

Abstract
Background and Purpose
Hypoxic conditions favour the reduction of nitrite to nitric oxide (NO) to elicit vasodilatation, but the mechanism(s) responsible for bioconversion remains ill defined. In the present study, we assess the role of aldehyde dehydrogenase 2 (ALDH2) in nitrite bioactivation under normoxia and hypoxia in the rat and human vasculature.
Experimental Approach
The role of ALDH2 in vascular responses to nitrite was studied using rat thoracic aorta and gluteal subcutaneous fat resistance vessels from patients with heart failure (HF; 16 patients) in vitro and by measurement of changes in forearm blood flow (FBF) during intra-arterial nitrite infusion (21 patients) in vivo. Specifically, we investigated the effects of (i) ALDH2 inhibition by cyanamide or propionaldehyde and the (ii) tolerance-independent inactivation of ALDH2 by glyceryl trinitrate (GTN) on the vasodilator activity of nitrite. In each setting, nitrite effects were measured via evaluation of the concentration–response relationship under normoxic and hypoxic conditions in the absence or presence of ALDH2 inhibitors.
Key Results
Both in rat aorta and human resistance vessels, dilatation to nitrite was diminished following ALDH2 inhibition, in particular under hypoxia. In humans there was a non-significant trend towards attenuation of nitrite-mediated increases in FBF.
Conclusions and Implications
In human and rat vascular tissue in vitro, hypoxic nitrite-mediated vasodilatation involves ALDH2. In patients with HF in vivo, the role of this enzyme in nitrite bioactivation is at the most, modest, suggesting the involvement of other more important mechanisms.
Tables of Links
| LIGANDS | |
|---|---|
| ACh | Nitric oxide (NO) |
| Aspirin | PGF2α |
| Cyanamide | Phenylephrine (PE) |
| Glyceryltrinitrate (GTN) | Spironolactone |
| Lidocaine |
These Tables list key protein targets and ligands in this article which are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Pawson et al., 2014) and are permanently archived in the Concise Guide to PHARMACOLOGY 2013/14 (a,bAlexander et al., 2013a,b).
Introduction
Nitrite can be chemically reduced in vivo to nitric oxide (NO) to elicit vasodilatation (Lundberg et al., 2011; Totzeck et al., 2012; Bailey et al., 2014). However, in vitro this conversion process is relatively slow, perhaps explaining why relatively high, supraphysiological nitrite concentrations are required to relax pre-constricted isolated blood vessels (Furchgott and Bhadrakom, 1953; Maher et al., 2008; Ormerod et al., 2011). We and others have demonstrated that infused nitrite acts as a vasodilator in healthy volunteers (Gladwin et al., 2000; Cosby et al., 2003; Larsen et al., 2006; Dejam et al., 2007; Maher et al., 2008), with greater potency as an arteriolar vasodilator in forearm resistance vessels of patients with heart failure (HF) compared with those of normal subjects (Maher et al., 2013). Although several candidate mechanisms have been postulated, the exact mechanism(s) responsible for bioactivation of nitrite in man remains ill defined.
Under conditions of profound hypoxia and/or acidosis, the conversion of nitrite to NO may occur via acid disproportionation. Under less extreme conditions, various factors have been reported to convert nitrite to NO. These include xanthine oxidoreductase (XOR), aldehyde oxidase, endothelial NOS (eNOS) and haem proteins (Doyle et al., 1981; Basu et al., 2007; Li et al., 2008; Pinder et al., 2009; Lundberg et al., 2011; Tiso et al., 2011; Totzeck et al., 2012). Previous studies have demonstrated that mitochondrial aldehyde dehydrogenase 2 (ALDH2) may be an important additional source of nitrite-derived NO in rat heart (Perlman et al., 2009) and the vasculature (Golwala et al., 2009), but whether the oxidative environment in the vasculature of HF patients may alter the role for ALDH2 remains unknown. Interestingly, ALDH2 is also known to act as a reductase of organic nitrates, such as glyceryl trinitrate (GTN; Chen et al., 2002), and currently GTN is used to treat HF patients. However, prolonged exposure of the vasculature to GTN leads to inactivation of ALDH2. Although it has been argued that ALDH2 inactivation is the principal mechanism for the development of nitrate tolerance (Chen et al., 2002; DiFabio et al., 2003), other investigators have demonstrated hysteresis between the onset/offset of ALDH2 inactivation by GTN and the time course of GTN tolerance (D'Souza et al., 2011), potentially permitting the utilization of GTN pre-exposure as an ALDH2-inactivating manoeuvre without induction of GTN tolerance.
Herein, we first explored the putative role of ALDH2 as a contributor to nitrite bioactivation under normoxic and hypoxic conditions, utilizing in vitro isolated rat vessels. To translate our findings into the clinical setting, we explored the role of ALDH2 in a proof-of-principle study in HF patients in vitro (isolated vessel) and in vivo forearm blood flow (FBF; measured by venous occlusion plethysmography). We chose to evaluate HF patients, first because of the potential therapeutic relevance, and second because we have previously shown that in vivo arteriolar responses to nitrite are increased in patients with HF versus healthy controls (Maher et al., 2013).
Methods
The animal studies were approved by the UK Home Office and conducted according to the Animals (Scientific Procedures) Act of 1986 and European Commission guidelines. A total of 35 animals were used in the experiments described here. The human investigations conformed to the Declaration of Helsinki and were approved by the South Birmingham Research Ethics Committee (10/H1207/50) and registered with the UK Clinical Research Network (UK CRN 9587). All patients gave written informed consent after satisfying the inclusion and exclusion criteria as described below.
Tension myography
Male Sprague–Dawley rats (250–300 g) were killed by an overdose of anaesthetic by an i.p. injection of sodium pentobarbital (100 mg·kg−1). The thoracic aorta was carefully removed and placed immediately in cold Krebs bicarbonate buffer (pH 7.4, 95% O2; 5% CO2) of the following composition (mmol·L−1): NaCl (119), KCl (4.7), CaCl2 (2.5), KH2PO4 (1.18), MgSO4 (1.19), NaHCO3 (25.1), glucose (11). Thoracic aortas were cleaned from adhering connective tissue and cut into three to four ring segments. Aortic rings were mounted in a myograph (Multi-Myograph 610M, Danish Myotechnology, Aarhus, Denmark) containing 5 mL Krebs bicarbonate buffer (37°C, pH 7.4) and gassed with 95% O2/5% CO2. After an equilibration period of 45 min, vessels were normalized as previously described (Madhani et al., 2003). Following normalization, each vessel was primed with KCl (4.8 mmol·L−1) before a supramaximal concentration of phenylephrine (PE; 10 μmol·L−1; Sigma Aldrich, Dorset, UK) was added. Once the contractile tone had stabilized, ACh (1 μmol·L−1; Sigma Aldrich) was added to the organ bath to assess the integrity of the endothelium. If the constrictor responses to PE were not maintained or ACh elicited relaxations <50% of the PE-induced pre-contractile tone, the preparation was discarded. Tissues were then washed for 30 min (by addition of fresh Krebs bicarbonate buffer at 15 min intervals) after which cumulative concentrations of PE (0.001–1 μmol·L−1) were added to the myograph. These tissues were then washed over 60 min to restore basal tone before being contracted by PE, to approximately 80% of the maximum PE-induced response. Once a stable response to PE was achieved, a cumulative concentration–response curve to sodium nitrite (NaNO2; Sigma Aldrich) was constructed (0.001–100 μmol·L−1) under normoxic conditions (95% O2/5% CO2). Under hypoxic conditions (95% N2/5% CO2; resulting in ≈1% tissue bath O2; Maher et al., 2008), the vessels were incubated for 30 min before exposure to the EC80 of PE (to achieve similar tension observed at 95% O2) and the administration of NaNO2.
