

Abstract
Background and Purpose
Pro-inflammatory cytokines are important in rheumatoid arthritis (RA) and their production is mainly regulated by NF-κB and inflammasomes. Carboxyamidotriazole (CAI) exhibits potent anti-inflammatory activities by decreasing cytokines. Here, we have investigated NACHT, LRR and PYD domains-containing protein (NALP) inflammasomes in a rat model of RA and explored the therapeutic effects of CAI in this model and the involvement of NF-κB and inflammasomes in the actions of CAI.
Experimental Approach
The anti-arthritic effects of CAI were assessed in the adjuvant arthritis (AA) model in rats, using radiological and histological techniques. NALP1 and NALP3 inflammasomes, NF-κB pathway and pro-inflammatory cytokines levels were measured with Western blots, immunohistochemistry and elisa.
Key Results
CAI decreased the arthritis index, improved radiological and histological changes, and reduced synovial IL-1β, IL-6, IL-18 and TNF-α levels in rats with AA. Compared with normal rats, the 70 kDa NALP1 isoform was up-regulated, NALP3 was down-regulated, and levels of the 165 kDa NALP1 isoform and the adaptor protein ASC were unchanged in synovial tissue from AA rats. CAI reduced the 70 kDa NALP1 isoform and restored NALP3 levels in AA rats; CAI inhibited caspase-1 activation in AA synovial tissue, but not its enzymic activity in vitro. In addition, CAI reduced expression of p65 NF-κB subunit and IκBα phosphorylation and degradation in AA rats.
Conclusion and Implications
NALP1 inflammasomes were activated in synovial tissues from AA rats and appeared to be a novel therapeutic target for RA. CAI could have therapeutic value in RA by inhibiting activation of NF-κB and NALP1 inflammasomes and by decreasing pro-inflammatory cytokines.
Tables of Links
| Targets | |
|---|---|
| Catalytic receptorsa | |
| NALP1 (NLRP1) | |
| NALP3 (NLRP3) | |
| Enzymesb | |
| Caspase 1 | |
| Caspase 5 |
| Ligands |
|---|
| Dexamethasone |
| IL-1β |
| IL-6 |
| IL-18 |
| IL-33 |
| TNF-α |
These Tables list key protein targets and ligands in this article which are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Pawson et al., 2014) and are permanently archived in the Concise Guide to PHARMACOLOGY 2013/14 (a,bAlexander et al., 2013a,b).
Introduction
Rheumatoid arthritis (RA) is a chronic and autoimmune inflammatory disease characterized by persistent synovitis in many joints, eventually leading to articular destruction and functional disability. Overproduction of pro-inflammatory cytokines particularly IL-1β, TNF-α and IL-6, is known to play a major role in RA pathogenesis by initiating and propagating the synovitis, and driving joint damage (Choy and Panayi, 2001; Feldmann, 2001). Therefore, the pathways regulating the production of cytokines, including those involved with NF-κB and the inflammasomes, may be important therapeutic targets in RA.
The inflammasomes are the multi-protein complexes formed in the cytoplasm upon stimulation and are responsible for the maturation and activation of pro-inflammatory IL-1β, IL-18 and IL-33 (Schroder and Tschopp, 2010). In general, inflammasomes comprise the NOD-like receptors (NLRs), apoptosis-associated speck-like protein containing a caspase activation and recruitment domain (CARD) (ASC) and caspase-1. In addition, caspase-5 is found only in NALP1 inflammasomes. Upon detecting the stimuli, the receptor recruits the caspase-1 precursor (pro-caspase-1) either directly or through the adaptor molecule ASC and the inflammasome is formed, enabling the proteolytic activation of pro-caspase-1. The activated caspase-1 subsequently cleaves inactive pro-IL-1β, pro-IL-18 and pro-IL-33 into their active forms, leading to their secretion from the cells. To date, four inflammasomes have been identified, which are defined by the receptor they contain: NALP1 (NLRP1), NALP3 (NLRP3), IPAF (NLRC4) and AIM2 (Latz, 2010; Franchi et al., 2012). Diverse microbial and endogenous stimuli activate the different inflammasomes. The IPAF inflammasome senses the bacterial flagellin, and AIM2 inflammasome senses foreign double-stranded DNA. The NALP3 inflammasome is the best-known inflammasome and activated by diverse signals including bacteria, viruses, purified microbial products, endogenous danger signals released from damaged or dying cells, low MW immune activators, and crystalline or aggregated materials (Schroder and Tschopp, 2010; Schroder et al., 2010). Recent studies have implicated the inappropriate activation of NALP3 inflammasomes in the pathogenesis of a number of human diseases including type 2 diabetes, gout, silicosis and neurodegeneration (Martinon et al., 2006; Cassel et al., 2008; Halle et al., 2008; Masters et al., 2010). Although the NALP1 inflammasome was the first inflammasome to be identified, the mechanism of activation and its exact function remain relatively unclear. It has been proposed that muramyl dipeptide and ATP may stimulate its assembly (de Zoete and Flavell, 2012).
Clinical and experimental studies point to a key role for IL-1β and IL-18 in RA pathophysiology. However, the expression of and changes in the NALP inflammasomes, the key molecular platforms for IL-1β and IL-18 maturation, are less well understood in RA. Only the components of NALP1 and NALP3 inflammasomes were detected at the mRNA and protein levels in RA synovium, without the change patterns, in comparison with the normal control (Rosengren et al., 2005; Kolly et al., 2009), and a recent study found that NALP1 gene polymorphism is a risk factor for RA (Sui et al., 2012).
Carboxyamidotriazole (CAI) is a non-cytotoxic anticancer drug in development. It inhibits the proliferation and invasive behaviour of a wide array of tumour cell lines in vitro and in vivo (Kohn et al., 1994; Wasilenko et al., 1996; Moody et al., 2003). Recently we reported the anti-inflammatory effects of CAI in several animal models of acute and chronic inflammation, including croton oil-induced ear oedema, cotton-induced granuloma and dextran sodium sulphate (DSS)-induced colitis. Another noticeable finding was that CAI decreased pro-inflammatory cytokines, such as TNF-α and IL-1β, at the inflammatory sites and in the serum of the experimental animals (Guo et al., 2008; 2012). We therefore suggested that such down-regulation of pro-inflammatory cytokines might make CAI a potential candidate for the treatment of RA, a condition in which pro-inflammatory cytokines play an important pathological role. Our initial studies showed that CAI inhibited the paw swelling induced by adjuvant arthritis (AA) in rats and decreased the levels of TNF-α and IL-1β in homogenates of the inflamed paw (Guo et al., 2008). However, the therapeutic effects, especially its effectiveness against joint damage, and related mechanisms of CAI on RA had not been studied.
The AA model is widely used as an animal model to investigate the pathogenic mechanisms of RA and to evaluate anti-arthritic drugs (Whitehouse, 2007). Here we used this model to detect the expressions and activities of NALP inflammasomes in the synovial tissue and changes in their pattern during arthritis. We also investigated whether CAI could attenuate the joint inflammation and the progressive joint destruction in the rat model of AA. Moreover, the effects of CAI on NF-κB and NALP inflammasomes, which are the crucial pathways regulating the production of pro-inflammatory cytokines were assessed.
