

Abstract
Background and Purpose
Toll-interacting protein (Tollip) is an endogenous inhibitor of toll-like receptors, a superfamily that plays a pivotal role in various pathological conditions, including myocardial infarction (MI). However, the exact role of Tollip in MI remains unknown.
Experimental Approach
MI models were established in Tollip knockout (KO) mice, mice with cardiac-specific overexpression of human Tollip gene and in their Tollip+/+ and non-transgenic controls respectively. The effects of Tollip on MI were evaluated by mortality, infarct size and cardiac function. Hypoxia-induced cardiomyocyte damage was investigated in vitro to confirm the role of Tollip in heart damage.
Key Results
Tollip expression was dramatically up-regulated in human ischaemic hearts and infarcted mice hearts. MI-induced mortality, infarct size and cardiac dysfunction were decreased in Tollip-KO mice compared with Tollip+/+ controls. Ischaemic hearts from Tollip-KO mice exhibited decreased inflammatory cell infiltration and reduced NF-κB activation. Tollip depletion also alleviated myocardial apoptosis by down-regulating pro-apoptotic protein levels and up-regulating anti-apoptotic protein expressions in infarct border zone. Conversely, MI effects were exacerbated in mice with cardiac-specific Tollip overexpression. This aggravated MI injury by Tollip in vivo was confirmed with in vitro assays. Inhibition of Akt signalling was associated with the detrimental effects of Tollip on MI injury; activation of Akt largely reversed the deleterious effects of Tollip on MI-induced cardiomyocyte death.
Conclusions and Implications
Tollip promotes inflammatory and apoptotic responses after MI, leading to increased mortality and aggravated cardiac dysfunction. These findings suggest that Tollip may serve as a novel therapeutic target for the treatment of MI.
Tables of Links
| Targets | |||
|---|---|---|---|
| Catalytic receptorsa | Enzymesb | ||
| Toll-like receptors | Akt (PKB) | GSK3β | mTOR |
| Caspase 3 | JNK | p38 | |
| ERK (MAPK) | MEK |
| Ligands |
|---|
| Buprenorphine |
| MK-2206 |
These Tables list key protein targets and ligands in this article which are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Pawson et al., 2014) and are permanently archived in the Concise Guide to PHARMACOLOGY 2013/14 (a,bAlexander et al., 2013a,b).
Introduction
Myocardial infarction (MI) caused by coronary artery blockade is the major cause of death worldwide (Rafiq et al., 2014). The continuous inflammatory response and necrosis of ischaemic tissue are two of the most marked characteristics that could mutually enhance during the process of MI-induced heart damage and eventually lead to heart failure (Jones et al., 2014). An inflammatory response is triggered immediately after MI. Although the infiltration of inflammatory cells into the myocardium could scavenge necrotic myocytes and extracellular matrix (ECM) debris to promote the healing process (Frangogiannis, 2012), sustained inflammatory and immune infiltrate is directly linked to myocardial apoptosis and impairs cardiac function (Kin et al., 2006; Schofield et al., 2013). MI-induced myocardial apoptosis persists months beyond the acute phase after MI (Baldi et al., 2002). The loss of cardiomyocytes in both the infarct border zone and remote areas causes persistent structural and functional impairments in the heart, thus accounting for the progression of heart failure (Wencker et al., 2003; Lal et al., 2012). The essential roles of myocardial apoptosis and inflammation in the pathological progression of MI-related cardiac impairments underscore the importance of anti-apoptotic and anti-inflammatory strategies and are of great clinical relevance. A better understanding of the regulators involved in myocardial apoptosis and inflammation is important for the exploitation of novel therapeutic targets for MI.
Toll-interacting protein (Tollip), initially identified as an adaptor protein in IL-1 receptor signalling, contains a C2 domain, a coupling ubiquitin to the endoplasmic reticulum degradation domain and a Tom1 binding domain (Burns et al., 2000). This molecule is ubiquitously expressed, with particularly high expression levels in the heart (Liu et al., 2014). Studies from our group and others recently reported that Tollip negatively regulates pathological cardiac hypertrophy in vivo and in vitro (Hu et al., 2009; Liu et al., 2014), suggesting a critical role for Tollip in heart disease. However, despite pressure overload and myocardial ischaemia being major contributors to heart failure, distinct pathological processes participate in cardiac hypertrophy and MI (Heineke and Molkentin, 2006; Heusch et al., 2014). Thus, it is worth investigating whether Tollip plays a role in the pathogenesis of myocardial ischaemia-induced cardiac dysfunction. Notably, previous studies provide extensive evidence that Tollip plays a pivotal role in the immune response. Tollip can regulate inflammatory cytokine production and apoptotic responses in several cell types by suppressing the kinase activity of IRAK-1 and toll-like receptor signalling (Burns et al., 2000; Zhang and Ghosh, 2002), which are critically involved in the pathogenesis of MI (Shishido et al., 2003; Riad et al., 2008; Timmers et al., 2008). The regulatory effects of Tollip on inflammation and apoptosis provide clues to its involvement in MI-induced heart failure. However, no report on this topic is currently available.
In the current study, to determine the role of Tollip in MI, we examined the post-infarction outcomes in both global Tollip knockout (KO) mice and transgenic (TG) mice with cardiac-specific overexpression of the human Tollip gene, and found that Tollip-TG mice have a higher mortality rate and develop more severe cardiac dysfunction after MI compared with their non-transgenic (NTG) controls, whereas Tollip depletion ameliorates this pathological process. Furthermore, we provide evidence that the effect of Tollip on MI is mediated, at least in part, by a blockade of Akt signalling.
Methods
Human heart samples
All procedures in the current study were approved by the Ethics Committee of Renmin Hospital of Wuhan University (Wuhan, China) and conformed to the principles outlined in the Declaration of Helsinki. Patients with ischaemic heart disease (IHD) who intended to undergo heart transplantation were the main donors of left ventricular heart samples (Zhang et al., 2014). The normal left ventricular samples were obtained from donor hearts that failed donor–recipient matching (Chen et al., 2013; Jiang et al., 2014b,c). Additionally, written informed consent was obtained from all patients and donors.
Experimental animals
All of the animal protocols in this study were approved by the Animal Care and Use Committee of Renmin Hospital of Wuhan University and conformed to the Guidelines for the Care and Use of Laboratory Animals prepared by the National Academy of Sciences and published by the National Institutes of Health. Experiments involving animals consulted the ARRIVE guidelines (Kilkenny et al., 2010; McGrath et al., 2010). A total of 226 male mice with C57BL/6J background aged 8–10 weeks (24–27 g) were used in the present study. Mice were housed with four to five mice in a cage and maintained under a 12/12 h light/dark cycle (lights on 07:00 a.m.) with the ambient humidity at 50–80% and the temperature at 21 ± 2°C. Food and water were provided ad libitum. The Tollip-KO mice were purchased from the European Mouse Mutant Archive (B6.Cg-Tolliptm1Kbns/Cnrm). The α-MHC-Tollip TG mice were established by microinjecting an α-MHC-Tollip construct into fertilized mouse embryos as previously described (Liu et al., 2014). The KO mice and their Tollip+/+ littermates as well as TG and NTG mice were randomized for subsequent experiments. Experimental subjects/preparations were randomized to groups and a randomized double-blind study was employed.
Mouse MI model
The MI model was established by ligating the left anterior descending (LAD) coronary artery as previously described (Xiao et al., 2012; Sun et al., 2013). Briefly, mice (aged 8–10 weeks, 23–28 g body weight) were i.p. injected with 0.3% pentobarbital sodium (90 mg·kg−1; P3761, Sigma-Aldrich, St. Louis, MO, USA) and ventilated with room air using a small animal ventilator (model VFA-23-BV; Kent Scientific, Litchfield, CT, USA). Adequate anaesthesia was characterized by slow but regular breathing and no pedal withdrawal reflex. The hearts were exposed through the third or fourth intercostal spaces after thoracotomy, and the LAD was ligated under the tip of the left atrial appendage with a 7-0 silk suture. Paleness around and below the ligation point was recognized as a sign of successful ligation. For the sham operation, a silk suture was placed around the LAD, but no ligation was performed. After the surgery, the chest of the mouse was closed, and the animal received buprenorphine (0.1 mg·kg−1, s.c.) for post-operative analgesia. Finally, the animals were placed back into independently ventilated cages after they had fully recovered. During the 4 week follow-up, animal deaths were recorded. All of the operators and the personnel involved in the subsequent data analysis were blinded to the study groups.
