

Abstract
Background and Purpose
cAMP as a second messenger stimulates expression of microphthalmia-associated transcription factor (MITF) or the tyrosinase gene in UVB-induced skin pigmentation. Diphenylmethylene hydrazinecarbothioamide (QNT 3-80) inhibits α-melanocyte-stimulating hormone (α-MSH)-induced melanin production in B16 murine melanoma cells but its molecular basis remains to be defined. Here, we investigated the mechanism underlying the amelioration of skin hyperpigmentation by QNT 3-80.
Experimental Approach
We used melanocyte cultures with raised levels of cAMP and UVB-irradiated dorsal skin of guinea pigs for pigmentation assays. Immunoprecipitation, kemptide phosphorylation, fluorescence analysis and docking simulation were applied to elucidate a molecular mechanism of QNT 3-80.
Key Results
QNT 3-80 inhibited melanin production in melanocyte cultures with elevated levels of cAMP, including those from human foreskin. This compound also ameliorated hyperpigmentation in vivo in UVB-irradiated dorsal skin of guinea pigs. As a mechanism, QNT 3-80 directly antagonized cAMP binding to the regulatory subunit of PKA, nullified the dissociation and activation of inactive PKA holoenzyme in melanocytes and fitted into the cAMP-binding site on the crystal structure of human PKA under the most energetically favourable simulation. QNT 3-80 consequently inhibited cAMP- or UVB-induced phosphorylation (activation) of cAMP-responsive element-binding protein in vitro and in vivo, thus down-regulating expression of genes for MITF or tyrosinase in the melanogenic process.
Conclusions and Implications
Our data suggested that QNT 3-80 could contribute significantly to the treatment of skin disorders with hyperpigmented patches with the cAMP-binding site of PKA as its molecular target.
Tables of Links
| Ligands | |
|---|---|
| α-MSH, α-melanocyte-stimulating hormone | H-89 |
| cAMP | IBMX |
| db-cAMP, N6,2′-O-dibutyryl-cAMP (bucladesine) | Rp-cAMPS, Rp-adenosine 3′,5′-cyclic monophosphorothioate |
| Forskolin |
These Tables list key protein targets and ligands in this article which are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Pawson et al., 2014) and are permanently archived in the Concise Guide to PHARMACOLOGY 2013/14 (a,bAlexander et al., 2013a,2013b).
Introduction
UVB in sunlight stimulates skin pigmentation through a complex process of melanogenesis that synthesizes melanin biopolymers in the melanosomes of melanocytes at the basal layer of skin epidermis followed by the transfer of the pigmented melanosomes to keratinocytes at the overlying epidermis (Miyamura et al., 2007; Ando et al., 2012). Skin cells communicate with each other via the secretion of auto- or paracrine factors including α-melanocyte-stimulating hormone (α-MSH) in the generation of heavily pigmented melanosomes under UVB stimulation (Imokawa, 2004; Slominski et al., 2004). The pigmented melanosomes contain a mixture of yellow-red pheomelanins and black-brown eumelanins (Ito and Wakamatsu, 2008). Biosynthetic pathways of both melanin pigments are initiated through the tyrosinase-catalyzed rate-limiting step, L-tyrosine to dopaquinone, and branched as a function of other melanogenic enzymes and an availability of cysteine or glutathione in the melanosomes (Olivares and Solano, 2009; Simon et al., 2009).
α-MSH specifically binds to the melanocortin MC1 receptor on the surface of melanocytes, which leads to G protein-coupled activation of adenylate cyclase increasing intracellular cAMP levels (Le Pape et al., 2008). In turn, cAMP stimulates the activation of PKA holoenzyme via dissociating two catalytic subunits from a regulatory subunit dimer (Kim et al., 2007). The catalytic subunits of PKA are diverse, as there are three isomers (Cα, Cβ and Cγ) and four regulatory subunits (RIα, RIβ, RIIα and RIIβ) (Brandon et al., 1997; Skalhegg and Tasken, 2000). Each regulatory subunit of PKA has tandem cAMP-binding A and B sites and dissociates from the catalytic subunits upon binding with cAMP molecules in a cooperative affinity mode (Diller et al., 2001; Zawadzki and Taylor, 2004). The catalytic subunit alone of PKA phosphorylates the cAMP-responsive element-binding protein (CREB) at Ser133, which up-regulates the microphthalmia-associated transcription factor (MITF) gene, and MITF is a key transcription factor enhancing the expression of tyrosinase or other melanogenic genes in melanocytes (Sands and Palmer, 2008; Vachtenheim and Borovansky, 2010). α-MSH also increases melanin biosynthesis in an MC1 receptor /cAMP-independent mechanism, directly removing an endogenous allosteric inhibitor 6(R)-L-erythro-5,6,7,8-tetrahydrobiopterin from tyrosinase (Schallreuter et al., 2008).
Abnormal accumulation and biosynthesis of melanin pigments are responsible for skin disorders such as melasma, freckles and senile lentigo (Ortonne and Passeron, 2005; Grimes et al., 2006). We previously reported that diphenylmethylene hydrazinecarbothioamide (QNT 3-80, Supporting Information Fig. S1A) and its derivatives decrease α-MSH-induced melanin production in cultured B16 cells with structure–activity relationships suggesting an important role of the double bond in thiosemicarbazone moiety and the hydrophobic property of the diphenylmethylene moiety (Lee et al., 2010; Thanigaimalai et al., 2011). However, their antimelanogenic mechanisms remain to be defined.
In the experiments described here we found that QNT 3-80 directly antagonized cAMP binding to the regulatory subunit of PKA and inhibited the dissociation and activation of inactive PKA holoenzyme, thus suppressing the expression of MITF or tyrosinase gene in vitro under cAMP-induced melanin production in cultured melanocytes and in vivo under UVB-induced skin pigmentation in guinea pigs.
Methods
Cell culture
B16 cells were purchased from ATCC (American Type Culture Collection; VA, USA) and cultured in DMEM supplemented with 10% FBS, 100 U·mL−1 penicillin and 100 μg·mL−1 streptomycin under an atmosphere of 5% CO2 at 37°C. Melan-a mouse melanocyte cells were obtained from LG Biotech (Daejeon, Korea) and cultured in RPMI 1640 supplemented with 10% FBS, 200 nM 12-O-tetradecanoylphorbol-13-acetate, 100 mM 2-mercaptoethanol, 100 U·mL−1 penicillin and 100 μg·mL−1 streptomycin under 5% CO2 at 37°C. Human epidermal melanocytes from moderately pigmented neonatal foreskins were purchased from Cascade Biologics (Portland, OR, USA) and cultured in Medium 254 (GIBCO M-254-500; Gibco BRL, Paisley, UK) containing growth supplement (GIBCO S-002-5) without antibiotics under 5% CO2 at 37°C.
Animals
All animal care and experimental procedures were in accordance with the institutional guidelines and were approved by the Ethics Committee on Experimental Animal Welfare of the Chungbuk National University. All studies involving animals are reported in accordance with the ARRIVE guidelines for reporting experiments involving animals (Kilkenny et al., 2010; McGrath et al., 2010). The total number of guinea pigs used in this study was 5. Brownish guinea pigs (male, 6–7 weeks old) were purchased from Daehan Biolink (Eumsung, Korea).
UVB-induced skin pigmentation in vivo
After shaving, the dorsal skin of each guinea pig was separated into four areas (2 cm × 2 cm each). QNT 3-80 was dissolved in a vehicle (propyleneglycol : ethanol: H2O = 5: 3: 2). As shown in Supporting Information Fig. S1B, the allocated skin areas were topically treated with QNT 3-80 (50 μL each, twice daily for 4 weeks) and concurrently irradiated with UVB (350 mJ·cm−2 per exposure, once every 2 days during 2 weeks). Skin pigmentation was then measured at 1 week after ending UVB radiation.
