are localized; INL, inner nuclear layer, where the post-photoreceptor neurons are localized. (E) Another source of problems is likely to be the release of the C-tail, which contains the AP2-binding site (shown as red oval in A). Arrestin-1 binds AP2 with ∼30-fold lower affinity than non-visual subtypes. Due to its very high expression in rods, even this low affinity matters: the association of full-length arrestin-1 with the constitutively active rhodopsin-K296E mutant causes photoreceptor death, whereas the replacement of the WT protein with arrestin-1 lacking the C-tail (this deletion pre-activates it similar to the 3A mutation) prevents photoreceptor loss (as reflected in the preservation of ONL thickness) and supports the functionality of photoreceptors expressing rhodopsin-K296E (data from Moaven et al., 2013).
Genome editing
Until recently, there were no methods for correcting the mutations underlying congenital disorders. Three approaches for targeted genome editing have recently emerged (Figure 2). Zinc finger nucleases (ZFNs) were developed first (Bibikova et al., 2003). ZFNs consist of several zinc finger domains, each recognizing a particular three-nucleotide sequence, and the nuclease domain from the Fok I restrictase. Since the Fok I nuclease must dimerize to cleave DNA, monomeric ZFNs bind two adjacent half-sites separated by 5–7 bp, where the Fok I dimer cleaves (Figure 2A). Since WT Fok I can dimerize and cleave DNA when only one ZFN is bound, leading to off-target activity, the dimeric interface of Fok I was modified to make heterodimers obligatory, greatly increasing ZFN specificity (Miller et al., 2007; Szczepek et al., 2007). However, not every sequence can be targeted by ZFNs, and many ZFNs are cytotoxic, probably due to off-target activity (Kim and Kim, 2014).
Transcription activator-like effector nucleases (TALENs) also target DNA and were successfully used to modify genes in many species, including mammals (Joung and Sander, 2013; Sun and Zhao, 2013). Structurally, TALENs are similar to ZFNs: a series of nucleotide-binding domains followed by the Fok I nuclease. Each 33–35 amino acid transcription activator-like effector (TALE) recognizes a single base pair in the major groove. Since TALEs that bind each of the four bases are known, TALENs can target virtually any DNA sequence. The only known limitation is the requirement of thymine at the 5′-end of the target sequence (recognized by the amino-terminal cryptic repeat in TALENs). Since the Fok I nuclease is used, two TALENs recognizing adjacent half-sites are necessary for DNA cleavage (Figure 2A).
The third genome-editing tool is based on the RNA-guided DNA cleavage system that bacteria and archaea use against foreign DNA: clustered regularly interspaced short palindromic repeats (CRISPR) and associated proteins (Cas). The active endonuclease in the CRISPR-Cas system consists of target-specific RNA, target-independent trans-activating RNA and the Cas9 protein (Jiang et al., 2013; Figure 2A). The two RNAs can be combined into a single-chain guide. This endonuclease recognizes a 23 bp target, where 20 bp represent the guide sequence, whereas Cas9 itself recognizes the 3′-trinucleotide. Cas9 proteins from different bacteria have distinct specificities for this three-base motif, which is the only limitation for targeting DNA sequences. CRISPR-Cas9 does not require dimerization, so it can target any 20 bp DNA element (when Cas9 recognizes the downstream three-bp sequence). The key advantage of the CRISPR-Cas9 system is the simplicity of targeting desired sites: Cas9 remains the same, so only the 20 bp guide DNA needs to be cloned into the vector that encodes the single-chain guide (Cho et al., 2013). Thus, the CRISPR-Cas9 system is as versatile as TALENs, easier to construct, but it is potentially less selective because it uses a single targeting DNA sequence, whereas TALENs and ZFNs use two.
Each system has its advantages and problems, and all three have the potential to be further developed and adapted for mammalian in vivo genome editing. They produce double-strand breaks in DNA, but nucleases can be re-engineered to generate single-strand breaks. Genome editing can achieve gene knockout, gene or tag insertion, and gene correction using donor DNA or single-strand oligonucleotides (Figure 2B). Thus, these tools open the possibility of correcting genetic errors. One obvious therapeutic application is genome editing of human pluripotent stem cells for stem cell-based therapy (Hockemeyer et al., 2011; Yusa et al., 2011; Park et al., 2014; Smith et al., 2014). These technologies enable the production of patient-specific ‘designer’ cells with a corrected genome (Soldner et al., 2011; Yusa et al., 2011) for replacement in neurodegenerative and other diseases. Furthermore, ZFNs, TALENs or CRISPR can be employed for in vivo genomic editing in somatic or stem/progenitor cells. One example is anti-HIV therapy, where genome editing was used to excise viral DNA integrated into the genome of latently infected cells (Hu et al., 2014) or disable HIV co-receptor CCR5 (Perez et al., 2008; Holt et al., 2010). Correction of mutations in genetic disorders (Li et al., 2011) and therapeutically beneficial genetic modifications (Ding et al., 2014) are also possible. The problem with in vivo targeting of somatic cells is that it would require efficient gene transfer and genome editing in a large number of cells. Virus-mediated delivery of ZFNs, Cas9/guiding RNAs or TALENs can accomplish that (Li et al., 2011; Ellis et al., 2013; Ding et al., 2014). Sometimes, therapeutic benefits are achieved with a relatively small proportion of cells within the targeted area undergoing genomic rearrangement. One example is when the objective of genetic editing was to reduce the concentration in the blood of a secreted protein by introducing a loss-of-function mutation into a fraction of the cells that secrete it (Ding et al., 2014). In neurodegenerative disorders, the introduction of neuroprotective mutations into a fraction of vulnerable neurons can conceivably also produce a positive functional outcome due to the high compensatory potential of the neural networks.
Although gene therapy and genome editing are designed to achieve similar objectives, they are not entirely interchangeable. When loss-of-function is associated with the loss of protein (deletion, certain nonsense mutations), both methods are likely to be successful. Genome editing with CRISPR can also enhance the activity of endogenous genes using inactive mutant Cas9 and guide RNA for targeting transcription activation elements to specific promoters (Qi et al., 2013; Sander and Joung, 2014). Similarly, catalytically inactive Cas9 targeted to promoter sites represses transcription by blocking transcription initiation or elongation (Maeder et al., 2013; Perez-Pinera et al., 2013; Qi et al., 2013; Sander and Joung, 2014; Figure 2C), which can substitute for siRNA-mediated gene therapy. When the mutant protein is more active than normal, genome editing has an advantage over classic gene therapy. If the mutant protein misfolds, like rhodopsin Pro23His (Olsson et al., 1992), or numerous parkin mutants causing Parkinson's disease (Sriram et al., 2005; Lim et al., 2006), its synthesis must be stopped. Correction or silencing of the damaged gene via genome editing can achieve this, but not gene replacement. Safety is another issue. The expression of WT protein is usually harmless, with the exception of special cases like rod photoreceptors, where the level of over-expression of perfectly normal rhodopsin correlates with cell death (Tan et al., 2001). Thus, the delivery of a normal gene is highly unlikely to cause problems, and its continuous expression after genome integration is desirable. In contrast, DNA editing enzymes cause off-target damage with a certain probability. To minimize unwanted side effects, it is imperative to express these enzymes transiently. This is easy to achieve in proliferating cells, where non-genomic DNA is lost after a few cell divisions. However, in non-dividing neurons, episomes would remain in the cell, driving the expression of gene editing enzymes after their job is already done, thereby increasing the chances of their off-target activity. In these cases, termination of the expression of gene editing proteins must be assured.