Methods
Animals
All animal care and experimental procedures were approved by the Institutional Animal Care and Use Committee of Chinese Academy of Medical Science. Studies involving animals are reported in accordance with the ARRIVE guidelines for reporting experiments involving animals (Kilkenny et al., 2010; McGrath et al., 2010). A total of 98 male Lewis rats (180–210 g, Beijing Vital River Laboratory Animal Co., Beijing, China) were used in this study. They were housed under controlled environmental conditions (22 ± 2°C temperature, 40–60% humidity, 08:00–20:00 h alternate light–dark cycles, food and water ad libitum).
AA induction and drug treatment
Rats were randomly divided into seven groups (n = 14 per group) before the experiment. AA was induced in six groups on day 0 by injecting 0.1 mL of complete Freund's adjuvant (CFA), prepared by suspending heat-killed Mycobacterium tuberculosis in mineral oil (10 mg·mL−1), s.c. at the base of the tail. The remaining normal control group received 0.1 mL of mineral oil alone (Montesinos et al., 2000). Each group of rats with AA received one of the following treatments once daily from day 12 when the signs of arthritis began to appear, and continued to day 26: CAI (10, 20 and 40 mg·kg−1, p.o ), dexamethasone (0.2 mg·kg−1, i.p.), vehicle polyethylene glycol 400 (PEG 400, p.o.) and vehicle normal saline (N.S., i.p.).
Arthritis assessment
The arthritic severity in each paw was evaluated by two observers unaware of the experiment using a semi-quantitative scoring system: 0 = normal, 1 = erythema or swelling of one joint, 2 = swelling of two or more joints, 3 = swelling of all joints, 4 = ankylosis or deformity. The cumulative score for all four paws of each rat was used as arthritis index with a maximum value of 16.
Radiological assessment
On day 27, rats were anaesthetized with 1% sodium pentobarbital. The radiographs of both hind limbs were obtained and evaluated by two observers unaware of the experiment on a 0∼3 scale (0 = normal, 1 = mild, 2 = moderate, 3 = severely affected joint), as described previously. The following three parameters were scored: osteoporosis, cartilage loss and erosion. Total radiological score was calculated from the sum of both hind limbs, with a maximum possible score of 6 for each radiological parameter per rat (Ahmed et al., 2010).
Histological assessment
After the X-ray check, rats were killed with ether anaesthesia and the ankle joints were removed for histological examination. For semiquantitative analysis, two sections of every joint (n = 4 per group) were taken at different depths, with six fields in each section. Histological changes were scored on a 0∼3 scale (0 = normal, 1 = mild, 2 = moderate, 3 = severely affected joint) for each of the following parameters: cartilage destruction, bone erosion and synovial inflammation (Ahmed et al., 2010).
Immunohistochemistry
Immunohistochemistry was performed as described previously (Ahmed et al., 2010). Six fields in each section from four rats of each group were inspected. Immunohistochemical score was calculated based on the sum of a proportion score (percent of positive-stained cells: 0, none; 1, <30%; 2, 30–70%; and 3, <70%) and intensity score (0, none; 1, weak; 2, intermediate; and 3, strong). And the numerical rating score were as follows: 0, negative; 2–3, weak positive; 4, positive; 5–6, strong positive (Allred et al., 1998).
Determination of cytokines in synovium homogenates
Synovial tissues were isolated from rat joints and homogenized in ice-cold saline solution. The levels of IL-1β, IL-6, IL-18 and TNF-α were measured using the appropriate elisa kits according to the manufacturer's instructions.
Western blot analysis
Synovial tissues were homogenized in ice-cold RIPA lysis buffer. Equivalent amounts of protein (60 μg) were separated on SDS-PAGE and were then transferred to PVDF membranes. Membranes were incubated with individual primary antibodies at 4°C overnight and then incubated with HRP-coupled secondary antibody. The immunoreactive proteins were detected with Immobilon Western Chemiluminescent HRP Substrate and quantified using Kodak 1D image analysis software (Eastman Kodak Company, Rochester, NY, USA).
Caspase-1 activity assay
The direct effects of CAI on caspase-1 activity in vitro were determined by a caspase-1 inhibitor drug screening kit. The assay utilized the synthetic caspase-1-specific peptide substrate YVAD-AFC. Active caspase-1 cleaved the substrate YVAD-AFC to release free AFC which can be quantified by fluorometry. The specific caspase-1 inhibitor Z-VAD-FMK provided by the kit was used as the positive control.
In addition, caspase-1 activity was measured in synovial tissues of rats from different groups using a caspase-1 fluorometric assay kit. Synovial tissues were homogenized and the peptide substrate YVAD-AFC was added to the lysates. Caspase-1 activity was measured by detecting fluorescent AFC and expressed as fluorescence intensity per mg protein.
Data analysis
Results are presented as mean ± SEM. Statistical significance was determined by one-way anova for multiple comparisons with Bonferroni's post hoc test; P < 0.05 was considered statistically significant.
Materials
CAI was synthesized by the Institute of Materia Medica, Chinese Academy of Medical Sciences. It was dissolved in PEG 400. Dexamethasone sodium phosphate injection was purchased from Tianjin Jinyao Amino Acid Company (Tianjing, China) and was diluted in N.S. M. tuberculosis was from the National Vaccine and Serum Institute (China). TNF-α, IL-1β, IL-6 and IL-18 elisa kits were from R&D Systems (Minneapolis, MN, USA). Caspase-1 fluorometric assay kit and caspase-1 inhibitor drug screening kit were from Biovision (Mountain View, CA, USA). The following antibodies were used: NALP1 (Cell Signaling Technology, Beverly, MA, USA); IL-1β (Abcam, Cambridge, MA, USA); IL-18 (R&D Systems); NALP3, ASC, p65, p-IkBα, IkBα, caspase-1 and β-actin (Santa Cruz Biotechnology, Santa Cruz, CA, USA); caspase-5 (Santa Cruz Biotechnology; BioVision, Mountain View, CA, USA).
Results
CAI suppressed adjuvant-induced arthritis in rats
In rats inoculated with CFA, signs of inflammation, including paw swelling, redness and warmth, appeared on about day 12, then gradually increased and reached a peak on day 22, with a maximum value of the arthritis index. The administration of CAI (10, 20, 40 mg·kg−1) dose-dependently decreased the arthritis index. Meanwhile, dexamethasone as the positive control almost completely inhibited the development of the arthritis from day 14 (Figure 1A).
Figure 1.