Echocardiography and cardiac catheterization
The echocardiography and haemodynamic evaluations have been described in detail elsewhere (Jiang et al., 2013; 2014a,b). Briefly, the mice were anaesthetized by inhalation of isoflurane at a concentration of 1.5–2.0%, and echocardiography was performed using a MyLab30CV ultrasound (Biosound Esaote, Inc., Indianapolis, IN, USA) with a 15 MHz linear array ultrasound transducer. The left ventricular end-diastolic diameter and left ventricular end-systolic diameter were obtained and analysed to obtain the left ventricular ejection fraction and fractional shortening (FS).
Cardiac catheterization was conducted using a catheter conducer (SPR-839, Millar Instruments, Houston, TX, USA) to evaluate the haemodynamic status of the left ventricle. The catheter was inserted into the left ventricular cavity via the right carotid artery under pressure control. Pressure signals and dP/dT were recorded continuously using an ARIA pressure–volume conductance system coupled with a Powerlab/4SP A/D converter after stabilization for 15 min. For subsequent analysis of pressure–volume loops, the PVAN software (Millar Instruments) was used.
Morphological analysis and infarct size measurement
Mice were killed at the indicated time points via an overdose of anaesthesia to access the morphology and infarction area of hearts. To obtain pathological samples, the excised hearts were immediately placed in 10% KCl solution to induce cardiac arrest during the diastolic phase and then injected with 10% neutral formalin solution improved fixation to prevent the collapse of the infarct site. Subsequently, the left and right atria of the heart were cut off, leaving the double ventricle for serial dehydration and embedding. The heart was then continuously sectioned from the bottom to the apex along the short axis. Slices, stained with haematoxylin and eosin, were collected with an interval of 300 μm between each section to determine the infarct size. Image-Pro Plus 6.0 (Media Cybernetics, Inc. Rockville, MD, USA) was used to calculate the length of the midline measured as midline circumference, while midline infarct length was taken as the midline of the length of infarct. Infarct size derived from midline length measurement was calculated by dividing the sum of midline infarct lengths by the sum of midline circumferences from all sections and multiplying by 100%. All measurements were made in a blinded fashion.
Immunofluorescence staining
To determine the presence of immune cell infiltration and quantify its magnitude, the paraffin sections were stained using standard immunofluorescence staining techniques. In brief, the sections were prepared, dried, dewaxed, hydrated and repaired. Subsequently, the sections were washed in PBS, sealed with 10% sheep serum for 1 h and incubated with primary antibodies at 4°C overnight. And then, the sections were washed in PBS and incubated with the corresponding secondary antibodies for 1 h at 37°C, followed by DAPI (S36939, Invitrogen, Carlsbad, CA, USA) staining to mark the nuclei. The qualitative expression of CD3, MAC1 and LY6G was assessed using a special Olympus DP72 fluorescence microscope (model BX51TRF, Olympus Corporation, Shinjuku, Tokyo, Japan), whereas quantitative assessment and analysis were performed by counting the number of positive cells in at least five randomly selected fields of the infarct border zone for each heart using Image-Pro Plus 6.0.
Neonatal rat cardiomyocytes (NRCMs) culture and infection with recombinant adenoviral vectors
Primary cultured NRCMs were prepared as previously described (Jiang et al., 2013; 2014a,b). In brief, the hearts of 1- to 2-day-old Sprague–Dawley rats were excised and digested with PBS containing 0.03% trypsin and 0.04% collagenase type II to isolate the cardiomyocytes from the fibroblasts. The cardiomyocytes were then seeded at a density of 1 × 106 cells per well in 6-well culture plates coated with fibronectin in plating medium, which consisted of F10 medium supplemented with 10% FCS and penicillin/streptomycin. To silence or overexpress Tollip, we infected the NRCMs with adenoviral short hairpin Tollip (AdshTollip) or adenoviral Tollip (AdTollip) as described previously (Liu et al., 2014). Additionally, to examine the role of Akt in the regulatory effects of Tollip on cardiomyocyte damage, the AdshTollip- or AdTollip-infected NRCMs were co-infected with AddnAkt (a dominant negative mutant of Akt) or AdcaAkt (constitutively activation of Akt) respectively. The AdshRNA- or AdGFP-infected NRCM cells were used as controls.
NRCM hypoxia treatment and viability assay
For the hypoxia treatment of NRCMs, after 24 h of normal culture, the medium was replaced with F10 medium containing 0.1% FCS and 0.1 mM BrdU (to inhibit the proliferation of fibroblasts). The cultured cardiomyocytes were exposed to hypoxia stimulation in a BioSpherix C-Chamber (model C-274, BioSpherix, Redfield, NY, USA) with a standard cell culture chamber. Using a ProOx 110 oxygen controller and a ProCO2 CO2 controller (BioSpherix, Redfield, NY, USA), the oxygen (O2) concentration was maintained at 5% and the CO2 concentration was maintained at 5% inside the C-Chamber by injecting N2 and CO2. For the controls, the C-Chamber was maintained at 37°C and filled with 5% CO2 and 95% air. Cell viability was assessed by colorimetric LDH cytotoxicity assay (G1782, Promega, Madison, WI, USA) and cell counting kit-8 (CK04, Dojindo, Tokyo, Japan) assay according to the manufacturer's instruction.
Determination of cell death
As previously described (Xiao et al., 2012; Wang et al., 2013), TUNEL was used to determine cell death due to apoptosis using the ApopTag® Plus In Situ Apoptosis Fluorescein Detection Kit (#S7111, Millipore, Billerica, MA, USA). The TUNEL-positive cells (%) in the infarcted border zone were assessed using the quantitative software Image-Pro Plus 6.0 under high magnification (×400).
Hoechst 33258/propidium iodide (PI) double staining was used to detect cell death as previously described (Zhang et al., 2014). Briefly, the NRCMs were exposed to hypoxia stimulation for 24 h and fixed with 4% paraformaldehyde for 15 min at 37°C. After washing in PBS, the cells were successively stained with Hoechst 33258 (H3569, Invitrogen) and PI (P4864, Sigma-Aldrich) for 10 min respectively. Finally, cells were washed in PBS, and the slides were sealed with glycerin. To observe the extent of cardiomyocyte apoptosis, the cells were photographed using a fluorescence microscope.
Western blot analysis
Total protein was extracted from the left ventricular samples and primary cultured cells. The protein concentration was determined using the Pierce BCA protein assay kit (23225, Thermo Scientific, Waltham, MA, USA). Protein extracts (30 μg) were separated using SDS-PAGE, transferred to a PVDF membrane (IPVH00010, Millipore) and incubated with indicated primary antibodies at 4°C overnight. After incubation with a peroxidase-conjugated secondary antibody – that is, peroxidase-affinipure goat anti-mouse IgG (H + L) (115-035-003) or peroxidase-affinipure goat anti-rabbit IgG (H + L) (111-035-003) (Jackson ImmunoResearch, West Grove, PA, USA) – the signals were visualized using a Bio-Rad ChemiDocTM XRS+ (Bio-Rad, Hercules, CA, USA). The specific protein expression levels, expressed as the grey values of the bands, were normalized to the GAPDH (loading control) values of the corresponding samples on the same nitrocellulose membrane.
Design and statistical analysis
The data are presented as the mean ± SD. Survival analysis was performed employing Kaplan–Meier curves and the survival rates of the indicated groups were compared by log-rank test. Independent sample t-tests and one-way anova were used to respectively compare the data between two groups and among multiple groups using Statistical Package for the Social Sciences (SPSS, IBM Corporation, Endicott, NY, USA) 13.0. P < 0.05 was considered statistically significant.
