

SUMMARY
Complete virions of hepatitis B virus (HBV) contain a DNA genome that is enclosed in a capsid composed of the HBV core antigen (HBcAg), which is in turn surrounded by a lipid envelope studded with viral surface antigens (HBsAg). In addition, HBV-infected cells release subviral particles composed of HBsAg only (HBsAg ‘spheres’ and ‘filaments’) or HBsAg enveloping HBcAg but devoid of viral DNA (‘empty virions’). The hepatitis B e antigen (HBeAg), a soluble antigen related to HBcAg, is also secreted in some HBV-infected patients. The goals of this study were to explore the levels of empty virions in HBV-infected patients before and during therapy with the nucleotide analog tenofovir disoproxil fumarate (TDF) that inhibits HBV DNA synthesis and the relationships of empty virions to complete virions, HBsAg and HBeAg. HBV DNA, HBcAg and HBsAg levels were determined in serum samples from 21 patients chronically infected with HBV and enrolled in clinical TDF studies. Serum levels of empty virions were found to exceed levels of DNA-containing virions, often by ≥100-fold. Levels of both empty and complete virions varied and were related to the HBeAg status. When HBV DNA replication was suppressed by TDF, empty virion levels remained unchanged in most but were decreased (to the limit of detection) in some patients who also experienced significant decrease or loss of serum HBsAg. In conclusion, empty virions are present in the serum of chronic hepatitis B patients at high levels and may be useful in monitoring response to antiviral therapy.
Keywords: cccDNA, empty hepatitis B virion, hepatitis B core antigen, hepatitis B e antigen, nucleotide analog
INTRODUCTION
More than 350 million people worldwide are infected with hepatitis B virus (HBV), which remains a major cause of chronic hepatitis, cirrhosis and hepatocellular carcinoma [1]. HBV is a member of the hepadnaviridae, a family of retroid viruses harbouring partially double-stranded, relaxed circular DNA (rcDNA) genomes replicated through an RNA intermediate called pregenomic RNA (pgRNA) [2,3]. In infectious virions, rcDNA is enclosed within a protein capsid composed of a single viral protein, the hepatitis B core antigen (HBcAg) [4,5], which is in turn surrounded by an outer envelope consisting of a host-derived lipid bilayer studded with three viral envelope or surface proteins (HBsAg) [6–8]. During the viral replication cycle, reverse transcription takes place after packaging of pgRNA and the viral reverse transcriptase by HBcAg into nucleocapsids (NCs). Mature NCs, which contain reverse transcribed rcDNA genomes, are then selected for envelopment by viral envelope proteins and subsequently secreted into the bloodstream [3,9–11].
In addition to rcDNA-containing (i.e. complete) virions, HBV-infected cells also secrete two additional classes of subviral particles: HBsAg particles and ‘empty virions’. HBsAg particles appear as spheres or filaments and are typically present in 10- to 100 000-fold excess over complete virions [1]. HBsAg particles are composed of host lipids and viral envelope proteins but no viral capsid or nucleic acids. ‘Empty virions’ contain viral envelope and capsid proteins but no nucleic acids and are secreted at ≥100-fold excess relative to complete virions in both cell culture and infected chimpanzees [12]. The prevalence of empty HBV virions in patients and their role in chronic viral infection remain to be elucidated.
HBV-infected cells also secrete a soluble, dimeric protein antigen called hepatitis B e antigen (HBeAg) [13], which shares most of its amino acid residues with HBcAg but has an N-terminal extension and C-terminal truncation relative to HBcAg. Independent of capsid assembly or viral replication, HBeAg is released through the secretory pathway and may exert immunoregulatory effects [14]. Historically, serum HBeAg has been widely used to monitor viral infection and treatment response as it is usually associated with high levels of viral replication [14].
HBV replication and secretion of all viral particles and proteins are ultimately driven by the HBV covalently closed circular DNA (cccDNA), which is synthesized from rcDNA in the nuclei of infected hepatocytes and serves as the viral transcriptional template [15,16]. As any cure of HBV infection requires the elimination of cccDNA (or stable silencing of its transcriptional activity), it is critical to monitor the levels of transcriptionally active cccDNA in the liver during treatment [17,18]. As the production of empty virions is uncoupled from viral DNA replication, they may serve as an effective biomarker for transcriptionally active cccDNA during antiviral therapy with nucleos (t)ide analog reverse transcriptase inhibitors, which can reduce serum HBV DNA (i.e. complete virions) to undetectable levels but has minimal impact on cccDNA levels or transcriptional activity [18–20]. A minority of treated patients do experience significant decreases in hepatic cccDNA, presumably due to host-mediated elimination of infected cells or cccDNA and the drug-mediated elimination of the cccDNA precursor (rcDNA) [18,21–23]. Defining reliable methods to monitor levels of transcriptionally active cccDNA will maximize the efficacy of response-guided treatment algorithms. The goals of this study were to characterize levels of empty HBV virions in infected patients and to explore their utility as a readily measurable peripheral biomarker for cccDNA.