To delineate the role of ALDH2 in nitrite-mediated vasorelaxation, concentration–response curves to NaNO2 were constructed before and after 30 min of incubation with cyanamide (1 mmol·L−1; ALDH2 inhibitor; DiFabio et al., 2003; Sigma Aldrich) or propionaldehyde (1 mmol·L−1; ALDH2 substrate that acts as a competitive inhibitor; DiFabio et al., 2003; Sigma Aldrich) under normoxic and hypoxic conditions.
To determine whether GTN pre-exposure of tissues leads to an attenuation of the response to nitrite (via inactivation of ALDH2), aortic rings were exposed to hypoxic conditions as described earlier and then incubated in the presence or absence of 30 or 100 μmol·L−1 of GTN for a period of 1 h (Keith et al., 1982). Thereafter, blood vessels were washed every 15 min for 1 h. After pre-contraction to the EC80 of PE, a concentration–response curve to NaNO2 was constructed.
In vitro and in vivo analysis in HF patients
The effect of ALDH2 inhibition on nitrite-mediated vasorelaxation was investigated in HF patients: (i) in vitro in isolated resistance vessels obtained from gluteal subcutaneous fat tissue and (ii) in vivo by measuring changes in FBF during intra-arterial infusion of sodium nitrite with and without GTN pretreatment (to decrease ALDH2 activity).
Patient demographics
Patients were grouped as follows: (i) biopsy group (in vitro myography; n = 16); (ii) plethysmography study: saline group (n = 8) and GTN group (n = 13); Table 1). Inclusion criteria: systolic HF [left ventricular ejection fraction (LVEF) <40], aged between 40 and 80 years, and non-smoker. Exclusion criteria: treatment with long-acting nitrates, past history of adverse reactions to organic nitrates, hypotension (systolic BP <110 mmHg), concomitant warfarin/clopidogrel therapy or with bleeding diathesis, and obstructive sleep apnoea. All subjects were asked to refrain from alcohol, and foods with a high nitrate/nitrite content for 24 h, and caffeine for 12 h before the study.
Table 1.
Patient demographics
| In vitro analysis | In vivo analysis | ||
|---|---|---|---|
| Biopsy-only group (n = 16) | Saline group (n = 8) | GTN group (n = 13) | |
| Age (years) | 64.5 ± 3.8 | 62 ± 4.0 | 66 ± 3.3 |
| Male gender, n (%) | 13 (81) | 7 (88) | 12 (92) |
| Mean weight (kg) | 78.3 ± 3.8 | 74.6 ± 2.5 | 82.3 ± 3.3 |
| Body mass index (kg·m–2) | 27.8 ± 1.5 | 25.4 ± 0.5 | 27.1 ± 0.9 |
| Ejection fraction (%) | 26.4 ± 2.3 | 25.1 ± 2.5 | 27 ± 2.1 |
| NYHA class | |||
| I | 2 | 1 | 1 |
| II | 6 | 4 | 10 |
| III | 8 | 3 | 2 |
| Heart rate (beats·min-1) | 72 ± 2.8 | 62 ± 4.4 | 62 ± 2.0 |
| MABP (mmHg) | 95 ± 4.1 | 89 ± 2.9 | 88 ± 2.2 |
| Aetiology, n (%) | |||
| Dilated cardiomyopathy | 8 (50) | 5 (62) | 6 (46) |
| Ischaemic cardiomyopathy | 6 (38) | 3 (38) | 6 (46) |
| Other | 2 (13) | 0 | 1 (8) |
| Medication, n (%) | |||
| ACEI/AT2 receptor antagonists | 15 (94) | 8 (1 00) | 12 (92) |
| β-Blockers | 10 (63) | 5 (62) | 10 (77) |
| Spironolactone/eplerenone | 10 (63) | 3 (38) | 3 (23) |
| Loop diuretic | 12 (75) | 4 (50) | 8 (62) |
| Aspirin | 12 (75) | 4 (50) | 10 (77) |
Data expressed as mean ± SEM.
ACEI, ACE inhibitors; MABP, mean arterial BP; NYHA, New York Heart Association classification.
Effect of ALDH2 inhibition in isolated resistance vessels
In a subgroup of nitrite/nitrate naïve HF patients (i.e. no infusions or treatment of NaNO2 and/or GTN), subcutaneous gluteal fat biopsies were obtained under local anaesthetic (2% lidocaine) and placed in cold Krebs bicarbonate buffer as previously described (Greenstein et al., 2009). Immediately after harvesting the subcutaneous fat biopsies, small resistance arteries (∼250 μm ID; 2 mm long) were isolated from the fat, and mounted onto a myograph (Greenstein et al., 2009). In most cases, two vessels from each biopsy were studied. The myograph protocol for normalization, priming of vessels with KCl, the assessment of supramaximal concentration of PE and the endothelial integrity, was assessed as above. Cumulative concentration–response curves to NaNO2 were constructed (0.001–100 μmol·L−1) under normoxic and hypoxic conditions as described earlier.
To investigate whether attenuation of nitrite efficacy during hypoxia was caused by the effects of ALDH2 inhibition on downstream signalling in the NO cascade, concentration–response curves to the NO donor, spermine-NONOate [N-(2-aminoethyl)-N-(2-hydroxy-2-nitrosohydrazino)-1,2-ethylenediamine (Sper/NO); 0.001–1 μmol·L−1; Calbiochem (EMD Millipore)], were constructed in the absence and presence of the ALDH2 inhibitor, cyanamide, under hypoxic conditions.
The effect of ALDH2 inactivation by GTN on FBF
To determine the effects of ALDH2 inactivation by GTN on arteriolar responsiveness in patients from HF, forearm vascular responsiveness was assessed using the plethysmography. The studies on HF patients were performed in a quiet temperature-controlled (22–24°C) laboratory. All subjects were placed in a semi-recumbent position enabling the administration of hypoxia to participants. Changes in FBF were determined in both arms by venous occlusion plethysmography (DE Hokanson, Bellevue, WA, USA) and BP, heart rate (HR) and oxygen saturation were monitored as previously described (Maher et al., 2008). A 27 gauge arterial needle (Coopers Engineering, Birmingham, UK) mounted onto a 16 gauge epidural catheter and sealed with dental wax was then inserted aseptically into the brachial artery of the non-dominant arm and was kept patent by the continuous infusion of normal saline (Baxter Healthcare, Deerfield, IL, USA; 0.9%). An i.v. cannula (20 gauge) was inserted in the contralateral arm for venous blood sampling. Saline was infused via the intra-arterial line at 1 mL·min-1 for 10 min for baseline measurements before infusion of NaNO2. Data were acquired from both arms, and any changes observed were corrected for those occurring in the contralateral control arm and presented as a ratio of FBF (FBF-R) during infusion compared with baseline (Maher et al., 2008).