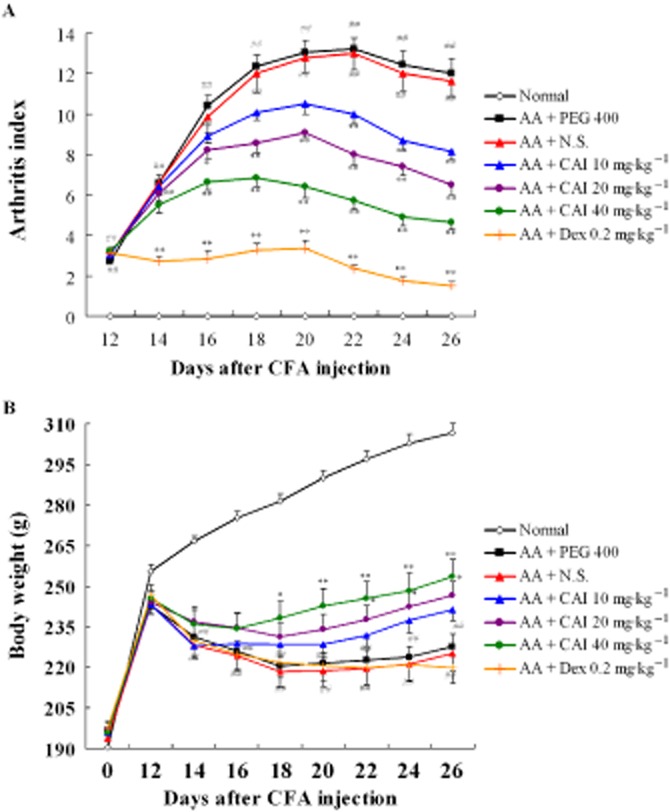
Anti-arthritis effects of CAI in rat model of AA. AA was induced by a single s.c. injection of 0.1 mL of CFA at the base of the tail on day 0. CAI (10, 20, 40 mg·kg−1) and dexamethasone (Dex; 0.2 mg·kg−1) were given from days 12 to 26. (A) Arthritis index was evaluated as described. (B) The body weight of each rat was measured over 26 days after injection of CFA. Data shown are means ± SEM of 14 rats. ##P < 0.01 versus normal group, *P < 0.05, **P < 0.01 versus vehicle-treated AA group. N.S., normal saline.
As AA is a systemic disease, body weight loss is a major clinical finding. Changes in body weight were measured during the period of drug treatment. In comparison with the normal rats, which showed a progressive increase of body weight, the AA rats started to lose body weight on day 12, and this trend continued throughout day 26. Treatment with CAI (20 mg·kg−1) increased the body weight of AA rats on days 22, 24 and 26, and, at the higher dose (40 mg·kg−1), did so from days 18 to 26. However, dexamethasone had no effect on the body weight of AA rats (Figure 1B).
CAI reduced the radiological signs of joint destruction in AA rats
The radiographic results from the vehicle-treated AA rats manifested soft tissue swelling around the joint and almost complete loss of the normal joint architecture. The increased radiological scores resulted from osteoporosis, joint space narrowing and bone erosion, in contrast to the normal rats. Administration of CAI (20 or 40 mg·kg−1) ameliorated the joint destruction and reduced all three radiological scores. However, the positive control dexamethasone failed to affect osteoporosis, and only decreased the joint space narrowing score and the bone erosion score (Figure 2).
Figure 2.
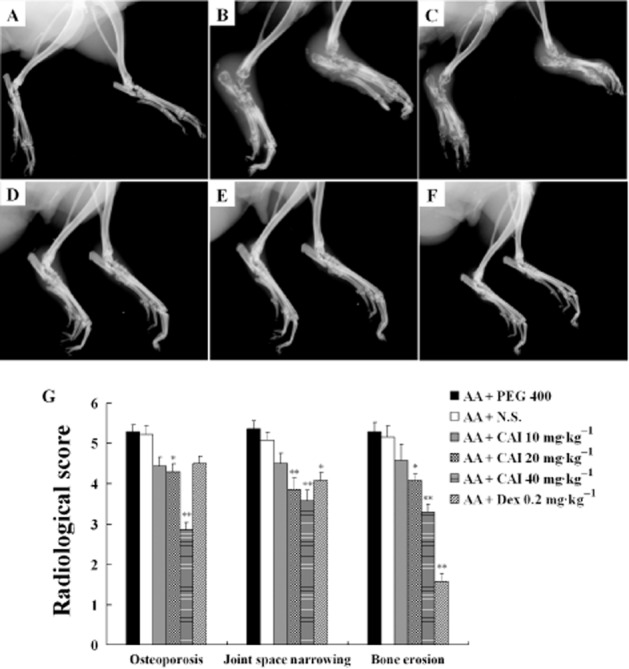
Effects of CAI on the radiological joint damage in AA rats. A–F: representative radiographs of the ankle joints of normal rats (A), vehicle PEG 400-treated AA rats (B), vehicle N.S.-treated AA rats (C), CAI 20 mg·kg−1-treated AA rats (D), CAI 40 mg·kg−1-treated AA rats (E), dexamethasone (Dex)-treated AA rats (F). (G) Three radiographical parameters – osteoporosis, joint space narrowing and bone erosion – were scored separately. Data shown are means ± SEM of 14 rats. *P < 0.05, **P < 0.01 versus vehicle-treated AA group.
CAI improved histopathological damage in AA rats
Histological analysis confirmed the clinical and radiological findings. In contrast to normal rats, the sections from vehicle-treated AA rats revealed markedly thickened and expanded synovium with intense infiltration of inflammatory cells, pannus formation, prominent cartilage and bone erosion. Treatment with CAI (20 mg·kg−1) diminished the infiltration of inflammatory cells, synovial hyperplasia, pannus formation, and cartilage and bone degradation. In AA rats treated with the higher dose of CAI (40 mg·kg−1) or dexamethasone, the joint architecture remained clear with little cartilage and bone erosion, with only a low level of inflammatory cell infiltration and synovial thickening (Figure 3A–F). These histopathological findings matched the results of histological scoring (Figure 3G).
Figure 3.
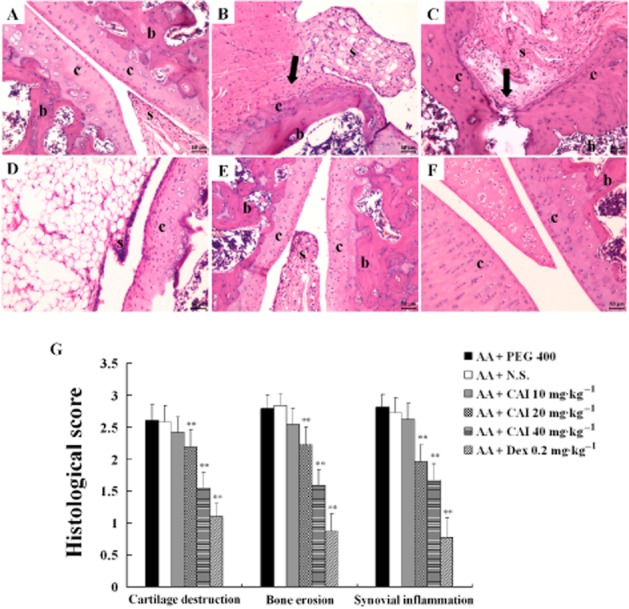
Effects of CAI on histopathology of AA rats. A–F: representative photomicrographs of haematoxylin and eosin staining sections from normal rats (A), vehicle PEG 400-treated AA rats (B), vehicle N.S.-treated AA rats (C), CAI 20 mg·kg−1-treated AA rats (D), CAI 40 mg·kg−1-treated AA rats (E), dexamethasone (Dex)-treated AA rats (F). Note the changes of synovial membrane (s), articular cartilage (c), subchondral bone (b). Black arrow shows cartilage and bone erosions by pannus invasion. (G) Histopathological changes were scored for three parameters – cartilage destruction, bone erosion and synovial inflammation. Data shown are means ± SEM of four rats. **P < 0.05 vs. vehicle-treated AA group.
CAI decreased pro-inflammatory cytokines in AA rats
The pro-inflammatory cytokines play an important role in RA pathogenesis and we therefore measured the effects of CAI on these mediators. The concentrations of IL-1β, IL-6, IL-18 and TNF-α in synovial homogenates from the vehicle-treated AA rats were significantly higher than those in normal rats. CAI (20, 40 mg·kg−1) and dexamethasone clearly decreased all these cytokines. Particularly, CAI at the lowest dose (10 mg·kg−1) also attenuated IL-1β, IL-18 and TNF-α levels in AA rats (Figure 4A–D).