Reagents
Antibodies against the following proteins were purchased from Cell Signaling Technology (Danvers, MA, USA) (1:1000 dilution): Bax (#2772), Bcl-2 (#2870), cleaved caspase-3 (#9661), caspase-3 (#9662), P-MEK (#9154), MEK (#9122), P-ERK (#4370), ERK (#4695), P-JNK (#4668), JNK (#9258), P-p38 (#4511), p38 (#9212), P-Akt (#4060), Akt (#4691), P-mTOR (#2971), mTOR (#2983), P-S6 (#2215), S6 (#2217), P-FOXO1 (#9461), FOXO1 (#2880), P-GSK3β (#9322), GSK3β (#9315), P-p65 (#3033), p65 (#4764), P-IκBα (#9246), IκBα (#4814) and GAPDH (#2118). The Tollip (SC27315) antibody was purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). The MAC1 (ab75476) antibody was purchased from Abcam (Cambridge, UK). CD3 antibody (GA045204) was obtained from GeneTech (Shanghai, China). The LY6G (551459) antibody was purchased from BD Biosciences (Franklin Lakes, NJ, USA). The Akt-specific inhibitor MK-2206 (S1078) was purchased from Selleck Chemicals LLC (Houston, TX, USA).
Results
Tollip expression is elevated in human and murine ischaemic hearts
As the alteration in the expression of target proteins might provide initial evidence for the involvement of certain factors in diseases; the protein expressions Tollip were first tested in the left ventricles of IHD patients undergoing heart transplantation due to end-stage heart failure. The Western blotting results indicated that the Tollip protein levels were increased by 40.14% in failing human hearts compared with that of donors (Figure 1A). Consistently, in murine hearts subjected to MI, the Tollip expression levels significantly increased in the myocardium derived from the infarct border zone after 1 week of LAD coronary artery ligation, and this increase progressively achieved to a higher level in 4 week post-MI injury (Figure 1B). The dramatic alterations in Tollip protein levels suggest that Tollip might be involved in and exert crucial roles in the progression of MI-induced heart failure.
Figure 1.
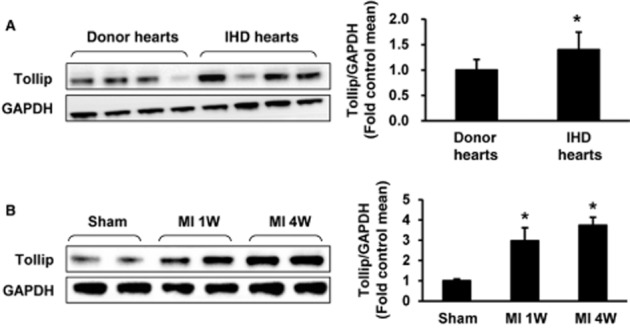
Up-regulation of Tollip in hearts subjected to MI. (A) Representative Western blotting of Tollip in the heart samples from normal donors (n = 6) and patients with IHD (n = 7). (B) Western blotting results for Tollip in the infarcted border zone of C57BL/6J mice at the indicated time points post-MI (n = 5). Left, representative Western blotting. Right, quantitative results. *P < 0.05 versus normal donors or sham-operated hearts.
Tollip exacerbates mortality, infarct size and cardiac dysfunction following MI
The elevated Tollip expression in ischaemic hearts promotes us to investigate the potential mediatory functions of Tollip in MI. Thus, the in vivo MI model was established in Tollip-KO and cardiac-specific Tollip-TG mice as well as their Tollip+/+ and NTG controls. Post MI operations, survival rates were monitored in the indicated groups and a significantly higher survival was observed in Tollip-KO mice compared with Tollip+/+ controls. As Figure 2A shows, only 19 out of 35 Tollip+/+ mice (54.29%) survived up to 4 weeks post-MI, while in the Tollip−/− group 19 out of 24 mice (79.17%) survived. Additionally, after MI injury, obvious infarction was observed in cross-sections of heart tissue from Tollip+/+ mice at 3 days and 4 weeks, which was dramatically reduced by Tollip depletion at 3 days (6.41 ± 0.12% in Tollip−/−/MI group vs. 13.78 ± 0.6% in Tollip+/+/MI group) and at 4 weeks (18.03 ± 3.75% in Tollip−/−/MI group vs. 31.55 ± 4.61% in Tollip+/+/MI group, Figure 2B,C). Concomitant with the diminished infract size, Tollip deficiency greatly ameliorated MI-induced cardiac dysfunction, as evidenced by the promoted FS percentage (FS%), ejection fraction percentage (EF%), dP/dtmax and dP/dtmin in Tollip−/− mice compared with those of Tollip+/+ controls post-MI (Figure 2D).
Figure 2.

Tollip increases MI-induced mortality, infarct size and cardiac dysfunction. (A) Kaplan–Meier survival curves for Tollip+/+ and Tollip KO mice after MI (*P < 0.05 vs. Tollip+/+/MI group). (B) Representative images of haematoxylin and eosin staining in heart cross-sections of Tollip+/+ and Tollip−/− mice at 3 days and 4 weeks after MI surgery (n = 6 mice per experimental group). (C) Measurement of infarct size in the heart tissue of mice from Tollip+/+ and Tollip−/− groups. *P < 0.05 versus Tollip+/+/MI group at 3 days; #P < 0.05 versus Tollip+/+/MI group at 4 weeks. (D) Echocardiographic and haemodynamic assessment of cardiac function in Tollip+/+ and Tollip−/− mice after MI (n = 8 mice per experimental group). *P < 0.05 versus Tollip+/+/sham operated group; #P < 0.05 versus Tollip+/+/MI group. (E) Comparison of mortality between NTG mice and Tollip-TG mice at 3 days and 4 weeks post-MI injury (*P < 0.05 vs. NTG/MI group). (F,G) Representative images and evaluation of infarct size in NTG and Tollip-TG mice at 3 days and 4 weeks after MI surgery (n = 6 mice per experimental group). *P < 0.05 versus NTG/MI group at 3 days; #P < 0.05 versus NTG/MI group at 4 weeks. (H) The parameters of cardiac function in NTG and Tollip-TG mice after 4 weeks of MI surgery (n = 8 for each group). *P < 0.05 versus NTG/sham operated group; #P < 0.05 versus NTG/MI group.
To provide further support for the potential roles of Tollip in MI-induced heart damage, the influence of Tollip up-regulation on MI outcomes was assayed and compared in Tollip-TG mice and their NTG controls subjected to MI. In contrast to Tollip-deficient mice, the Tollip-TG mice presented a significantly increased mortality rate (66.07% in Tollip-TG/MI group vs. 45.71% in NTG/MI group), while the infarct size in Tollip-TG heart sections strikingly increased from 11.07 to 24.03% at 3 days, and from 32.29 to 45.71% at 4 weeks after MI in NTG controls (Figure 2E–G). Additionally, compared with NTG mice, the reduced FS%, EF%, dP/dtmax and dP/dtmin in Tollip-TG further confirmed the aggravated effect of Tollip overexpression on MI injury (Figure 2H). However, interestingly, at baseline, neither global KO nor cardiac overexpression of Tollip manifested abnormalities in cardiac structure or function compared with wild-type controls. Collectively, these results obtained from Tollip-KO and Tollip-TG mice indicate that during MI-induced heart damage, Tollip could deteriorate mortality rate, infarct size and cardiac dysfunction.
Tollip augments immune cell infiltration following MI
Given that inflammatory response is a key component of ischaemic cardiac injury (Schofield et al., 2013), we determined the effects of Tollip on inflammatory and immune cell infiltration in the infarct border zone of different experimental groups. As shown in Figure 3A, the Mac-1-positive macrophages, LY6G-positive neutrophiles and CD3-positive T cells accumulated in the infarct border zone at 3 days and 4 weeks after MI, while Tollip depletion led to profoundly reduced inflammatory cell infiltration (Figure 3A). In contrast, overexpression of Tollip in the heart significantly promoted inflammatory and immune cells infiltration to infarct border zone at 3 days and 4 weeks after MI (Figure 3B), indicating the potently exacerbated effects of Tollip on MI-related migration and accumulation of inflammatory and immune cells.
Figure 3.