As depicted in Figure 1, following 10 min of rest, two doses of NaNO2 (Martindale Pharmaceuticals, Buckingham, UK) were infused into the brachial artery of the non-dominant arm (784 nmol·min−1 then 7.84 μmol·min−1 for 20 min each; the infusion rate was 1 mL·min−1 as described previously; Maher et al., 2008). FBF was measured in both arms. Following the second dose of NaNO2 (7.84 μmol·min−1), the patients switched from breathing room air to inspiring 12% oxygen via a facemask connected to a two-way valve. Upon achieving target oxygen saturation of 83–88%, the study proceeded with the 7.84 μmol·min−1 infusion of nitrite for a further 10 min. Thereafter, patients were randomized to receive either an i.v. infusion of 10 μg·min GTN (to decrease ALDH2 activity; Merck-Lipha Pharmaceuticals Ltd, Middlesex, UK) as described previously (Philpott et al., 2007) or saline (the placebo group; active ALDH2) for 4 h (15 mL·h−1). Following a 30 min washout period (saline at 1 mL·min−1), repeat infusions of NaNO2 were administered during normoxia and hypoxia as described earlier.
Figure 1.

Schematic protocol of the plethysmography study.
Upon completion of the plethysmography protocol, the intra-arterial needle was removed. Participants were allowed to rest for 10 min following which a subcutaneous fat biopsy was obtained (Figure 1) as described earlier. Philpott et al. (2007) have previously reported that low-dose GTN infusion rapidly inactivates ALDH2 and that this inactivation occurs before the development of significant nitrate tolerance. Therefore, to determine whether the infusion of GTN had induced significant nitrate tolerance, GTN vasodilator concentration–response curves were compared in resistance vessels taken via gluteal biopsy in patients from the saline (placebo group; active ALDH2) and GTN (inactive ALDH2) infusion group. Briefly, the isolated tissue resistance vessels were mounted in a myograph, the vascular rings were assessed for endothelial integrity and then contracted sub-maximally with PE and cumulative concentration–response curves were constructed for GTN (0.001–1 μmol·L−1) from the saline and GTN infusion HF patient group respectively.
Blood samples
Venous blood samples were taken at eight time points during the plethysmography study (Figure 4) for determination of venous methaemoglobin (MetHb) and pH (blood gas analyser Bayer Rapidlab 865, Siemens, NY, USA), and determination of total plasma 8-iso prostaglandin F2α (8-isoprostane EIA assay kit protocol, Cayman Chemical, Ann Arbor, MI, USA).
Figure 4.
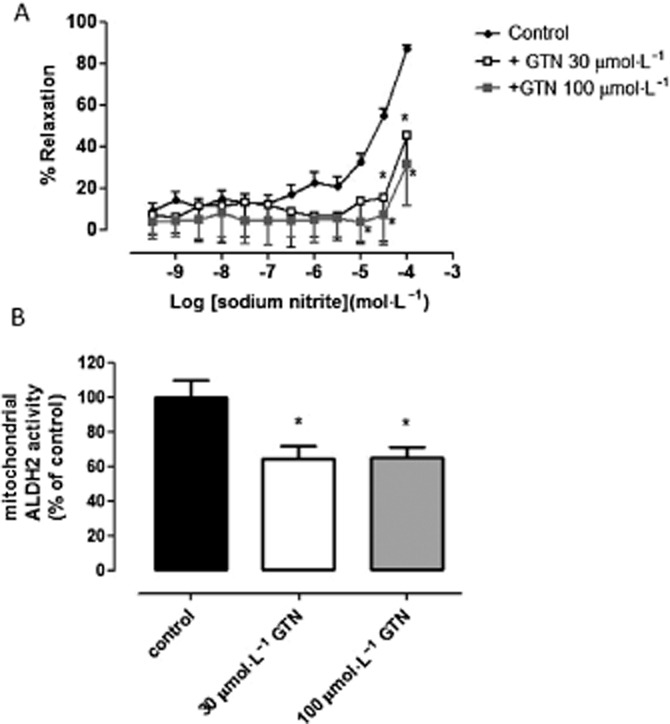
Tolerance-independent inactivation of ALDH2 attenuates vasorelaxation in rat aortae. (A) Concentration–response curve to NaNO2 in the presence or absence of GTN during hypoxic conditions. Relaxation is expressed as mean ± SEM percentage reversal of PE-induced tone (n = 10); *P < 0.05, ***P < 0.001 versus control, two-way anova. (B) The effect of sodium nitrite (control), in the presence or absence of GTN during hypoxic conditions, on mitochondrial ALDH2 activity (mean ± SEM from n = 4–6 animals; *P < 0.05 vs. control by one-way anova).
Isolation of mitochondrial fraction
Rat aortic vessels that were treated as described earlier in the tension myography studies were immediately snap frozen at the end of the protocol for isolation of the mitochondrial fraction. Frozen thoracic aorta was suspended in the mitochondrial buffer containing 10 mmol·L−1 MOPS (pH 7.2), 10 mmol·L−1 KCl, 1.5 mM MgCl2, 1 mmol·L−1 EDTA, 10 μg·mL−1 leupeptin, 10 μg·mL−1 aprotinin and 0.25 mol·L−1 sucrose, and gently homogenized with a Dounce homogenizer (30 strokes) as previously described (Paneni et al., 2013). The homogenate was centrifuged at 750× g for 10 min at 4°C to remove nuclei and unbroken cells, and the supernatant was subsequently centrifuged at 10 000× g for 15 min. The resultant mitochondrial pellet was used for the ALDH2 assay kit (see ALDH2 activity assay for details).
Mitochondrial ALDH2 activity assay
ALDH2 activity was determined in mitochondria isolated from rat thoracic aorta following solubilization and extraction as specified in the manufacturer's recommendations (mitochondrial ALDH2 activity assay kit; Abcam, Cambridge, UK). The homogenate was then incubated on ice for 20 min and centrifuged at 16 000× g for 20 min at 4°C. Protein concentration of the supernatant was determined and 20 μg of protein was used to detect ALDH2 activity. In this assay, the generation of NADH is coupled to the 1:1 reduction of a reporter dye to yield reaction product concentration, which was monitored by measuring the absorbance increase at 450 nm.
Statistical analysis
All data are expressed as mean ± SEM, and significance was accepted with P < 0.05. For the myography analysis, concentration–response curves were analysed using two-way anova. For the in vivo FBF analysis, one-way anova repeated measures coupled with a Bonferroni post hoc test was used to compare the effects of pre-saline/GTN or post-saline/GTN infusion treatment. A paired non-parametric test (Wilcoxon signed-rank test) was used to compare FBF following hypoxia between pre-GTN infusion and post-GTN infusion. Statistical analysis was undertaken using Prism software (version 4.0, GraphPad Software, La Jolla, CA, USA).
Results
Evaluation of the role of ALDH2 in nitrite-mediated bioactivation in rat aorta
As depicted in Figure 2, the vasorelaxant response to NaNO2 displays a biphasic concentration–response relationship. As shown in Figure 2A, following incubation of vessels with the ALDH2 inhibitor cyanamide during normoxic conditions, vasorelaxation by NaNO2 was paradoxically enhanced at nanomolar concentrations and significantly inhibited at higher NaNO2 concentrations (10 and 30 μM) when compared with controls (P < 0.05; by two-way anova). Hypoxia enhanced nitrite-induced relaxation but pretreatment with cyanamide during hypoxia caused a significant attenuation of nitrite's efficiency to relax blood vessels at all concentrations tested when compared with the control (P < 0.05; by two-way anova; Figure 2B). Similar results were obtained using the ALDH2 substrate, propionaldehyde. Incubation of vessels with propionaldehyde under normoxic conditions blunted the vasorelaxant effect of low concentrations of nitrite without affecting its efficacy at higher concentrations, but these differences did not reach statistical significance (P < 0.05; by two-way anova; Figure 2C). However, under hypoxic conditions, propionaldehyde significantly attenuated the response to nitrite in the nano- to micromolar concentrations range when compared with control (P < 0.05; by two-way anova; Figure 2D) without affecting maximal relaxation induced by the highest concentration of nitrite.