Figure 4.

Effects of CAI on the levels of pro-inflammatory cytokines in AA rats. The concentrations of IL-1β (A), IL-6 (B), IL-18 (C) and TNF-α (D) in homogenates of synovial tissue were measured by elisa. Data shown are means ± SEM of four rats. (E) Pro-IL-1β, mature IL-1β p17, pro-IL-18 and mature IL-18 p18 in extracts of synovial tissue were detected by Western blot analysis. (F) Densitometric analysis of immunoblot bands. β-actin was used as the internal control. Data shown are means ± SEM of three rats. ##P < 0.01 versus normal group, *P < 0.05, **P < 0.01 versus vehicle-treated AA group.
IL-1β and IL-18 are synthesized as the inactive precursor molecules. They must be cleaved into their biologically active mature forms by inflammasomes before they are released. Both the precursor and mature forms of IL-1β and IL-18 were detected in extracts of synovial tissues by Western blot. The expressions of pro-IL-1β, mature IL-1β p17, pro-IL-18 and mature IL-18 p18 in the synovial tissues of the vehicle-treated AA rats were all markedly increased, compared with those in the samples from normal rats. Treatment with CAI (20, 40 mg·kg−1) suppressed the expression of pro-IL-1β and pro-IL-18, and CAI at all three doses (10, 20, 40 mg·kg−1) decreased the mature forms of IL-1β p17 and IL-18 p18. Dexamethasone also inhibited levels of both the precursor and mature forms of IL-1β and IL-18 (Figure 4E,F).
CAI inhibited NF-κB activation in AA rats
Given the importance of NF-κB in the transcription of the cytokines and the results observed in Figure 4 that the pro-IL-1β, pro-IL-18, IL-6 and TNF-α were up-regulated in AA synovium and profoundly decreased by CAI treatment, the effect of CAI on the activation state of NF-κB in the synovium of AA rats was investigated. The p65 subunit of NF-κB was expressed at a very low level in normal synovium and was mainly located in the cytoplasm, as detected by immunohistochemistry (Figure 5A). In the vehicle-treated AA rats, the synovium was hyperplastic and the expression of p65 NF-κB subunit was much higher than that of the normal control. Furthermore, the p65 staining was particularly located in the cell nucleus (Figure 5B, C). Treatment with CAI (20, 40 mg·kg−1) decreased the inflammatory reaction in AA synovium and, at the same time, decreased p65 staining (Figure 5D, E). Dexamethasone also significantly decreased immunohistochemical staining for the p65 subunit (Figure 5F).
Figure 5.
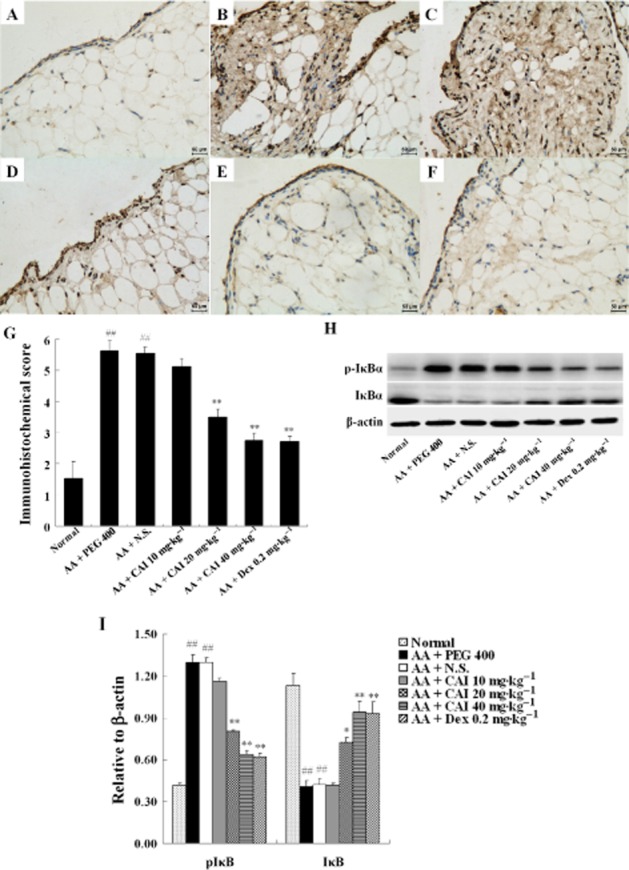
Effects of CAI on NF-κB activation in AA rats. Immunohistochemistry of the p65 NF-κB subunit in the synovium of normal rats (A), vehicle PEG 400-treated AA rats (B), vehicle N.S.-treated AA rats (C), CAI 20 mg·kg−1-treated AA rats (D), CAI 40 mg·kg−1-treated AA rats (E), and dexamethasone (Dex)-treated AA rats (F). (G) Semiquantitative analysis of the immunohistochemical p65 NF-κB subunit expression was performed. Data shown are means ± SEM of four rats. H: IκBα expression and phosphorylation were detected by Western blot analysis. I: Densitometric analysis of immunoblot bands. β-actin was used as the internal control. Data shown are means ± SEM of three rats. ##P < 0.01 versus normal group, *P < 0.05, **P < 0.01 versus vehicle-treated AA group.
As NF-κB activity is regulated by the phosphorylation of IκBα, the levels of IκBα and its phosphorylated form p-IκBα were analysed by Western blot. Levels of p-IκBα were markedly increased and those of IκBα were significantly decreased in the synovial tissue from vehicle-treated AA rats, compared with the normal rats. CAI treatment (20, 40 mg·kg−1) inhibited the phosphorylation and degradation of IκBα in AA rats, with an efficacy similar to that of dexamethasone (Figure 5H, I). These findings indicated that CAI inhibited the activation of NF-κB in arthritic synovium.
Detection of components of NALP inflammasomes in the synovium and the effects of CAI
Precursors of IL-1β and IL-18 have to be cleaved by the inflammasomes before they were released as mature cytokines (Franchi et al., 2009). As the inflammsome-dependent active forms of IL-1β and IL-18 in AA synovium were up-regulated and significantly decreased by CAI treatment (Figure 4), we postulated that the NALP inflammasomes in the synovial tissues might be activated in AA rats. The expression of the inflammasome component proteins, NALP1, NALP3, ASC, caspase-1 and caspase-5, in the synovium were analysed by Western blot. As shown in Figure 6, two NALP1 isoforms with apparent molecular sizes of 165 kDa and 70 kDa were detected, but they were expressed in very low levels in the synovium of normal rats. In the vehicle-treated AA rats, the 165 kDa isoform protein level did not differ from that in normal rats, while the 70 kDa isoform was significantly increased. In the synovium of AA rats, treatment with CAI (20, 40 mg·kg−1) or dexamethasone had no effects on the level of the 165 kDa isoform,but clearly decreased levels of the 70 kDa isoform (Figure 6A, B).
Figure 6.