MATERIALS AND METHODS
Patient samples
Serum samples were evaluated from patients enrolled in Gilead Sciences clinical studies GS-US-174-0106 [24,25] and GS-US-174-0123 [26]. Study GS-US-174-0106 evaluated tenofovir disoproxil fumarate (TDF) monotherapy compared to emtricitabine (FTC) plus TDF in patients with incomplete response to adefovir dipivoxil (ADV), while Study GS-US-174-0123 evaluated the use of TDF in Asian patients. These studies conformed to the ethical guidelines of the Declaration of Helsinki, as approved by each participating institutional review committee. Informed consent was obtained from patients prior to any study procedures. Serum HBV DNA was measured using the COBAS TaqMan polymerase chain reaction (PCR) assay (Roche Molecular Systems, Pleasanton, CA, USA) that has a lower limit of quantitation of 169 copies/mL (29 IU/mL). Serum HBsAg levels were measured using the Architect HBsAg quantitative assay (Abbott Laboratories, Abbott Park, IL, USA), which has a lower limit of quantification of 0.05 IU/mL.
Native agarose gel electrophoresis assay for virions
Serum samples (5–10 μL) from HBV-infected patients were resolved on native agarose gels, as described previously [12,27]. HBV virions and naked NCs were concentrated from culture supernatant of HepG2 cells transfected with the wild-type HBV or a mutant defective in expressing the reverse transcriptase or polymerase (Pol−) by polyethylene glycol (PEG) precipitation and digested with DNase I (1 mg/mL at 37 °C for 1 h) to eliminate residual plasmid DNA. Virions were further separated from naked NCs by isopycnic CsCl gradient ultracentrifugation [10,12,28]. Purified virion or NC fractions or PEG-concentrated and DNase-digested medium samples were analysed in parallel by native agarose gel electrophoresis. Following Southern blot transfer of viral particles from the gels to nitrocellulose membranes, encapsidated DNA in viral particles was detected using a 32P-labelled HBV DNA probe, followed by detection of core proteins associated with virions or naked NCs on the same membrane using a rabbit polyclonal anti-HBc antibody (Dako, Carpinteria, CA, USA) as previously described [12]. Goat polyclonal anti-HBs antibody (Dako) was used to detect the viral envelope proteins after stripping the membrane. DNA levels were quantified using phosphoimaging following Southern blot analysis. Southern blot detection had a limit of detection of approximately 1 pg per 5–10 μL sample loaded (i.e. 100 pg/mL), which was less sensitive than PCR. Thus, only the quantitative data obtained using PCR are reported.
Sodium dodecyl sulphate (SDS)–polyacrylamide gel electrophoresis (PAGE)
HBcAg and HBsAg were also analysed by Western blot analysis as described above following resolution by SDS-PAGE [12]. To estimate levels of HBcAg or HBsAg, purified recombinant HBcAg expressed in bacteria [12,29] or HBsAg (eEnzyme, Gaithersburg, MD, USA) was used as quantitative standard. HBcAg or HBsAg quantification was performed using a mouse monoclonal antibody against the highly conserved N-terminal sequence of HBcAg [12] or a polyclonal rabbit antibody against HBsAg (Virostat, Portland, ME, USA) [12], respectively. Protein levels were estimated using densitometry or chemiluminescence imaging after Western blot analysis. HBsAg quantification results obtained using the gel-based assay were in agreement with those obtained using the Architect assay, although the gel-based assay was less sensitive than the Architect assay. Thus, only the Architect HBsAg results are reported.
RESULTS
Selection of patient sera for analysis of empty HBV virions
To investigate levels of empty HBV virions in chronic hepatitis B patients, we selected a subset of serum samples from HBeAg+ and HBeAg− patients enrolled in one of the two clinical studies described above. Twenty-one patients with a range of clinical outcomes were selected for evaluation: 8 HBeAg+ patients who remained HBeAg+ (patients 001–008), 5 HBeAg+ patients who achieved HBeAg loss during therapy (patients 009–013), 5 HBeAg− patients who remained HBeAg− and did not achieve HBsAg loss during therapy (patients 014–018) and 3 HBeAg+ patients who achieved HBeAg and HBsAg loss during therapy (patients 019–021) (Table 1). Serum samples from pre-treatment and on-treatment time points were evaluated to examine the relationship between serum empty virions, serum HBsAg and serum HBV DNA levels over a range of different clinical outcomes associated with TDF-containing treatment.
Table 1.
Patient demographics and baseline clinical characteristics
| HBeAg+ | HBeAg− | |
|---|---|---|
| No. of patients | 16 | 5 |
| Sex | ||
| M | 13 | 4 |
| F | 3 | 1 |
| Age, Median (Range) | 33 (18–48) | 41 (26–48) |
| Ethnicity | ||
| Asian | 7 | 2 |
| Caucasian | 7 | 3 |
| Other | 2 | 0 |
| Previous NA Treatment | ||
| ADV | 3 | 2 |
| LAM and ADV | 9 | 3 |
| None | 4 | 0 |
| HBV Genotype | ||
| A | 4 | 0 |
| B | 1 | 0 |
| C | 6 | 1 |
| D | 5 | 4 |
| BL HBV DNA (log10 cp/mL) (median, range) | 6.42 (3.51–9.90) | 5.83 (4.28–6.57) |
| Treatment | ||
| FTC/TDF | 6 | 3 |
| TDF | 10 | 2 |
| HBeAg Loss | 8 | N/A |
| HBsAg Loss | 3 | 0 |
ADV, adefovir dipivoxil; BL, baseline; FTC, emtricitabine; HBeAg, hepatitis B e antigen; HBsAg, hepatitis B surface antigen; HBV, hepatitis B virus; LAM, lamivudine; N/A, nucleos(t)ide analog; TDF, tenofovir disoproxil fumarate; N/A, not applicable.