Rebalancing signalling with re-engineered proteins
With the exception of infectious diseases and very few others, virtually all human ailments are caused by signalling imbalances (Gurevich and Gurevich, 2012). Signal transduction controlling every aspect of cell behaviour involves numerous protein–protein interactions (Elowitz and Lim, 2010). Disease-associated signalling deregulations could be multifaceted, dynamic, occurring in many cells throughout the body, and often not traceable to specific genetic defects, making straightforward genetic approaches impractical or impossible. Disorders with a genetic component are often caused by an unfortunate combination of polymorphisms in many genes, producing predisposition rather than causing the disease. Furthermore, signalling proteins are not necessarily missing or defective, but certain aspects of their activity in the disease context are either too strong or insufficient. To achieve therapeutic benefits, it is often necessary to dampen the signalling or rebalance it, rather than completely shut down a pathway. So far, signalling abnormalities have been treated with small molecules or biologics with limited success: these tools temporarily alleviate symptoms, rather than treat the disease.
Extracellular and internal signals act by binding specific receptors and changing their interactions with downstream partners. GPCRs are versatile signalling proteins. Humans have more than 1000 different GPCRs (SEVENS database, http://sevens.cbrc.jp/), more than all other types of receptors combined, and GPCRs are targeted by <30% of clinically used drugs (Hopkins and Groom, 2002). GPCR activation by agonists increases their affinity for cognate hetero-trimeric G proteins (Samama et al., 1993). GPCRs facilitate the release of bound GDP and subsequent binding of GTP. This triggers the dissociation of G protein α- and βγ-subunits, whereupon both bind various effectors (Dessauer et al., 1996). GTP-liganded α-subunits of most G proteins bind cognate RGS proteins, which speed up G protein self-inactivation via GTPase activity of their α-subunits (Berman and Gilman, 1998). GRKs preferentially phosphorylate active GPCRs (Gurevich et al., 2011). Arrestins selectively bind active, phosphorylated GPCRs (Gurevich and Gurevich, 2004). The formation of the arrestin-receptor complex stops G protein-activation, facilitates GPCR internalization (Carman and Benovic, 1998), and initiates G protein-independent signalling (Gurevich and Gurevich, 2003; DeWire et al., 2007). This example shows that every step in activation and inactivation of these signalling cascades involves specific protein–protein interactions. GPCR allosteric modulators are actively developed as ‘the next generation’ of GPCR-targeting drugs (Rask-Andersen et al., 2011; Nickols and Conn, 2014). Other potentially druggable proteins in this cascade include enzymes, such as GRKs and G proteins, although at present, there are no marketed drugs engaging these targets. The proteins that act exclusively through protein–protein interactions, like arrestins (Gurevich and Gurevich, 2010), are not considered druggable even theoretically. Yet targeted disruption or enhancement of particular protein–protein interactions is the most straightforward method of compensating for genetic or acquired imbalances in cell signalling. Many proteins, such as GRKs, that have a ‘day job’ as enzymes often ‘moonlight’ as scaffolds (Gurevich et al., 2011). Attempts are being made to modulate GRK/arrestin functions indirectly by designing biased agonists that engage G proteins or arrestins, but not both (DeWire and Violin, 2011; Kenakin, 2012; Wisler et al., 2014).
Protein–protein interactions are fundamental in all signalling processes. When protein complexes are assembled following a specific signalling event, such as receptor activation, their formation can be modulated by small molecules acting at the receptor. The most obvious example is the GPCR signalling cascade, where the formation of multiple signalling protein complexes (active G protein effectors; GPCR – arrestin, etc.) is modulated by orthosteric or allosteric receptor ligands. Allosteric activators of receptor tyrosine kinases seem to be able to induce the receptor dimerization required for full activation (Massa et al., 2010). Many protein–protein interactions involved in cell signalling are not triggered by any specific stimuli, but are governed by the concentrations of the interacting partners. Such protein–protein interactions are virtually never targeted by small molecules for many reasons (Gurevich and Gurevich, 2014), which leaves the proteins involved as the only tools to regulate these interactions. Unfortunately, most proteins have multiple functions. In many pathological conditions, some functions are beneficial, whereas others are harmful. If the elements fulfilling individual functions are known, the protein can be re-engineered by targeted mutations to suppress or enhance certain functions without affecting others. Proteins with specific modifications can serve as therapeutic tools, channeling cell signalling in the desired direction (Gurevich and Gurevich, 2010; 2012). Unfortunately, the knowledge of the inner workings of the proteins required for their targeted re-engineering is rarely available. This approach has not yet been used in human patients. Because of its huge therapeutic potential, we discuss proof-of-concept experiments performed in vivo in animals.
Mutations in GPCRs underlie numerous congenital disorders (Schöneberg et al., 2004). While loss-of-function mutations can be corrected by gene replacement, there are no generally accepted strategies for gain-of-functions mutations causing excessive signalling. When genome editing progresses from experimental science to therapy, such problems could be solved using genomic tools. An alternative idea is compensational gene therapy: push the system closer to normal by reducing the signalling by an overactive GPCR using ‘enhanced’ arrestin that suppresses it more than WT (Song et al., 2009). This approach was tested in rod photoreceptors where rhodopsin and cognate visual arrestin-1 are expressed at very high levels (Pugh and Lamb, 2000; Song et al., 2011). Structure-function studies yielded many ‘pre-activated’ forms of arrestin-1 that bind unphosphorylated light-activated rhodopsin (Gray-Keller et al., 1997; Gurevich, 1998; Vishnivetskiy et al., 1999; 2013a,b). The ability of one of these mutants to compensate for the defect of rhodopsin phosphorylation was tested. The good news is that, in principle, the compensational approach works: the phosphorylation-independent arrestin-1 mutant improved photoreceptor survival and morphology and facilitated shutoff of unphosphorylated light-activated rhodopsin (Song et al., 2009; Figure 3). However, the rate of signalling shutoff in ‘compensated’ rods was much slower than in WT rods with normal rhodopsin phosphorylation, suggesting that more powerful mutants are needed (Song et al., 2009; Vishnivetskiy et al., 2013b). In addition, two unexpected side effects were uncovered. First, WT arrestin-1 robustly self-associates, forming dimers and tetramers (Hanson et al., 2007; 2008a,b), whereas the activating mutation used in this study significantly impeded its self-association (Song et al., 2013; Figure 3). This turned out to be important: high expression of oligomerization-deficient arrestin-1 caused photoreceptor death (Song et al., 2013). Second, arrestin-1 possesses a functional binding site for clathrin adaptor AP2 (Moaven et al., 2013), despite significant divergence between arrestin-1 and non-visual subtypes (Hanson et al., 2006) that bind AP2 with high affinity (Laporte et al., 1999). Even this low-affinity site was biologically relevant: WT arrestin-1 did not prevent degeneration of rods expressing the constitutively active rhodopsin mutant, whereas truncated arrestin-1 lacking the AP2 site protected these rods (Moaven et al., 2013; Figure 3). These findings show that to engineer a therapeutically usable mutant, we need to know every function of the WT protein and modify just the right combination of those.