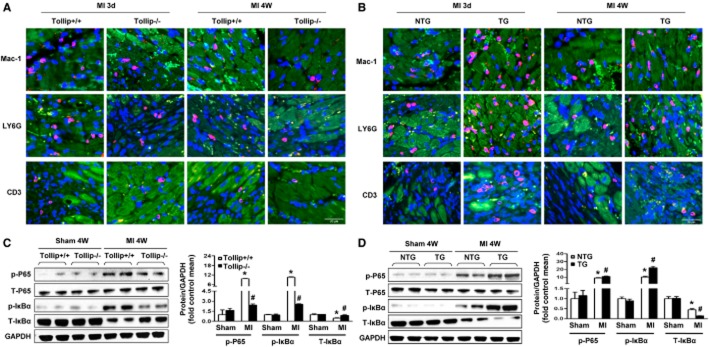
Tollip increases inflammatory and immune cell infiltration and NF-κB activation after MI. (A,B) Representative immunofluorescence staining images of macrophage (Mac-1), neutrophil (LY6G) and T cell (CD3) accumulation in the infarct border zone of Tollip+/+ and Tollip-KO mice (A) and in NTG and Tollip-TG mice (B) at 3 days and 4 weeks after MI. (C,D) Representative Western blotting and quantitative results showing the phosphorylation levels of p65 and IκBα in the infarct border zone of hearts from (C) Tollip+/+ and Tollip-KO mice and from (D) NTG and Tollip-TG mice at 4 weeks after MI (n = 13 mice per experimental group). Left, representative Western blotting. Right, quantitative results. *P < 0.05 versus Tollip+/+/sham or NTG/sham-operated group; #P < 0.05 versus Tollip+/+/MI or NTG/MI group.
NF-κB signalling is one of the most critical molecular programmes that modulate the initiation and persistence of inflammatory response. Thus, to explore the mechanism underlying Tollip-induced alterations in inflammatory and immune cell infiltrations, we examined the regulatory effect of Tollip on NF-κB pathways. In the ischaemic heart samples (infarct border zone), NF-κB signalling was significantly activated, which was indicated by the down-regulated IκBα expression and up-regulated protein expression of phosphorylated p65 and IκBα in the heart from Tollip+/+ and NTG mice (Figure 3C,D). As expected, the degradation of IκBα in the myocardium was attenuated in Tollip-KO mice compared with Tollip+/+ mice after MI; consistently, the phosphorylation levels of the NF-κB family member p65 and IκBα were significantly lower in Tollip-KO mice (Figure 3C). Conversely, the hearts of TG mice exhibited increased IκBα degradation and p65 phosphorylation when compared with their NTG controls (Figure 3D). Taken together, these data imply that Tollip could promote inflammatory response in the context of MI through enhancing the activation of the NF-κB pathway.
Tollip mediates cardiomyocyte apoptosis following MI
During MI-induced heart damage, cardiomyocyte apoptosis contributes to the formation of infract size, and thus lead to cardiac dysfunction or even heart rupture after MI (Pu et al., 2013). Therefore, the possible role of Tollip in myocardial apoptosis was investigated applying TUNEL staining assay in the infarct border zone. After MI, significantly lower percentage of TUNEL-positive apoptotic cardiomyocytes was observed in Tollip-KO hearts (9.98% at 3 days, and 4.89% at 4 weeks) compared with that of Tollip+/+ hearts (19.38% at 3 days, and 10.98% at 4 weeks) (Figure 4A,B). In contrast, Tollip overexpression dramatically increased the percentage of TUNEL-positive cardiomyocytes to 32.99% at 3 days and to 19.94% at 4 weeks respectively (Figure 4A,C). To gain a deeper insight into the mechanisms underlying the pro-apoptotic property of Tollip, we evaluated pro- and anti-apoptotic protein levels. After MI injury, the anti-apoptotic protein Bcl-2 and pro-apoptotic protein Bax were significantly decreased and increased respectively in Tollip+/+ and NTG mice (Figure 4D,E). The results of Western blotting showed that Tollip KO maintained the high protein expression level of Bcl-2 and down-regulated the expression of Bax compared with Tollip+/+ controls (Figure 4D). However, hearts from the Tollip-TG mice showed up-regulation of Bax and down-regulation of Bcl-2 compared with the hearts from NTG controls (Figure 4E). Correspondingly, a significant decrease in caspase-3 cleavage was detected in Tollip-KO hearts compared with Tollip+/+ controls (Figure 4D), whereas Tollip-TG mice showed a significantly increased caspase-3 cleavage level compared with those of NTG controls (Figure 4E).
Figure 4.
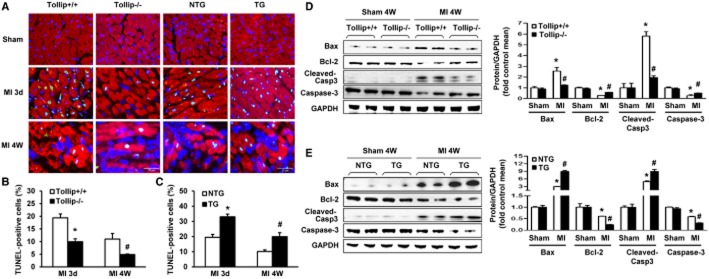
Tollip promotes apoptosis and regulates the expression of apoptosis-related genes in response to MI. (A–C) Representative TUNEL staining images (A) and quantitative results (B,C) TUNEL-positive cells in heart sections from (B) Tollip+/+ and Tollip−/− mice and from (C) NTG and Tollip-TG mice at 3 days and 4 weeks after MI (n = 6 mice per experimental group, *P < 0.05 vs. Tollip+/+/MI or NTG/MI group at 3 days; #P < 0.05 vs. Tollip+/+/MI or NTG/MI group at 4 weeks). (D,E) Representative Western blotting and quantitative analysis showing the protein levels of Bax, Bcl-2 and cleaved caspase-3 in (D) Tollip+/+ and Tollip-KO mice and in (E) NTG and Tollip-TG mice at 4 weeks after MI induction (n = 13 mice per experimental group). Left, representative Western blotting. Right, quantitative results. *P < 0.05 versus Tollip+/+/sham or NTG/sham-operated group; #P < 0.05 versus Tollip+/+/MI or NTG/MI group.
To further confirm that Tollip is directly linked to cardiomyocyte apoptosis, we tested the effect of Tollip in hypoxia-stimulated cytotoxicity of primary cardiomyocytes. Cultured NRCMs were infected with AdshTollip or AdTollip to silence or overexpress Tollip respectively. The hypoxia-induced cell death was visualized by Hoechst/PI double staining. Consistent with the in vivo results, Tollip promoted hypoxia-elicited cell death in cardiomyocytes, as AdshTollip infection led to fewer PI-positive cells, whereas much abundant PI-positive cells were observed in the AdTollip-infected cardiomyocytes (Figure 5A,C). Additionally, the exacerbated cytotoxicity induced by Tollip overexpression was evidenced by the decreased cell viability and increased LDH release in AdshTollip-infected cardiomyocytes, while AdshTollip infection cause the opposite results (Figure 5B,D). Based on these results, we further tested the effect of Tollip on apoptosis-related protein expression in NRCMs exposed to hypoxia. AdshTollip-infected cells presented significantly higher levels of the anti-apoptotic protein Bcl-2 and markedly lower levels of the pro-apoptotic protein Bax and cleaved caspase-3 compared with AdshRNA-infected cells exposed to hypoxia (Figure 5E). In contrast, AdTollip-infected NRCMs exhibited significantly increased protein levels of Bax and cleaved caspase-3 as well as lower expression of Bcl-2 compared with AdGFP-infected NRCMs (Figure 5F). Taken together, these results suggest that Tollip mediates apoptosis-related protein expression and thus promotes cardiomyocytes apoptosis.
Figure 5.

Tollip enhances hypoxia-induced cardiomyocyte death and regulates the expression of apoptosis-related genes in vitro. (A,C) Representative photomicrographs of (A) AdshTollip- or (C) AdTollip-infected primary cardiomyocytes double labelled with Hoechst 33258 for nuclei (blue) and PI for dead cells (red) after 24 h of hypoxia stimulation (n = 5). (B,D) Determination of cell viability and cell toxicity by cell counting kit-8 (CCK-8) and LDH release assay in NRCMs with (B) Tollip knockdown or (D) overexpression after exposure to hypoxia for 24 h (n = 5). *P < 0.05 versus AdshRNA/normoxia or AdGFP/normoxia; #P < 0.05 versus AdshRNA/hypoxia or AdGFP/hypoxia. (E,F) Western blotting results for the protein levels of Bax, Bcl-2 and cleaved caspase-3 in NRCMs infected with (E) AdshTollip or (F) AdTollip after hypoxia treatment for 24 h (n = 5 samples). Top, representative Western blotting. Bottom, quantitative results. *P < 0.05 versus AdshRNA/normoxia or AdGFP/normoxia; #P < 0.05 versus AdshRNA/hypoxia or AdGFP/hypoxia.