Pre-TDF treatment levels of empty HBV virions in serum samples varied widely and greatly outnumbered DNA-containing virions
Virion-associated HBcAg as well as virion DNA was detected in serum samples using native agarose gel electrophoresis followed by Western and Southern blot analyses, together with control virion samples collected from the supernatant of HBV-transfected hepatoma cells in culture (Fig. 1). To further verify the identity of HBV virions, the baseline and week 48 post-TDF treatment serum samples from one patient (003) were also fractionated by CsCl gradient centrifugation (Fig. 2). As we reported before [12], on the agarose gel, the virions containing HBcAg and HBsAg (with or without viral DNA) migrated much slower than naked NCs containing no HBsAg, which were detected in hepatoma culture supernatant but absent from any of the serum samples examined (Figs 1 and 2). As we reported before [12], exclusively genome-free (empty) virions harvested from the polymerase-defective HBV mutant (unable to package viral RNA or synthesize viral DNA) migrated at the same position as DNA-containing virions on the agarose gel (Fig. 2, lane 17). The HBV virions in the samples from patient 002 appeared to migrate slightly slower than in the other samples. The significance, if any, of this altered migration remains unclear. As shown in Fig. 1a, top and middle panels, both the virion DNA and capsid signals migrated slower and they did coincide, indicating they likely represent authentic virions instead of nonspecific background signals. The HBsAg signal detected in the bottom panel of Fig. 1a, which co-migrated with the other samples, was mostly derived from the HBsAg spheres and filaments, as they exceeded the virions (enveloped capsids with or without DNA) by ca. 100-fold in these samples (usually by 1000-fold) (Figs 5 and 6). The HBsAg contributed by the virions in these samples, which would have migrated slightly above the HBsAg signal in the other samples, most likely was below the limit of detection (ca. 1 μg/mL) by our gel-based assay.
Fig. 1.

Analysis of complete and empty HBV virions by native agarose gel electrophoresis. Serum samples (5 μL/well) from four HBeAg+ patients who remained HBeAg+ during TDF therapy (a) and three who experienced HBeAg and HBsAg loss during TDF therapy (b) were resolved on native agarose gels and detected following Southern blot transfer using a 32P-labelled HBV DNA probe to detect the virion-associated DNA genome (rcDNA-containing virions; top row), an HBc-specific antibody to detect virion-associated capsids (rcDNA-containing and empty virions; middle row) and an HBsAg-specific antibody to detect envelope proteins associated with virions as well as HBsAg spheres and filaments (bottom row). Patient numbers are indicated on the top, as are the time points evaluated: baseline (BL) and the indicated weeks (W) after starting TDF-containing therapy. HBV virions and naked NCs concentrated from the culture supernatant of HBV-transfected hepatoma (HepG2) cells (medium or M, lane 17) and HBV virions (10 ng of virion capsid protein) purified by CsCl density gradient fractionation from the supernatant (virion or V, lane 18) were loaded in parallel. The boxes in lane 17 denote the naked NCs present in the supernatant that were removed by the gradient fractionation. The * symbol denotes unknown cross-reacting materials in the serum samples. The diagrams on the left depict schematically the complete or empty virions (V, large circles (envelope) with an inner diamond shell (capsid), with or without rcDNA inside, respectively), HBsAg spheres (S, small circle; HBsAg filaments omitted for brevity), empty and single-stranded DNA (straight line) or viral RNA (wavy line) containing naked (nonenveloped) capsids (C, hexagonal shells).
Fig. 2.

Analysis of HBV virions in human sera by CsCl density gradient fractionation. Two serum samples, at baseline (lanes 1–8) and week 48 (lanes 9–16) post-treatment, from patient 003 were fractionated by CsCl gradient centrifugation. The indicated fractions were resolved on a native agarose gel. HBV DNA, HBcAg and HBsAg were detected as described in Fig. 1. HBV virions purified from culture supernatant of HepG2 cells transfected with wild-type (WT; lane 18) HBV DNA or a mutant defective in polymerase expression (P−; lane 17) were included as controls. A virion (V) and naked capsid (NC) fraction harvested from the wild-type HBV-transfected HepG2 cell supernatant by CsCl gradient fractionation were loaded respectively in lanes 19 and 20 to show the migration of naked capsids relative to virions. Other labels are the same as in Fig. 1.
Fig. 5.
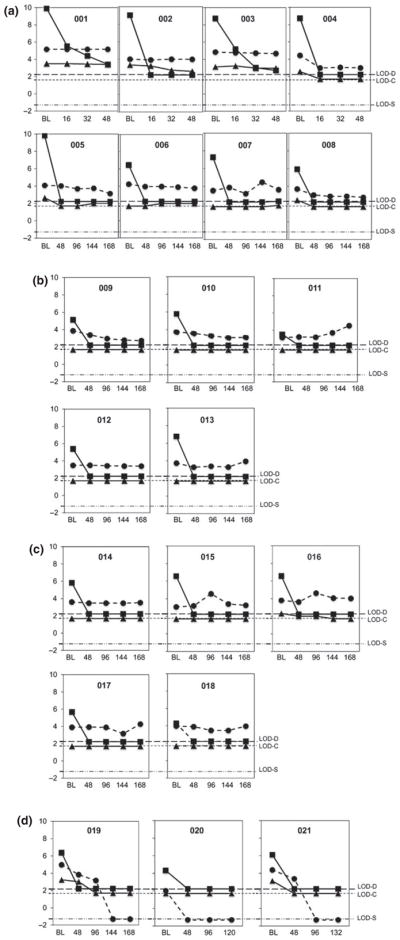
HBV DNA, HBcAg and HBsAg levels at baseline and during TDF therapy. Patients are grouped based on HBeAg status: HBeAg+ (a), HBeAg loss on TDF (b), HBeAg− pre-TDF therapy (c), and HBeAg and HBsAg loss on TDF therapy (d). Patient numbers and time points of serum collection (BL, baseline; and weeks after the start of therapy) are indicated. Y-axis values are Log10: DNA in copies/mL (squares), HBcAg in ng/mL (triangles) and HBsAg in IU/mL (circles). Dashed lines show limits of detection (LOD) for viral DNA (D), HBcAg (C) and HBsAg (S). Time points of HBeAg loss were weeks 120, 84, 96, 108 and 120 for patients 009–013, respectively (b), and weeks 108, 48 and 84 for patients 019–021, respectively (d).