Melanin quantification
B16 cells were stimulated with 100 nM α-MSH for 3 days, Melan-a cells for 4 days and primary human melanocytes for 6 days. The cells were counted (Trypan blue exclusion) and then disrupted in 0.85 N NaOH and 20% DMSO by heating at 80°C. Absorbance values were measured at 405 nm with synthetic melanin as a standard and normalized to the cell numbers. The dorsal skin of guinea pigs was topically treated with QNT 3-80 and concurrently irradiated with UVB. Skin pigmentation was measured by using a chromometer and is represented as a relative whitening (L) index with normal skin L = 10. Skin tissues were biopsied and then incubated with 2 M NaBr at 37°C for 5 h to separate the epidermis from the dermis. The epidermis was disrupted in 1 N NaOH and 10% DMSO under heating at 80°C. Absorbance values were measured at 405 nm with synthetic melanin as a standard and normalized to the protein contents. Skin tissues were freshly fixed in 10% formaldehyde, serially sectioned with 5 μm thickness and then stained with Fontana-Masson silver nitrate (Scytek, Logan, UT, USA).
3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay
B16 cells were incubated with QNT 3-80 for 3 days and then cell viability determined by MTT assay. In brief, the cells were incubated with 50 μg·mL−1 MTT for 2–4 h. Formazan crystals were dissolved in 50% DMSO and absorbance values measured at 590 nm.
Fluorescence analysis
B16 cells were incubated with 500 nM TAMRA-NDP-α-MSH, the fluorescence probe for α-MSH, (Anaspec, San Jose, CA, USA) at 37°C for 2 h. After washing, the cells were subjected to flow cytometry to determine TAMRA-NDP-α-MSH binding to its receptor. The fluorescence probe for cAMP, 8-[φ-575]-cAMP (1 μM), was pre-incubated with 4 μg·mL−1 PKA-RIIβ polypeptide in cell-free reactions for 2 h and incubated with non-fluorescent QNT 3-80 for another 2 h. 8-[φ-575]-cAMP binding to PKA-RIIβ was measured as relative fluorescence units using excitation at 575 nm and emission at 590–650 nm.
cAMP quantification
B16 cells were stimulated with 10 μM forskolin for 10–20 min. Cell extracts were used to determine cAMP levels using an elisa kit (R&D Systems, Minneapolis, MN, USA).
Immunoprecipitation
Cell extracts were incubated at 4°C overnight with 2 μg antibody against the catalytic subunit α of PKA (PKA-Cα) and then precipitated with protein G sepharose.
Western blot analysis
Cell extracts were resolved on SDS-PAGE and transferred to a PVDF membrane. Either 5% non-fat milk in PBS containing Tween 20 or 5% BSA in Tris-buffered saline (TBS) containing Tween 20 was used as the blocking buffer. The blots were reacted at 4°C overnight with primary antibody and then incubated with HRP-conjugated secondary antibody at room temperature for 2–5 h. Immune complexes on the blots were visualized after reaction with enhanced chemiluminescence reagent (GE Healthcare, Chalfont St. Giles, UK).
Kemptide phosphorylation
Enzyme sources of PKA were incubated with 83 μM kemptide (Leu-Arg-Arg-Ala-Ser-Leu-Gly) as an exogenous substrate and 10 μCi [γ-32P]ATP as a probe at 30°C for 15 min. Aliquots of the reaction mixture were spotted onto P81 phosphocellulose papers, washed three times with 0.75% H3PO4 followed by one wash with 100% acetone and then the radioactivity was measured, as counts per min.
Molecular docking
Crystallographic structure of human PKA-RIIβ was cited from Protein Data Bank (PDB code 1CX4). The structure of QNT 3-80 was drawn with the Sybyl package and minimized with the Tripos force field and Gasteiger-Huckel charge. QNT 3-80 docking to the PKA-RIIβ was carried out using the Surflex-Dock in Sybyl version 8.1.1 by Tripos Associates (St. Louis, MO, USA), operating under Red Hat Linux 4.0.
Confocal microscopy
Skin tissues were serially sectioned with 5 μm thickness, mounted onto slide glasses, immersed in 10 mM sodium citrate and then heated in a microwave oven for 3–5 min. The specimens were treated with 3% H2O2 for 30 min and washed twice with H2O followed by once with TBS. For immunostaining, the specimens were reacted with anti-phospho (p)-CREB antibody and then incubated with Alexa Fluor 568-labelled secondary antibody. After nuclei staining with 3 μM DAPI, the specimens were examined by confocal fluorescence microscopy.
RT-PCR analysis
Total RNAs were subjected to RT-PCR with an RNA PCR kit (Bioneer, Daejeon, Korea), determining mRNA levels of MITF or tyrosinase gene with the internal control β-actin. Nucleotide sequences of the PCR primers were previously described (Roh et al., 2013). In brief, total RNAs were reversely transcribed at 42°C for 1 h and then subjected to 25–30 cycles of PCR consisting of 30 s denaturation at 94°C, 60 s annealing at 50–55°C and 90 s extension at 72°C. RT-PCR products were resolved on agarose gels by electrophoresis and then stained with 1% ethidium bromide.
Luciferase assay
B16 cells were transfected with each luciferase reporter construct of MITF (−2200/+95)-Luc or tyrosinase (−2236/+59)-Luc in combination with Renilla control vector using a lipofectamine kit (Invitrogen, Camarillo, CA, USA). The transfected cells were stimulated with 100 nM α-MSH for 20 h. Cell extracts were subjected to a dual-luciferase assay (Promega, Madison, WI, USA). Firefly luciferase activity as a reporter was normalized to the Renilla activity as a reference of transfection efficiency.
Measurement of tyrosinase activity
Catalytically active mushroom tyrosinase or human tyrosinase (EnzoLife Sci, Laussen, Switzerland) was reacted with 5 mM L-tyrosine in cell-free reactions. Tyrosinase activity was determined by the initial rate increasing absorbance values at 475 nm as previously described (Kim et al., 2002). One unit of the enzyme activity was defined as the oxidation of 1 nmole L-tyrosine per minute.
Data analysis
Data are represented as mean ± SD from at least three independent experiments. Differences between means using the average value of triplicate in each experiment after the anova followed by the Dunnett's test. Values of P < 0.05 were considered as significantly different.
Materials
QNT 3-80 (<97% purity) was synthesized as previously described (Thanigaimalai et al., 2011). Primary and secondary antibodies were purchased from Abcam (Cambridge, UK), Cell Signaling Technology (Danvers, MA, USA) or Santa Cruz Biotech (Paso Robles, CA, USA). The regulatory subunit IIβ of PKA (PKA-RIIβ) and rhPKA were purchased from Proteintech group (Chicago, IL, USA), and 8-[φ-575]-cAMP from BioLog (Bremen, Germany). All other chemicals including α-MSH were purchased from Sigma-Aldrich (St. Louis, MO, USA).
Results
QNT 3-80 inhibits melanin production in vitro and in vivo
We first measured cAMP-induced melanin production in cultured melanocytes. Upon exposure to α-MSH alone, B16 cells markedly increased melanin production over unstimulated levels (Figure 1A). Treatment with QNT 3-80 in vitro decreased α-MSH-induced melanin levels in a dose-dependent manner, as did Rp-adenosine 3′,5′-cyclic monophosphorothioate (Rp-cAMPS) or H-89 (Figure 1A). Rp-cAMPS is a cell-permeable cAMP antagonist interrupting the dissociation and activation of inactive PKA holoenzyme and H-89 is an ATP-competitive inhibitor of the catalytic subunit PKA-mediated kinase activity after dissociation from the holoenzyme complex (Wu et al., 2004; Lochner and Moolman, 2006). QNT 3-80 also inhibited α-MSH-induced melanin production in Melan-a cells or human epidermal melanocytes-derived from the neonatal foreskins (Figure 1B and C), showing it could exert antimelanogenic activity in different types of melanocytes. We next examined whether QNT 3-80 could affect melanin production in B16 cells stimulated with other agents know to raise cAMP levels. Treatment with QNT 3-80 inhibited forskolin-, IBMX-, or N6,2′-O-dibutyryl (db)-cAMP-induced melanin production, as did Rp-cAMPS, H-89 or arbutin (Supporting Information Fig. S2A–D). Forskolin is a stimulator of AC, IBMX is an inhibitor of cAMP phosphodiesterase, db-cAMP (bucladesine) is a cell-permeable cAMP agonist and arbutin is known to be a skin whitener targeting the substrate-binding site for tyrosinase activity (Maeda and Fukuda, 1996; Busca and Ballotti, 2000). Finally, QNT 3-80 at these concentrations showing antimelanogenic activity did not affect the viability of B16 cells (Supporting Information Fig. S2E), thus excluding non-specific cytotoxicity.
Figure 1.