Tollip regulates MI outcomes in an Akt-dependent manner
In an attempt to characterize the downstream effectors responsible for the deleterious effects of Tollip after MI, we first examined whether the MAPK cascade, a signalling pathway that could greatly regulate MI-induced inflammatory and apoptotic responses (Xuan et al., 2011), was disrupted by Tollip in hearts subjected to MI. As shown in Figure 6A,B, the phosphorylation of mitogen-activated protein kinase kinase (MEK)1/2, ERK1/2, JNK and p38 was increased in response to ischaemic stress. Nevertheless, no significant difference was observed between the Tollip-KO or Tollip-TG mice and their littermate controls in the infarct border zone (Figure 6A,B). These data indicate that the MAPK cascade might be not involved in Tollip-regulated MI injury. Therefore, we further examined the effect of Tollip on another important molecular pathway, Akt signalling, in MI progression. Interestingly, the phosphorylation levels of proteins of Akt signalling including Akt, mammalian target of rapamycin (mTOR), glycogen synthase kinase 3-beta (GSK3β), S6 and Forkhead box protein O1 (FOXO1) were significantly higher in the Tollip-KO mice than in the Tollip+/+ controls, whereas the phosphorylation expressions of these proteins were dramatically reduced in Tollip-TG hearts compared with the NTG controls (Figure 6C–E).
Figure 6.
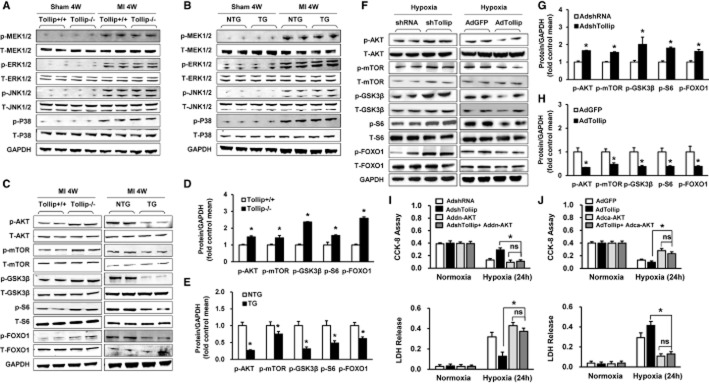
Tollip exacerbates cardiac injury post-MI by inactivating the Akt signalling pathway. (A,B) Representative Western blotting showing phosphorylated and total MEK1/2, ERK1/2, JNK1/2 and p38 protein levels from (A) Tollip+/+ and Tollip−/− mice and from (B) NTG and Tollip-TG mice at 4 weeks after MI (n = 13 mice per experimental group). (C– E) The (C) representative Western blotting and (D and E) quantitative results showing the levels of phosphorylated and total Akt, mTOR, S6, FOXO1 and GSK3β protein from heart tissue of mice in the indicated groups at 4 weeks after MI (n = 13 mice per experimental group). *P < 0.05 versus Tollip+/+/MI or NTG/MI group. (F–H) Representative Western blotting results showing the expression levels of proteins in Akt signalling in NRCMs infected with AdshTollip or AdTollip after hypoxia treatment for 24 h (n = 6 samples). *P < 0.05 versus AdshRNA/hypoxia or AdGFP/hypoxia. (I) The cell viability and LDH release of cardiomyocytes infected with AdshRNA, AdshTollip, AddnAkt or AdshTollip + AddnAkt after exposure to hypoxia for 24 h (n = 6 samples). (J) The cell viability and LDH release of cardiomyocytes in the indicated groups after treatment of hypoxia for 24 h (n = 6 samples). ns, no significant difference.
To further support the regulatory role of Tollip in Akt phosphorylation, we performed gain- and loss-of-function studies in cultured NRCMs. As shown in Figure 6F–H, consistent with the in vivo data, obvious increased expressions of P-Akt, P-mTOR, P-GSK3β, P-S6 and P-FOXO1 were detected in the AdshTollip-infected NRCMs compared with AdshRNA-transfected NRCMs; nevertheless, the AdTollip infected strikingly suppressed the phosphorylation of Akt signalling compared with the AdGFP-transfected cells. Moreover, to evaluate the influence of Akt signalling in the Tollip-regulated MI progression, the AdshTollip- and AdTollip-infected cardiomyocytes were co-infected with AddnAkt and AdcaAkt, respectively, and exposed to hypoxia for 24 h. Notably, the ameliorated and aggravated cell damage induced by artificial down-regulation and up-regulation of Tollip were largely abolished by AddnAkt and AdcaAkt, respectively (Figure 6I–J), which suggest the Akt-dependent manner of Tollip-mediated cardiomyocyte damage in vitro. Furthermore, Tollip+/+ and Tollip−/− mice were treated with the Akt-specific inhibitor MK-2206 (120 mg·kg−1, p.o., three times a week) for 1 week to evaluate whether the detrimental role of Tollip on MI-induced injury is mediated by AKT in vivo. Our results showed that compared with vehicle (DMSO treatment), MK-2206 treatment significantly reduces the phosphorylation levels of Akt, increases infarct size and aggravates cardiac dysfunction (Figure 7A–D). Notably, Tollip−/−+MK-2206 mice had a comparable infarct size and cardiac function with Tollip+/++MK-2206 mice, which indicated that the detrimental effects of Tollip on cardiac injury in response to MI is majorly dependent on Akt inhibition (Figure 7B–D).
Figure 7.
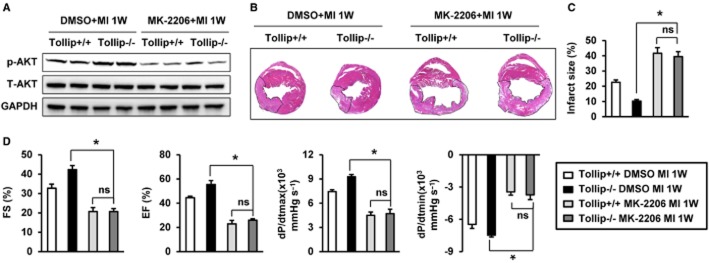
Akt inhibition was responsible for the detrimental role of Tollip on MI-induced cardiac damage. (A) Representative Western blotting showing phosphorylated and total Akt protein levels from Tollip+/+ and Tollip−/− mice treated with Akt inhibitor MK-2206 at 1 week after MI (n = 8 mice per experimental group). (B) Representative images of haematoxylin and eosin staining in heart cross-sections of Tollip+/+ and Tollip−/− mice treated with Akt inhibitor MK-2206 at 1 week after MI surgery (n = 6 mice per experimental group). (C) Measurement of infarct size in the heart tissue of mice from Tollip+/+ and Tollip−/− groups treated with Akt inhibitor MK-2206. (D) Echocardiographic and haemodynamic assessment of cardiac function in Tollip+/+ and Tollip−/− mice treated with Akt inhibitor MK-2206 after MI (n = 6 mice per experimental group). ns, no significant difference.
Discussion
In the current study, we identified a novel role for Tollip in mediating the pathological progression of MI-induced heart damage. Using gain- and loss-of-function approaches, an obvious elevation of survival, a diminished infarct size and a sustained cardiac function were found in Tollip-KO mice. In contrast, the forced expression of Tollip exacerbated myocardial apoptosis and immune cell infiltration, leading to an increased mortality rate and marked loss of cardiac function. Mechanistically, these deleterious effects of Tollip were largely dependent on the inhibition of Akt signalling.
Examining the expression alterations of target factor is one of easily achieved approaches for auxiliary prediction of its involvement in certain pharmacological conditions (Cravatt et al., 2007). In the present study, Tollip was dramatically up-regulated in human ischaemic heart and animal MI models. Interestingly, this finding is contrary to our previous finding that Tollip was down-regulated in pressure overload-induced cardiac hypertrophy (Liu et al., 2014). This disparity may be attributable to the different corresponding disease models. In fact, similar to the divergence observed in the heart, the expression of Tollip varies widely in the colon in response to different stress stimuli (Pimentel-Nunes et al., 2012; Xu et al., 2013). In the context of colitis, the expression of Tollip is elevated, while in a model of colon cancer, the Tollip level is decreased. Correspondingly, the different alterations of Tollip expressions might be also related to its diverse functions in disease models. Tollip preserved pressure overload-induced cardiac hypertrophy and delayed heart failure by inhibiting cardiomyocyte enlargement and cardiac fibrosis (Liu et al., 2014), whereas during MI injury, as shown in the current study, Tollip aggravated cardiac dysfunction and increased mortality through promoting inflammatory and apoptotic responses.