An obvious disadvantage of proteins as therapeutic tools is their complexity and multifunctionality. We need to know a lot about each protein to re-engineer it for our purposes. However, small molecules have the same problem: a search of the Psychoactive Drug Screening Program Ki database (http://pdsp.med.unc.edu/pdsp.php) with virtually any ‘selective’ small molecule ligand reveals sub-micromolar affinities for several targets. Importantly, these compounds were only tested for competition with known orthosteric ligands of some, but not all GPCRs, and no other proteins. Thus, there is no such thing as a small molecule targeting only one protein. Improved mechanistic understanding of proteins enables re-engineering to retain or enhance certain functions, while reducing or eliminating others. On the positive side, proteins respond to complex regulation, in contrast to small molecules that do not have an on-off switch and act regardless of the state of the body. This makes it less likely that an over-expressed protein, WT or engineered, would cause side effects via excessive interaction with its intended target (on-target side effects). Proteins also offer the option of controlling their target selectivity via specific subcellular localization that could also be artificially controlled by mutagenesis.
Thus, novel protein-based tools that can be used to rebalance cell signalling would be beneficial in many disorders. Re-engineered signalling proteins are also necessary to create special cells with therapeutic capacity.
Cell-based therapies
The idea of supplying lost cells or replacing damaged or diseased ones is not new. Some diseases appear perfect for cell replacement therapy. A good example is Parkinson's disease, where degeneration of a defined group of neurons causes the trouble. Attempts to replace lost nigral neurons with fetal dopaminergic neurons culminated in several clinical studies. The results of two NIH-funded clinical trials in late 1990s were disappointing: in both trials, the patients with grafts did not significantly improve in comparison with sham-operated patients, and some developed dyskinesia (Barker et al., 2013). Later analysis of the results suggested that many potentially confounding variables had been overlooked in the original conclusions, and that some patients actually received long-lasting benefits from the graft (Buttery and Barker, 2014). At that time, there were huge ethical and practical problems associated with the availability of fetal dopaminergic neurons for transplantation. Now dopaminergic neurons can be derived from induced pluripotent stem cells (iPSCs; Ambasudhan et al., 2014). Although there is great enthusiasm for this approach, serious conceptual problems remain: possible uncontrolled proliferation of iPSC-derived neurons, the necessity of tightly controlling their differentiation, and the possibility that the patient-derived cells contain the pathogenic factors that would affect the transplanted cells (Barker et al., 2013). A combination of protein engineering and gene transfer may allow the production of dopaminergic neurons controlled remotely via an expressed ‘designer’ receptor exclusively activated by a ‘designer’ drug (Dell'Anno et al., 2014).
Genetic modifications in patient-derived cells in culture, which are then introduced back into the patient, is often called ex vivo gene therapy, as opposed to in vivo gene therapy when gene transfer is carried out in the living organism. It has been successfully applied to a number of diseases (Hanna et al., 2007; Wang et al., 2012; Filareto et al., 2013). The obvious advantages are the relative ease of genetic manipulations in cultured cells and the use of the patient's own cells to avoid immunological reaction. The disadvantage is that iPSCs are used, which are differentiated and introduced back where they need to integrate for the therapy to succeed. The latter is easier in some tissues than in others.
Another emerging idea is to use specifically engineered microbial or human cells for therapeutic purposes. Cells have obvious advantages: they are much more complex then proteins, respond to numerous inputs, and are capable of sophisticated behaviour (Fischbach et al., 2013). The body uses immune cells to fight invaders and clean up after damage, so it appears natural to use cells for therapy. Moreover, cells self-replicate, so the therapeutic agent can self-perpetuate. However, the ability to reproduce is a double-edged sword. Uncontrolled proliferation of implanted mammalian cells could lead to cancer. Therefore, the life span of therapeutic cells needs to be carefully controlled by limiting cell divisions or inducing cell death after a certain number of divisions, which could be done via engineered signalling mechanisms (Di Stasi et al., 2011). It remains unclear, however, how to achieve effective control over retention of such mechanisms in implanted cells. There is always a probability that some cells would lose these mechanisms and proliferate. The microbiota-based therapies using a culture-derived mixture of bacterial strains introduced via faecal transplants have been successfully employed to treat gastrointestinal infectious diseases (van Nood et al., 2013; Petrof et al., 2013). Future attempts to use engineered bacterial cells to treat a wider range of human diseases probably associated with the microbiota functions (obesity, diabetes type II; Holmes et al., 2012; Kootte et al., 2012), while opening up enormous possibilities, will face the same problem of retention of transformed cells in the implanted cell population.
Future prospects
Each approach has its advantages and limitations. Small molecules are relatively easy to design, screen and optimize for selectivity, favourable pharmacokinetics and bioavailability. Many small molecule drugs can be taken orally, simplifying delivery. Small molecules targeting invader-specific enzymes or ribosomes are likely to remain the best tools against pathogenic microorganisms. Traditionally, GPCRs were targeted by orthosteric agonists and antagonists, which bind in the same site as endogenous ligands. Two new functional modalities of small molecules targeting GPCRs hold promise: allosteric modulators and biased agonists (Kenakin and Miller, 2010; Kenakin, 2010; 2012; Kenakin and Christopoulos, 2013). The action of positive or negative allosteric modulators is contingent on the presence of endogenous ligands, the effects of which they enhance or suppress (Kenakin and Miller, 2010; Kenakin, 2010). These are the only small molecule drugs that ‘listen’ to the body. GPCRs activated by endogenous agonists signal via G proteins and arrestins (DeWire et al., 2007; Kenakin, 2012; Kenakin and Christopoulos, 2013). One of these branches is desirable and the other harmful. Recent findings that some synthetic agonists preferentially activate one of these two branches of signalling (Wisler et al., 2014) pave the way to designing agonists with minimal or no ‘on-target’ side effects. While these new avenues should certainly be explored, small molecules will always remain fairly simple tools suitable for simple tasks. Antibodies, other extracellular proteins and peptides (insulin, other hormones, growth factors) can perform certain tasks that small molecules cannot, but they are essentially just as simple, receiving no feedback from the body.
The delivery of genome-editing enzymes and signalling proteins requires transfection or infection of targeted cells with plasmids or viruses encoding them. Gene delivery methods are being perfected (Bartel et al., 2012b; Nguyen and Szoka, 2012). These methods are more sophisticated than the delivery of small molecules or biologics, but the difficulties are not insurmountable (Bainbridge et al., 2008; Cideciyan et al., 2008; Hauswirth et al., 2008; Maguire et al., 2008). Genome editing has the potential to correct original errors in congenital disorders, whereas gene therapy only compensates for them. Both approaches have the same limitation: the magnitude of the correction will probably depend on the fraction of cells that received the delivered gene. Experiments in animals suggest that correction even in a relatively small fraction of these cells can significantly alleviate the consequences of molecular errors, although more studies are needed to address this issue. It is also important to determine the temporal window of opportunity: to use the retina as an example: after the photoreceptors have disappeared, it is too late to correct their signalling. Hence, this temporal window needs to be elucidated for each disorder.