Effect of QNT 3-80 on melanin pigmentation. B16 cells were stimulated with α-MSH for 3 days (A), Melan-a cells for 4 days (B) or primary human melanocytes for 6 days (C) in the presence of QNT 3-80 or other sample. Melanin contents were determined with synthetic melanin as a standard and then normalized to the cell numbers. Data are mean ± SD from three to five independent experiments. #P < 0.05 versus media alone. *P < 0.05 versus α-MSH alone. Dorsal skin of guinea pigs was topically treated with QNT 3-80 and concurrently irradiated with UVB as described. Skin tissues were biopsied. (D) Melanin pigments were extracted from the skin epidermis, quantified with synthetic melanin as a standard and then normalized to the protein contents. Data are mean ± SD from five separate specimens. #P < 0.05 versus normal skins without UVB radiation. *P < 0.05 versus UVB plus vehicle alone. (E) Skin tissues were serially sectioned (5 μm thick) and then stained with Fontana-Masson silver nitrate.
To assess the effects of QNT 3-80 in vivo, we carried out assays of UVB-induced skin pigmentation in guinea pigs. The dorsal skin of guinea pigs was topically treated with QNT 3-80 (0.03–0.1%) or arbutin (5%, a dose recommended by Korea Food and Drug Administration). These doses were based on the relative activity of QNT 3-80 and arbutin in vitro; the IC50 value for QNT 3-80 was more than 50-fold less than that of arbutin (Supporting Information Fig. S2D), and concurrently irradiated with UVB (350 mJ·cm−2 per exposure). The dose of UVB was used in our previous work (Roh et al., 2013; 2014) in order to induce facultative pigmentation without erythema or oedema in the dorsal skin of guinea pigs. Topical treatment with QNT 3-80 on the UVB-irradiated dorsal skin of guinea pigs decreased visual pigmentation in the absence of skin corrosion and increased the skin-whitening index (L), as did arbutin (Supporting Information Fig. S3A and B). Skin tissues were then biopsied and melanin pigments were extracted from the epidermis. Upon exposure to UVB alone, dorsal skin significantly increased melanin levels over the basal values (Figure 1D). Topical treatment with QNT 3-80 decreased UVB-induced melanin contents in the skin specimens (Figure 1D). Skin tissues were serially sectioned and then stained with Fontana-Masson silver nitrate. Topical treatment with QNT 3-80 consistently decreased UVB-induced melanin granules, more clearly visible in the basal layer of epidermis (Figure 1E). In another experiment, QNT 3-80 or arbutin absorbed very little UVB at 290–320 nm, compared with the well-known suncreen ensulizole (Supporting Information Fig. S3C), thus excluding a direct effect of QNT 3-80 on skin photoprotection.
QNT 3-80 interrupts cAMP-induced dissociation and activation of PKA holoenzyme
QNT 3-80 itself affected neither the fluorescent probe for α-MSH (TAMRA-NDP-α-MSH) binding to its receptor on B16 cells nor the increase of intracellular cAMP levels induced by forskolin (Supporting Information Fig. S4), thus excluding direct effects of QNT 3-80 on α-MSH antagonism or cAMP metabolism. We therefore tested the effects of QNT 3-80 on cAMP-induced proximal signalling events in the melanogenic process. B16 cells were stimulated with α-MSH, db-cAMP or forskolin in the presence of QNT 3-80. Cell extracts were subjected to immunoprecipitation (IP) with anti-PKA-Cα antibody and these IP complexes were then probed with another antibody against PKA-RIIβ to detect the co-precipitation. Upon exposure to each cAMP stimulator, B16 cells markedly dissociated PKA-Cα from PKA-RIIβ (Figure 2A and B). Treatment with QNT 3-80 inhibited α-MSH-induced dissociation of PKA-Cα from PKA-RIIβ in the cells, as did Rp-cAMPS (Figure 2A). QNT 3-80 also inhibited db-cAMP- or forskolin-induced dissociation of PKA-Cα from PKA-RIIβ (Figure 2B). In another experiment, B16 cells displayed similar protein levels of PKA-Cα or PKA-RIIβ even if they were stimulated with α-MSH or treated with QNT 3-80 (Supporting Information Fig. S5).
Figure 2.
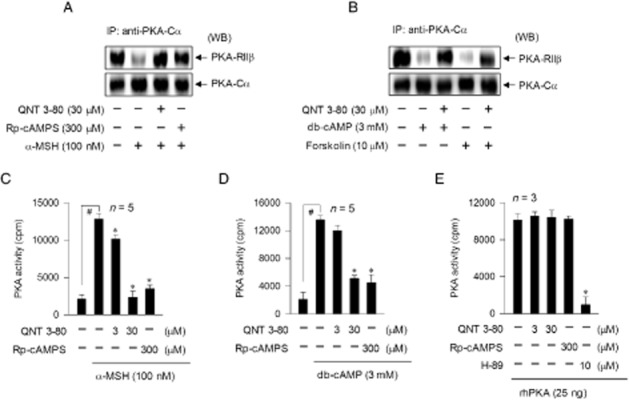
Effect of QNT 3-80 on the dissociation and activation of PKA holoenzyme. B16 cells were pretreated with QNT 3-80 or Rp-cAMPS for 2 h and stimulated with α-MSH (A, C), db-cAMP (B, D) or forskolin (B) for 20–30 min in the presence of QNT 3-80 or Rp-cAMPS. (A, B) Cell extracts were analysed with IP, using anti-PKA-Cα antibody, and the IP complexes were then probed with anti-PKA-RIIβ or anti-PKA-Cα antibody for Western blot analysis (WB). (C, D) Cell extracts were used to determine PKA activity. (E) rhPKA with catalytic subunit alone was treated with QNT 3-80 or other sample for 10 min in cell-free reactions. PKA activity was measured by monitoring the incorporation of [32P] from the probe [γ-32P]ATP into the exogenous substrate, kemptide. Data are mean ± SD from three to five independent experiments. #P < 0.05 versus media alone. *P < 0.05 versus α-MSH or db-cAMP alone (C, D) or rhPKA alone (E).
We next examined whether the dissociation of the catalytic subunit from inactive PKA holoenzyme could be correlated with its kinase activity. Upon exposure to α-MSH or db-cAMP alone, B16 cells markedly increased kemptide phosphorylation as an assay of kinase activity of PKA (Figure 2C and D). Treatment with QNT 3-80 inhibited α-MSH- or db-cAMP-induced PKA activity in the cells, as did Rp-cAMPS (Figure 2C and D). To understand whether QNT 3-80 could directly interact with the catalytic subunit of PKA, we carried out in vitro kinase assays. Samples of rhPKA with catalytic subunit only were treated with QNT 3-80 in cell-free conditions and then its kinase activity measured. Treatment with QNT 3-80 or Rp-cAMPS did not affect the catalytic subunit PKA-mediated kemptide phosphorylation, whereas H-89 did inhibit as expected (Figure 2E). Therefore, QNT 3-80 can interrupt cAMP-induced dissociation and activation of PKA holoenzyme but not catalytic subunit PKA-mediated kinase activity, a mechanism of action which is similar to the PKA-directed cAMP antagonist, Rp-cAMPS (Wu et al., 2004).
QNT 3-80 antagonizes the 8-[φ-575]-cAMP binding to PKA-RIIβ
To elucidate a target site for QNT 3-80 on PKA, we performed a fluorescence analysis with 8-[φ-575]-cAMP, specifically probing the cAMP-binding A and B sites on the regulatory subunit of PKA (Moll et al., 2008). Upon binding to PKA-RIIβ polypeptide in cell-free conditions, 8-[φ-575]-cAMP markedly increased its fluorescence intensity over the basal values (Figure 3A). Treatment with QNT 3-80 dose-dependently decreased the PKA-RIIβ polypeptide-enhanced fluorescence intensity of 8-[φ-575]-cAMP (Figure 3A), indicating displacement of 8-[φ-575]-cAMP from the complex with PKA-RIIβ. This mechanism of action was reversible because fluorescence intensity was recovered in the pretreatment with QNT 3-80 followed by competitive titration with excess 8-[φ-575]-cAMP, as also observed with Rp-cAMPS (Supporting Information Fig. S6). However, non-fluorescent QNT 3-80 itself did not affect basal fluorescence values of 8-[φ-575]-cAMP in the absence of PKA-RIIβ polypeptide (Figure 3B). Rp-cAMPS also inhibited 8-[φ-575]-cAMP binding to PKA-RIIβpolypeptide, as did QNT 3-80, while H-89 did not affect this binding (Figure 3B).
