

Abstract
Melilotus comprises 19 species, while the phylogenetic relationships between species remain unclear. In the present work, three chloroplast genes, rbcL, matK, trnL-F, and one nuclear region, ITS (internal transcribed spacer) belonging to 48 populations of 18 species of Melilotus were sequenced and phylogenetic trees were constructed to study their interspecific relationships. Based on the phylogenetic tree generated in this study using rbcL analysis, the Melilotus genus is clearly monophyletic in the legume family. Both Bayesian and maximum-parsimony approaches were used to analyze the data. The nrDNA ITS provided more informative characteristics (9.8%) than cpDNA (3.0%). Melilotus contains two closely related groups, clade I and clade II. M. spicatus, M. indicus and M. segetalis have a close relationship. M. infestus, M. siculus and M. sulcatus are closely related. The comparing between molecular phylogeny and flower color classification in Melilotus showed that the flower color is not much informative for phylogenetics of this genus.
Introduction
Melilotus (sweet clover) belongs to the tribe Trifolieae of the legume family and comprises 19 annual and biennial species [1]. All species are native to Eurasia or North Africa [2], and three species are cultivated: M. albus, M. officinalis and M. indicus [3]. M. albus and M. officinalis are mainly capable of self-pollination [4], but when the pistil is longer than the stamens, there is very little self-pollination [5]. Melilotus also has entomophilous flowers, which can lead to hybridization. Several species have invaded the Northwest Territories in Canada and the Midwestern USA, among which M. albus and M. officials are often studied [6, 7, 8]. Members of the Melilotus genus have high seed yields and, relative to most other forages, are more tolerant to extremes in environmental conditions, e.g. drought, cold and high salinity [9, 10]. Melilotus also has important medicinal value in addition to being an important forage crop [11]. Furthermore, the nitrogen fixation rate of Melilotus is higher than that of other legumes, making it beneficial for crop rotations [12].
Members of Melilotus exhibit wide variations in flower structure, flower color, seed, leaf and pod characteristics [13, 14]. The classification of Melilotus are more difficult based on morphological traits and growth habits [15, 16]. However, except for morphological studies, no other taxonomic assessments have been conducted on interspecific phylogenetic relationships among species. Analysis of DNA has been widely used in the phylogenetic and classification studies. These methods are more effective and specific than traditionally morphological methods in phylogenetic relationships and genetic variation involved in sibling species and morphologically intermediate species [17, 18].
Phylogenetic results that used a single gene may lead to misleading, especially in cpDNA, which is inherited maternally [19]. Hybridization between different species or genera may lead to reticulate evolution [20]. The employment of a different molecular marker could help to assess and to reduce this problem. Nuclear ribosomal genes with alternating gene and spacer regions and tandom repeat structures can provide this option [21, 22, 23]. The nrDNA internal transcribed spacer (ITS) region and chloroplast DNA have higher variability and are thus suitable for classifying lower taxonomic levels [24, 25, 26]. Accordingly, these regions are useful for inferring phylogenetic relationships at lower taxonomic levels and have been successfully used to analyze plant systematics [27, 28]. Here we selected three cpDNA termed the rbcL gene, matK gene and trnL-F gene and one nrDNA ITS to study the interspecific relationships [29, 30].
In this study, except for M. macrocarpus in Melilotus genus, plant samples from 48 populations of 18 Melilotus species were collected. To study the phylogenetic relationships among members of the Melilotus genus and to generate more accurate estimates of its genetic diversity, we constructed the molecular phylogenetic trees of single nrDNA ITS, 3-cpDNA and the concatenated sequences of all four genes. Finally the molecular phylogenetic classification was compared based on flower color and karyotype in Melilotus.
Materials and Methods
Sampling
Seeds from 48 populations representing 18 species were obtained from National Plant Germplasm System (NPGS, America) and planted at Yuzhong (35°57'N, 104°09'E) in Gansu Province, China (Table 1). Samples were collected from public land instead of protected areas in the northwest China, and no samples of endangered or protected species were included in our study.