Fig. 6.

Secretion of DNA-containing and empty virions, HBsAg spheres and filaments, and HBeAg during HBV replication and effects of antiviral therapy. The various particles are depicted as in Fig. 1, with their approximate titres indicated. The HBsAg filament is depicted as a cylinder. The soluble, dimeric HBeAg is depicted as grey double bars. The dashed arrow denotes the fact that HBeAg is not always secreted during viral replication. The wavy lines denote the viral RNAs: C, mRNA for HBc (and pgRNA); S and LS, mRNAs for HBsAg; PreC, mRNA for HBeAg. S/M/L, the small, middle and large envelope protein; NRTI, nucleoside analog RT inhibitor; rc, rcDNA; ccc, cccDNA; dsl, double-stranded linear DNA. See text for details.
Also as we reported [12], on the CsCl gradient, the virions were detected in fractions 15–17 (density: 1.21–1.28 g/cm3), with the virion DNA peak (i.e. complete virions; fraction 17) slightly denser than the virion HBcAg peak (i.e. empty virions; factions 15–16) (Fig. 2). Both the complete and empty virion fractions contained abundant amounts of HBsAg. The still lighter fraction 14 (density: 1.19 g/cm3) (Fig. 2, lanes 1 and 9) contained high levels of HBsAg but no viral DNA or HBcAg, representing HBsAg spheres and filaments with neither HBcAg nor DNA and thus lower density than virions. The density gradient analysis also confirmed that no naked NCs were detected in the serum samples, which would have been found in fractions 19–21 (density: 1.34–1.37 g/cm3) and would have migrated at the front of the agarose gel, as shown for the naked NCs in the hepatoma culture supernatant [12] (Fig. 1a, lane 17; Fig. 2, lane 20). It is interesting to note that the amount of HBsAg (spheres and filaments), relative to the amount of the virion-associated DNA or HBcAg, was much less in the hepatoma cell culture supernatant than in the patient sera (Figs 1 and 2).
Pre-TDF treatment levels of virion-associated HBcAg varied widely, ranging from below the limit of detection (approximately 50 ng/mL or 5 × 109 virion capsids/mL, assuming each virion capsid consists of 240 copies of HBc) to more than 3 μg/mL or 3 × 1011 virion capsids/mL (Figs 1 and 3a). Serum virion DNA (i.e. complete virion) levels also varied widely, ranging from 103 to approximately 1010 copies/mL (Figs 1 and 3b). We calculated the ratio of virion-associated HBcAg to serum HBV DNA in the patient samples [12]. Enveloped capsids (as estimated from the total amount of virion-associated capsid protein), when detectable, outnumbered virion DNA by approximately 50-fold (in patient 005) up to 100 000-fold (in patient 019) (Figs 1, 4a and 5). As complete virions (as measured by DNA copy number) accounted for a small fraction (maximal 2% but <1% in most cases) of the total virions (measured by capsid Western blotting), the vast majority of virions in patient sera were empty virions. Naked NCs were not detected in any of the patient sera tested, despite their abundance in hepatoma cell culture supernatant (Fig. 1). Although the absolute levels of complete and empty virions both varied widely pre-TDF treatment, the levels of these two serum HBV markers were positively correlated across patients prior to TDF therapy (Fig. 4a).
Fig. 3.

Distribution of serum HBcAg, serum HBV DNA and serum HBsAg levels in patients in relation to HBeAg status. Baseline (pre-TDF treatment) levels of serum HBcAg (Western Blot) (a), serum HBV DNA (COBAS TaqMan PCR) (b) and serum HBsAg (Architect HBsAg quantitative assay) (c) are plotted for each patient. Patients are grouped by HBeAg status: first column, 8 HBeAg+ patients that remained HBeAg+ on treatment; second column, 5 HBeAg− patients who remained HBeAg− on treatment but did not lose HBsAg; third column, 5 HBeAg+ patients who lost HBeAg on therapy, but who did not lose HBsAg; fourth column, 3 HBeAg+ patients who lost both HBeAg and HBsAg on therapy. LOD, limit of detection.
Fig. 4.
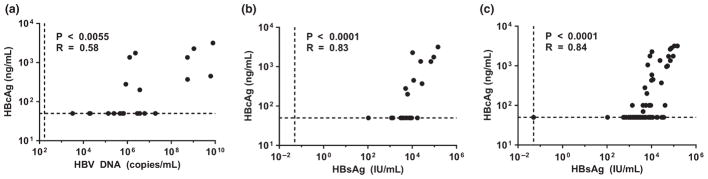
Correlation of serum HBcAg levels to those of serum HBV DNA and serum HBsAg at baseline and during therapy. Serum HBcAg levels (Western blot) plotted against serum HBV DNA levels (COBAS TaqMan PCR) at baseline (pre-TDF therapy) (a). Serum HBcAg levels plotted against serum HBsAg levels (Architect HBsAg quantitative assay) at baseline (b). Serum HBcAg levels plotted against serum HBsAg levels at baseline (n = 21) and during TDF therapy (n = 99) (c). Limits of detection are indicated by vertical and horizontal dotted lines. Two-tailed P values and R values (Pearson correlation coefficient test) are indicated.