Detection of the components of inflammasomes in the synovium and the effects of CAI. (A) Synovial tissues were homogenized and analysed by Western blot, with the antibodies as indicated. (B and C) Densitometric analysis of immunoblot bands. β-actin was used as the internal control. Data shown are means ± SEM of three rats. (D) Caspase-1 activity was measured in synovial tissues from rats from different groups. Data shown are means ± SEM of four rats. ##P < 0.01 versus normal group, *P < 0.05, **P < 0.01 versus vehicle-treated AA group. (E) The direct effects of CAI on the activity of caspase-1 in vitro were assayed. The specific caspase-1 inhibitor Z-VAD-FMK was used as positive controls. Data shown are means ± SEM from six independent experiments. **P < 0.01 versus the control without drug (0 μM).
Immunohistochemical analysis of the synovial sections for NALP1 showed that this protein was mainly expressed in the synoviocytes, vascular endothelial cells and adipocytes. In contrast to the weak expression in samples from normal rats, NALP1 immunostaining in the synovium of the vehicle-treated AA rats demonstrated a marked up-regulation, and these effects were suppressed by treatment with CAI (20, 40 mg·kg−1) or dexamethasone (Figure 7), which was consistent with Western blot data. As for NALP3, its expression level in AA synovium was significantly down-regulated, compared with its strong expression in the normal synovium. The higher dose of CAI (40 mg·kg−1) restored NALP3 expression levels in AA synovium. No differences were observed in the expression of the adaptor protein ASC between any of the groups (Figure 6).
Figure 7.
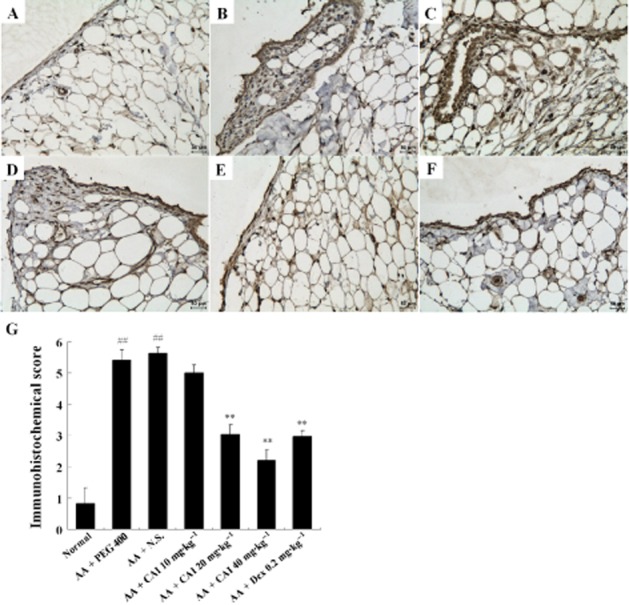
Immunohistochemistry for NALP1 in the synovium. A–F: representative images from normal rats (A), vehicle PEG 400-treated AA rats (B), vehicle N.S.-treated AA rats (C), CAI 20 mg·kg−1-treated AA rats (D), CAI 40 mg·kg−1-treated AA rats (E), and dexamethasone (Dex) -treated AA rats (F). (G) Semiquantitative analysis of immunohistochemical NALP1 expression was performed and data shown are means ± SEM of four rats. ##P < 0.01 versus normal group, **P < 0.01 versus vehicle-treated AA group.
Caspase-1 processing is the ultimate step in inflammasome activation. It is expressed as a proenzyme of 45 kDa and autocatalytically cleaved into p20 (20 kDa) and p10 (10 kDa) subunits in the inflammasome, and then assembles into its active form consisting of two heterodimers with a p20 and p10 subunit each (Franchi et al., 2009). Although the pro-caspase-1 levels in the synovium of AA rats did not differ from those of normal rats, the level of the activated p20 fragment was significantly higher in AA synovium (Figure 6A, C). In addition to the protein level, the activity of caspase-1 in the synovial tissues of AA rats was markedly increased, compared with those in normal rats (Figure 6D), suggesting that caspase-1 was activated in the AA rats. Administration of CAI (10, 20, 40 mg·kg−1) reduced both the level of the activated p20 fragment and the activity of caspase-1 in AA synovium. Similar effects were observed in AA rats treated with dexamethasone (Figure 6A, C and D). In order to exclude a direct inhibitory effect of CAI on the activity of caspase-1, a caspase-1 inhibitor drug screening kit in vitro was used to assess whether CAI could inhibit caspase-1. Our results showed that CAI, over a wide concentration range (1.56–100 μM) had no effects on caspase-1 activity in vitro, while the specific caspase-1 inhibitor Z-VAD-FMK significantly suppressed the caspase-1 activity in the same assays (Figure 6E). For caspase-5, although pro-caspase-5 was detectable in the synovial tissue and had no differences among all groups, the activated forms of caspase-5, p10 and p20, could not be detected (Figure 6A, C). These data suggested that NALP1 inflammasomes were activated in the inflamed synovium and that CAI treatment inhibited the activation of NALP1 inflammasomes.
Discussion
In the present study, the administration of CAI significantly suppressed the arthritis index and prevented body weight loss, as well as reducing the radiological and histological features of joint destruction in AA rats. These findings imply that CAI attenuated the arthritis and joint damage in rats with AA. The effects of CAI may be translated into a potential clinical benefit for the treatment of RA. What, then, is the mechanism by which CAI exerted its effects on arthritis?
CAI has no direct inhibitory effect on the activities of COX-1 and COX-2 in vitro (data not shown), which indicated that CAI exerted its anti-inflammatory effects through mechanisms differing from those of COX-inhibiting NSAIDs. The network of the cytokines is involved in the development and progression of RA (Choy and Panayi, 2001). Therefore, we focused on the effects of CAI on the cytokines in arthritis. IL-1β and TNF-α are considered to be of great importance among the pro-inflammatory factors in the inflamed synovium. In particular, they seem to initiate a cytokine cascade to induce the production of other inflammatory molecules (Tran et al., 2005). Overproduction of IL-1β and TNF-α in the synovium induces inflammatory cell infiltration, synovial angiogenesis and hyperplasia, cartilage matrix degradation and bone erosions. Anti-IL-1β and anti-TNF-α therapies have been effective in the treatment of RA (Szekanecz et al., 2000; Choy and Panayi, 2001). In addition to IL-1β and TNF-α, IL-6 and IL-18 are two other pleiotropic cytokines important in RA pathogenesis. IL-6 and IL-18 are known to be increased in the synovium, synovial fluid and serum of RA patients (Dayer and Choy, 2010; Volin and Koch, 2011). Therefore, blockade of the production of the cytokines may be an effective strategy for RA management. In our study, the levels of IL-1β, TNF-α, IL-6 and IL-18 were markedly elevated in synovial tissues from rats with AA but were decreased by CAI treatment. We previously reported that CAI improved DSS-induced colitis in mice accompanied by a reduction of IL-1β, TNF-α and IL-6 levels both in serum and inflamed colon tissues (Guo et al., 2012). These data suggested that the inhibition of cytokines might account for the anti-inflammatory effects of CAI and its beneficial effects in AA.