The delivery of cells engineered for therapeutic purposes appears attractive: these tools are even more sophisticated than proteins and can self-replicate. This is likely to be the greatest problem: there is a non-zero probability of mutation in every cell, and mutations that eliminate the intended beneficial functions will certainly create a selective advantage.
We do not know to what extent each of these new directions will lead to the development of therapeutic tools. It is safe to predict that the combination of several approaches will be necessary for curing various diseases. New tools will not replace old ones, but will be added to the toolbox to increase our therapeutic prowess.
Acknowledgments
Supported in part by NIH grants GM077561 and EY011500 (V. V. G.), NS045117 and NS065868 (E. V. G.).
Glossary
- CRISPR
clustered regularly interspaced short palindromic repeats
- GRK
GPCR kinase
- iPSCs
induced pluripotent stem cells
- TALE
transcription activator-like effector
- TALEN
transcription activator-like effector nuclease
- WT
wild type
- ZFN
zinc finger nuclease
Author contributions
E. V. G. and V. V. G. generated ideas and wrote the manuscript.
Conflict of interest
The authors declare no conflict of interest.
References
- Ahmed MR, Berthet A, Bychkov E, Porras G, Li Q, Bioulac BH, et al. Lentiviral overexpression of GRK6 alleviates L-dopa-induced dyskinesia in experimental Parkinson's disease. Sci Transl Med. 2010;2:28ra28. doi: 10.1126/scitranslmed.3000664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M, et al. The Concise Guide to PHARMACOLOGY 2013/14: G protein-coupled receptors. Br J Pharmacol. 2013a;170:1459–1581. doi: 10.1111/bph.12445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M, et al. The Concise Guide to PHARMACOLOGY 2013/14: Enzymes. Br J Pharmacol. 2013b;170:1797–1867. doi: 10.1111/bph.12451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ambasudhan R, Dolatabadi N, Nutter A, Masliah E, McKercher SR, Lipton SA. Potential for cell therapy in Parkinson's disease using genetically programmed human embryonic stem cell-derived neural progenitor cells. J Comp Neurol. 2014;522:2845–2856. doi: 10.1002/cne.23617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asokan A, Samulski RJ. An emerging adeno-associated viral vector pipeline for cardiac gene therapy. Hum Gene Ther. 2013;24:906–913. doi: 10.1089/hum.2013.2515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bainbridge JW, Smith AJ, Barker SS, Robbie S, Henderson R, Balaggan K, et al. Effect of gene therapy on visual function in Leber's congenital amaurosis. N Engl J Med. 2008;358:2231–2239. doi: 10.1056/NEJMoa0802268. [DOI] [PubMed] [Google Scholar]
- Barker RA, Barrett J, Mason S, Björklund A. Fetal dopaminergic transplantation trials and the future of neural grafting in Parkinson's disease. Lancet Neurol. 2013;12:84–91. doi: 10.1016/S1474-4422(12)70295-8. [DOI] [PubMed] [Google Scholar]
- Bartel MA, Weinstein JR, Schaffer DV. Directed evolution of novel adeno-associated viruses for therapeutic gene delivery. Gene Ther. 2012a;19:694–700. doi: 10.1038/gt.2012.20. [DOI] [PubMed] [Google Scholar]
- Bartel MA, Weinstein JR, Schaffer DV. Directed evolution of novel adeno-associated viruses for therapeutic gene delivery. Gene Ther. 2012b;19:694–700. doi: 10.1038/gt.2012.20. [DOI] [PubMed] [Google Scholar]
- Bartus RT, Baumann TL, Brown L, Kruegel BR, Ostrove JM, Herzog CD. Advancing neurotrophic factors as treatments for age-related neurodegenerative diseases: developing and demonstrating ‘clinical proof-of-concept’ for AAV-neurturin (CERE-120) in Parkinson's disease. Neurobiol Aging. 2013;34:35–61. doi: 10.1016/j.neurobiolaging.2012.07.018. [DOI] [PubMed] [Google Scholar]
- Berman DM, Gilman AG. Mammalian RGS proteins: barbarians at the gate. J Biol Chem. 1998;273:1269–1272. doi: 10.1074/jbc.273.3.1269. [DOI] [PubMed] [Google Scholar]
- Bibikova M, Beumer K, Trautman JK, Carroll D. Enhancing gene targeting with designed zinc finger nucleases. Science. 2003;300:764. doi: 10.1126/science.1079512. [DOI] [PubMed] [Google Scholar]
- Boado RJ, Hui EK, Lu JZ, Pardridge WM. Drug targeting of erythropoietin across the primate blood-brain barrier with an IgG molecular Trojan horse. J Pharmacol Exp Ther. 2010a;333:961–969. doi: 10.1124/jpet.109.165092. [DOI] [PubMed] [Google Scholar]
- Boado RJ, Hui EK, Lu JZ, Zhou QH, Pardridge WM. Selective targeting of a TNFR decoy receptor pharmaceutical to the primate brain as a receptor-specific IgG fusion protein. J Biotechnol. 2010b;146:84–91. doi: 10.1016/j.jbiotec.2010.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourdenx M, Dutheil N, Bezard E, Dehay B. Systemic gene delivery to the central nervous system using Adeno-associated virus. Front Mol Neurosci. 2014;7:50. doi: 10.3389/fnmol.2014.00050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burnett JC, Rossi JJ, Tiemann K. Current progress of siRNA/shRNA therapeutics in clinical trials. Biotechnol J. 2011;6:1130–1146. doi: 10.1002/biot.201100054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buttery PC, Barker RA. Treating Parkinson's disease in the 21st century: can stem cell transplantation compete? J Comp Neurol. 2014;522:2802–2816. doi: 10.1002/cne.23577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carman CV, Benovic JL. G-protein-coupled receptors: turn-ons and turn-offs. Curr Opin Neurobiol. 1998;8:335–344. doi: 10.1016/s0959-4388(98)80058-5. [DOI] [PubMed] [Google Scholar]
- Castanotto D, Rossi JJ. The promises and pitfalls of RNA-interference-based therapeutics. Nature. 2009;457:426–433. doi: 10.1038/nature07758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho SW, Kim S, Kim JM, Kim JS. Targeted genome engineering in human cells with the Cas9 RNA-guided endonuclease. Nat Biotechnol. 2013;31:230–232. doi: 10.1038/nbt.2507. [DOI] [PubMed] [Google Scholar]
- Christie MJ. Cellular neuroadaptations to chronic opioids: tolerance, withdrawal and addiction. Br J Pharmacol. 2008;154:384–396. doi: 10.1038/bjp.2008.100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cideciyan AV, Aleman TS, Boye SL, Schwartz SB, Kaushal S, Roman AJ, et al. Human gene therapy for RPE65 isomerase deficiency activates the retinoid cycle of vision but with slow rod kinetics. Proc Natl Acad Sci U S A. 2008;105:15112–15117. doi: 10.1073/pnas.0807027105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen P, Alessi DR. Kinase drug discovery – what's next in the field? ACS Chem Biol. 2013;8:96–104. doi: 10.1021/cb300610s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colella P, Auricchio A. Gene therapy of inherited retinopathies: a long and successful road from viral vectors to patients. Hum Gene Ther. 2012;23:796–807. doi: 10.1089/hum.2012.123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dacher M, Nugent FS. Opiates and plasticity. Neuropharmacology. 2011;61:1088–1096. doi: 10.1016/j.neuropharm.2011.01.028. [DOI] [PubMed] [Google Scholar]