Relationship between HBeAg status and pre-TDF treatment levels of empty and complete HBV virions
Six of the eight HBeAg+ patients who did not lose HBeAg or HBsAg during therapy had high pre-TDF treatment levels of both empty virions (3 × 1010–3 × 1011/mL) and complete virions (108–1010/mL) (Figs 1a and 3a,b). In contrast, four of five HBeAg− patients had low levels of empty virions (at the limit of detection [5 × 109/mL] or below detection), and all five had low levels of complete virions (104–106/mL) (Fig. 3a,b). All eight patients who experienced HBeAg loss during treatment had lower levels of complete virions (103–106/mL), and most (6/8) also had lower levels of empty virions at baseline (5 × 109/mL to below detection) (Fig. 3a,b). These include five patients who underwent HBeAg loss only, plus three patients who experienced HBeAg and HBsAg loss during TDF treatment (Figs 3a,b and 5b,d). Interestingly, two of the HBsAg loss patients (019 and 021) had high levels of empty (1.8 × 1011 and 1.4 × 1011/mL, respectively) but low levels of complete virions (ca 106/mL) pre-TDF therapy (Figs 1b, 3a,b and 5d).
Serum empty virion levels generally tracked with serum HBsAg levels
Pre-TDF treatment levels of serum empty virions and HBsAg were positively correlated (Fig. 4b), especially in HBeAg+ patients (Fig. 3), who tended to have high levels of both empty virions and HBsAg. In general, serum HBsAg levels were 100- to 1,000-fold greater than empty virions (Figs 3–5) (up to 1014/mL; 1 IU or 4 ng equals 8 × 108 spheres assuming 100 copies HBsAg subunits per sphere [30]). In HBeAg− patients and in those who experienced HBeAg loss, pre-TDF treatment serum HBsAg levels were high (Fig. 3c), in contrast to the low empty virion levels that were observed (Fig. 3a). Among the three patients (019, 020, 021) who experienced both HBeAg and HBsAg loss, two had high pre-TDF levels of both HBcAg and HBsAg (019, 021), and the other had low levels of both (020) (Figs 3 and 5d).
Similar to what was observed with empty virion levels, most patients showed no or little decrease in serum HBsAg levels during TDF therapy (Figs 1a and 5a–c). Both complete and empty virions were detected at high levels, and they changed little during TDF therapy for patients 001–003 (Figs 1a and 5a). There was also no significant decrease in serum HBsAg levels during therapy for patients 006, 007 and 011–018, who had barely detectable or undetectable levels of HBcAg at baseline and throughout therapy (Fig. 5a–c). For the five patients (004, 005, 008, 019 and 021) who experienced rapid and significant decreases in empty virion levels, all experienced significant decreases in HBsAg levels, with two patients (019 and 021) also experiencing HBeAg and HBsAg loss (Figs 1 and 5a,d). Three patients (009, 010 and 020) demonstrated substantial HBsAg decreases (by 5- to 10-fold); however, it was uncertain whether empty virion levels decreased as they were at or below the limit of detection at all time points (Figs 1b and 5b,d). Overall, serum HBcAg was positively correlated with serum HBsAg both pre-TDF and during TDF therapy (Fig. 4b,c).
Empty virion levels did not correlate with HBV DNA levels during TDF-containing treatment
Pre-TDF treatment, there was a significant positive correlation between the levels of serum HBcAg and those of serum HBV DNA (Fig. 4a). During TDF-containing therapy, 19/21 patients achieved undetectable HBV DNA by 48 weeks of treatment (Figs 1 and 5). In contrast, levels of serum empty virions were variable during TDF-containing treatment. For four patients, empty virions either remained stable (001, 003) or decreased with slower kinetics and a smaller magnitude (002 and 016) (Figs 1 and 5a,c). However, five patients (004, 005, 008, 019 and 021) experienced a rapid and significant decrease in empty virion levels (5- to 30-fold decrease from baseline to undetectable by week 16, 48 or 96) (Figs 1 and 5a,d). For the remaining patients (006, 007, 009–015, 017, 018 and 020), pre-TDF treatment serum HBcAg was at or below the limit of detection, so any decrease in HBcAg could not be reliably assessed (Fig. 5a–d). CsCl gradient centrifugation confirmed that the HBcAg signal remaining in patient 003 at 48 weeks post-treatment, following a 6-log reduction of viral DNA (Fig. 5a), represented enveloped capsids with the same density as (empty) virions (Fig. 2).