Figure 3.
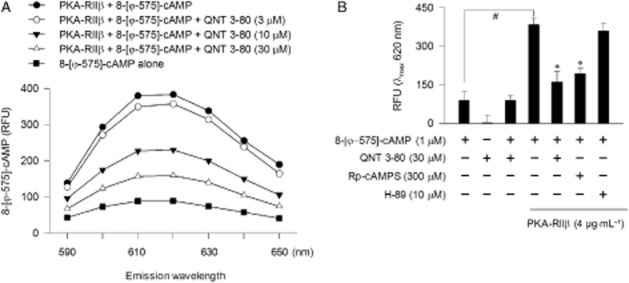
Effect of QNT 3-80 on the binding of 8-[φ-575]-cAMP to PKA-RIIβ polypeptide. (A) The fluorescent probe 8-[φ-575]-cAMP (1 μM) was pre-incubated with PKA-RIIβ polypeptide (4 μg·mL−1) in cell-free conditions for 2 h to reach stable fluorescence values under excitation at 575 nm and treated with QNT 3-80 for another 2 h. Emission spectra at 590–650 nm are represented as relative fluorescence units (RFU). (B) 8-[φ-575]-cAMP was pre-incubated in the absence or presence of PKA-RIIβ polypeptide in cell-free conditions for 2 h and treated with QNT 3-80 or other sample for another 2 h. Fluorescence values are represented as RFU under excitation at 575 nm and emission at 620 nm. Data are mean ± SD from three independent experiments. #P < 0.05 versus 8-[φ-575]-cAMP alone. *P < 0.05 versus 8-[φ-575]-cAMP plus PKA-RIIβ polypeptide alone.
On the basis of the experimental evidence that QNT 3-80 inhibited not only 8-[φ-575]-cAMP binding to PKA-RIIβ polypeptide but also cAMP-induced dissociation and activation of inactive PKA holoenzyme, we proposed a docking model of QNT 3-80, based on the crystal structure of human PKA-RIIβ (Diller et al., 2001; Zawadzki and Taylor, 2004). QNT 3-80 was well fitted into the cAMP-binding A site of PKA-RIIβ with close contacts to Val201, Tyr213 and Leu222 residues, under the most energetically favourable simulation (Figure 4). Hydrogen bonding could be achieved between the thioamide moiety of QNT 3-80 and the side chain of Tyr213 and hydrophobic interaction between the phenyl rings of QNT 3-80 and the side chains of Val201 or Leu222. Similarly, QNT 3-80 could also be docked to the cAMP-binding B site of PKA-RIIβ with close contacts with Ile302, Tyr347 and Tyr397 residues (Figure 4). Hydrogen bonding could be achieved between the thiourea moiety of QNT 3-80 and the peptidyl carbonyl backbone of Tyr347 or the side chain of Tyr397 and hydrophobic interaction between the phenyl rings of QNT 3-80 and the side chains of Ile302 or Tyr397. Moreover, the docking simulations of QNT 3-80 to PKA-RIIβ overlapped with those of the endogenous ligand cAMP (Supporting Information Fig. S7).
Figure 4.
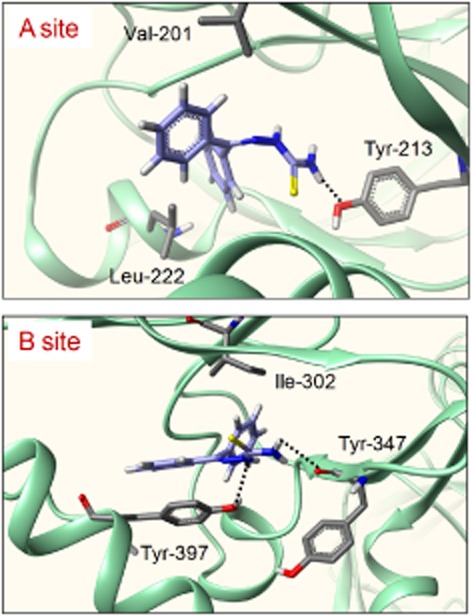
Molecular docking of QNT 3-80 to the crystal structure of PKA-RIIβ. Docking arrangements of QNT 3-80 to the crystal structure of cAMP-binding A or B site of human PKA-RIIβ were carried out using the Surflex-Dock program. QNT 3-80 or its interacting residues on PKA-RIIβ is represented by a grey colour and the other PKA-RIIβ backbone by a green colour. Hydrogen bonding between QNT 3-80 and PKA-RIIβ is shown as a black dotted line.
QNT 3-80 inhibits CREB phosphorylation
We asked whether QNT 3-80 could consequently affect the phosphorylation (activation) of CREB at Ser133, as CREB is a cellular substrate of PKA in the melanogenic process. B16 cells were stimulated with α-MSH or db-cAMP in the presence of QNT 3-80. Cell extracts were then subjected to Western blot analysis with anti-p-CREB or anti-CREB antibody. Treatment with QNT 3-80 or Rp-cAMPS in vitro decreased α-MSH- or db-cAMP-induced p-CREB levels in the cells, whereas no significant change was observed in total levels of CREB (Figure 5A and B). To understand whether QNT 3-80 could affect CREB phosphorylation in vivo, dorsal skin of guinea pigs was topically treated with QNT 3-80 and concurrently irradiated with UVB. Skin tissues were serially sectioned and then reacted with anti-p-CREB antibody for confocal microscopy. Topical treatment with QNT 3-80 in vivo consistently decreased UVB-induced p-CREB levels in the skin epidermis (Figure 5C).
Figure 5.
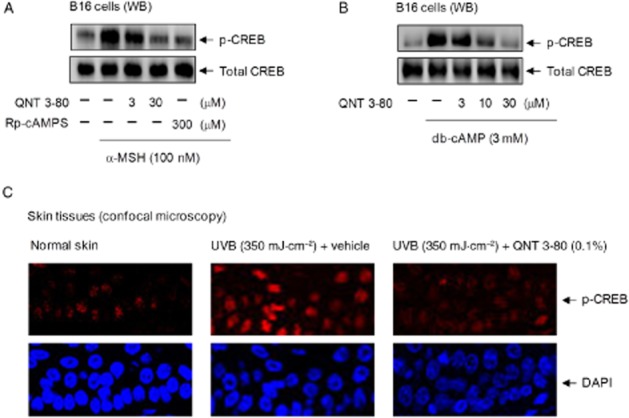
Effect of QNT 3-80 on CREB phosphorylation. B16 cells were pretreated with QNT 3-80 or Rp-cAMPS for 2 h and stimulated with α-MSH (A) or db-cAMP (B) for 20–30 min in the presence of QNT 3-80 or Rp-cAMPS. Cell extracts were analysed by Western blot (WB) with anti-p-CREB or anti-CREB antibody. (C) Dorsal skin of guinea pigs were topically treated with QNT 3-80 and concurrently irradiated with UVB as described. Skin tissues were biopsied and then serially sectioned (5 μm thick). The specimens were reacted with anti-p-CREB antibody for confocal fluorescence microscopy, displaying the p-CREB-stained with Alexa Fluor 568-labelled secondary antibody as a red colour and the nuclei-stained with DAPI as a blue colour.
QNT 3-80 down-regulates expression of MITF or tyrosinase gene
CREB-responsive CRE motifs appear to be essential for maximal MITF induction in cAMP-elevated melanocytes and MITF is a key transcription factor enhancing the melanocyte-specific expression of tyrosinase gene, even though the promoter region of MITF or tyrosinase gene contains several cis-acting elements (Sands and Palmer, 2008; Vachtenheim and Borovansky, 2010). Treatment with QNT 3-80 in vitro decreased α-MSH-induced protein and mRNA levels of MITF in B16 cells, as did Rp-cAMPS (Figure 6A and B). Transcriptional regulation of MITF by QNT 3-80 was further described by promoter activity-dependent reporter assays. B16 cells were transfected with MITF-Luc, a construct encoding MITF promoter (−2200/+95) fused to luciferase gene as a reporter (Udono et al., 2000). Upon exposure to α-MSH alone, B16 cells containing the MITF-Luc construct significantly increased luciferase expression over the basal levels (Figure 6C). Treatment with QNT 3-80 in vitro inhibited α-MSH-induced luciferase expression, reporting the promoter activity of MITF gene (Figure 6C). We then examined whether QNT 3-80 could affect MITF expression in vivo. Dorsal skin of guinea pigs were topically treated with QNT 3-80 and concurrently irradiated with UVB. Topical treatment with QNT 3-80 in vivo suppressed UVB-induced protein and mRNA levels of MITF in the skin tissues (Figure 6D and E).