Figure 2.
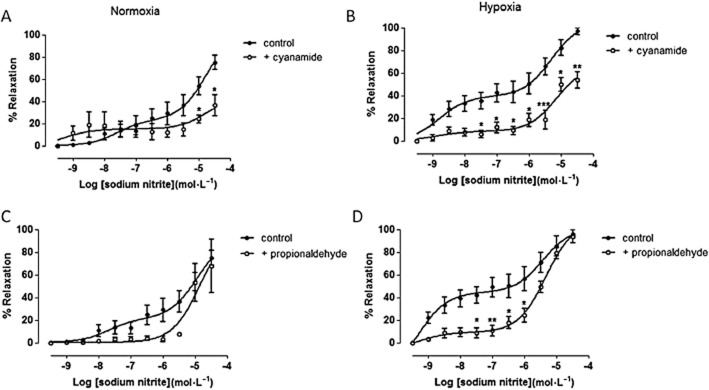
ALDH2 inhibition attenuates nitrite-induced vasorelaxation in rat aortae. Concentration–response curves to sodium nitrite in the presence or absence of ALDH2 inhibitor, cyanamide, during (A) normoxia (n ≥ 5), (B) hypoxia (n ≥ 5), and ALDH2 substrate, propionaldehyde, during (C) normoxia (n ≥ 5) and (D) hypoxia (n ≥ 5). Relaxation is expressed as mean ± SEM percentage reversal of PE-induced tone. *P < 0.05, **P < 0.01 and ***P < 0.001 control versus cyanamide or propionaldehyde by two-way anova.
Effect of ALDH2 inhibition on nitrite-mediated vasorelaxation in resistance vessels from HF patients
Under both normoxic and hypoxic conditions, NaNO2 caused concentration-dependent relaxation of resistance vessels from HF patients (Figures 3A and B). During normoxic conditions, pretreatment with cyanamide tended to shift the NaNO2 concentration–response curve to the right but this was not statistically significant (Figure 3A). Under hypoxic conditions, pretreatment with cyanamide caused a marked and concentration-dependent attenuation of the relaxation responses to NaNO2 when compared with control (P < 0.05; by two-way anova; Figure 3B). To assess whether this effect was nitrite specific, concentration–response curves to the NO donor Sper/NO were constructed in the presence or absence of cyanamide. Cyanamide had no inhibitory activity on the vasorelaxant responses to Sper/NO under hypoxic conditions (Figure 3C).
Figure 3.

ALDH2 inhibition decreases nitrite-induced vasorelaxation in resistance vessels from HF patients. Concentration–response curve to sodium nitrite in the presence or absence of cyanamide during (A) normoxia (n = 7) and (B) hypoxia (n = 9). (C) Concentration–response curve to Sper/NO in the presence or absence of cyanamide during hypoxic conditions (n ≥ 6). Relaxation is expressed as mean ± SEM percentage reversal of PE-induced tone. *P < 0.05 and **P < 0.01 control versus cyanamide by two-way anova.
Effect of GTN pretreatment on nitrite-mediated vasorelaxation in rat aorta
In addition to the aforementioned inhibitor/substrate experiments, we also assessed the effects of GTN (used as a tool to decrease ALDH2 activity) on nitrite-mediated vasorelaxation. Using a well-established in vitro model of nitrate tolerance in rat isolated aorta (Keith et al., 1982; Irvine et al., 2007), blood vessels were pretreated with GTN (30 or 100 μmol·L−1) for 1 h followed by construction of a concentration–response curve to NaNO2 under hypoxic conditions. Pretreatment with GTN (30 and 100 μmol·L−1) significantly attenuated the response to NaNO2 compared with controls (P < 0.05; by two-way anova; Figure 4A), particularly at higher concentrations. As shown in Figure 4B, mitochondrial ALDH2 activity was significantly decreased following pretreatment with GTN (30 and 100 μmol·L−1) when compared with control (NaNO2; P < 0.05; by one-way anova).
Effects of GTN-induced ALDH2 inactivation on nitrite-mediated vasodilatation in the human forearm
FBF corrected for changes in the non-infused arm (FBF ratio) increased dose-dependently with NaNO2 in the saline (placebo) and GTN groups respectively (Figure 5A and B). As depicted in Figure 5A, one-way anova repeated measures showed that 7.84 μmol·min−1 nitrite significantly increased FBF in the pre-saline infusion group from 0.94 ± 0.09 at baseline to 1.69 ± 0.21 during normoxic (P < 0.05 compared with baseline) and 1.76 ± 0.22 during hypoxic conditions (P < 0.05 compared with baseline; Figure 5A). Following 4 h i.v. saline infusion (placebo group; active ALDH2), a similar profile in FBF-R response to NaNO2 was observed when compared with pre-saline infusion. Nitrite 7.84 μmol·min−1 significantly increased FBF in the post-saline infusion group from 1.02 ± 0.04 at baseline to 1.93 ± 0.14 during normoxic (P < 0.001 compared with baseline) and 2.05 ± 0.18 during hypoxic conditions (P < 0.001 compared with baseline).
Figure 5.

GTN infusion attenuates nitrite-induced vasorelaxation in the resistance vasculature of HF patients. Forearm vasodilator measurements following nitrite infusion in HF patients subjected to 4 h infusion of (A) saline (n = 8 patients) or (B) GTN (n = 11 patients) treatment. *P < 0.05, **P < 0.01, ***P < 0.001 compared with baseline. (C) Comparison of pre- and post-GTN infusion following 7.84 μmol·min−1 sodium nitrite during hypoxic conditions (n = 8 and n = 11 patients, respectively; P = 0.08). (D) Concentration–response curve to GTN in isolated resistance vessels from HF patients (saline n = 7; GTN n = 11 patients).
In the GTN group (Figure 5B), prior to administration of i.v. GTN, one-way anova repeated measures showed that 7.84 μmol·min−1 nitrite infusion significantly increased FBF from 1.24 ± 0.08 at baseline to 2.34 ± 0.29 during normoxic (P < 0.05 compared with baseline) and 2.48 ± 0.36 (P < 0.01 compared with baseline) during hypoxic conditions. Following 4 h infusion of GTN to inactivate ALDH2, although 7.84 μmol·min−1 nitrite infusion significantly increased FBF from 1.20 ± 0.11 (baseline) to 1.95 ± 0.15 during normoxic conditions (P < 0.001 when compared with baseline) and 1.72 ± 0.15 during hypoxia (P < 0.05 when compared with baseline). However, GTN treatment tended to attenuate the increase in FBF following nitrite infusion (7.84 μmol·min–1) when compared with the pre-GTN infusion. Using a paired non-parametric test (Wilcoxon signed-rank test), we compared pre- and post-GTN infusion for each of baseline and 784 nmol and 7.84 μmol at normoxic conditions and at hypoxic conditions respectively. There was no significant difference between pre-GTN versus post-GTN infusion at baseline (1.24 ± 0.09 and 1.20 ± 0.11, respectively; P = 0.79), pre-GTN versus post-GTN infusion with 784 nmol·min−1 (2.16 ± 0.72 and 1.35 ± 0.10, respectively; P = 0.34), or at pre-GTN versus post-GTN infusion with 7.84 μmol·min−1 (2.34 ± 0.29 and 1.95 ± 0.15, respectively; P = 0.28). During hypoxic conditions, GTN treatment attenuated the increase in FBF following nitrite infusion (7.84 μmol·min−1) but this effect did not reach statistical significance (2.48 ± 0.36 and 1.72 ± 0.15, pre- and post-GTN infusion, respectively; P = 0.08; Figure 5C).