In the early stage of MI injury, inflammatory response is triggered rapidly to confront the interrupted blood supply-induced cellular damage, which is characterized by the inflammatory and immune cells infiltrating into the myocardium and scavenging ECM debris (Frangogiannis, 2012). However, sustained immune cell infiltration is directly linked to myocardial apoptosis and impairs cardiac function (Kin et al., 2006; Schofield et al., 2013). Our results demonstrated that Tollip overexpression promoted the migration and accumulation of macrophages, neutrophiles and CD3+-T lymphocytes at 3 days and 4 weeks after MI. Further, our analysis revealed that Tollip enhanced MI-induced activation of NF-κB signalling at 4 weeks post-infarction, which profoundly contributes to inflammatory and immune cell infiltration (Ma et al., 2012). Thus, the increased acute and chronic inflammatory response may, at least partially, explain the detrimental effect of Tollip on apoptosis and the subsequent cardiac dysfunction after MI. However, inconsistent with our study, previous reports suggest that in RAW264.7 cells and human dermal endothelial cells, Tollip up-regulation inhibited NF-κB activation in response to LPS (Bulut et al., 2001; Zhang and Ghosh, 2002). The discrepancy between our study and others may stem from the different cell types and models used, reflecting the complex nature of Tollip-dependent regulation of NF-κB activity. In fact, similar disparities exist for other proteins that are involved in regulating inflammation. For example, class A scavenger receptor alleviated the inflammatory response in an endotoxic shock model (Amiel et al., 2009) but exacerbated inflammation during pulmonary infection (Usui et al., 2007).
Concomitant with the chronic inflammatory response, myocardial apoptosis occurs during the acute phase of MI and persists for a long term after MI, which directly affects cardiac function and is associated with a higher mortality rate (Zhang et al., 2011; Lal et al., 2012). Thus, apoptosis inhibition is linked to a reduced infarct size after MI (Zhao et al., 2003). In the current study, our findings demonstrated that Tollip mediates apoptosis of cardiomyocytes: Tollip depletion reduced the number of apoptotic cells, whereas overexpression of Tollip augmented cell death. Additionally, the imbalance between pro-apoptotic protein (such as Bax and cleaved caspase-3) and anti-apoptotic protein (Bcl-2) has long been established as a key determinant of myocardial apoptosis (Chandrashekhar et al., 2004; Pu et al., 2013). In support of the pro-apoptotic function of Tollip, we observed that Tollip down-regulated the protein expression of Bcl-2, but elevated the levels of Bax and cleaved caspase-3. Taken together, these results indicate that Tollip exacerbates cardiac dysfunction and increases infarct size after MI by promoting cell death through regulating apoptosis-related proteins.
In the pathogenesis of MI, the MAPK signalling is greatly involved in and regulates the inflammatory and apoptotic responses based on the activation and/or inactivation of the three critical subfamily, ERK, JNK and p38, upon extracellular stimuli (Xuan et al., 2011). However, unexpectedly, Tollip exhibited no significant effect on MI-activated MAPK signalling in the current study. Consistent with our results, Didierlaurent et al. (2006) has demonstrated that LPS-induced MAPK activation in mouse embryonic fibroblasts failed to be disrupted by Tollip. Except for MAPK cascades, the Akt signalling is a well-established molecular programme involved in MI progression that could direct cardiomyocyte fate by influencing cell survival and other cellular events (Jonassen et al., 2001; Yamashita et al., 2001; Hausenloy and Yellon, 2004; Suarez et al., 2014). Our findings suggest that the function of Tollip in the ischaemic heart is associated with the manipulation of Akt signalling, as the results demonstrated that Tollip deficiency dramatically increased the phosphorylated levels of Akt, mTOR, GSK3β, S6 and FOXO1 proteins in both mice heart tissue post-MI injury in vivo and hypoxia-stimulated cardiomyocytes in vitro. Importantly, the exacerbated cardiomyocyte damage induced by Tollip overexpression could be almost completely abolished by Akt up-regulation, suggesting an Akt-dependent manner of Tollip-regulated MI injury. The inactivation of Akt signalling by Tollip also provides support for the discrepancy of Tollip on MI- and hypertrophy-induced heart damage as Akt promotes cardiac hypertrophy and cardiomyocyte survival concomitantly (Fujio et al., 2000; Yan et al., 2011). Taken together, these data suggest that the effect of Tollip on signalling pathways during MI-induced cardiac dysfunction is relatively selective.
During cardiac remodelling post-MI, myocyte apoptosis in the infarct zone and border zone directly affects cardiac function and is associated with a higher mortality rate (Zhang et al., 2011; Lal et al., 2012). Thus, strategies to suppress cardiomyocyte death are important for mitigating post ischaemic heart injury. Using the cardiac-specific Tollip overexpression mice and isolated neonatal myocytes, our present study clearly demonstrated that Tollip significantly promotes cardiomyocyte apoptosis. However, Tollip is also expressed in other cell types, including fibroblasts and endothelial cells (Lee and Chung, 2012; Byun et al., 2014). It is possible that Tollip may have effects in these cells contributing to the cardiac remodelling process post-MI, which needs further investigation. Aside from cell death, inflammation as another critical response was greatly involved in the progression of post-MI remodelling (Schofield et al., 2013). Although the initial inflammatory response is indispensable for wound healing, sustained immune cell infiltration is directly linked to myocardial apoptosis and impairs cardiac function. Our current study demonstrated a promoted function of Tollip on immune cell infiltration, which partly explained the detrimental effect of Tollip on post-MI remodelling. Additionally, our previous study showed that Tollip also plays a critical role in pressure overload-induced cardiac fibrosis and hypertrophy, important contributors to cardiac remodelling (Liu et al., 2014). Notably, post-MI angiogenesis is not a process to be ignored that affects cardiomyocyte death via improving blood supply (Cochain et al., 2013), but the extent of this process has not been found to be influenced by the Tollip gene disturbance in our present study. Taken together, multiple capacities of Tollip to regulate post-MI apoptotic and inflammatory permit its profound function in MI-induced cardiac remodelling.
Although genetically engineered mice are robust tools for determining the role of endogenous Tollip in the ischaemic heart, genetic intervention as a clinical treatment remains challenging (Dykxhoorn and Lieberman, 2005). Thus, from a clinical point of view, studies exploring the exogenous blockade of Tollip or activation of Akt through pharmacological approaches are needed. Furthermore, considering that Tollip and Akt signalling exert opposing effects in MI and cardiac hypertrophy, future studies on exogenous inhibition or activation of certain proteins should focus on the counterbalance of their expression to rescue ischaemic injury with little or no augment on cardiac hypertrophy.
In conclusion, our study is the first to demonstrate that Tollip promotes the inflammatory response and cell apoptosis, leading to aggravated cardiac dysfunction and increased mortality after MI, and that these effects are largely dependent on the inhibition of Akt signalling. Our findings provide a novel insight into the pathogenesis of MI and highlight the potential therapeutic value of Tollip inhibition in the treatment of MI.
Acknowledgments
This work was supported by grants from the National Science Fund for Distinguished Young Scholars (no. 81425005), the National Natural Science Foundation of China (no. 81170086, no. 81270184), National Science and Technology Support Project (no. 2011BAI15B02; no. 2012BAI39B05; no. 2013YQ030923-05, 2014BAI02B01 and 2015BAI08B01), the Key Project of the National Natural Science Foundation (no. 81330005), the National Basic Research Program China (no. 2011CB503902) and Natural Science Foundation of Hubei Province (2013CFB259).
Glossary
- FS
fractional shortening
- IHD
ischaemic heart disease
- KO
knockout
- LAD
left anterior descending
- MI
myocardial infarction
- NRCMs
neonatal rat cardiomyocytes
- NTG
non-transgenic
- PI
propidium iodide
- TG
transgenic
- Tollip
toll-interacting protein
Author contributions
N. W., X. L. and X.-J. Z. performed experiments and data analysis, and wrote the manuscript; Y. Z., G. H. and F. W. performed data analysis and wrote the manuscript; R. Z. and X. Z. performed experiments; H. X. and H. L. overall designed the research, analysed data and wrote the manuscript.