Figure 6.
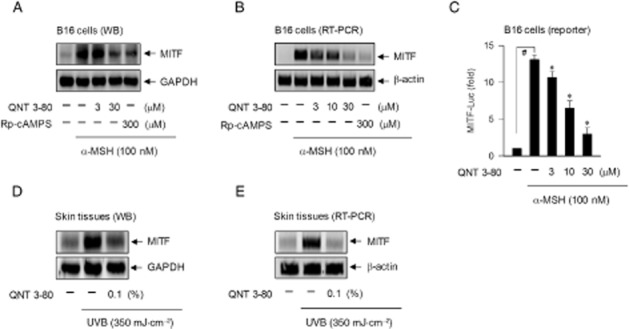
Effect of QNT 3-80 on MITF expression. B16 cells were pretreated with QNT 3-80 or Rp-cAMPS for 2 h and stimulated with α-MSH for 4 h (A) or 2 h (B) in the presence of QNT 3-80 or Rp-cAMPS. (A) Cell extracts were analysed by Western blot with anti-MITF or anti-GAPDH antibody. (B) Total RNAs were subjected to RT-PCR analysis of MITF with the internal control β-actin. (C) B16 cells were transfected with MITF (−2200/+95)-Luc reporter construct in combination with Renilla control vector and then stimulated with α-MSH for 20 h in the presence of QNT 3-80. Cell extracts were analysed by a dual-luciferase assay. Firefly luciferase activity is represented as relative fold change after normalizing to the Renilla activity. Data are mean ± SD from three independent experiments. #P < 0.05 versus media alone. *P < 0.05 versus α-MSH alone. Dorsal skins of guinea pigs were topically treated with QNT 3-80 and concurrently irradiated with UVB as described. Skin tissues were biopsied. (D) Cell extracts from the tissues were analysed by Western blotting with anti-MITF or anti-GAPDH antibody. (E) Total RNAs were analysed by RT-PCR of MITF with the internal control β-actin.
In another experiment, treatment with QNT 3-80 in vitro decreased α-MSH-induced protein and mRNA levels of tyrosinase in B16 cells, as did Rp-cAMPS, as well as inhibited α-MSH-induced promoter activity of tyrosinase gene (Supporting Information Fig. S8A–C), in which the tyrosinase-Luc construct encodes a firefly luciferase gene fused to tyrosinase promoter (−2236/+59) (Bertolotto et al., 1996). These results indicate that QNT 3-80 could down-regulate α-MSH-induced expression of tyrosinase gene at the transcription level. Moreover, topical treatment with QNT 3-80 in vivo decreased protein and mRNA levels of tyrosinase in UVB-irradiated dorsal skin of guinea pigs (Supporting Information Fig. S8D and E).
We finally examined whether QNT 3-80 could act as a copper ion chelator and inhibit the catalytic activity of tyrosinase, as reported for the paeonol thiosemicarbazone derivatives (Zhu et al., 2013). The maximal peak of QNT 3-80 was shifted from 275 nm to 230 nm upon reacting with CuSO4 (Supporting Information Fig. S9A), indicating a complex formation between QNT 3-80 and copper ions. Mushroom or human tyrosinase was then treated with QNT 3-80 in cell-free conditioin and the velocityof L-tyrosine oxidation was measured. QNT 3-80 did not directly affect the catalytic activity of tyrosinase, whereas arbutin did inhibit (Supporting Information Fig. S9B and C). These results suggest that the thiourea moiety in thiosemicarbazone plays an important role in the metal ion chelation but the diphenylmethylene moiety in QNT 3-80 instead of paeonol may be too bulky to access the copper-binding catalytic center of tyrosinase, with L-tyrosine as the substrate.
Discussion and conclusions
Chemical-based regulation of skin disorders with hyperpigmented patches has been a long-standing goal for cosmetic and pharmaceutical applications. Numerous approaches have been taken to find chemicals that inhibit the tyrosinase activity or disturb the synthesis or release of melanin pigments in cell-based assays (Solano et al., 2006; Ando et al., 2007; Gillbro and Olsson, 2011). However, a large number of the chemicals have met with limited or no success in the translation from in vitro outcomes to patients. This discrepancy suggests that antimelanogenic chemicals with clinical effectiveness must overcome the challenge of penetrating the skin barrier and be stable in the active conformation without toxicity to the skin cells. Guinea pig skin displays similar kinetic parameters to human skin in the transdermal absorption of numerous chemicals and can thus serve as a surrogate for human skin, in terms of their cutaneous pharmacology (Barbero and Frasch, 2009).
In the current study, treatment with QNT 3-80 in vitro inhibited cAMP-induced melanin production in melanocytes. Moreover, topical treatment with QNT 3-80 in vivo ameliorated UVB-induced skin pigmentation in guinea pigs. As a molecular mechanism, QNT 3-80 directly antagonized the binding of the fluorescent probe for cAMP to the regulatory subunit of PKA as well as inhibiting the dissociation and activation of inactive PKA holoenzyme, following raised levels of cAMP (using α-MSH, forskolin or db-cAMP). Moreover, in molecular modelling, QNT 3-80 was well fitted into the cAMP-binding A and B sites on the crystal structure of PKA under the most energetically favourable simulation. This mechanism of QNT 3-80 consequently affected PKA-catalyzed CREB phosphorylation in the melanogenic process, thus suppressing the expression of MITF or tyrosinase gene in vitro and in vivo. However, QNT 3-80 did not inhibit binding of the fluorescent probe for α-MSH to its receptor, forskolin-induced cAMP production, catalytic subunit rhPKA-mediated kemptide phosphorylation and tyrosinase-catalyzed L-tyrosine oxidation activity. The safety profile of QNT 3-80 remains to be clarified but it affected neither the viability of B16 cells nor the skin corrosion in guinea pigs under topical application. Similarly, bisindolylmaleimide and haginin A have been reported as modifiers of PK activity, ameliorating UVB-induced skin pigmentation in guinea pigs without toxicity to epidermal melanocytes (Park et al., 2004; Kim et al., 2008).
The PKA-directed cAMP antagonist Rp-cAMPS decreased melanin production in α-MSH- or db-cAMP-stimulated B16 cells, as did QNT 3-80. Similarly, bisabolangelone (BISA) and 4-hydroxy-3-methoxycinnamaldehyde (4H3MC) have been identified as inhibitors of cAMP-induced dissociation and activation of inactive PKA holoenzyme in melanocytes and their topical application ameliorated UVB-induced skin pigmentation in guinea pigs (Roh et al., 2013; 2014). Therefore, QNT 3-80 mimicked Rp-cAMPS, BISA or 4H3MC in terms of its antimelanogenic mechanism. Advantages of QNT 3-80 include a simpler synthetic chemistry than BISA, which has chiral isomers, presumably lowering cost for clinical application, and has greater drug-like property than 4H3MC, according to Lipinski's rule.
In conclusion, the cAMP-binding site of PKA is a molecular target of QNT 3-80 in its antimelanogenic activity. This compound down-regulated expression of MITF or tyrosinase gene in vitro in assays of melanin production in cAMP-elevated melanocytes and in vivo using skin pigmentation in UVB-irradiated guinea pigs. Finally, this study suggests a potential application of QNT 3-80 in the cutaneous treatment of hyperpigmented skin disorders.
Acknowledgments
This work was financially supported by a grant from Chungbuk National University in 2013. We appreciate S. Shibahara (Tohoku University School of Medicine, Japan) and R. Ballotti (INSERM, France) for kind supply of MITF-Luc or tyrosinase-Luc construct.