GTN infusion does not induce tolerance in resistance vessels
To confirm that our low-dose GTN infusion protocol did not induce systemic nitrate tolerance, a gluteal biopsy was obtained at the end of the plethysmography protocol and isolated resistance vessels were used for myography experiments. There was no attenuation of the vasodilator response to GTN compared with that in the control vessels isolated from the saline infusion group (P = 0.87; Figure 5D).
Assessment of haemodynamics, venous pH, 8-isoprostanes and methaemoglobin formation during nitrite infusion in HF patients
HR and mean arterial BP (MABP) did not alter significantly from baseline following administration of NaNO2 in the saline and GTN groups (Table 2). Arterial oxygen saturations remained stable throughout the normoxia period in the saline group, but fell significantly following administration of 12% oxygen in both the saline and GTN groups (from 97 ± 0.5% to 87 ± 1.1%, P < 0.001 and from 97 ± 0.3 to 87 ± 0.6, P < 0.001 respectively). There was no significant change in either venous pH (saline group n = 5–7; GTN group n = 9–11; P < 0.05) or 8-isoprostane levels (saline group n = 4–6; GTN group n = 7–9; P < 0.05) in both the saline and GTN groups.
Table 2.
Assessment of haemodynamic, venous pH, 8-isoprostane and methaemoglobin
| Saline group | GTN group | |||||||
|---|---|---|---|---|---|---|---|---|
| BL | 784 nmol·min−1 | 7.84 μmol·min−1 | Hypoxia + 7.84 μmol·min−1 | BL | 784 nmol·min−1 | 7.84 μmol·min−1 | Hypoxia + 7.84 μmol·min−1 | |
| HR (bpm) | 62 ± 4.1 | 64 ± 4.1 | 65 ± 4.8 | 68 ± 5.7 | 62 ± 2.3 | 62 ± 2.2 | 60 ± 1.6 | 6.4 ± 0.9 |
| MABP (mmHg) | 84 ± 1.9 | 85 ± 2.9 | 78 ± 2.5 | 80 ± 2.6 | 83 ± 2.8 | 83 ± 2.6 | 86 ± 2.5 | 85 ± 2.6 |
| Arterial O2 Sat (%) | 97 ± 0.5 | 97 ± 0.5 | 97 ± 0.5 | 87 ± 1.1* | 97 ± 0.3 | 98 ± 0.3 | 98 ± 0.3 | 87 ± 0.6* |
| pH | 7.37 ± 0.02 | 7.39 ± 0.01 | 7.40 ± 0.01 | 7.41 ± 0.01 | 7.37 ± 0.01 | 7.38 ± 0.01 | 7.38 ± 0.01 | 7.39 ± 0.01 |
| Isoprostanes (ng·L−1) | 5.96 ± 0.55 | 7.42 ± 0.93 | 7.13 ± 0.39 | 7.29 ± 0.88 | 7.61 ± 1.00 | 9.14 ± 1.75 | 7.54 ± 0.83 | 9.45 ± 2.45 |
| MetHb (% of total Hb) | 0.31 ± 0.04 | 0.61 ± 0.08 | 1.39 ± 0.21* | 1.63 ± 0.17* | 0.37 ± 0.06 | 0.58 ± 0.07 | 1.51 ± 0.18* | 1.57 ± 0.15* |
Data expressed as mean ± SEM.
P < 0.0001.
BL, baseline; GTN, glyceryl trinitrate; HR, heart rate; MABP, mean arterial blood pressure; MetHb%, methaemoglobin measurements following sodium nitrite infusion in the saline or GTN group; O2, arterial oxygen saturation; pH, plasma 8-isoprostane.
Following 4 h saline infusion, venous MetHb levels increased during escalating NaNO2 dose infusions from 0.31 ± 0.04% to 1.39 ± 0.21% (baseline and 7.84 μmol·min−1 nitrite, normoxia) of total Hb. Highest levels were reached following 7.84 μmol·min−1 nitrite infusion during hypoxic conditions (1.63 ± 0.17%; P < 0.001; n = 5–7). In the GTN group, a similar trend was observed, MetHb levels increased from 0.37 ± 0.06% to 1.51 ± 0.18% (baseline and 7.84 μmol·min−1 nitrite, respectively, normoxia) with the highest levels of MetHb (1.57 ± 0.15%; P < 0.001; n = 9–11) reached following 7.84 μmol·min−1 nitrite infusion during hypoxic conditions. There was no significant difference between the two groups. Pre-infusion data were also similar for the haemodynamics and MetHb pre-saline/GTN (data not shown).
Discussion
We previously demonstrated that intra-brachial artery infusion of nitrite causes forearm vasodilatation in both healthy subjects and in HF patients, with greater potency in the latter (Maher et al., 2013), but the mechanism(s) underlying these effects remains incompletely understood. Based on these observations, we here explored the role of ALDH2 in nitrite bioactivation under normoxia and hypoxia in both rats and human vasculature. Our findings suggest that ALDH2 plays a major role in nitrite-mediated vasorelaxation in vitro. However, this effect was clearly less marked in vivo, and we postulate that this could be due to the presence of multiple in vivo mechanisms.
Several nitrite reductases have been identified, including eNOS, XOR, aldehyde oxidase and haem proteins such as deoxyhaemoglobin and myoglobin, to bioactivate nitrite under hypoxia (Doyle et al., 1981; Basu et al., 2007; Pinder et al., 2009; Lundberg et al., 2011; Tiso et al., 2011; Baliga et al., 2012; Ghosh et al., 2013). All of these agents alone and/or in combination are capable of bioconverting nitrite to NO in vitro, but their physiological role remains incompletely understood. Previous studies have reported that ALDH2 may be an important source of nitrite-derived NO in the heart (Perlman et al., 2009) and vasculature (Badejo et al., 2010), but whether the oxidative environment in the vasculature alters the effects of ALDH2 on nitrite-mediated vasorelaxation remains unclear. Therefore, in the present study we first explored the role of ALDH2 in nitrite-mediated vasorelaxation in vitro in rat isolated conduit vessels (aorta) during normoxic and hypoxic conditions. Our in vitro data showed that the ALDH2 inhibitor, cyanamide and the ALDH2 substrate, propionaldehyde significantly reduced the potency of nitrite under hypoxic conditions. These data indicate the potential for ALDH2 to be involved in hypoxic nitrite vasodilator responses.