DISCUSSION
Our analyses have verified that empty HBV virions are secreted into the blood of HBV-infected patients. Notably, empty virions were present at levels up to 1011/mL and at more than 50- to 100 000-fold excess compared to rcDNA-containing virions; these findings are consistent with recent observations in HBV-infected chimpanzees and in the supernatants of HBV-replicating hepatoma cells [12]. Together, these results indicate that empty virions, similar to HBsAg spheres and filaments, are a distinct class of subviral particles produced during HBV infection (Fig. 6). Moreover, the results reported here confirm that antiviral therapy with nucleos(t)ide analogs (TDF in this case), which can dramatically reduce serum HBV DNA [31], had a minimal impact on serum HBcAg (empty virion) levels in most patients even after years of potent HBV DNA suppression. These results were expected because most nucleos(t)ide analog-treated patients do not clear hepatic cccDNA, which drives the HBcAg and HBsAg expression sufficient for empty virion secretion, even in the absence of viral DNA synthesis (Fig. 6). Dramatic reductions in serum empty virions were observed in a minority of treated patients (including those who achieved HBsAg loss), as anticipated based on previous observations that a minority of patients experience significant reductions in hepatic cccDNA levels during nucleos(t)ide analog therapy [18,23].
The observation that serum empty virion levels correlate with serum HBsAg levels is not unexpected as both proteins are expressed from cccDNA. Interestingly, patients who expressed low pre-TDF treatment serum HBcAg were frequently associated with HBeAg negativity (patients 014–018) or HBeAg loss (patients 009–013). Although HBsAg levels were lower than in HBeAg+ patients, they were still quite high and detectable in patients who were HBeAg− or underwent HBeAg loss, possibly because HBsAg expression, but unlike HBcAg expression, can result from integrated HBV DNA that accumulates steadily (by 10- to 100-fold) during the later stages of infection (Fig. 6) [32–34]. The decline of both HBcAg and HBsAg levels in a minority of treated patients likely reflects either the loss of infected hepatocytes (killing), loss of cccDNA from the infected cells (curing) or suppression of cccDNA transcriptional activity (silencing). This is also supported by the fact that HBsAg loss, the current standard for viral clearance, was accompanied by the significant decline (and likely loss) of serum HBcAg.
Currently, both HBeAg and HBsAg are used as biomarkers for cccDNA; however, both have drawbacks. HBV frequently mutates to reduce or eliminate HBeAg expression under immune pressure (Fig. 6) [17]. Thus, HBeAg cannot be used as a biomarker for cccDNA levels in HBeAg− patients [17]. Also, serum HBsAg levels may not always correlate with cccDNA levels due to the accumulation of integrated HBV DNA (Fig. 6) [17,18]. In principle, the level of empty virions in the serum may be a more reliable marker for hepatic cccDNA levels and its transcriptional activity than serum HBsAg levels. It is unlikely that HBV DNA integrations will lead to a functional HBcAg gene because integrated HBV DNA is derived predominantly from a minor form of viral DNA called double-stranded linear [32,35] in which the HBcAg coding sequence is disconnected from the core promoter. In contrast, integrated HBV DNA is well documented to direct HBsAg expression [36,37] as the HBsAg gene is maintained in the double-stranded linear DNA and the integrants (Fig. 6). As empty virion secretion requires both HBcAg and HBsAg expression, the level of empty virions in the serum may reflect the level of HBcAg expression driven by cccDNA (Fig. 6).
The decision of when to stop antiviral therapy remains an important issue in the treatment of chronic hepatitis B [38]. For patients remaining HBsAg+, a significant decline and loss of serum HBcAg during treatment may indicate cccDNA pool reduction (or silencing). A recent estimate suggested that it may require lifelong treatment (average 52 years) with current antiviral chemotherapy options, even in the absence of drug resistance, to clear HBsAg [39]. However, this may be an overly pessimistic view, in particular for patients with low levels of serum HBsAg, where residual HBsAg could be produced exclusively from integrated HBV DNA. In such cases, continued nucleos(t)ide analog treatment is unlikely to improve patient outcomes. The measurement of empty virions in the serum may distinguish whether residual HBsAg is being driven by cccDNA or integrated HBV DNA.
Future studies with larger sample sizes and more sensitive assays will be needed to confirm these initial observations to determine the role, as well as the potential diagnostic and prognostic significance, of serum HBcAg (empty virions). Also, direct measurements of cccDNA levels will be required to more firmly establish the correlation between levels of hepatic cccDNA and serum empty virions. In combination with classical peripheral HBV markers (e.g. HBV DNA, HBsAg and HBeAg), HBcAg may provide a more accurate view of hepatic cccDNA levels and transcriptional activity.
Acknowledgments
K.M. Kitrinos and W.E. Delaney are employees of Gilead Sciences and own stock in Gilead Sciences. This work was supported by Public Health Service grant (R01 AI074982 to J.H.) from the National Institutes of Health. The preparation of this article was funded, in part, by Gilead Sciences. We thank the students and postdoctoral fellows in J. Hu’s laboratory at Penn State University and Eduardo Martins (Gilead Sciences) for critical review of this manuscript. Writing support was provided by Becky Norquist and funded by Gilead Sciences.