Glossary
- α-MSH
α-melanocyte-stimulating hormone
- CREB
cAMP-responsive element-binding protein
- db-cAMP
N6,2′-O-dibutyryl-cAMP (bucladesine)
- MITF
microphthalmia-associated transcription factor
- PKA-Cα
catalytic subunit α of PKA
- PKA-RIIβ
regulatory subunit IIβ of PKA
- QNT 3-80
diphenylmethylene hydrazinecarbothioamide
- Rp-cAMPS
Rp-adenosine 3′,5′-cyclic monophosphorothioate
Authorship contribution statement
H. S., S. D. H., W.-J. C., S. H. P., D. Y. Y. and S. M. K. performed the research. H. S., E. R., J. T. H. and Y. K. designed the research study. S.-H. J. and B. Y. H. contributed essential reagents. H. S., T.-Y. H., S.-B. H. and Y. K. analysed the data. H. S. and Y. K. wrote the paper.
Conflict of interest
The authors declare no conflict of interest.
Supporting Information
Figure S1 Experimental protocol of UVB-induced skin pigmentation in guinea pigs. (A) Chemical structure of QNT 3-80. (B) Dorsal skin of guinea pigs were topically treated with QNT 3-80 (0.03–0.1%) and concurrently irradiated with UVB (350 mJ·cm−2 per exposure). Skin pigmentation was measured at 1 week after ending UVB radiation.
Figure S2 Effect of QNT 3-80 on cAMP-induced melanin production. B16 cells were stimulated with forskolin (A), IBMX (B) or db-cAMP (C, D) for 3 days in the presence of QNT 3-80 or other sample. Melanin contents were measured with synthetic melanin as a standard and normalized to the cell numbers. Data are shown as pg melanin per cell (A–C) or inhibition % (D). (E) B16 cells were incubated with QNT 3-80 for 3 days in the absence or presence of α-MSH. Cell viability was measured by MTT method, representing as absorbance values at 590 nm (A). Data are mean ± SD from three to five independent experiments. #P < 0.05 versus media alone. *P < 0.05 versus α-MSH, IBMX or db-cAMP alone-.
Figure S3 Effect of QNT 3-80 on UVB-induced skin pigmentation. Dorsal skin of guinea pigs were topically treated with vehicle, QNT 3-80 or arbutin and concurrently irradiated with UVB as described. Groups of five animals were used in this experiment. (A) A representative photograph of the pigmented skins. (B) Skin pigmentation was measured using chromometer and is represented as a relative whitening (L) index with normal skin L = 10. #P < 0.05 versus normal skin without UVB radiation. *P < 0.05 versus UVB plus vehicle alone. (C) QNT 3-80 or other sample was dissolved in 100% ethyl alcohol and absorbance values at 200–400 nm were measured.
Figure S4 Effects of QNT 3-80 on α-MSH binding to its receptor and forskolin-induced cAMP production. (A) B16 cells were incubated with the fluorescent probe for α-MSH, TAMRA-NDP-α-MSH (500 nM), for 2 h in the presence of QNT 3-80. After washing, the cells were subjected to flow cytometric analysis. (B) B16 cells were pretreated with QNT 3-80 for 2 h and stimulated with forskolin for 10–20 min in the presence of QNT 3-80. Cell extracts were subjected to elisa to determine cAMP levels and normalized to the protein contents. Data are mean ± SD from three independent experiments. #P < 0.05 versus media alone.
Figure S5 Effect of QNT 3-80 on protein levels of PKA-Cα or PKA-RIIβ. B16 cells were pretreated with QNT 3-80 for 2 h and stimulated with α-MSH for 20–30 min in the presence of QNT 3-80. Cell extracts were analysed by Western blot analysis (WB) with anti-PKA-RIIβ, anti-PKA-Cα or anti-GAPDH antibody.
Figure S6 Competition between QNT 3-80 and 8-[ϕ-575]-cAMP in the binding to PKA-RIIβ. PKA-RIIβ polypeptide was pretreated with QNT 3-80 or Rp-cAMPS in cell-free conditions for 2 h and incubated with the fluorescent probe 8-[ϕ-575]-cAMP at excess concentrations of up to 10-fold (10X) starting from 1× (1 μM) for another 2 h. 8-[ϕ-575]-cAMP binding to PKA-RIIβ polypeptide was measured as relative fluorescence units (RFU) under excitation at 575 nm and emission at 620 nm. Data are mean ± SD from three independent experiments. *P < 0.05 versus 8-[ϕ-575]-cAMP (1X) plus QNT 3-80 alone or 8-[ϕ-575]-cAMP (1X) plus Rp-cAMPS.
Figure S7 Superimposed docking of QNT 3-80 onto the cAMP bound with PKA-RIIβ. Docking arrangements of QNT 3-80 to the crystal structure of cAMP-binding A or B site of human PKA-RIIβ were carried out using the Surflex-Dock program and superimposed onto those of the endogenous ligand cAMP. QNT 3-80 or its interacting residues on PKA-RIIβ is represented by a grey colour, cAMP by a pink colour and the other PKA-RIIβ backbone by a green colour. Hydrogen bonding between QNT 3-80 and PKA-RIIβ is shown as a black dotted line.
Figure S8 Effect of QNT 3-80 on tyrosinase expression. B16 cells were pretreated with QNT 3-80 or Rp-cAMPS for 2 h and stimulated with α-MSH for 48 h (A) or 12 h (B) in the presence of QNT 3-80 or Rp-cAMPS. (A) Cell extracts were subjected to Western blot analysis (WB) with anti-tyrosinase or anti-GAPDH antibody. (B) Total RNAs were subjected to RT-PCR analysis of tyrosinase with the internal control β-actin. (C) B16 cells were transfected with tyrosinase (−2236/+59)-Luc reporter construct in combination with Renilla control vector and then stimulated with α-MSH for 20 h in the presence of QNT 3-80. Cell extracts were analysed by a dual-luciferase assay. Firefly luciferase activity is represented as relative fold change after normalizing to the Renilla activity. Data are mean ± SD from three independent experiments. #P < 0.05 versus media alone. *P < 0.05 versus α-MSH alone. Dorsal skin of guinea pigs were topically treated with QNT 3-80 and concurrently irradiated with UVB as described. Skin tissues were biopsied. (D) Cell extracts were subjected to Western blots with anti-tyrosinase or anti-GAPDH antibody. (E) Total RNAs were subjected to RT-PCR analysis of tyrosinse with the internal control β-actin.
Figure S9 Effects of QNT 3-80 on the copper ion chelation and the tyrosinase activity. (A) QNT 3-80 (50 μM) was dissolved in 100% ethyl alcohol, reacted with variable concentrations (0–50 μM) of CuSO4 and then measured absorbance values at 200–400 nm. Mushroom tyrosinase (B) or human tyrosinase (C) was treated with QNT 3-80 or arbutin in cell-free reactions for 10 min and its catalytic activity was then determined as L-tyrosine oxidation velocity. Data are mean ± SD from three independent experiments. *P < 0.05 versus tyrosinase alone.