Much of our current understanding about nitrite's mode of action as a vasodilator is based on animal experimental work and considerably less information is available on the mechanism of vasodilatation by nitrite in human tissue. Nevertheless, studies in healthy human subjects have shown that nitrite causes marked venodilatation and moderate dose-dependent arteriolar dilatation, and these effects are augmented by hypoxia or exercise in healthy subjects (Cosby et al., 2003; Maher et al., 2008). We have previously shown that in vivo arteriolar responses to nitrite are increased in patients with HF versus healthy controls (Maher et al., 2013). Because of the potential therapeutic relevance of these findings, we now chose to translate these observations to the clinical setting by evaluating the role of ALDH2 in HF patients in vitro and in vivo. Because the vascular responses to intra-arterial nitrite are most accurately assessed by measuring changes in FBF (i.e. resistance vessel effects), we first investigated whether the effects of ALDH2 inhibition on nitrite-mediated vasorelaxation observed in the rat isolated conduit vessels were replicated in vitro in resistance vessels from patients with HF. We confirmed that the inhibition of ALDH2 significantly attenuates nitrite-mediated vasorelaxation during hypoxic conditions. Importantly, cyanamide did not alter the potency of the NO donor Sper/NO, ruling out non-specific effects of this inhibitor on NO bioactivity. These observations concur with functional data obtained by Huellner et al. (2008), where pharmacological inhibition of ALDH2 did not alter the potency to the NO donor DEA/NO in isolated human veins. To our knowledge, this is the first study in man to assess the effects of NaNO2 in isolated resistance vessels from HF patients. Interestingly, the magnitude difference in vasorelaxation response to NaNO2 observed in the normoxic versus hypoxic group of blood vessels from HF patients was similar. Moreover, the response to NaNO2 when comparing rat thoracic aorta versus HF patient resistance vessels was also different. This might be related to different vascular beds, species differences or the heterogeneity in vascular response between healthy and pathophysiology tissues. Regarding the latter, based on our previous findings (Maher et al., 2013), we believe that the reason why the magnitude difference in nitrite-induced relaxation is not that different between normoxia versus hypoxia in isolated resistance vessels from HF patients when compared with conduit vessels (thoracic aorta) from healthy rats is because, first, the vasomotor response to nitrite is altered and enhanced and therefore the magnitude is not that different when compared with hypoxia as suggested from our previous in vivo studies (Maher et al., 2013).
Previous studies have reported that prior GTN exposure induces nitrate tolerance in association with attenuated ALDH2 activity in the vasculature (DiFabio et al., 2003; Huellner et al., 2008; D'Souza et al., 2011). Therefore, using a well-established in vitro model of nitrate tolerance in rat isolated aorta (Keith et al., 1982; Irvine et al., 2007), we demonstrated attenuation of nitrite-mediated vasorelaxation following pretreatment with GTN during hypoxic conditions. To substantiate the role of ALDH2 as a key effector in nitrite-mediated vasorelaxation, we here show that this attenuation in vascular response is associated with reduced mitochondrial ALDH2 activity.
It is important to recognize that there are 19 known human ALDH isoenzymes, of which only a few, such as ALDH2 (Koppaka et al., 2012), have been thoroughly characterized biochemically and by susceptibility to pharmacological inhibition. To date, no antagonists have been developed to specifically inhibit each ALDH isoenzyme without affecting others. Moreover, clear sensitivity differences to different ALDH2 inhibitors have been reported, for example, GTN bioactivation in rat liver was sensitive to chloral hydrate but not to daidzin (Kollau et al., 2005). In the current study, we chose to use cyanamide (an irreversible inhibitor) and propionaldehyde (a reversible competitive inhibitor) as both are capable of inhibiting ALDH and have been used to examine the role of ALDH2 in organic nitrate-induced vasorelaxation in rat aorta (Chen et al., 2002; DiFabio et al., 2003). Notwithstanding possible species differences in sensitivity to both inhibitors and vasodilators (including further differences in bioactivation mechanisms), we further complemented these results by a mechanism-based ALDH2 inhibition approach employing an in vitro tolerance model and have reached the same conclusion, that is, that ALDH2 inhibition exerts inhibitory actions of NaNO2-mediated vasorelaxation in vitro.
Assessing the involvement of specific enzymes in mediating the vasodilator effects of nitrite in man in vivo remains a challenge. The ALDH2 inhibitor disulfiram is commonly used in clinical studies without ill effect (Johansson, 1992; Mackenzie et al., 2005), but its use in HF patients is contra-indicated (Huffman and Stern, 2003). Therefore, in the present study we elected to use a low-dose GTN infusion as it has been previously reported to rapidly inactivate vascular ALDH2, and that this inactivation occurs prior to the development of significant nitrate tolerance and prior to detectable impairment of GTN bioconversion (Philpott et al., 2007). We measured the vasodilator activity of nitrite in HF patients and tested whether pre-infusion of GTN (in order to decrease ALDH2 activity) attenuates forearm vasodilator responses to nitrite during normoxic and hypoxic conditions. Despite the well-recognized presence of both endothelial dysfunction and NO resistance in HF patients (Marti et al., 2012), adequate vascular activity in response to nitrite was observed in our study. During normoxic conditions, we found an approximate twofold increase in forearm vasodilator responses following 7.84 μmol·min−1 of NaNO2 in both the saline and GTN group compared with their baseline responses. These results are similar to our previous findings in healthy volunteers (Maher et al., 2008). Under hypoxic conditions, inhibition of ALDH2 with GTN (post-GTN infusion group) tended to reduce the forearm vasodilator response but was not significant when compared with the pre-GTN infusion group. Our observation in gluteal resistance vessels taken at the end of the in vivo study confirmed that the low-dose GTN regime had not induced systemic tolerance, which is consistent with a previous study demonstrating that the same regime inhibited ALDH2 without inducing GTN tolerance (Philpott et al., 2007).
Study limitations
Despite strict inclusion/exclusion criteria to standardize experimental procedures, a degree of heterogeneity will exist in the population studied in terms of cause and severity of HF. Furthermore, we did not measure plasma levels of nitrite/nitrate as the results are likely to have been affected by the local and systemic metabolism of GTN to nitrite and nitrate, masking any changes to circulating endogenous nitrite/nitrate levels that may have occurred as a result of ALDH2 inhibition. Finally, we were unable to confirm in our in vivo studies whether ALDH2 activity was actually attenuated by GTN.
Conclusions
Our in vitro data confirm a role for ALDH2 in nitrite-mediated vasorelaxation during hypoxic conditions in both rat aorta and human resistance vessels. Because nitrite is under consideration as a therapeutic agent, the role for ALDH2 in the bioactivation of nitrite during hypoxic vasodilatation is of importance. Our in vitro experiments demonstrate that nitrite could directly affect vasorelaxation without necessitating interaction with blood constituents. However, we cannot discount other non-enzymatic and/or nitrite reductase species (e.g. haem proteins, eNOS, XOR and aldehyde oxidase), which may contribute to the reduction in nitrite during hypoxic conditions in vitro (Li et al., 2008; Pinder et al., 2009; Baliga et al., 2012; Totzeck et al., 2012; Ghosh et al., 2013). Furthermore, although our in vivo results demonstrate a (non-significant) trend towards attenuation of the forearm vasodilator response to nitrite following GTN treatment, we suggest that this is probably due to a decrease in ALDH2 activity. However, no firm conclusion can be drawn about the mechanism sub-serving this effect in vivo. The fact that this effect was non-significant (P = 0.08) may reflect the presence of multiple in vivo mechanisms. In particular, we cannot exclude the role for deoxyhemoglobin in vivo as blood-borne NO species derived from nitrite may contribute to its vasodilator effects (Angelo et al., 2006). In addition, previous work by Gladwin's group has demonstrated that the inhibition of eNOS or XOR does not attenuate nitrite-induced vasodilatation in healthy volunteers (Cosby et al., 2003; Dejam et al., 2007). However, although it seems that XOR may not contribute to nitrite reduction in health, a recent work by Ghosh et al. (2013) reported that XOR does mediate nitrite reduction during pathological conditions (hypertensive animals and patients). Thus, their relative contribution to the hypoxic vasodilator effect of nitrite in man in vivo, in particular in patients with HF, remains unresolved and is yet to be fully explored.