Abbreviations
- ADV
adefovir dipivoxil
- cccDNA
covalently closed circular DNA
- FTC
emtricitabine
- HBc
HBV core
- HBcAg
HBV core antigen
- HBeAg
hepatitis B e antigen
- HBsAg
HBV surface antigen
- HBV
hepatitis B virus
- NC
nucleocapsid
- PCR
polymerase chain reaction
- PEG
polyethylene glycol
- pgRNA
pregenomic RNA
- rcDNA
relaxed circular DNA
- SDS
sodium dodecyl sulphate
- TDF
tenofovir disoproxil fumarate
References
- 1.Seeger C, Zoulim F, Mason WS. Hepadnaviruses. In: Knipe DM, Howley PM, editors. Fields Virology. Philadelphia: Lippincott, Williams & Wilkins; 2007. pp. 2977–3030. [Google Scholar]
- 2.Summers J, O’Connell A, Millman I. Genome of hepatitis B virus: restriction enzyme cleavage and structure of DNA extracted from Dane particles. Proc Natl Acad Sci USA. 1975;72:4597–4601. doi: 10.1073/pnas.72.11.4597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Summers J, Mason WS. Replication of the genome of a hepatitis B–like virus by reverse transcription of an RNA intermediate. Cell. 1982;29(2):403–415. doi: 10.1016/0092-8674(82)90157-x. [DOI] [PubMed] [Google Scholar]
- 4.Conway JF, Cheng N, Zlotnick A, Wingfield PT, Stahl SJ, Steven AC. Visualization of a 4-helix bundle in the hepatitis B virus capsid by cryo-electron microscopy. Nature. 1997;386(6620):91–94. doi: 10.1038/386091a0. [DOI] [PubMed] [Google Scholar]
- 5.Wynne SA, Crowther RA, Leslie AG. The crystal structure of the human hepatitis B virus capsid. Mol Cell. 1999;3(6):771–780. doi: 10.1016/s1097-2765(01)80009-5. [DOI] [PubMed] [Google Scholar]
- 6.Bruss V, Ganem D. The role of envelope proteins in hepatitis B virus assembly. Proc Natl Acad Sci USA. 1991;88(3):1059–1063. doi: 10.1073/pnas.88.3.1059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Seitz S, Urban S, Antoni C, Bottcher B. Cryo-electron microscopy of hepatitis B virions reveals variability in envelope capsid interactions. EMBO J. 2007;26(18):4160–4167. doi: 10.1038/sj.emboj.7601841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dryden KA, Wieland SF, Whitten-Bauer C, Gerin JL, Chisari FV, Yeager M. Native hepatitis B virions and capsids visualized by electron cryomicroscopy. Mol Cell. 2006;22 (6):843–850. doi: 10.1016/j.molcel.2006.04.025. [DOI] [PubMed] [Google Scholar]
- 9.Gerelsaikhan T, Tavis JE, Bruss V. Hepatitis B virus nucleocapsid envelopment does not occur without genomic DNA synthesis. J Virol. 1996;70(7):4269–4274. doi: 10.1128/jvi.70.7.4269-4274.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Perlman D, Hu J. Duck hepatitis B virus virion secretion requires a double-stranded DNA genome. J Virol. 2003;77(3):2287–2294. doi: 10.1128/JVI.77.3.2287-2294.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Seeger C, Hu J. Why are hepadnaviruses DNA and not RNA viruses? Trends Microbiol. 1997;5(11):447–450. doi: 10.1016/s0966-842x(97)01141-4. [DOI] [PubMed] [Google Scholar]
- 12.Ning X, Nguyen D, Mentzer L, et al. Secretion of genome-free hepatitis B virus–single strand blocking model for virion morphogenesis of pararetrovirus. PLoS Pathog. 2011;7(9):e1002255. doi: 10.1371/journal.ppat.1002255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.DiMattia MA, Watts NR, Stahl SJ, et al. Antigenic switching of hepatitis B virus by alternative dimerization of the capsid protein. Structure. 2013;21(1):133–142. doi: 10.1016/j.str.2012.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Milich D, Liang TJ. Exploring the biological basis of hepatitis B e antigen in hepatitis B virus infection. Hepatology. 2003;38(5):1075–1086. doi: 10.1053/jhep.2003.50453. [DOI] [PubMed] [Google Scholar]
- 15.Tuttleman JS, Pourcel C, Summers J. Formation of the pool of covalently closed circular viral DNA in hepadnavirus-infected cells. Cell. 1986;47:451–460. doi: 10.1016/0092-8674(86)90602-1. [DOI] [PubMed] [Google Scholar]
- 16.Gao W, Hu J. Formation of hepatitis B virus covalently closed circular DNA: removal of genome-linked protein. J Virol. 2007;81(12):6164–6174. doi: 10.1128/JVI.02721-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Thompson AJ, Nguyen T, Iser D, et al. Serum hepatitis B surface antigen and hepatitis B e antigen titers: disease phase influences correlation with viral load and intrahepatic hepatitis B virus markers. Hepatology. 2010;51(6):1933–1944. doi: 10.1002/hep.23571. [DOI] [PubMed] [Google Scholar]
- 18.Zoulim F, Testoni B, Lebosse F. Kinetics of intrahepatic covalently closed circular DNA and serum hepatitis B surface antigen during antiviral therapy for chronic hepatitis B: lessons from experimental and clinical studies. Clin Gastroenterol Hepatol. 2013;11 (8):1011–1013. doi: 10.1016/j.cgh.2013.04.010. [DOI] [PubMed] [Google Scholar]
- 19.Guo JT, Pryce M, Wang X, Barrasa MI, Hu J, Seeger C. Conditional replication of duck hepatitis B virus in hepatoma cells. J Virol. 2003;77(3):1885–1893. doi: 10.1128/JVI.77.3.1885-1893.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Litwin S, Toll E, Jilbert AR, Mason WS. The competing roles of virus replication and hepatocyte death rates in the emergence of drug-resistant mutants: theoretical considerations. J Clin Virol. 2005;34 (Suppl 1):S96–S107. doi: 10.1016/s1386-6532(05)80018-6. [DOI] [PubMed] [Google Scholar]