References
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M, et al. The Concise Guide to PHARMACOLOGY 2013/14: Enzymes. Br J Pharmacol. 2013a;170:1797–1867. doi: 10.1111/bph.12451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M, et al. The Concise Guide to PHARMACOLOGY 2013/14: G Protein-Coupled Receptors. Br J Pharmacol. 2013b;170:1459–1581. doi: 10.1111/bph.12445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ando H, Kondoh H, Ichihashi M, Hearing VJ. Approaches to identify inhibitors of melanin biosynthesis via the quality control of tyrosinase. J Invest Dermatol. 2007;127:751–761. doi: 10.1038/sj.jid.5700683. [DOI] [PubMed] [Google Scholar]
- Ando H, Niki Y, Ito M, Akiyama K, Matsui MS, Yarosh DB, et al. Melanosomes are transferred from melanocytes to keratinocytes through the processes of packaging, release, uptake, and dispersion. J Invest Dermatol. 2012;132:1222–1229. doi: 10.1038/jid.2011.413. [DOI] [PubMed] [Google Scholar]
- Barbero AM, Frasch HF. Pig and guinea pig skin as surrogates for human in vitro penetration studies: a quantitative review. Toxicol In Vitro. 2009;23:1–13. doi: 10.1016/j.tiv.2008.10.008. [DOI] [PubMed] [Google Scholar]
- Bertolotto C, Bille K, Ortonne JP, Ballotti R. Regulation of tyrosinase gene expression by cAMP in B16 melanoma cells involves two CATGTG motifs surrounding the TATA box: implication of the microphthalmia gene product. J Cell Biol. 1996;134:747–755. doi: 10.1083/jcb.134.3.747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandon EP, Idzerda RL, McKnight GS. PKA isoforms, neural pathways, and behaviour: making the connection. Curr Opin Neurobiol. 1997;7:397–403. doi: 10.1016/s0959-4388(97)80069-4. [DOI] [PubMed] [Google Scholar]
- Busca R, Ballotti R. Cyclic AMP a key messenger in the regulation of skin pigmentation. Pigment Cell Res. 2000;13:60–69. doi: 10.1034/j.1600-0749.2000.130203.x. [DOI] [PubMed] [Google Scholar]
- Diller TC, Madhusudan, Xuong NH, Taylor SS. Molecular basis for regulatory subunit diversity in cAMP-dependent protein kinase: crystal structure of the type IIβ regulatory subunit. Structure. 2001;9:73–82. doi: 10.1016/s0969-2126(00)00556-6. [DOI] [PubMed] [Google Scholar]
- Gillbro JM, Olsson MJ. The melanogenesis and mechanisms of skin-lightening agents: existing and new approaches. Int J Cosmet Sci. 2011;33:210–221. doi: 10.1111/j.1468-2494.2010.00616.x. [DOI] [PubMed] [Google Scholar]
- Grimes P, Nordlund JJ, Pandya AG, Taylor S, Rendon M, Ortonne JP. Increasing our understanding of pigmentary disorders. J Am Acad Dermatol. 2006;54:S255–S261. doi: 10.1016/j.jaad.2005.12.042. [DOI] [PubMed] [Google Scholar]
- Imokawa G. Autocrine and paracrine regulation of melanocytes in human skin and in pigmentary disorders. Pigment Cell Res. 2004;17:96–110. doi: 10.1111/j.1600-0749.2003.00126.x. [DOI] [PubMed] [Google Scholar]
- Ito S, Wakamatsu K. Chemistry of mixed melanogenesis: pivotal roles of dopaquinone. Photochem Photobiol. 2008;84:582–592. doi: 10.1111/j.1751-1097.2007.00238.x. [DOI] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG. Animal research: reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1577–1579. doi: 10.1111/j.1476-5381.2010.00872.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim C, Cheng CY, Saldanha SA, Taylor SS. PKA-I holoenzyme structure reveals a mechanism for cAMP-dependent activation. Cell. 2007;130:1032–1043. doi: 10.1016/j.cell.2007.07.018. [DOI] [PubMed] [Google Scholar]
- Kim JH, Baek SH, Kim DH, Choi TY, Yoon TJ, Hwang JS, et al. Downregulation of melanin synthesis by haginin A and its application to in vivo lightening model. J Invest Dermatol. 2008;128:1227–1235. doi: 10.1038/sj.jid.5701177. [DOI] [PubMed] [Google Scholar]
- Kim YM, Yun J, Lee CK, Lee H, Min KR, Kim Y. Oxyresveratrol and hydroxystilbene compounds: inhibitory effect on tyrosinase and mechanism of action. J Biol Chem. 2002;277:16340–16344. doi: 10.1074/jbc.M200678200. [DOI] [PubMed] [Google Scholar]
- Le Pape E, Wakamatsu K, Ito S, Wolber R, Hearing VJ. Regulation of eumelanin/pheomelanin synthesis and visible pigmentation in melanocytes by ligands of the melanocortin 1 receptor. Pigment Cell Melanoma Res. 2008;21:477–486. doi: 10.1111/j.1755-148X.2008.00479.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee KC, Thanigaimalai P, Sharma VK, Kim MS, Roh E, Hwang BY, et al. Structural characteristics of thiosemicarbazones as inhibitors of melanogenesis. Bioorg Med Chem Lett. 2010;20:6794–6796. doi: 10.1016/j.bmcl.2010.08.114. [DOI] [PubMed] [Google Scholar]
- Lochner A, Moolman JA. The many faces of H89: a review. Cardiovasc Drug Rev. 2006;24:261–274. doi: 10.1111/j.1527-3466.2006.00261.x. [DOI] [PubMed] [Google Scholar]
- Maeda K, Fukuda M. Arbutin: mechanism of its depigmenting action in human melanocyte culture. J Pharmacol Exp Ther. 1996;276:765–769. [PubMed] [Google Scholar]
- McGrath J, Drummond G, McLachlan E, Kilkenny C, Wainwright C. Guidelines for reporting experiments involving animals: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1573–1576. doi: 10.1111/j.1476-5381.2010.00873.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyamura Y, Coelho SG, Wolber R, Miller SA, Wakamatsu K, Zmudzka BZ, et al. Regulation of human skin pigmentation and responses to ultraviolet radiation. Pigment Cell Res. 2007;20:2–13. doi: 10.1111/j.1600-0749.2006.00358.x. [DOI] [PubMed] [Google Scholar]
- Moll D, Prinz A, Brendel CM, Berrera M, Guske K, Zaccolo M, et al. Biochemical characterization and cellular imaging of a novel, membrane permeable fluorescent cAMP analog. BMC Biochem. 2008;9:18. doi: 10.1186/1471-2091-9-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olivares C, Solano F. New insights into the active site structure and catalytic mechanism of tyrosinase and its related proteins. Pigment Cell Melanoma Res. 2009;22:750–760. doi: 10.1111/j.1755-148X.2009.00636.x. [DOI] [PubMed] [Google Scholar]
- Ortonne JP, Passeron T. Melanin pigmentary disorders: treatment update. Dermatol Clin. 2005;23:209–226. doi: 10.1016/j.det.2005.01.001. [DOI] [PubMed] [Google Scholar]
- Park HY, Lee J, Gonzalez S, Middelkamp-Hup MA, Kapasi S, Peterson S, et al. Topical application of a protein kinase C inhibitor reduces skin and hair pigmentation. J Invest Dermatol. 2004;122:159–166. doi: 10.1046/j.0022-202X.2003.22134.x. [DOI] [PubMed] [Google Scholar]
- Pawson AJ, Sharman JL, Benson HE, Faccenda E, Alexander SP, Buneman OP, et al. NC-IUPHAR. The IUPHAR/BPS Guide to PHARMACOLOGY: an expert-driven knowledge base of drug targets and their ligands. Nucl Acids Res. 2014;42(Database Issue):D1098–D1106. doi: 10.1093/nar/gkt1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roh E, Yun CY, Yun JY, Park D, Kim ND, Hwang BY, et al. cAMP-binding site of PKA as a molecular target of bisabolangelone against melanocyte-specific hyperpigmented disorder. J Invest Dermatol. 2013;133:1072–1079. doi: 10.1038/jid.2012.425. [DOI] [PubMed] [Google Scholar]
- Roh E, Jeong IY, Shin H, Song S, Kim ND, Jung SH, et al. Downregulation of melanocyte-specific facultative melanogenesis by 4-hydroxy-3-methoxycinnamaldehyde acting as a cAMP antagonist. J Invest Dermatol. 2014;134:551–553. doi: 10.1038/jid.2013.341. [DOI] [PubMed] [Google Scholar]
- Sands WA, Palmer TM. Regulating gene transcription in response to cyclic AMP elevation. Cell Signal. 2008;20:460–466. doi: 10.1016/j.cellsig.2007.10.005. [DOI] [PubMed] [Google Scholar]
- Schallreuter KU, Kothari S, Chavan B, Spencer JD. Regulation of melanogenesis: controversies and new concepts. Exp Dermatol. 2008;17:395–404. doi: 10.1111/j.1600-0625.2007.00675.x. [DOI] [PubMed] [Google Scholar]
- Simon JD, Peles D, Wakamatsu K, Ito S. Current challenges in understanding melanogenesis: bridging chemistry, biological control, morphology, and function. Pigment Cell Melanoma Res. 2009;22:563–579. doi: 10.1111/j.1755-148X.2009.00610.x. [DOI] [PubMed] [Google Scholar]