Moreover, the observations of the current study suggest that pre-exposure to GTN (even without inducing tolerance) might attenuate nitrite vasodilator effects. This finding may be of clinical importance when considering nitrite as a therapeutic intervention in patients who may have received recent therapy with organic nitrates. Further studies are required to elucidate the extent to which ALDH2 is involved in nitrite reduction to NO in vivo in man.
Acknowledgments
This work was supported by the British Heart Foundation (FS/10/030/28261).
Glossary
- ALDH
aldehyde dehydrogenase
- eNOS
endothelial NOS
- FBF
forearm blood flow
- GTN
glyceryl trinitrate
- HF
heart failure
- MABP
mean arterial BP
- MetHb
methaemoglobin
- NaNO2
sodium nitrite
- NO2−
nitrite
- Sper/NO
Spermine-NONOate
- XOR
xanthine oxidoreductase
Author contributions
S. A., A. B., E. L.-S. L. and A. G. O. performed the research, data acquisition and analysis. V. S., N. E. D., A. M. and J. M. performed the research. P. N. was responsible for data analysis. R. S. B. and J. D. H. performed the design of the work and interpretation; M. F., M. P. F. and M. M. design the work, data interpretation, article drafting and revision. M. P. F. and M. M. carried out supervision of the study concept, article drafting and final revision.
Conflict of interest
The authors disclose no conflict of interest.
References
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M, et al. The Concise Guide to PHARMACOLOGY 2013/14: G protein-coupled receptors. Br J Pharmacol. 2013a;170:1459–1581. doi: 10.1111/bph.12445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M, et al. The Concise Guide to PHARMACOLOGY 2013/14: enzymes. Br J Pharmacol. 2013b;170:1797–1867. doi: 10.1111/bph.12451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angelo M, Singel DJ, Stamler JS. An S-nitrosothiol (SNO) synthase function of hemoglobin that utilizes nitrite as a substrate. Proc Natl Acad Sci U S A. 2006;103:8366–8371. doi: 10.1073/pnas.0600942103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Badejo AM, Jr, Hodnette C, Dhaliwal JS, Casey DB, Pankey E, Murthy SN, et al. Mitochondrial aldehyde dehydrogenase mediates vasodilator responses of glyceryl trinitrate and sodium nitrite in the pulmonary vascular bed of the rat. Am J Physiol Heart Circ Physiol. 2010;299:H819–H826. doi: 10.1152/ajpheart.00959.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey JC, Feelisch M, Horowitz JD, Frenneaux MP, Madhani M. Pharmacology and therapeutic role of inorganic nitrite and nitrate in vasodilatation. Pharmacol Ther. 2014;144:303–320. doi: 10.1016/j.pharmthera.2014.06.009. [DOI] [PubMed] [Google Scholar]
- Baliga RS, Milsom AB, Ghosh SM, Trinder SL, MacAllister RJ, Ahluwalia A, et al. Dietary nitrate ameliorates pulmonary hypertension: cytoprotective role for endothelial nitric oxide synthase and xanthine oxidoreductase. Circulation. 2012;125:2922–2932. doi: 10.1161/CIRCULATIONAHA.112.100586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basu S, Grubina R, Huang J, Conradie J, Huang Z, Jeffers A, et al. Catalytic generation of N2O3 by the concerted nitrite reductase and anhydrase activity of hemoglobin. Nat Chem Biol. 2007;3:785–794. doi: 10.1038/nchembio.2007.46. [DOI] [PubMed] [Google Scholar]
- Chen Z, Zhang J, Stamler JS. Identification of the enzymatic mechanism of nitroglycerin bioactivation. Proc Natl Acad Sci U S A. 2002;99:8306–8311. doi: 10.1073/pnas.122225199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cosby K, Partovi KS, Crawford JH, Patel RP, Reiter CD, Martyr S, et al. Nitrite reduction to nitric oxide by deoxyhemoglobin vasodilates the human circulation. Nat Med. 2003;9:1498–1505. doi: 10.1038/nm954. [DOI] [PubMed] [Google Scholar]
- Dejam A, Hunter CJ, Tremonti C, Pluta RM, Hon YY, Grimes G, et al. Nitrite infusion in humans and nonhuman primates: endocrine effects, pharmacokinetics, and tolerance formation. Circulation. 2007;116:1821–1831. doi: 10.1161/CIRCULATIONAHA.107.712133. [DOI] [PubMed] [Google Scholar]
- DiFabio J, Ji Y, Vasiliou V, Thatcher GR, Bennett BM. Role of mitochondrial aldehyde dehydrogenase in nitrate tolerance. Mol Pharmacol. 2003;64:1109–1116. doi: 10.1124/mol.64.5.1109. [DOI] [PubMed] [Google Scholar]
- Doyle MP, Pickering RA, DeWeert TM, Hoekstra JW, Pater D. Kinetics and mechanism of the oxidation of human deoxyhemoglobin by nitrites. J Biol Chem. 1981;256:12393–12398. [PubMed] [Google Scholar]
- D'Souza Y, Dowlatshahi S, Bennett BM. Changes in aldehyde dehydrogenase 2 expression in rat blood vessels during glyceryl trinitrate tolerance development and reversal. Br J Pharmacol. 2011;164:632–643. doi: 10.1111/j.1476-5381.2011.01448.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furchgott RF, Bhadrakom S. Reactions of strips of rabbit aorta to epinephrine, isopropylarterenol, sodium nitrite and other drugs. J Pharmacol Exp Ther. 1953;108:129–143. [PubMed] [Google Scholar]
- Ghosh SM, Kapil V, Fuentes-Calvo I, Bubb KJ, Pearl V, Milsom AB, et al. Enhanced vasodilator activity of nitrite in hypertension: critical role for erythrocytic xanthine oxidoreductase and translational potential. Hypertension. 2013;61:1091–1102. doi: 10.1161/HYPERTENSIONAHA.111.00933. [DOI] [PubMed] [Google Scholar]
- Gladwin MT, Shelhamer JH, Schechter AN, Pease-Fye ME, Waclawiw MA, Panza JA, et al. Role of circulating nitrite and S-nitrosohemoglobin in the regulation of regional blood flow in humans. Proc Natl Acad Sci U S A. 2000;97:11482–11487. doi: 10.1073/pnas.97.21.11482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golwala NH, Hodenette C, Murthy SN, Nossaman BD, Kadowitz PJ. Vascular responses to nitrite are mediated by xanthine oxidoreductase and mitochondrial aldehyde dehydrogenase in the rat. Can J Physiol Pharmacol. 2009;87:1095–1101. doi: 10.1139/Y09-101. [DOI] [PubMed] [Google Scholar]
- Greenstein AS, Khavandi K, Withers SB, Sonoyama K, Clancy O, Jeziorska M, et al. Local inflammation and hypoxia abolish the protective anticontractile properties of perivascular fat in obese patients. Circulation. 2009;119:1661–1670. doi: 10.1161/CIRCULATIONAHA.108.821181. [DOI] [PubMed] [Google Scholar]