- 21.Colonno RJ, Genovesi EV, Medina I, et al. Long-term entecavir treatment results in sustained antiviral efficacy and prolonged life span in the woodchuck model of chronic hepatitis infection. J Infect Dis. 2001;184 (10):1236–1245. doi: 10.1086/324003. [DOI] [PubMed] [Google Scholar]
- 22.Zhu Y, Yamamoto T, Cullen J, et al. Kinetics of hepadnavirus loss from the liver during inhibition of viral DNA synthesis. J Virol. 2001;75(1):311–322. doi: 10.1128/JVI.75.1.311-322.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Werle-Lapostolle B, Bowden S, Locarnini S, et al. Persistence of cccDNA during the natural history of chronic hepatitis B and decline during adefovir dipivoxil therapy. Gastroenterology. 2004;126(7):1750–1758. doi: 10.1053/j.gastro.2004.03.018. [DOI] [PubMed] [Google Scholar]
- 24.Berg T, Marcellin P, Zoulim F, et al. Tenofovir is effective alone or with emtricitabine in adefovir-treated patients with chronic-hepatitis B virus infection. Gastroenterology. 2010;139(4):1207–1217. doi: 10.1053/j.gastro.2010.06.053. [DOI] [PubMed] [Google Scholar]
- 25.Berg T, Zoulim F, Moeller B, et al. Long-term efficacy and safety of emtricitabine plus tenofovir DF vs. tenofovir DF monotherapy in adefovir-experienced chronic hepatitis B patients. J Hepatol. 2014;60(4):715–722. doi: 10.1016/j.jhep.2013.11.024. [DOI] [PubMed] [Google Scholar]
- 26.Pan CQ, Trinh H, Yao A, et al. Efficacy and safety of tenofovir disoproxil fumarate in asian-americans with chronic hepatitis B in community settings. PLoS ONE. 2014;9(3):e89789. doi: 10.1371/journal.pone.0089789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lenhoff RJ, Summers J. Coordinate regulation of replication and virus assembly by the large envelope protein of an avian hepadnavirus. J Virol. 1994;68(7):4565–4571. doi: 10.1128/jvi.68.7.4565-4571.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Perlman DH, Berg EA, O’Connor PB, Costello CE, Hu J. Reverse transcription-associated dephosphorylation of hepadnavirus nucleocapsids. Proc Natl Acad Sci USA. 2005;102(25):9020–9025. doi: 10.1073/pnas.0502138102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ludgate L, Ning X, Nguyen DH, Adams C, Mentzer L, Hu J. Cyclin-dependent kinase 2 phosphorylates s/t-p sites in the hepadnavirus core protein C-terminal domain and is incorporated into viral capsids. J Virol. 2012;86(22):12237–12250. doi: 10.1128/JVI.01218-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Deguchi M, Yamashita N, Kagita M, et al. Quantitation of hepatitis B surface antigen by an automated chemiluminescent microparticle immunoassay. J Virol Methods. 2004;115(2):217–222. doi: 10.1016/j.jviromet.2003.10.002. [DOI] [PubMed] [Google Scholar]
- 31.Seto W-K, Yuen M-F, Fung J, Lai CL. Tenofovir disoproxil fumarate for the treatment of chronic hepatitis B monoinfection. Hepatol Int. 2013;7:327. doi: 10.1007/s12072-011-9282-y. [DOI] [PubMed] [Google Scholar]
- 32.Yang W, Summers J. Integration of hepadnavirus DNA in infected liver: evidence for a linear precursor. J Virol. 1999;73(12):9710–9717. doi: 10.1128/jvi.73.12.9710-9717.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Summers J, Mason WS. Residual integrated viral DNA after hepadnavirus clearance by nucleoside analog therapy. Proc Natl Acad Sci USA. 2004;101(2):638–640. doi: 10.1073/pnas.0307422100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hu J, Nguyen D. Therapy for chronic hepatitis B: the earlier, the better? Trends Microbiol. 2004;12 (10):431–433. doi: 10.1016/j.tim.2004.08.007. [DOI] [PubMed] [Google Scholar]
- 35.Staprans S, Loeb DD, Ganem D. Mutations affecting hepadnavirus plus-strand DNA synthesis dissociate primer cleavage from translocation and reveal the origin of linear viral DNA. J Virol. 1991;65(3):1255–1262. doi: 10.1128/jvi.65.3.1255-1262.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.MacNab GM, Alexander JJ, Lecatsas G, Bey EM, Urbanowicz JM. Hepatitis B surface antigen produced by a human hepatoma cell line. Br J Cancer. 1976;34(5):509–515. doi: 10.1038/bjc.1976.205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Knowles BB, Howe CC, Aden DP. Human hepatocellular carcinoma cell lines secrete the major plasma proteins and hepatitis B surface antigen. Science. 1980;209(4455):497–499. doi: 10.1126/science.6248960. [DOI] [PubMed] [Google Scholar]
- 38.Scaglione SJ, Lok AS. Effectiveness of hepatitis B treatment in clinical practice. Gastroenterology. 2012;142(6):1360–8.e1. doi: 10.1053/j.gastro.2012.01.044. [DOI] [PubMed] [Google Scholar]
- 39.Chevaliez S, Hezode C, Bahrami S, Grare M, Pawlotsky JM. Long-term hepatitis B surface antigen (HBsAg) kinetics during nucleoside/nucleotide analogue therapy: finite treatment duration unlikely. J Hepatol. 2013;58(4):676–683. doi: 10.1016/j.jhep.2012.11.039. [DOI] [PubMed] [Google Scholar]