NF-κB is a key transcriptional factor consisting of homo- or heterodimers of Rel-related proteins and activates the gene expression of inflammatory mediators. Under resting conditions, NF-κB, usually a dimer of p50 and p65 subunits, resides in the cytoplasm and forms an inactive complex with the inhibitory protein IκB. In a canonical activating pathway induced by various stimuli, such as cytokines (e.g. IL-1β, TNF-α), LPS and oxidants, IκBα is rapidly phosphorylated and degraded. In turn, the liberated NF-κB enters the nucleus and activates the gene transcription of a variety of cytokines, chemokines, angiogenic factors and MMPs (Bonizzi and Karin, 2004; Oeckinghaus et al., 2011). The positive feedback regulation between NF-κB and the inflammatory molecules contributes to the constitutive activation of inflammation as well as the cartilage and bone erosion in RA. NF-κB activation has previously been found in several arthritic animal models and in the synovium of patients with RA (Han et al., 1998). Consistent with these reports, we observed NF-κB activation in AA synovium as shown by overexpression of its p65 subunit, increased phosphorylation level and decreased expression of IκBα. Administration of CAI down-regulated the expression of the p65 subunit, and inhibited the phosphorylation and degradation of IκBα in AA rats. These results were similar to those showing inhibition of NF-κB activation by CAI, in the DSS-induced colitis model (Guo et al., 2012). NF-κB is recognized as a central ‘switch’ in the crucial initial steps of inflammation as well as in its persistence, in RA and inhibition of NF-κB reduced the severity of arthritis (He et al., 2008; Lee et al., 2009). Therefore, our results suggested that the inhibition of NF-κB activation could be a key mechanism for the anti-arthritic effects of CAI.
In contrast to the other cytokines, the sequence of events culminating in IL-1β and IL-18 secretion is complex, but it can be summarized in two steps: induction of pro-IL-1β and pro-IL-18, and their subsequent processing into active IL-1β and IL-18 by the inflammasomes (Schroder and Tschopp, 2010). We found increased levels of both the precursor and mature forms of IL-1β and IL-18 and the activation of NF-κB in AA synovium. Next, we detected the NALP inflammasomes in synovial tissue, which is involved in the key second step in the production of IL-1β and IL-18. The data presented here showed that NALP1 inflammasomes were activated in AA synovium and CAI treatment inhibited this activation, which might be another mechanism of CAI, in addition to the suppression of NF-κB activation, underlying its anti-arthritic effects.
NALP1 is expressed in long and short forms, both of which have biological activity (Kummer et al., 2007; Li et al., 2009). Accordingly, we detected both the two forms of NALP1 in the synovium with apparent molecular sizes of 165 and 70 kDa, which were expressed at very low levels in the normal rats. Although the protein levels of the 165 kDa long form did not change, the 70 kDa short form was increased markedly in AA synovium. Moreover, our immunohistochemical data showed that NALP1 was mainly expressed in the synoviocytes, vascular endothelial cells and adipocytes, and that this immunostaining was significantly enhanced in AA rats. Furthermore, both the protein level of the activated caspase-1 p20 fragment and the activity of caspase-1 in AA synovium was markedly increased, compared with the normal rats. Taken together, our results demonstrated that NALP1 inflammasomes were activated in AA synovium, which was further supported by the increased level of activated IL-1β and IL-18. It seems that the NALP1 inflammasome might be a novel, potential target for RA treatment. Consequently, identification of drugs that inhibit the activated NALP1 inflammasome is important for understanding its mechanism of activation and developing effective therapies for inflammation-related diseases such as RA. In this study, we found that CAI inhibited activation of the NALP1 inflammasome, as shown by decreased expression of the NALP 70 kDa isoform and of caspase-1 p20, as well as decreased caspase-1 activity in AA synovium.
In contrast, the evidence for the importance of the NALP3 inflammasome to RA is scant and contradictory. Rosengren et al. showed that NALP3 mRNA levels were increased in the synovium of RA, in comparison with osteoarthritis (OA) (Rosengren et al., 2005). However, Kolly et al. found that there were no differences in NALP3 protein expression between RA and OA synovial tissues (Kolly et al., 2009). In this study, we demonstrated that the synovial tissues from normal rats expressed a high level of NALP3, which was significantly down-regulated in synovial tissues from rats with AA. So, at present, the mechanism(s) whereby NALP1 and NALP3 inflammasomes could be differently regulated in arthritic synovium are largely unknown. Moreover, how expression of the components of NALP1 inflammasomes is regulated and how NALP1 inflammasomes are activated in arthritis are still to be elucidated. Nevertheless, our results highlight the need for further investigation of such mechanisms.
Conventional anti-inflammatory agents including steroids and NSAIDs have long been used as the major therapeutic strategy for RA. However, they have limited use because of their serious side effects and because they are only partly effective (Seibert et al., 1994; Kleiman and Tuckermann, 2007). In the last decade, clinical use of biological agents against cytokines, such as TNF-α and IL-1β, has increased the therapeutic options for RA patients (Bathon et al., 2000; Breedveld et al., 2006). However, their therapeutic efficiency are not ideal (Bathon et al., 2000; Fleischmann et al., 2004; Rubbert-Roth and Finckh, 2009) with the disadvantages of high cost, hypersensitivity, administration via repeated injections and possibility of serious infections (Bongartz et al., 2006; Wolfe and Michaud, 2007). Based on this situation in the context of RA, it is proposed that neutralizing the cytokines already produced is not an ideal strategy but an inhibition of their production might provide better clinical results. In addition, a decreased production of TNF-α and IL-1β might be more effective and have less disadvantages than a complete inhibition of the effects of TNF-α and IL-1β (Cascão et al., 2012). Thus, the development of novel therapies with different mode of action and favourable safety and tolerability profile is currently in high demand. Our results revealed that CAI could inhibit the dysregulated activation of both NF-κB and the NALP1 inflammasomes, which are two of the most important pathways regulating the production of cytokines. Furhtermore, oral administration of CAI did not produce systemic toxic effects, as reflected in the fact that the CAI-treated arthritic rats gained more body weight than did the vehicle-treated AA rats. In addition, a series of clinical trials of CAI as an anti-cancer agent showed that the adverse effects of CAI were slight and most commonly consisted of grades 1–2 gastrointestinal and neurological side effects such as nausea, vomiting and dizziness (Figg et al., 1995; Kohn et al., 1996; Stadler et al., 2005). Taken together, CAI might be an effective and relatively safe therapeutic strategy for the treatment of RA.
In conclusion, we have demonstrated that NALP1 inflammasomes were activated in synovial tissues of rats with AA, which could represent a novel therapeutic target for RA treatment and merits further investigation. We also showed that oral administration of CAI ameliorated the joint inflammation and destruction in AA rats. Inhibition of the activation of both NF-κB and NALP1 inflammasomes and subsequent suppression of the production of pro-inflammatory cytokines contributed to this anti-inflammatory effect. Collectively, our data suggested that CAI might serve as a promising therapeutic strategy for RA.
Acknowledgments
This study was supported by the National Natural Science Foundation of China (81102454) and by the Special Program for Key Basic Research of the Ministry of Science and Technology, China (2014ZX09507003-003).
Glossary
- AA
adjuvant arthritis
- ASC
apoptosis-associated speck-like protein
- CAI
carboxyamidotriazole
- CARD
caspase activation and recruitment domain
- CFA
complete Freund's adjuvant
- DSS
dextran sodium sulphate
- PEG 400
polyethylene glycol 400
- NLR
NOD-like receptor
- N.S
normal saline
- RA
rheumatoid arthritis
Author contributions
Le. Z. designed and performed the experiments, analysed the data, and wrote the paper; J. L., L. G., X. Y. and D. W. performed the experiments; L. L., Li. Z., W. C., and C. C. analysed the data; and C. Y. and D. Z. conceived and designed the experiments, and wrote the paper.
Conflict of interest
None.