- Skalhegg BS, Tasken K. Specificity in the cAMP/PKA signaling pathway: differential expression, regulation, and subcellular localization of subunits of PKA. Front Biosci. 2000;5:D678–D693. doi: 10.2741/skalhegg. [DOI] [PubMed] [Google Scholar]
- Slominski A, Tobin DJ, Shibahara S, Wortsman J. Melanin pigmentation in mammalian skin and its hormonal regulation. Physiol Rev. 2004;84:1155–1228. doi: 10.1152/physrev.00044.2003. [DOI] [PubMed] [Google Scholar]
- Solano F, Briganti S, Picardo M, Ghanem G. Hypopigmenting agents: an updated review on biological, chemical and clinical aspects. Pigment Cell Res. 2006;19:550–571. doi: 10.1111/j.1600-0749.2006.00334.x. [DOI] [PubMed] [Google Scholar]
- Thanigaimalai P, Lee KC, Sharma VK, Roh E, Kim Y, Jung SH. Ketonethiosemicarbazones: structure-activity relationships for their melanogenesis inhibition. Bioorg Med Chem Lett. 2011;21:3527–3530. doi: 10.1016/j.bmcl.2011.04.146. [DOI] [PubMed] [Google Scholar]
- Udono T, Yasumoto K, Takeda K, Amae S, Watanabe K, Saito H, et al. Structural organization of the human microphthalmia-associated transcription factor gene containing four alternative promoters. Biochim Biophys Acta. 2000;1491:205–219. doi: 10.1016/s0167-4781(00)00051-8. [DOI] [PubMed] [Google Scholar]
- Vachtenheim J, Borovansky J. ‘Transcription physiology’ of pigment formation in melanocytes: central role of MITF. Exp Dermatol. 2010;19:617–627. doi: 10.1111/j.1600-0625.2009.01053.x. [DOI] [PubMed] [Google Scholar]
- Wu J, Jones JM, Nguyen-Huu X, Ten Eyck LF, Taylor SS. Crystal structures of RIα subunit of cyclic adenosine 5′-monophosphate (cAMP)-dependent protein kinase complexed with (Rp)-adenosine 3′,5′-cyclic monophosphothioate and (Sp)-adenosine 3′,5′-cyclic monophosphothioate, the phosphothioate analogues of cAMP. Biochemistry (Mosc) 2004;43:6620–6629. doi: 10.1021/bi0302503. [DOI] [PubMed] [Google Scholar]
- Zawadzki KM, Taylor SS. cAMP-dependent protein kinase regulatory subunit type IIβ: active site mutations define an isoform-specific network for allosteric signaling by cAMP. J Biol Chem. 2004;279:7029–7036. doi: 10.1074/jbc.M310804200. [DOI] [PubMed] [Google Scholar]
- Zhu TH, Cao SW, Yu YY. Synthesis, characterization and biological evaluation of paeonol thiosemicarbazone analogues as mushroom tyrosinase inhibitors. Int J Biol Macromol. 2013;62:589–595. doi: 10.1016/j.ijbiomac.2013.09.056. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 Experimental protocol of UVB-induced skin pigmentation in guinea pigs. (A) Chemical structure of QNT 3-80. (B) Dorsal skin of guinea pigs were topically treated with QNT 3-80 (0.03–0.1%) and concurrently irradiated with UVB (350 mJ·cm−2 per exposure). Skin pigmentation was measured at 1 week after ending UVB radiation.
Figure S2 Effect of QNT 3-80 on cAMP-induced melanin production. B16 cells were stimulated with forskolin (A), IBMX (B) or db-cAMP (C, D) for 3 days in the presence of QNT 3-80 or other sample. Melanin contents were measured with synthetic melanin as a standard and normalized to the cell numbers. Data are shown as pg melanin per cell (A–C) or inhibition % (D). (E) B16 cells were incubated with QNT 3-80 for 3 days in the absence or presence of α-MSH. Cell viability was measured by MTT method, representing as absorbance values at 590 nm (A). Data are mean ± SD from three to five independent experiments. #P < 0.05 versus media alone. *P < 0.05 versus α-MSH, IBMX or db-cAMP alone-.
Figure S3 Effect of QNT 3-80 on UVB-induced skin pigmentation. Dorsal skin of guinea pigs were topically treated with vehicle, QNT 3-80 or arbutin and concurrently irradiated with UVB as described. Groups of five animals were used in this experiment. (A) A representative photograph of the pigmented skins. (B) Skin pigmentation was measured using chromometer and is represented as a relative whitening (L) index with normal skin L = 10. #P < 0.05 versus normal skin without UVB radiation. *P < 0.05 versus UVB plus vehicle alone. (C) QNT 3-80 or other sample was dissolved in 100% ethyl alcohol and absorbance values at 200–400 nm were measured.
Figure S4 Effects of QNT 3-80 on α-MSH binding to its receptor and forskolin-induced cAMP production. (A) B16 cells were incubated with the fluorescent probe for α-MSH, TAMRA-NDP-α-MSH (500 nM), for 2 h in the presence of QNT 3-80. After washing, the cells were subjected to flow cytometric analysis. (B) B16 cells were pretreated with QNT 3-80 for 2 h and stimulated with forskolin for 10–20 min in the presence of QNT 3-80. Cell extracts were subjected to elisa to determine cAMP levels and normalized to the protein contents. Data are mean ± SD from three independent experiments. #P < 0.05 versus media alone.
Figure S5 Effect of QNT 3-80 on protein levels of PKA-Cα or PKA-RIIβ. B16 cells were pretreated with QNT 3-80 for 2 h and stimulated with α-MSH for 20–30 min in the presence of QNT 3-80. Cell extracts were analysed by Western blot analysis (WB) with anti-PKA-RIIβ, anti-PKA-Cα or anti-GAPDH antibody.
Figure S6 Competition between QNT 3-80 and 8-[ϕ-575]-cAMP in the binding to PKA-RIIβ. PKA-RIIβ polypeptide was pretreated with QNT 3-80 or Rp-cAMPS in cell-free conditions for 2 h and incubated with the fluorescent probe 8-[ϕ-575]-cAMP at excess concentrations of up to 10-fold (10X) starting from 1× (1 μM) for another 2 h. 8-[ϕ-575]-cAMP binding to PKA-RIIβ polypeptide was measured as relative fluorescence units (RFU) under excitation at 575 nm and emission at 620 nm. Data are mean ± SD from three independent experiments. *P < 0.05 versus 8-[ϕ-575]-cAMP (1X) plus QNT 3-80 alone or 8-[ϕ-575]-cAMP (1X) plus Rp-cAMPS.
Figure S7 Superimposed docking of QNT 3-80 onto the cAMP bound with PKA-RIIβ. Docking arrangements of QNT 3-80 to the crystal structure of cAMP-binding A or B site of human PKA-RIIβ were carried out using the Surflex-Dock program and superimposed onto those of the endogenous ligand cAMP. QNT 3-80 or its interacting residues on PKA-RIIβ is represented by a grey colour, cAMP by a pink colour and the other PKA-RIIβ backbone by a green colour. Hydrogen bonding between QNT 3-80 and PKA-RIIβ is shown as a black dotted line.
Figure S8 Effect of QNT 3-80 on tyrosinase expression. B16 cells were pretreated with QNT 3-80 or Rp-cAMPS for 2 h and stimulated with α-MSH for 48 h (A) or 12 h (B) in the presence of QNT 3-80 or Rp-cAMPS. (A) Cell extracts were subjected to Western blot analysis (WB) with anti-tyrosinase or anti-GAPDH antibody. (B) Total RNAs were subjected to RT-PCR analysis of tyrosinase with the internal control β-actin. (C) B16 cells were transfected with tyrosinase (−2236/+59)-Luc reporter construct in combination with Renilla control vector and then stimulated with α-MSH for 20 h in the presence of QNT 3-80. Cell extracts were analysed by a dual-luciferase assay. Firefly luciferase activity is represented as relative fold change after normalizing to the Renilla activity. Data are mean ± SD from three independent experiments. #P < 0.05 versus media alone. *P < 0.05 versus α-MSH alone. Dorsal skin of guinea pigs were topically treated with QNT 3-80 and concurrently irradiated with UVB as described. Skin tissues were biopsied. (D) Cell extracts were subjected to Western blots with anti-tyrosinase or anti-GAPDH antibody. (E) Total RNAs were subjected to RT-PCR analysis of tyrosinse with the internal control β-actin.
Figure S9 Effects of QNT 3-80 on the copper ion chelation and the tyrosinase activity. (A) QNT 3-80 (50 μM) was dissolved in 100% ethyl alcohol, reacted with variable concentrations (0–50 μM) of CuSO4 and then measured absorbance values at 200–400 nm. Mushroom tyrosinase (B) or human tyrosinase (C) was treated with QNT 3-80 or arbutin in cell-free reactions for 10 min and its catalytic activity was then determined as L-tyrosine oxidation velocity. Data are mean ± SD from three independent experiments. *P < 0.05 versus tyrosinase alone.