Table 1. Information for 48 populations of 18 Melilotus species.
| Species | Population number | Origin | Latitude | Longitude |
|---|---|---|---|---|
| M. albus | PI 90557 | China, Manchuria | 45°19' | 124°29' |
| Ames 21597 | Italy | 41°52' | 12°34' | |
| M. altissimus | Ames 18376 | United States, Nebraska | 41°29' | -99°54' |
| PI 275975 | - | - | - | |
| PI 420163 | France | 46°13' | -2°12' | |
| M. dentatus | PI 108656 | Armenia | 40°4' | -45°2' |
| PI 90753 | China | 35°51' | -104°11' | |
| M. elegans | PI 250873 | Iran | 32°25' | -53°41' |
| PI 260271 | Ethiopia, Shewa | 9°9' | -37°48' | |
| M. hirsutus | Ames 22882 | Russian Federation | 61°31' | 105°19' |
| PI 129697 | Sweden | 60°7' | -18°38' | |
| M. indicus | Ames 24055 | Egypt | 26°49' | 30°48' |
| PI 107562 | Uzbekistan | 41°22' | 64°35' | |
| PI 260756 | Turkey | 38°57' | 35°14' | |
| PI 43595 | - | - | - | |
| Ames 21619 | United States, Nebraska | 41°29' | -99°54' | |
| PI 308524 | Peru | -9°11' | -75°0' | |
| M. infestus | PI 306327 | Italy | 41°52' | 12°34' |
| PI 306328 | Hungary | 47°9' | 19°30' | |
| PI 306326 | Algeria | 27°13' | 2°29' | |
| M. italicus | PI 317638 | Israel | 31°2' | 34°51' |
| PI 317635 | Czechoslovakia | 14°28' | 121°2' | |
| M. officinalis | PI 304530 | Turkey | 38°57' | -35°14' |
| M. polonicus | PI 314386 | Former Soviet Union | 24°47' | 120°60' |
| PI 108647 | Former Soviet Union | 24°47' | 120°60' | |
| M. segetalis | PI 317633 | Algeria | 27°13' | 2°29' |
| PI 43597 | - | - | - | |
| PI 317649 | Czechoslovakia | 14°28' | 121°2' | |
| M. siculus | PI 129703 | Malta | 35°56' | 14°22' |
| PI 318508 | Greece | 39°4' | 21°49' | |
| PI 33366 | Former Soviet Union | 24°46' | 120°59' | |
| M. speciosus | PI 317650 | Canada, Manitoba | 53°45' | -98°48' |
| M. spicatus | PI 317644 | Algeria | 27°13' | 2°29' |
| Ames 25647 | Ukraine, Krym | 44°57' | 34°6' | |
| Ames 18402 | United States, Nebraska | 41°29' | -99°54' | |
| PI 314466 | Uzbekistan | 41°22' | 64°35' | |
| M. suaveolens | Ames 18444 | United States, Nebraska | 41°29' | -99°54' |
| Ames 23793 | Mongolia | 46°51' | 103°50' | |
| PI 593408 | United States, South Dakota | 43°58' | -99°54' | |
| PI 595395 | United States, lowa | 41°52' | -93°5' | |
| M. sulcatus | PI 198090 | Morocco | 31°47' | -7°5' |
| PI 227595 | Tunisia | 33°53' | -9°32' | |
| M. tauricus | PI 67510 | Ukraine, Krym | 44°57' | 34°6' |
| Ames 18446 | United States, Nebraska | 41°29' | -99°54' | |
| Ames 25789 | Ukraine, Krym | 44°57' | 34°6' | |
| M. wolgicus | PI 317665 | Denmark | 56°15' | 9°30' |
| PI 502547 | Russian Federation | 61°31' | 105°19' | |
| PI 317666 | Czechoslovakia | 14°28' | 121°2' |
Young leaves from 2 to 12 individuals of each population were sampled (totaling 406 individuals). Leaves were frozen in liquid nitrogen and stored at -80°C.
DNA extraction, amplification and sequencing
Four genes were amplified and sequenced: three chloroplast genes (cpDNA), trnL-F, rbcL and matK, and one nuclear region (nrDNA), ITS (Table 2). For each population, 2 to 12 independent DNA samples were obtained to check for sequencing errors. Total genomic DNA was extracted using an SDS (sodium dodecyl sulfate) method [31]. Polymerase chain reactions were then conducted in a 25-μL tube containing 1 μL genomic DNA (50 ng / mL), 1 μL of each primer (5 pmol / mL), 12.5 μL Takara Taq DNA polymerase master mix and 9.5 μL deionized water. For nuclear DNA ITS, the region was amplified using a PCR protocol of 94°C for 3 min, followed by 35 cycles of denaturation at 94°C for 30 s, annealing at 50°C for 30 s, and extension at 72°C for 1 min, and a final extension at 72°C for 10 min. For trnL-F gene using a PCR protocol of 94°C for 3 min, then 30 cycles at 94°C for 45 s, annealing at 50°C for 45 s, extension at 72°C for 1min and a final extension step at 72°C for 7 min. The PCR temperature protocol of the matK gene was: 94°C for 3 min then 35 cycles of denaturation at 94°C for 45 s, annealing at 58°C for 45 s, extension at 72°C for 1 min and a final extension step at 72°C for 10 min. Finally, for rbcL gene the following PCR conditions were used: initial denaturation at 94°C for 3 min, followed by 36 cycles of denaturation at 94°C for 30 s, annealing at 56°C for 30 s, and extension at 72°C for 1 min, and a final extension at 72°C for 10 min. The sequencing reactions were performed by Shanghai Shenggong Biotechnological, Ltd. (Shanghai, China).