References
- Ahmed AS, Li J, Ahmed M, Hua L, Yakovleva T, Ossipov MH, et al. Attenuation of pain and inflammation in adjuvant-induced arthritis by the proteasome inhibitor MG132. Arthritis Rheum. 2010;62:2160–2169. doi: 10.1002/art.27492. [DOI] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M, et al. The Concise Guide to PHARMACOLOGY 2013/14: Catalytic Receptors. Br J Pharmacol. 2013a;170:1676–1705. doi: 10.1111/bph.12449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M, et al. The Concise Guide to PHARMACOLOGY 2013/14: Enzymes. Br J Pharmacol. 2013b;170:1797–1867. doi: 10.1111/bph.12451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allred DC, Harvey JM, Berardo M, Clark GM. Prognostic and predictive factors in breast cancer by immunohistochemical analysis. Mod Pathol. 1998;11:155–168. [PubMed] [Google Scholar]
- Bathon JM, Martin RW, Fleischmann RM, Tesser JR, Schiff MH, Keystone EC, et al. A comparison of etanercept and methotrexate in patients with early rheumatoid arthritis. N Engl J Med. 2000;343:1586–1593. doi: 10.1056/NEJM200011303432201. [DOI] [PubMed] [Google Scholar]
- Bongartz T, Sutton AJ, Sweeting MJ, Buchan I, Matteson EL, Montori V. Anti-TNF antibody therapy in rheumatoid arthritis and the risk of serious infections and malignancies: systematic review and meta-analysis of rare harmful effects in randomized controlled trials. JAMA. 2006;295:2275–2285. doi: 10.1001/jama.295.19.2275. [DOI] [PubMed] [Google Scholar]
- Bonizzi G, Karin M. The two NF-kappaB activation pathways and their role in innate and adaptive immunity. Trends Immunol. 2004;25:280–288. doi: 10.1016/j.it.2004.03.008. [DOI] [PubMed] [Google Scholar]
- Breedveld FC, Weisman MH, Kavanaugh AF, Cohen SB, Pavelka K, van Vollenhoven R, et al. The PREMIER study: a multicenter, randomized, double-blind clinical trial of combination therapy with adalimumab plus methotrexate versus methotrexate alone or adalimumab alone in patients with early, aggressive rheumatoid arthritis who had not had previous methotrexate treatment. Arthritis Rheum. 2006;54:26–37. doi: 10.1002/art.21519. [DOI] [PubMed] [Google Scholar]
- Cascão R, Vidal B, Raquel H, Neves-Costa A, Figueiredo N, Gupta V, et al. Effective treatment of rat adjuvant-induced arthritis by celastrol. Autoimmun Rev. 2012;11:856–862. doi: 10.1016/j.autrev.2012.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cassel SL, Eisenbarth SC, Iyer SS, Sadler JJ, Colegio OR, Tephly LA, et al. The NALP3 inflammasome is essential for the development of silicosis. Proc Natl Acad Sci U S A. 2008;105:9035–9040. doi: 10.1073/pnas.0803933105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choy EH, Panayi GS. Cytokine pathways and joint inflammation in rheumatoid arthritis. N Engl J Med. 2001;344:907–916. doi: 10.1056/NEJM200103223441207. [DOI] [PubMed] [Google Scholar]
- Dayer JM, Choy E. Therapeutic targets in rheumatoid arthritis: the interleukin-6 receptor. Rheumatology (Oxford) 2010;49:15–24. doi: 10.1093/rheumatology/kep329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feldmann M. Pathogenesis of arthritis: recent research progress. Nat Immunol. 2001;2:771–773. doi: 10.1038/ni0901-771. [DOI] [PubMed] [Google Scholar]
- Figg WD, Cole KA, Reed E, Steinberg SM, Piscitelli SC, Davis PA, et al. Pharmacokinetics of orally administered carboxyamido-triazole, an inhibitor of calcium-mediated signal transduction. Clin Cancer Res. 1995;1:797–803. [PubMed] [Google Scholar]
- Fleischmann R, Stern R, Iqbal I. Anakinra: an inhibitor of IL-1 for the treatment of rheumatoid arthritis. Expert Opin Biol Ther. 2004;4:1333–1344. doi: 10.1517/14712598.4.8.1333. [DOI] [PubMed] [Google Scholar]
- Franchi L, Eigenbrod T, Muñoz-Planillo R, Nuñez G. The inflammasome: a caspase-1-activation platform that regulates immune responses and disease pathogenesis. Nat Immunol. 2009;10:241–247. doi: 10.1038/ni.1703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franchi L, Muñoz-Planillo R, Núñez G. Sensing and reacting to microbes through the inflammasomes. Nat Immunol. 2012;13:325–332. doi: 10.1038/ni.2231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo L, Ye CY, Chen WY, Ye H, Zheng R, Li J, et al. Anti-inflammatory and analgesic potency of carboxyamidotriazole, a tumoristatic agent. J Pharmacol Exp Therap. 2008;325:10–16. doi: 10.1124/jpet.107.131888. [DOI] [PubMed] [Google Scholar]
- Guo L, Ye C, Hao X, Zheng R, Ju R, Wu D, et al. Carboxyamidotriazole ameliorates experimental colitis by inhibition of cytokine production, nuclear factor-κB activation, and colonic fibrosis. J Pharmacol Exp Ther. 2012;342:356–365. doi: 10.1124/jpet.112.192849. [DOI] [PubMed] [Google Scholar]
- Halle A, Hornung V, Petzold GC, Stewart CR, Monks BG, Reinheckel T, et al. The NALP3 inflammasome is involved in the innate immune response to amyloid-beta. Nat Immunol. 2008;9:857–865. doi: 10.1038/ni.1636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han Z, Boyle DL, Manning AM, Firestein GS. AP-1 and NF-kappaB regulation in rheumatoid arthritis and murine collagen-induced arthritis. Autoimmunity. 1998;28:197–208. doi: 10.3109/08916939808995367. [DOI] [PubMed] [Google Scholar]
- He Y, Xu H, Liang L, Zhan Z, Yang X, Yu X, et al. Antiinflammatory effect of Rho kinase blockade via inhibition of NF-κB activation in rheumatoid arthritis. Arthritis Rheum. 2008;58:3366–3376. doi: 10.1002/art.23986. [DOI] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG. Animal research: reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1577–1579. doi: 10.1111/j.1476-5381.2010.00872.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleiman A, Tuckermann JP. Glucocorticoid receptor action in beneficial and side effects of steroid therapy: lessons from conditional knockout mice. Mol Cell Endocrinol. 2007;275:98–108. doi: 10.1016/j.mce.2007.05.009. [DOI] [PubMed] [Google Scholar]
- Kohn EC, Felder CC, Jacobs W, Holmes KA, Day A, Freer R, et al. Structure-function analysis of signal and growth inhibition by carboxyamido-triazole, CAI. Cancer Res. 1994;54:935–942. [PubMed] [Google Scholar]
- Kohn EC, Reed E, Sarosy G, Christian M, Link CJ, Cole K, et al. Clinical investigation of a cytostatic calcium influx inhibitor in patients with refractory cancers. Cancer Res. 1996;56:569–573. [PubMed] [Google Scholar]