- Day TP, Byrne LC, Schaffer DV, Flannery JG. Advances in AAV vector development for gene therapy in the retina. Adv Exp Med Biol. 2014;801:687–693. doi: 10.1007/978-1-4614-3209-8_86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dell'Anno MT, Caiazzo M, Leo D, Dvoretskova E, Medrihan L, Colasante G, et al. Remote control of induced dopaminergic neurons in Parkinsonian rats. J Clin Invest. 2014;124:3215–3229. doi: 10.1172/JCI74664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng Y, Wang CC, Choy KW, Du Q, Chen J, Wang Q, et al. Therapeutic potentials of gene silencing by RNA interference: principles, challenges, and new strategies. Gene. 2014;538:217–227. doi: 10.1016/j.gene.2013.12.019. [DOI] [PubMed] [Google Scholar]
- Dessauer CW, Posner BA, Gilman AG. Visualizing signal transduction: receptors, G-proteins, and adenylate cyclases. Clin Sci (Lond) 1996;91:527–537. doi: 10.1042/cs0910527. [DOI] [PubMed] [Google Scholar]
- DeWire SM, Violin JD. Biased ligands for better cardiovascular drugs: dissecting G-protein-coupled receptor pharmacology. Circ Res. 2011;109:205–216. doi: 10.1161/CIRCRESAHA.110.231308. [DOI] [PubMed] [Google Scholar]
- DeWire SM, Ahn S, Lefkowitz RJ, Shenoy SK. Beta-arrestins and cell signaling. Annu Rev Physiol. 2007;69:483–510. doi: 10.1146/annurev.physiol.69.022405.154749. [DOI] [PubMed] [Google Scholar]
- Di Stasi A, Tey SK, Dotti G, Fujita Y, Kennedy-Nasser A, Martinez C, et al. Inducible apoptosis as a safety switch for adoptive cell therapy. N Engl J Med. 2011;365:1673–1683. doi: 10.1056/NEJMoa1106152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding Q, Strong A, Patel KM, Ng SL, Gosis BS, Regan SN, et al. Permanent alteration of PCSK9 with in vivo CRISPR-Cas9 genome editing. Circ Res. 2014;115:488–492. doi: 10.1161/CIRCRESAHA.115.304351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eglen R, Reisine T. Drug discovery and the human kinome: recent trends. Pharmacol Ther. 2011;130:144–156. doi: 10.1016/j.pharmthera.2011.01.007. [DOI] [PubMed] [Google Scholar]
- Ellis BL, Hirsch ML, Porter SN, Samulski RJ, Porteus MH. Zinc-finger nuclease-mediated gene correction using single AAV vector transduction and enhancement by Food and Drug Administration-approved drugs. Gene Ther. 2013;20:35–42. doi: 10.1038/gt.2011.211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elowitz M, Lim WA. Build life to understand it. Nature. 2010;468:889–890. doi: 10.1038/468889a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esteves AR, Swerdlow RH, Cardoso SM. LRRK2, a puzzling protein: insights into Parkinson's disease pathogenesis. Exp Neurol. 2014;261:206–216. doi: 10.1016/j.expneurol.2014.05.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ezan E. Pharmacokinetic studies of protein drugs: past, present and future. Adv Drug Deliv Rev. 2013;65:1065–1073. doi: 10.1016/j.addr.2013.03.007. [DOI] [PubMed] [Google Scholar]
- Ezan E, Becher F, Fenaille F. Assessment of the metabolism of therapeutic proteins and antibodies. Expert Opin Drug Metab Toxicol. 2014;10:1079–1091. doi: 10.1517/17425255.2014.925878. [DOI] [PubMed] [Google Scholar]
- Fang Z, Grütter C, Rauh D. Strategies for the selective regulation of kinases with allosteric modulators: exploiting exclusive structural features. ACS Chem Biol. 2013;8:58–70. doi: 10.1021/cb300663j. [DOI] [PubMed] [Google Scholar]
- Filareto A, Parker S, Darabi R, Borges L, Iacovino M, Schaaf T, et al. An ex vivo gene therapy approach to treat muscular dystrophy using inducible pluripotent stem cells. Nat Commun. 2013;4:1549. doi: 10.1038/ncomms2550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischbach MA, Bluestone JA, Lim WA. Cell-based therapeutics: the next pillar of medicine. Sci Transl Med. 2013;5:179ps177. doi: 10.1126/scitranslmed.3005568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gelfand Y, Kaplitt MG. Gene therapy for psychiatric disorders. World Neurosurg. 2013;80:e11–e18. doi: 10.1016/j.wneu.2012.12.028. [DOI] [PubMed] [Google Scholar]
- Gherardini L, Bardi G, Gennaro M, Pizzorusso T. Novel siRNA delivery strategy: a new ‘strand’ in CNS translational medicine? Cell Mol Life Sci. 2014;71:1–20. doi: 10.1007/s00018-013-1310-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gold SJ, Hoang CV, Potts BW, Porras G, Pioli E, Kim KW, et al. RGS9-2 negatively modulates L-3,4-dihydroxyphenylalanine-induced dyskinesia in experimental Parkinson's disease. J Neurosci. 2007;27:14338–14348. doi: 10.1523/JNEUROSCI.4223-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray SJ. Gene therapy and neurodevelopmental disorders. Neuropharmacology. 2013;68:136–142. doi: 10.1016/j.neuropharm.2012.06.024. [DOI] [PubMed] [Google Scholar]
- Gray-Keller MP, Detwiler PB, Benovic JL, Gurevich VV. Arrestin with a single amino acid substitution quenches light-activated rhodopsin in a phosphorylation independent fashion. Biochemistry. 1997;36:7058–7063. doi: 10.1021/bi963110k. [DOI] [PubMed] [Google Scholar]
- Grunberger G. The need for better insulin therapy. Diabetes Obes Metab. 2013;15(Suppl. 1):1–5. doi: 10.1111/dom.12061. [DOI] [PubMed] [Google Scholar]
- Guigoni C, Aubert I, Li Q, Gurevich VV, Benovic JL, Ferry S, et al. Pathogenesis of levodopa-induced dyskinesia: focus on D1 and D3 dopamine receptors. Parkinsonism Relat Disord. 2005;11:S25–S29. doi: 10.1016/j.parkreldis.2004.11.005. [DOI] [PubMed] [Google Scholar]
- Dopamine receptors and the treatment of Parkinson's disease. In: Gurevich EV, Gurevich VV, editors; Neve K, editor. Dopamine Receptors. New York: Humana Press; 2010. pp. 525–584. In: (ed.) [Google Scholar]
- Gurevich EV, Gurevich VV. Therapeutic potential of small molecules and engineered proteins. Handb Exp Pharmacol. 2014;219:1–12. doi: 10.1007/978-3-642-41199-1_1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurevich EV, Tesmer JJ, Mushegian A, Gurevich VV. G protein-coupled receptor kinases: more than just kinases and not only for GPCRs. Pharmacol Ther. 2011;133:40–69. doi: 10.1016/j.pharmthera.2011.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurevich EV, Ahmed MR, Carl YT. In vivo gene silencing by virally delivered microRNA. In: Brambilla R, editor. Viral Vector Approaches in Neurobiology and Brain Diseases. New York: Humana Press; 2014. pp. 245–268. In: (ed.). [Google Scholar]