Conflict of interest
None declared.
References
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M, et al. The Concise Guide to PHARMACOLOGY 2013/14: catalytic receptors. Br J Pharmacol. 2013a;170:1676–1705. doi: 10.1111/bph.12449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M, et al. The Concise Guide to PHARMACOLOGY 2013/14: enzymes. Br J Pharmacol. 2013b;170:1797–1867. doi: 10.1111/bph.12451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amiel E, Acker JL, Collins RM, Berwin B. Uncoupling scavenger receptor A-mediated phagocytosis of bacteria from endotoxic shock resistance. Infect Immun. 2009;77:4567–4573. doi: 10.1128/IAI.00727-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baldi A, Abbate A, Bussani R, Patti G, Melfi R, Angelini A, et al. Apoptosis and post-infarction left ventricular remodeling. J Mol Cell Cardiol. 2002;34:165–174. doi: 10.1006/jmcc.2001.1498. [DOI] [PubMed] [Google Scholar]
- Bulut Y, Faure E, Thomas L, Equils O, Arditi M. Cooperation of Toll-like receptor 2 and 6 for cellular activation by soluble tuberculosis factor and Borrelia burgdorferi outer surface protein A lipoprotein: role of Toll-interacting protein and IL-1 receptor signaling molecules in Toll-like receptor 2 signaling. J Immunol. 2001;167:987–994. doi: 10.4049/jimmunol.167.2.987. [DOI] [PubMed] [Google Scholar]
- Burns K, Clatworthy J, Martin L, Martinon F, Plumpton C, Maschera B, et al. Tollip, a new component of the IL-1RI pathway, links IRAK to the IL-1 receptor. Nat Cell Biol. 2000;2:346–351. doi: 10.1038/35014038. [DOI] [PubMed] [Google Scholar]
- Byun EB, Mi S, Kim JH, Song DS, Lee BS, Park JN, et al. Epigallocatechin-3-gallate-mediated Tollip induction through the 67-kDa laminin receptor negatively regulating TLR4 signaling in endothelial cells. Immunobiology. 2014;219:866–872. doi: 10.1016/j.imbio.2014.07.010. [DOI] [PubMed] [Google Scholar]
- Chandrashekhar Y, Sen S, Anway R, Shuros A, Anand I. Long-term caspase inhibition ameliorates apoptosis, reduces myocardial troponin-I cleavage, protects left ventricular function, and attenuates remodeling in rats with myocardial infarction. J Am Coll Cardiol. 2004;43:295–301. doi: 10.1016/j.jacc.2003.09.026. [DOI] [PubMed] [Google Scholar]
- Chen K, Gao L, Liu Y, Zhang Y, Jiang DS, Wei X, et al. Vinexin-beta protects against cardiac hypertrophy by blocking the Akt-dependent signalling pathway. Basic Res Cardiol. 2013;108:338. doi: 10.1007/s00395-013-0338-0. [DOI] [PubMed] [Google Scholar]
- Cochain C, Channon KM, Silvestre JS. Angiogenesis in the infarcted myocardium. Antioxid Redox Signal. 2013;18:1100–1113. doi: 10.1089/ars.2012.4849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cravatt BF, Simon GM, Yates JR., 3rd The biological impact of mass-spectrometry-based proteomics. Nature. 2007;450:991–1000. doi: 10.1038/nature06525. [DOI] [PubMed] [Google Scholar]
- Didierlaurent A, Brissoni B, Velin D, Aebi N, Tardivel A, Kaslin E, et al. Tollip regulates proinflammatory responses to interleukin-1 and lipopolysaccharide. Mol Cell Biol. 2006;26:735–742. doi: 10.1128/MCB.26.3.735-742.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dykxhoorn DM, Lieberman J. The silent revolution: RNA interference as basic biology, research tool, and therapeutic. Annu Rev Med. 2005;56:401–423. doi: 10.1146/annurev.med.56.082103.104606. [DOI] [PubMed] [Google Scholar]
- Frangogiannis NG. Regulation of the inflammatory response in cardiac repair. Circ Res. 2012;110:159–173. doi: 10.1161/CIRCRESAHA.111.243162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujio Y, Nguyen T, Wencker D, Kitsis RN, Walsh K. Akt promotes survival of cardiomyocytes in vitro and protects against ischemia-reperfusion injury in mouse heart. Circulation. 2000;101:660–667. doi: 10.1161/01.cir.101.6.660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hausenloy DJ, Yellon DM. New directions for protecting the heart against ischaemia-reperfusion injury: targeting the Reperfusion Injury Salvage Kinase (RISK)-pathway. Cardiovasc Res. 2004;61:448–460. doi: 10.1016/j.cardiores.2003.09.024. [DOI] [PubMed] [Google Scholar]
- Heineke J, Molkentin JD. Regulation of cardiac hypertrophy by intracellular signalling pathways. Nat Rev Mol Cell Biol. 2006;7:589–600. doi: 10.1038/nrm1983. [DOI] [PubMed] [Google Scholar]
- Heusch G, Libby P, Gersh B, Yellon D, Bohm M, Lopaschuk G, et al. Cardiovascular remodelling in coronary artery disease and heart failure. Lancet. 2014;383:1933–1943. doi: 10.1016/S0140-6736(14)60107-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Y, Li T, Wang Y, Li J, Guo L, Wu M, et al. Tollip attenuated the hypertrophic response of cardiomyocytes induced by IL-1beta. Front Biosci (Landmark Ed) 2009;14:2747–2756. doi: 10.2741/3411. [DOI] [PubMed] [Google Scholar]
- Jiang DS, Bian ZY, Zhang Y, Zhang SM, Liu Y, Zhang R, et al. Role of interferon regulatory factor 4 in the regulation of pathological cardiac hypertrophy. Hypertension. 2013;61:1193–1202. doi: 10.1161/HYPERTENSIONAHA.111.00614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang DS, Li L, Huang L, Gong J, Xia H, Liu X, et al. Interferon regulatory factor 1 is required for cardiac remodeling in response to pressure overload. Hypertension. 2014a;64:77–86. doi: 10.1161/HYPERTENSIONAHA.114.03229. [DOI] [PubMed] [Google Scholar]
- Jiang DS, Wei X, Zhang XF, Liu Y, Zhang Y, Chen K, et al. IRF8 suppresses pathological cardiac remodelling by inhibiting calcineurin signalling. Nat Commun. 2014b;5:3303. doi: 10.1038/ncomms4303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang DS, Zhang XF, Gao L, Zong J, Zhou H, Liu Y, et al. Signal regulatory protein-alpha protects against cardiac hypertrophy via the disruption of toll-like receptor 4 signaling. Hypertension. 2014c;63:96–104. doi: 10.1161/HYPERTENSIONAHA.113.01506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonassen AK, Sack MN, Mjos OD, Yellon DM. Myocardial protection by insulin at reperfusion requires early administration and is mediated via Akt and p70s6 kinase cell-survival signaling. Circ Res. 2001;89:1191–1198. doi: 10.1161/hh2401.101385. [DOI] [PubMed] [Google Scholar]
- Jones BM, Kapadia SR, Smedira NG, Robich M, Tuzcu EM, Menon V, et al. Ventricular septal rupture complicating acute myocardial infarction: a contemporary review. Eur Heart J. 2014;35:2060–2068. doi: 10.1093/eurheartj/ehu248. [DOI] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG Group NCRRGW. Animal research: reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1577–1579. doi: 10.1111/j.1476-5381.2010.00872.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kin H, Wang NP, Halkos ME, Kerendi F, Guyton RA, Zhao ZQ. Neutrophil depletion reduces myocardial apoptosis and attenuates NFkappaB activation/TNFalpha release after ischemia and reperfusion. J Surg Res. 2006;135:170–178. doi: 10.1016/j.jss.2006.02.019. [DOI] [PubMed] [Google Scholar]