- Kolly L, Busso N, Palmer G, Talabot-Ayer D, Chobaz V, So A. Expression and function of the NALP3 inflammasome in rheumatoid synovium. Immunology. 2009;129:175–185. doi: 10.1111/j.1365-2567.2009.03174.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kummer JA, Broekhuizen R, Everett H, Agostini L, Kuijk L, Martinon F, et al. Inflammasome components NALP 1 and 3 show distinct but separate expression profiles in human tissues suggesting a site-specific role in the inflammatory response. J Histochem Cytochem. 2007;55:443–452. doi: 10.1369/jhc.6A7101.2006. [DOI] [PubMed] [Google Scholar]
- Latz E. The inflammasomes: mechanisms of activation and function. Curr Opin Immunol. 2010;22:28–33. doi: 10.1016/j.coi.2009.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee YR, Kweon SH, Kwon KB, Park JW, Yoon TR, Park BH. Inhibition of IL-1β- mediated inflammatory responses by the IκB alpha super-repressor in human fibroblast-like synoviocytes. Biochem Biophys Res Commun. 2009;378:90–94. doi: 10.1016/j.bbrc.2008.11.002. [DOI] [PubMed] [Google Scholar]
- Li WW, Guo TZ, Liang D, Shi X, Wei T, Kingery WS, et al. The NALP1 inflammasome controls cytokine production and nociception in a rat fracture model of complex regional pain syndrome. Pain. 2009;147:277–286. doi: 10.1016/j.pain.2009.09.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinon F, Pétrilli V, Mayor A, Tardivel A, Tschopp J. Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature. 2006;440:237–241. doi: 10.1038/nature04516. [DOI] [PubMed] [Google Scholar]
- Masters SL, Dunne A, Subramanian SL, Hull RL, Tannahill GM, Sharp FA, et al. Activation of the NLRP3 inflammasome by islet amyloid polypeptide provides a mechanism for enhanced IL-1β in type 2 diabetes. Nat Immunol. 2010;11:897–904. doi: 10.1038/ni.1935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGrath J, Drummond G, McLachlan E, Kilkenny C, Wainwright C. Guidelines for reporting experiments involving animals: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1573–1576. doi: 10.1111/j.1476-5381.2010.00873.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montesinos MC, Yap JS, Desai A, Posadas I, McCrary CT, Cronstein BN. Reversal of the antiinflammatory effects of methotrexate by the nonselective adenosine receptor antagonists theophylline and caffeine: evidence that the antiinflammatory effects of methotrexate are mediated via multiple adenosine receptors in rat adjuvant arthritis. Arthritis Rheum. 2000;43:656–663. doi: 10.1002/1529-0131(200003)43:3<656::AID-ANR23>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- Moody TW, Chiles J, Moody E, Sieczkiewicz GJ, Kohn EC. CAI inhibits the growth of small cell lung cancer cells. Lung Cancer. 2003;39:279–288. doi: 10.1016/s0169-5002(02)00525-1. [DOI] [PubMed] [Google Scholar]
- Oeckinghaus A, Hayden MS, Ghosh S. Crosstalk in NF-κB signaling pathways. Nat Immunol. 2011;12:695–708. doi: 10.1038/ni.2065. [DOI] [PubMed] [Google Scholar]
- Pawson AJ, Sharman JL, Benson HE, Faccenda E, Alexander SP, Buneman OP, et al. NC-IUPHAR. The IUPHAR/BPS Guide to PHARMACOLOGY: an expert-driven knowledge base of drug targets and their ligands. Nucl. Acids Res. 2014;42:D1098–1106. doi: 10.1093/nar/gkt1143. (Database Issue): [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosengren S, Hoffman HM, Bugbee W, Boyle DL. Expression and regulation of cryopyrin and related proteins in rheumatoid arthritis synovium. Ann Rheum Dis. 2005;64:708–714. doi: 10.1136/ard.2004.025577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubbert-Roth A, Finckh A. Treatment options in patients with rheumatoid arthritis failing initial TNF inhibitor therapy: a critical review. Arthritis Res Ther. 2009;11(Suppl. 1):S1. doi: 10.1186/ar2666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroder K, Tschopp J. The inflammasomes. Cell. 2010;140:821–832. doi: 10.1016/j.cell.2010.01.040. [DOI] [PubMed] [Google Scholar]
- Schroder K, Zhou R, Tschopp J. The NLRP3 inflammasome: a sensor for metabolic danger? Science. 2010;327:296–300. doi: 10.1126/science.1184003. [DOI] [PubMed] [Google Scholar]
- Seibert K, Zhang Y, Leahy K, Hauser S, Masferrer J, Perkins W, et al. Pharmacological and biochemical demonstration of the role of cyclooxygenase 2 in inflammation and pain. Proc Natl Acad Sci U S A. 1994;91:12013–12017. doi: 10.1073/pnas.91.25.12013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stadler WM, Rosner G, Small E, Hollis D, Rini B, Zaentz SD, et al. Successful implementation of the randomized discontinuation trial design: an application to the study of the putative antiangiogenic agent carboxyaminoimidazole in renal cell carcinoma – CALGB 69901. J Clin Oncol. 2005;23:3726–3732. doi: 10.1200/JCO.2005.44.150. [DOI] [PubMed] [Google Scholar]
- Sui J, Li H, Fang Y, Liu Y, Li M, Zhong B, et al. NLRP1 gene polymorphism influences gene transcription and is a risk factor for rheumatoid arthritis in han Chinese. Arthritis Rheum. 2012;64:647–654. doi: 10.1002/art.33370. [DOI] [PubMed] [Google Scholar]
- Szekanecz Z, Halloran MM, Volin MV, Woods JM, Strieter RM, Kenneth Haines G, 3rd, et al. Temporal expression of inflammatory cytokines and chemokines in rat adjuvant-induced arthritis. Arthritis Rheum. 2000;43:1266–1277. doi: 10.1002/1529-0131(200006)43:6<1266::AID-ANR9>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- Tran CN, Lundy SK, Fox DA. Synovial biology and T cells in rheumatoid arthritis. Pathophysiology. 2005;12:183–189. doi: 10.1016/j.pathophys.2005.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volin MV, Koch AE. Interleukin-18: a mediator of inflammation and angiogenesis in rheumatoid arthritis. J Interferon Cytokine Res. 2011;31:745–751. doi: 10.1089/jir.2011.0050. [DOI] [PubMed] [Google Scholar]
- Wasilenko WJ, Palad AJ, Somers KD, Blackmore PF, Kohn EC, Rhim JS, et al. Effects of the calcium influx inhibitor carboxyamido-triazole on the proliferation and invasiveness of human prostate tumor cell lines. Int J Cancer. 1996;68:259–264. doi: 10.1002/(SICI)1097-0215(19961009)68:2<259::AID-IJC20>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- Whitehouse MW. Adjuvant arthritis 50 years on: the impact of the 1956 article by C. M. Pearson, ‘Development of arthritis, periarthritis and periostitis in rats given adjuvants. Inflamm Res. 2007;56:133–138. doi: 10.1007/s00011-006-6117-8. [DOI] [PubMed] [Google Scholar]
- Wolfe F, Michaud K. Biologic treatment of rheumatoid arthritis and the risk of malignancy: analyses from a large US observational study. Arthritis Rheum. 2007;56:2886–2895. doi: 10.1002/art.22864. [DOI] [PubMed] [Google Scholar]
- de Zoete MR, Flavell RA. NLRP1 joins the dark side? Immunity. 2012;37:950–952. doi: 10.1016/j.immuni.2012.11.008. [DOI] [PubMed] [Google Scholar]