- Huellner MW, Schrepfer S, Weyand M, Weiner H, Wimplinger I, Eschenhagen T, et al. Inhibition of aldehyde dehydrogenase type 2 attenuates vasodilatory action of nitroglycerin in human veins. FASEB J. 2008;22:2561–2568. doi: 10.1096/fj.07-098830. [DOI] [PubMed] [Google Scholar]
- Huffman JC, Stern TA. Disulfiram use in an elderly man with alcoholism and heart disease: a discussion. Prim Care Companion J Clin Psychiatry. 2003;5:41–44. doi: 10.4088/pcc.v05n0107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irvine JC, Favaloro JL, Widdop RE, Kemp-Harper BK. Nitroxyl anion donor, Angeli's salt, does not develop tolerance in rat isolated aortae. Hypertension. 2007;49:885–892. doi: 10.1161/01.HYP.0000259328.04159.90. [DOI] [PubMed] [Google Scholar]
- Johansson B. A review of the pharmacokinetics and pharmacodynamics of disulfiram and its metabolites. Acta Psychiatr Scand Suppl. 1992;369:15–26. doi: 10.1111/j.1600-0447.1992.tb03310.x. [DOI] [PubMed] [Google Scholar]
- Keith RA, Burkman AM, Sokoloski TD, Fertel RH. Vascular tolerance to nitroglycerin and cyclic GMP generation in rat aortic smooth muscle. J Pharmacol Exp Ther. 1982;221:525–531. [PubMed] [Google Scholar]
- Kollau A, Hofer A, Russwurm M, Koesling D, Keung WM, Schmidt K, et al. Contribution of aldehyde dehydrogenase to mitochondrial bioactivation of nitroglycerin: evidence for the activation of purified soluble guanylate cyclase through direct formation of nitric oxide. Biochem J. 2005;385:769–777. doi: 10.1042/BJ20041354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koppaka V, Thompson DC, Chen Y, Ellermann M, Nicolaou KC, Juvonen RO, et al. Aldehyde dehydrogenase inhibitors: a comprehensive review of the pharmacology, mechanism of action, substrate specificity, and clinical application. Pharmacol Rev. 2012;64:520–539. doi: 10.1124/pr.111.005538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsen FJ, Ekblom B, Sahlin K, Lundberg JO, Weitzberg E. Effects of dietary nitrate on blood pressure in healthy volunteers. N Engl J Med. 2006;355:2792–2793. doi: 10.1056/NEJMc062800. [DOI] [PubMed] [Google Scholar]
- Li H, Cui H, Kundu TK, Alzawahra W, Zweier JL. Nitric oxide production from nitrite occurs primarily in tissues not in the blood: critical role of xanthine oxidase and aldehyde oxidase. J Biol Chem. 2008;283:17855–17863. doi: 10.1074/jbc.M801785200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lundberg JO, Carlstrom M, Larsen FJ, Weitzberg E. Roles of dietary inorganic nitrate in cardiovascular health and disease. Cardiovasc Res. 2011;89:525–532. doi: 10.1093/cvr/cvq325. [DOI] [PubMed] [Google Scholar]
- Mackenzie IS, Maki-Petaja KM, McEniery CM, Bao YP, Wallace SM, Cheriyan J, et al. Aldehyde dehydrogenase 2 plays a role in the bioactivation of nitroglycerin in humans. Arterioscler Thromb Vasc Biol. 2005;25:1891–1895. doi: 10.1161/01.ATV.0000179599.71086.89. [DOI] [PubMed] [Google Scholar]
- Madhani M, Scotland RS, MacAllister RJ, Hobbs AJ. Vascular natriuretic peptide receptor-linked particulate guanylate cyclases are modulated by nitric oxide-cyclic GMP signalling. Br J Pharmacol. 2003;139:1289–1296. doi: 10.1038/sj.bjp.0705365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maher AR, Milsom AB, Gunaruwan P, Abozguia K, Ahmed I, Weaver RA, et al. Hypoxic modulation of exogenous nitrite-induced vasodilation in humans. Circulation. 2008;117:670–677. doi: 10.1161/CIRCULATIONAHA.107.719591. [DOI] [PubMed] [Google Scholar]
- Maher AR, Arif S, Madhani M, Abozguia K, Ahmed I, Fernandez BO, et al. Impact of chronic congestive heart failure on pharmacokinetics and vasomotor effects of infused nitrite. Br J Pharmacol. 2013;169:659–670. doi: 10.1111/bph.12152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marti CN, Gheorghiade M, Kalogeropoulos AP, Georgiopoulou VV, Quyyumi AA, Butler J. Endothelial dysfunction, arterial stiffness, and heart failure. J Am Coll Cardiol. 2012;60:1455–1469. doi: 10.1016/j.jacc.2011.11.082. [DOI] [PubMed] [Google Scholar]
- Ormerod JO, Ashrafian H, Maher AR, Arif S, Steeples V, Born GV, et al. The role of vascular myoglobin in nitrite-mediated blood vessel relaxation. Cardiovasc Res. 2011;89:560–565. doi: 10.1093/cvr/cvq299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paneni F, Osto E, Costantino S, Mateescu B, Briand S, Coppolino G, et al. Deletion of the activated protein-1 transcription factor JunD induces oxidative stress and accelerates age-related endothelial dysfunction. Circulation. 2013;127:1229–1240. doi: 10.1161/CIRCULATIONAHA.112.000826. [DOI] [PubMed] [Google Scholar]
- Pawson AJ, Sharman JL, Benson HE, Faccenda E, Alexander SP, Buneman OP, et al. NC-IUPHAR. The IUPHAR/BPS Guide to PHARMACOLOGY: an expert-driven knowledgebase of drug targets and their ligands. Nucl Acids Res. 2014;42(Database Issue):D1098–D1106. doi: 10.1093/nar/gkt1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perlman DH, Bauer SM, Ashrafian H, Bryan NS, Garcia-Saura MF, Lim CC, et al. Mechanistic insights into nitrite-induced cardioprotection using an integrated metabolomic/proteomic approach. Circ Res. 2009;104:796–804. doi: 10.1161/CIRCRESAHA.108.187005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Philpott A, Sage P, Stafford I, Murphy G, de la Lande IS, Stuklis R, et al. 2007. Dissociation of aldehyde dehydrogenase activity from nitrate effect, bioconversion, and tolerance in humans.
- Pinder AG, Pittaway E, Morris K, James PE. Nitrite directly vasodilates hypoxic vasculature via nitric oxide-dependent and -independent pathways. Br J Pharmacol. 2009;157:1523–1530. doi: 10.1111/j.1476-5381.2009.00340.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiso M, Tejero J, Basu S, Azarov I, Wang X, Simplaceanu V, et al. Human neuroglobin functions as a redox-regulated nitrite reductase. J Biol Chem. 2011;286:18277–18289. doi: 10.1074/jbc.M110.159541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Totzeck M, Hendgen-Cotta UB, Luedike P, Berenbrink M, Klare JP, Steinhoff HJ, et al. Nitrite regulates hypoxic vasodilation via myoglobin-dependent nitric oxide generation. Circulation. 2012;126:325–334. doi: 10.1161/CIRCULATIONAHA.111.087155. [DOI] [PMC free article] [PubMed] [Google Scholar]