- Gurevich VV. The selectivity of visual arrestin for light-activated phosphorhodopsin is controlled by multiple nonredundant mechanisms. J Biol Chem. 1998;273:15501–15506. doi: 10.1074/jbc.273.25.15501. [DOI] [PubMed] [Google Scholar]
- Gurevich VV, Gurevich EV. The new face of active receptor bound arrestin attracts new partners. Structure. 2003;11:1037–1042. doi: 10.1016/s0969-2126(03)00184-9. [DOI] [PubMed] [Google Scholar]
- Gurevich VV, Gurevich EV. The molecular acrobatics of arrestin activation. Trends Pharmacol Sci. 2004;25:105–111. doi: 10.1016/j.tips.2003.12.008. [DOI] [PubMed] [Google Scholar]
- Gurevich VV, Gurevich EV. The structural basis of arrestin-mediated regulation of G protein-coupled receptors. Pharmacol Ther. 2006;110:465–502. doi: 10.1016/j.pharmthera.2005.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurevich VV, Gurevich EV. Custom-designed proteins as novel therapeutic tools? The case of arrestins. Expert Rev Mol Med. 2010;12:e13. doi: 10.1017/S1462399410001444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurevich VV, Gurevich EV. Synthetic biology with surgical precision: targeted reengineering of signaling proteins. Cell Signal. 2012;24:1899–1908. doi: 10.1016/j.cellsig.2012.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanna J, Wernig M, Markoulaki S, Sun CW, Meissner A, Cassady JP, et al. Treatment of sickle cell anemia mouse model with iPS cells generated from autologous skin. Science. 2007;318:1920–1923. doi: 10.1126/science.1152092. [DOI] [PubMed] [Google Scholar]
- Hanson SM, Francis DJ, Vishnivetskiy SA, Klug CS, Gurevich VV. Visual arrestin binding to microtubules involves a distinct conformational change. J Biol Chem. 2006;281:9765–9772. doi: 10.1074/jbc.M510738200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanson SM, Van Eps N, Francis DJ, Altenbach C, Vishnivetskiy SA, Arshavsky VY, et al. Structure and function of the visual arrestin oligomer. EMBO J. 2007;26:1726–1736. doi: 10.1038/sj.emboj.7601614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanson SM, Vishnivetskiy SA, Hubbell WL, Gurevich VV. Opposing effects of inositol hexakisphosphate on rod arrestin and arrestin2 self-association. Biochemistry. 2008a;47:1070–1075. doi: 10.1021/bi7021359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanson SM, Dawson ES, Francis DJ, Van Eps N, Klug CS, Hubbell WL, et al. A model for the solution structure of the rod arrestin tetramer. Structure. 2008b;16:924–934. doi: 10.1016/j.str.2008.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hauswirth WW, Aleman TS, Kaushal S, Cideciyan AV, Schwartz SB, Wang L, et al. Treatment of leber congenital amaurosis due to RPE65 mutations by ocular subretinal injection of adeno-associated virus gene vector: short-term results of a phase I trial. Hum Gene Ther. 2008;19:979–990. doi: 10.1089/hum.2008.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hockemeyer D, Wang H, Kiani S, Lai CS, Gao Q, Cassady JP, et al. Genetic engineering of human pluripotent cells using TALE nucleases. Nat Biotechnol. 2011;29:731–734. doi: 10.1038/nbt.1927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmes E, Kinross J, Gibson GR, Burcelin R, Jia W, Pettersson S, et al. Therapeutic modulation of microbiota-host metabolic interactions. Sci Transl Med. 2012;4:137rv136. doi: 10.1126/scitranslmed.3004244. [DOI] [PubMed] [Google Scholar]
- Holt N, Wang J, Kim K, Friedman G, Wang X, Taupin V, et al. Human hematopoietic stem/progenitor cells modified by zinc-finger nucleases targeted to CCR5 control HIV-1 in vivo. Nat Biotechnol. 2010;28:839–847. doi: 10.1038/nbt.1663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hopkins AL, Groom CR. The druggable genome. Nat Rev Drug Discov. 2002;1:727–730. doi: 10.1038/nrd892. [DOI] [PubMed] [Google Scholar]
- Hu W, Kaminski R, Yang F, Zhang Y, Cosentino L, Li F, et al. RNA-directed gene editing specifically eradicates latent and prevents new HIV-1 infection. Proc Natl Acad Sci U S A. 2014;111:11461–11466. doi: 10.1073/pnas.1405186111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imming P, Sinning C, Meyer A. Drugs, their targets, and the nature and number of drug targets. Nat Rev Drug Discov. 2006;5:821–834. doi: 10.1038/nrd2132. [DOI] [PubMed] [Google Scholar]
- Jiang W, Bikard D, Cox D, Zhang F, Marraffini LA. RNA-guided editing of bacterial genomes using CRISPR–Cas systems. Nat Biotechnol. 2013;31:233–239. doi: 10.1038/nbt.2508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joung JK, Sander JD. TALENs: a widely applicable technology for targeted genome editing. Nat Rev Mol Cell Biol. 2013;14:49–55. doi: 10.1038/nrm3486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenakin T, Christopoulos A. Signalling bias in new drug discovery: detection, quantification and therapeutic impact. Nat Rev Drug Discov. 2013;12:205–216. doi: 10.1038/nrd3954. [DOI] [PubMed] [Google Scholar]
- Kenakin T, Miller LJ. Seven transmembrane receptors as shapeshifting proteins: the impact of allosteric modulation and functional selectivity on new drug discovery. Pharmacol Rev. 2010;62:265–304. doi: 10.1124/pr.108.000992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenakin TP. Ligand detection in the allosteric world. J Biomol Screen. 2010;15:119–130. doi: 10.1177/1087057109357789. [DOI] [PubMed] [Google Scholar]
- Kenakin TP. Biased signalling and allosteric machines: new vistas and challenges for drug discovery. Br J Pharmacol. 2012;165:1659–1669. doi: 10.1111/j.1476-5381.2011.01749.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H, Kim JS. A guide to genome engineering with programmable nucleases. Nat Rev Genet. 2014;15:321–334. doi: 10.1038/nrg3686. [DOI] [PubMed] [Google Scholar]
- Kootte RS, Vrieze A, Holleman F, Dallinga-Thie GM, Zoetendal EG, de Vos WM, et al. The therapeutic potential of manipulating gut microbiota in obesity and type 2 diabetes mellitus. Diabetes Obes Metab. 2012;14:112–120. doi: 10.1111/j.1463-1326.2011.01483.x. [DOI] [PubMed] [Google Scholar]