Table 2. Sequences of primers used to amplify genes.
| Primer | Sequence | Reference |
|---|---|---|
| ITS_F | GGAAGKARAAGTCGTAACAAGG | - |
| ITS_R | RGTTTCTTTTCCTCCGCTTA | - |
| rbcL_F | AGACCTWTTTGAAGAAGGTTCWGT | [48] |
| rbcL_R | TCGGTYAGAGCRGGCATRTGCCA | [48] |
| matK_F | CCCRTYCATCTGGAA ATCTTGGTTC | [49] |
| matK_R | GCTRTRATAATGAGAAAGATTCTGC | [49] |
| trnL-F_F | CGAAATCGGTAGACGCTACG | [50] |
| trnL-F_R | ATTTGAACTGGTGACACGAG | [50] |
To analyze the phylogenetic relationship between Melilotus and other Legume forage and confirm the monophyly of genus Melilotus, we downloaded the only one available gene rbcL for most of legumes close to Melilotus from NCBI, including Medicago, Trifolium, Caragana, Lathyrus and Vicia. These rbcL sequences were used to construct a phylogenetic tree together with the sequences of 18 species of Melilotus obtained in the present study.
Phylogenetic analyses
Phylogenetic analyses were performed using Bayesian and maximum-parsimony approaches. Sequence alignment was initially performed using ClustalX [32] and manually adjusted using MEGA5.0 [33]. The maximum-parsimony analyses involved a heuristic search strategy with 1000 replicates of random sequence addition in combination with TBR branch swapping in MEGA5.0. All character states were treated as unordered and equally weighted. Informative insertions and deletions (indels) were coded as binary characters (0, 1) according to Graham et al. (2000). A strict consensus tree was constructed from the most parsimonious trees. Bayesian analyses were conducted using MrBayes version 3.1 [34]. A model of sequence evolution for the combined dataset was selected using the program ModelTest version 3.6 [35] as implemented in MrMTgui [36] and based on the Akaike information criterion (AIC) [37]. The dataset was analyzed as a single partition using the GTR + I + G model. Four chains were run, beginning with a random tree and saving a tree every 100 generations for one million generations. Finally, the ITS region, three cpDNAs and the dataset of the four genes ITS, rbcL, matK and trnL-F were combined for phylogenetic analyses. One sequence from each population was used to construct phylogenetic trees for the genus Melilotus.
Results
Alignments and DNA sequence data
A total of 96 haplotypes were identified for the ITS region, and 31, 83 and 106 haplotypes were identified for the rbcL, matK, and trnL-F genes, respectively. One sequence from each population was used for constructing phylogenetic trees. Accession numbers of rbcL, matK, trnL-F and ITS respectively are KP987625—KP987627, KP987673—KP987720, KP987577—KP987624 and KP987721—KP987768. The 755-bp fragment of the rbcL gene yielded the most parsimonious trees (length = 138 steps; CI = 0.667; RI = 0.928). The nrDNA tree was based on an alignment of 714 bp (length = 159 steps; CI = 0.765; RI = 0.929). The combined dataset of 3 cpDNAs comprised 2284 bp (length = 241 steps; CI = 0.838; RI = 0.916) and the 4-gene dataset 2998 bp, with maximum-parsimony analyses resulting in the most parsimonious trees (length = 252 steps; CI = 0.625; RI = 0.852). The aligned sequence information for the phylogenetic analysis is presented in Table 3.
Table 3. Dataset and tree statistics from separate maximum parsimony analyses.