- Lal H, Zhou J, Ahmad F, Zaka R, Vagnozzi RJ, Decaul M, et al. Glycogen synthase kinase-3alpha limits ischemic injury, cardiac rupture, post-myocardial infarction remodeling and death. Circulation. 2012;125:65–75. doi: 10.1161/CIRCULATIONAHA.111.050666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee HJ, Chung KC. PINK1 positively regulates IL-1beta-mediated signaling through Tollip and IRAK1 modulation. J Neuroinflammation. 2012;9:271. doi: 10.1186/1742-2094-9-271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Jiang XL, Jiang DS, Zhang Y, Zhang R, Chen Y, et al. Toll-interacting protein (Tollip) negatively regulates pressure overload-induced ventricular hypertrophy in mice. Cardiovasc Res. 2014;101:87–96. doi: 10.1093/cvr/cvt232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma J, Wei M, Wang Q, Li J, Wang H, Liu W, et al. Deficiency of Capn4 gene inhibits nuclear factor-kappaB (NF-kappaB) protein signaling/inflammation and reduces remodeling after myocardial infarction. J Biol Chem. 2012;287:27480–27489. doi: 10.1074/jbc.M112.358929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGrath J, Drummond G, McLachlan E, Kilkenny C, Wainwright C. Guidelines for reporting experiments involving animals: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1573–1576. doi: 10.1111/j.1476-5381.2010.00873.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pawson AJ, Sharman JL, Benson HE, Faccenda E, Alexander SP, Buneman OP, et al. NC-IUPHAR. The IUPHAR/BPS Guide to PHARMACOLOGY: an expert-driven knowledgebase of drug targets and their ligands. Nucl Acids Res. 2014;42:D1098–D1106. doi: 10.1093/nar/gkt1143. (Database Issue): [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pimentel-Nunes P, Goncalves N, Boal-Carvalho I, Afonso L, Lopes P, Roncon-Albuquerque R, Jr, et al. Decreased Toll-interacting protein and peroxisome proliferator-activated receptor gamma are associated with increased expression of Toll-like receptors in colon carcinogenesis. J Clin Pathol. 2012;65:302–308. doi: 10.1136/jclinpath-2011-200567. [DOI] [PubMed] [Google Scholar]
- Pu J, Yuan A, Shan P, Gao E, Wang X, Wang Y, et al. Cardiomyocyte-expressed farnesoid-X-receptor is a novel apoptosis mediator and contributes to myocardial ischaemia/reperfusion injury. Eur Heart J. 2013;34:1834–1845. doi: 10.1093/eurheartj/ehs011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rafiq K, Kolpakov MA, Seqqat R, Guo J, Guo X, Qi Z, et al. c-Cbl inhibition improves cardiac function and survival in response to myocardial ischemia. Circulation. 2014;129:2031–2043. doi: 10.1161/CIRCULATIONAHA.113.007004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riad A, Jager S, Sobirey M, Escher F, Yaulema-Riss A, Westermann D, et al. Toll-like receptor-4 modulates survival by induction of left ventricular remodeling after myocardial infarction in mice. J Immunol. 2008;180:6954–6961. doi: 10.4049/jimmunol.180.10.6954. [DOI] [PubMed] [Google Scholar]
- Schofield ZV, Woodruff TM, Halai R, Wu MC, Cooper MA. Neutrophils – a key component of ischemia-reperfusion injury. Shock. 2013;40:463–470. doi: 10.1097/SHK.0000000000000044. [DOI] [PubMed] [Google Scholar]
- Shishido T, Nozaki N, Yamaguchi S, Shibata Y, Nitobe J, Miyamoto T, et al. Toll-like receptor-2 modulates ventricular remodeling after myocardial infarction. Circulation. 2003;108:2905–2910. doi: 10.1161/01.CIR.0000101921.93016.1C. [DOI] [PubMed] [Google Scholar]
- Suarez J, Wang H, Scott BT, Ling H, Makino A, Swanson E, et al. In vivo selective expression of thyroid hormone receptor alpha1 in endothelial cells attenuates myocardial injury in experimental myocardial infarction in mice. Am J Physiol Regul Integr Comp Physiol. 2014;307:R340–R346. doi: 10.1152/ajpregu.00449.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Y, Yi W, Yuan Y, Lau WB, Yi D, Wang X, et al. C1q/tumor necrosis factor-related protein-9, a novel adipocyte-derived cytokine, attenuates adverse remodeling in the ischemic mouse heart via protein kinase A activation. Circulation. 2013;128(11 Suppl. 1):S113–S120. doi: 10.1161/CIRCULATIONAHA.112.000010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Timmers L, Sluijter JP, van Keulen JK, Hoefer IE, Nederhoff MG, Goumans MJ, et al. Toll-like receptor 4 mediates maladaptive left ventricular remodeling and impairs cardiac function after myocardial infarction. Circ Res. 2008;102:257–264. doi: 10.1161/CIRCRESAHA.107.158220. [DOI] [PubMed] [Google Scholar]
- Usui HK, Shikata K, Sasaki M, Okada S, Matsuda M, Shikata Y, et al. Macrophage scavenger receptor-a-deficient mice are resistant against diabetic nephropathy through amelioration of microinflammation. Diabetes. 2007;56:363–372. doi: 10.2337/db06-0359. [DOI] [PubMed] [Google Scholar]
- Wang L, Lu Y, Guan H, Jiang D, Guan Y, Zhang X, et al. Tumor necrosis factor receptor-associated factor 5 is an essential mediator of ischemic brain infarction. J Neurochem. 2013;126:400–414. doi: 10.1111/jnc.12207. [DOI] [PubMed] [Google Scholar]
- Wencker D, Chandra M, Nguyen K, Miao W, Garantziotis S, Factor SM, et al. A mechanistic role for cardiac myocyte apoptosis in heart failure. J Clin Invest. 2003;111:1497–1504. doi: 10.1172/JCI17664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao J, Moon M, Yan L, Nian M, Zhang Y, Liu C, et al. Cellular FLICE-inhibitory protein protects against cardiac remodelling after myocardial infarction. Basic Res Cardiol. 2012;107:239. doi: 10.1007/s00395-011-0239-z. [DOI] [PubMed] [Google Scholar]
- Xu N, Yu ZH, Yao QS, Wang ZQ, Qu HL, Sun Y, et al. PPAR-gamma and tollip are associated with toll-like receptors in colitis rats. J Immunoassay Immunochem. 2013;34:219–231. doi: 10.1080/15321819.2012.699490. [DOI] [PubMed] [Google Scholar]
- Xuan W, Liao Y, Chen B, Huang Q, Xu D, Liu Y, et al. Detrimental effect of fractalkine on myocardial ischaemia and heart failure. Cardiovasc Res. 2011;92:385–393. doi: 10.1093/cvr/cvr221. [DOI] [PubMed] [Google Scholar]
- Yamashita K, Kajstura J, Discher DJ, Wasserlauf BJ, Bishopric NH, Anversa P, et al. Reperfusion-activated Akt kinase prevents apoptosis in transgenic mouse hearts overexpressing insulin-like growth factor-1. Circ Res. 2001;88:609–614. doi: 10.1161/01.res.88.6.609. [DOI] [PubMed] [Google Scholar]
- Yan L, Wei X, Tang QZ, Feng J, Zhang Y, Liu C, et al. Cardiac-specific mindin overexpression attenuates cardiac hypertrophy via blocking AKT/GSK3beta and TGF-beta1-Smad signalling. Cardiovasc Res. 2011;92:85–94. doi: 10.1093/cvr/cvr159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang G, Ghosh S. Negative regulation of toll-like receptor-mediated signaling by Tollip. J Biol Chem. 2002;277:7059–7065. doi: 10.1074/jbc.M109537200. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Kohler K, Xu J, Lu D, Braun T, Schlitt A, et al. Inhibition of p53 after acute myocardial infarction: reduction of apoptosis is counteracted by disturbed scar formation and cardiac rupture. J Mol Cell Cardiol. 2011;50:471–478. doi: 10.1016/j.yjmcc.2010.11.006. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Liu X, She ZG, Jiang DS, Wan N, Xia H, et al. Interferon regulatory factor 9 is an essential mediator of heart dysfunction and cell death following myocardial ischemia/reperfusion injury. Basic Res Cardiol. 2014;109:434. doi: 10.1007/s00395-014-0434-9. [DOI] [PubMed] [Google Scholar]
- Zhao ZQ, Morris CD, Budde JM, Wang NP, Muraki S, Sun HY, et al. Inhibition of myocardial apoptosis reduces infarct size and improves regional contractile dysfunction during reperfusion. Cardiovasc Res. 2003;59:132–142. doi: 10.1016/s0008-6363(03)00344-4. [DOI] [PubMed] [Google Scholar]