- Kwon I, Schaffer DV. Designer gene delivery vectors: molecular engineering and evolution of adeno-associated viral vectors for enhanced gene transfer. Pharm Res. 2008;25:489–499. doi: 10.1007/s11095-007-9431-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laporte SA, Oakley RH, Zhang J, Holt JA, Ferguson SG, Caron MG, et al. The 2-adrenergic receptor/arrestin complex recruits the clathrin adaptor AP-2 during endocytosis. Proc Natl Acad Sci U S A. 1999;96:3712–3717. doi: 10.1073/pnas.96.7.3712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee VM, Trojanowski JQ. Mechanisms of Parkinson's disease linked to pathological alpha-synuclein: new targets for drug discovery. Neuron. 2006;52:33–38. doi: 10.1016/j.neuron.2006.09.026. [DOI] [PubMed] [Google Scholar]
- Li H, Haurigot V, Doyon Y, Li T, Wong SY, Bhagwat AS, et al. In vivo genome editing restores haemostasis in a mouse model of haemophilia. Nature. 2011;475:217–221. doi: 10.1038/nature10177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim KL, Dawson VL, Dawson TM. Parkin-mediated lysine 63-linked polyubiquitination: a link to protein inclusions formation in Parkinson's and other conformational diseases? Neurobiol Aging. 2006;27:524–529. doi: 10.1016/j.neurobiolaging.2005.07.023. [DOI] [PubMed] [Google Scholar]
- Lopes VS, Diemer T, Williams DS. Assessment of different virus-mediated approaches for retinal gene therapy of Usher 1B. Adv Exp Med Biol. 2014;801:725–731. doi: 10.1007/978-1-4614-3209-8_91. [DOI] [PubMed] [Google Scholar]
- Maeder ML, Linder SJ, Cascio VM, Fu Y, Ho QH, Joung JK. CRISPR RNA-guided activation of endogenous human genes. Nat Methods. 2013;10:977–979. doi: 10.1038/nmeth.2598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maguire AM, Simonelli F, Pierce EA, Pugh ENJ, Mingozzi F, Bennicelli J, et al. Safety and efficacy of gene transfer for Leber's congenital amaurosis. N Engl J Med. 2008;358:2240–2248. doi: 10.1056/NEJMoa0802315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marques O, Outeiro TF. Alpha-synuclein: from secretion to dysfunction and death. Cell Death Dis. 2012;3:e350. doi: 10.1038/cddis.2012.94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massa SM, Yang T, Xie Y, Shi J, Bilgen M, Joyce JN, et al. Small molecule BDNF mimetics activate TrkB signaling and prevent neuronal degeneration in rodents. J Clin Invest. 2010;120:1774–1785. doi: 10.1172/JCI41356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller JC, Holmes MC, Wang J, Guschin DY, Lee YL, Rupniewski I, et al. An improved zinc-finger nuclease architecture for highly specific genome editing. Nat Biotechnol. 2007;25:778–785. doi: 10.1038/nbt1319. [DOI] [PubMed] [Google Scholar]
- Moaven H, Koike Y, Jao CC, Gurevich VV, Langen R, Chen J. Visual arrestin interaction with clathrin adaptor AP-2 regulates photoreceptor survival in the vertebrate retina. Proc Natl Acad Sci U S A. 2013;110:9463–9468. doi: 10.1073/pnas.1301126110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen J, Szoka FC. Nucleic acid delivery: the missing pieces of the puzzle? Acc Chem Res. 2012;45:1153–1162. doi: 10.1021/ar3000162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nickols HH, Conn PJ. Development of allosteric modulators of GPCRs for treatment of CNS disorders. Neurobiol Dis. 2014;61:55–71. doi: 10.1016/j.nbd.2013.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Nood E, Vrieze A, Nieuwdorp M, Fuentes S, Zoetendal EG, de Vos WM, et al. Duodenal infusion of donor feces for recurrent Clostridium difficile. N Engl J Med. 2013;368:407–415. doi: 10.1056/NEJMoa1205037. [DOI] [PubMed] [Google Scholar]
- Olsson JE, Gordon JW, Pawlyk BS, Roof D, Hayes A, Molday RS, et al. Transgenic mice with a rhodopsin mutation (Pro23His): a mouse model of autosomal dominant retinitis pigmentosa. Neuron. 1992;9:815–830. doi: 10.1016/0896-6273(92)90236-7. [DOI] [PubMed] [Google Scholar]
- Pardridge WM, Boado RJ. Reengineering biopharmaceuticals for targeted delivery across the blood–brain barrier. Methods Enzymol. 2012;503:269–292. doi: 10.1016/B978-0-12-396962-0.00011-2. [DOI] [PubMed] [Google Scholar]
- Park CY, Kim J, Kweon J, Son JS, Lee JS, Yoo JE, et al. Targeted inversion and reversion of the blood coagulation factor 8 gene in human iPS cells using TALENs. Proc Natl Acad Sci U S A. 2014;111:9253–9258. doi: 10.1073/pnas.1323941111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parkes DG, Mace KF, Trautmann ME. Discovery and development of exenatide: the first antidiabetic agent to leverage the multiple benefits of the incretin hormone, GLP-1. Expert Opin Drug Discov. 2013;8:219–244. doi: 10.1517/17460441.2013.741580. [DOI] [PubMed] [Google Scholar]
- Pawson AJ, Sharman JL, Benson HE, Faccenda E, Alexander SP, Buneman OP, et al. NC-IUPHAR. The IUPHAR/BPS Guide to PHARMACOLOGY: an expert-driven knowledgebase of drug targets and their ligands. Nucl Acids Res. 2014;42(Database Issue):D1098–D1106. doi: 10.1093/nar/gkt1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez EE, Wang J, Miller JC, Jouvenot Y, Kim KA, Liu O, et al. Establishment of HIV-1 resistance in CD4+ T cells by genome editing using zinc-finger nucleases. Nat Biotechnol. 2008;26:808–816. doi: 10.1038/nbt1410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez-Pinera P, Ousterout DG, Brunger JM, Farin AM, Glass KA, Guilak F, et al. Synergistic and tunable human gene activation by combinations of synthetic transcription factors. Nat Methods. 2013;10:239–242. doi: 10.1038/nmeth.2361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrocca F, Lieberman J. Promise and challenge of RNA interference-based therapy for cancer. J Clin Oncol. 2011;29:747–754. doi: 10.1200/JCO.2009.27.6287. [DOI] [PubMed] [Google Scholar]
- Petrof EO, Gloor GB, Vanner SJ, Weese SJ, Carter D, Daigneault MC, et al. Stool substitute transplant therapy for the eradication of Clostridium difficile infection: ‘RePOOPulating’ the gut. Microbiome. 2013;1:3. doi: 10.1186/2049-2618-1-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrs-Silva H, Linden R. Advances in gene therapy technologies to treat retinitis pigmentosa. Clin Ophthalmol. 2014;8:127–136. doi: 10.2147/OPTH.S38041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pugh EN, Jr, Lamb TD. Phototransduction in vertebrate rods and cones: molecular mechanisms of amplification, recovery and light adaptation. In: Stavenga DG, DeGrip WJ, Pugh EN Jr, editors. Handbook of Biological Physics. Molecular Mechanisms in Visual Transduction. Amsterdam: Elsevier; 2000. pp. 183–255. In: (eds). [Google Scholar]
- Qi LS, Larson MH, Gilbert LA, Doudna JA, Weissman JS, Arkin AP, et al. Repurposing CRISPR as an RNA-guided platform for sequence-specific control of gene expression. Cell. 2013;152:1173–1183. doi: 10.1016/j.cell.2013.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramaswamy S, Kordower JH. Gene therapy for Huntington's disease. Neurobiol Dis. 2012;48:243–254. doi: 10.1016/j.nbd.2011.12.030. [DOI] [PubMed] [Google Scholar]
- Rask-Andersen M, Sällman Almén MS, Schiöth HB. Trends in the exploitation of novel drug targets. Nat Rev Drug Discov. 2011;10:579–590. doi: 10.1038/nrd3478. [DOI] [PubMed] [Google Scholar]
- Remy S, Beck H. Molecular and cellular mechanisms of pharmacoresistance in epilepsy. Brain. 2006;129:18–35. doi: 10.1093/brain/awh682. [DOI] [PubMed] [Google Scholar]
- Samama P, Cotecchia S, Costa T, Lefkowitz RJ. A mutation-induced activated state of the beta 2-adrenergic receptor. Extending the ternary complex model. J Biol Chem. 1993;268:4625–4636. [PubMed] [Google Scholar]
- Sander JD, Joung JK. CRISPR-Cas systems for editing, regulating and targeting genomes. Nat Biotechnol. 2014;32:343–355. doi: 10.1038/nbt.2842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitz O, Brock B, Rungby J. Amylin agonists: a novel approach in the treatment of diabetes. Diabetes. 2004;53(Suppl. 3):S233–S238. doi: 10.2337/diabetes.53.suppl_3.s233. [DOI] [PubMed] [Google Scholar]
- Schöneberg T, Schulz A, Biebermann H, Hermsdorf T, Römpler H, Sangkuhl K. Mutant G-protein-coupled receptors as a cause of human diseases. Pharmacol Ther. 2004;104:173–206. doi: 10.1016/j.pharmthera.2004.08.008. [DOI] [PubMed] [Google Scholar]
- Sheridan C. Gene therapy finds its niche. Nat Biotechnol. 2011;29:121–128. doi: 10.1038/nbt.1769. [DOI] [PubMed] [Google Scholar]
- Smith C, Gore A, Yan W, Abalde-Atristain L, Li Z, He C, et al. Whole-genome sequencing analysis reveals high specificity of CRISPR/Cas9 and TALEN-based genome editing in human iPSCs. Cell Stem Cell. 2014;15:12–13. doi: 10.1016/j.stem.2014.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soldner F, Laganière J, Cheng AW, Hockemeyer D, Gao Q, Alagappan R, et al. Generation of isogenic pluripotent stem cells differing exclusively at two early onset Parkinson point mutations. Cell. 2011;146:318–331. doi: 10.1016/j.cell.2011.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song X, Vishnivetskiy SA, Gross OP, Emelianoff K, Mendez A, Chen J, et al. Enhanced arrestin facilitates recovery and protects rod photoreceptors deficient in rhodopsin phosphorylation. Curr Biol. 2009;19:700–705. doi: 10.1016/j.cub.2009.02.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song X, Vishnivetskiy SA, Seo J, Chen J, Gurevich EV, Gurevich VV. Arrestin-1 expression in rods: balancing functional performance and photoreceptor health. Neuroscience. 2011;174:37–49. doi: 10.1016/j.neuroscience.2010.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song X, Seo J, Baameur F, Vishnivetskiy SA, Chen Q, Kook S, et al. Rapid degeneration of rod photoreceptors expressing self-association-deficient arrestin-1 mutant. Cell Signal. 2013;25:2613–2624. doi: 10.1016/j.cellsig.2013.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sriram SR, Li X, Ko HS, Chung KK, Wong E, Lim KL, et al. Familial-associated mutations differentially disrupt the solubility, localization, binding and ubiquitination properties of parkin. Hum Mol Genet. 2005;14:2571–2586. doi: 10.1093/hmg/ddi292. [DOI] [PubMed] [Google Scholar]
- Sun N, Zhao H. Transcription activator-like effector nucleases (TALENs): a highly efficient and versatile tool for genome editing. Biotechnol Bioeng. 2013;110:1811–1821. doi: 10.1002/bit.24890. [DOI] [PubMed] [Google Scholar]
- Szczepek M, Brondani V, Büchel J, Serrano L, Segal DJ, Cathomen T. Structure-based redesign of the dimerization interface reduces the toxicity of zinc-finger nucleases. Nat Biotechnol. 2007;25:786–793. doi: 10.1038/nbt1317. [DOI] [PubMed] [Google Scholar]
- Tan E, Wang Q, Quiambao AB, Xu X, Qtaishat NM, Peachey NS, et al. The relationship between opsin overexpression and photoreceptor degeneration. Invest Ophthalmol Vis Sci. 2001;42:589–600. [PubMed] [Google Scholar]
- Thal DM, Yeow RY, Schoenau C, Huber J, Tesmer JJ. Molecular mechanism of selectivity among G protein-coupled receptor kinase 2 inhibitors. Mol Pharmacol. 2011;80:294–303. doi: 10.1124/mol.111.071522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vishnivetskiy SA, Paz CL, Schubert C, Hirsch JA, Sigler PB, Gurevich VV. How does arrestin respond to the phosphorylated state of rhodopsin? J Biol Chem. 1999;274:11451–11454. doi: 10.1074/jbc.274.17.11451. [DOI] [PubMed] [Google Scholar]
- Vishnivetskiy SA, Baameur F, Findley KR, Gurevich VV. Critical role of the central 139-loop in stability and binding selectivity of arrestin-1. J Biol Chem. 2013a;288:11741–11750. doi: 10.1074/jbc.M113.450031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vishnivetskiy SA, Chen Q, Palazzo MC, Brooks EK, Altenbach C, Iverson TM, et al. Engineering visual arrestin-1 with special functional characteristics. J Biol Chem. 2013b;288:11741–11750. doi: 10.1074/jbc.M112.445437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang D, Zhong L, Nahid MA, Gao G. The potential of adeno-associated viral vectors for gene delivery to muscle tissue. Expert Opin Drug Deliv. 2014;11:345–364. doi: 10.1517/17425247.2014.871258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Zheng CG, Jiang Y, Zhang J, Chen J, Yao C, et al. Genetic correction of β-thalassemia patient-specific iPS cells and its use in improving hemoglobin production in irradiated SCID mice. Cell Res. 2012;22:637–648. doi: 10.1038/cr.2012.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wisler JW, Xiao K, Thomsen AR, Lefkowitz RJ. Recent developments in biased agonism. Curr Opin Cell Biol. 2014;27:18–24. doi: 10.1016/j.ceb.2013.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan X, Naguib S, Wu Z. Recent advances of siRNA delivery by nanoparticles. Expert Opin Drug Deliv. 2011;8:521–536. doi: 10.1517/17425247.2011.559223. [DOI] [PubMed] [Google Scholar]
- Yusa K, Rashid ST, Strick-Marchand H, Varela I, Liu PQ, Paschon DE, et al. Targeted gene correction of α1-antitrypsin deficiency in induced pluripotent stem cells. Nature. 2011;478:391–394. doi: 10.1038/nature10424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeller SJ, Kumar P. RNA-based gene therapy for the treatment and prevention of HIV: from bench to bedside. Yale J Biol Med. 2011;84:3019–3309. [PMC free article] [PubMed] [Google Scholar]