| Sequence | Number of sequences | Aligned Length/bp | Conserved site/bp | Variable site /bp | Singleton site/bp | Parsimony informative /bp | Parsimony informative site/% | (G+C) Content /% |
|---|---|---|---|---|---|---|---|---|
| rbcL | 42 | 755 | 666 | 88 | 27 | 61 | 8.1 | 40.8 |
| nrDNA | 48 | 714 | 572 | 111 | 42 | 69 | 9.8 | 49.0 |
| 3—cpDNA | 48 | 2284 | 2058 | 117 | 29 | 88 | 3.0 | 35.0 |
| 4-genes | 48 | 2998 | 2611 | 236 | 73 | 163 | 5.4 | 38.4 |
Phylogenetic analyses
rbcL analysis in the legume family
The phylogenetic tree in the legume family shown in Fig 1 comprised two large clades, designated A and B. In phylogenetic tree, all the Melilotus species formed a monophyletic clade with a high bootstrap value of 100. Among them, the Melilotus was divided into two subclades, named clade I and clade II, with bootstrap values of 65 and 50, respectively. In clade II, M. siculus and M. sulcatus formed one subgroup, named clade IIa, and M. indicus and M. Segetalis formed another subgroup, named clade IIb. These Melilotus species clustering within the same subclade may have closer genetic relationships. All species of genera Medicago and Trifolium formed a clade named clade III which cluster together with genera Melilotus and can be used as outgroups in phylogenetic studies of Melilotus. All species of genera Caragana, Lathyrus and Vicia formed a big clade named clade IV, with members of Lathyrus and Vicia forming a subclade.
Fig 1. Topology resulting from maximum parsimony analysis of rbcL in the legume family dataset using MEGA5.0.
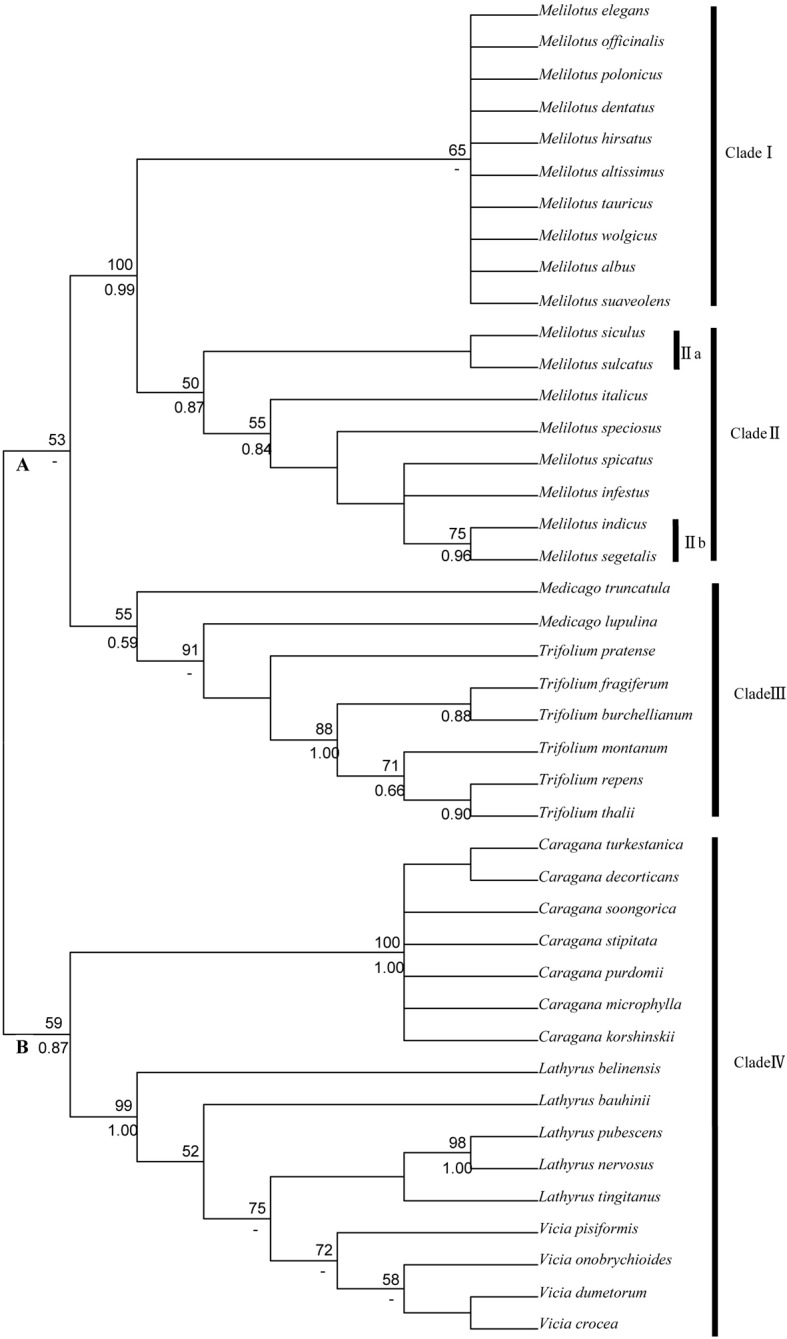
Bootstrap support values (> 50%) are indicated above the branches (posterior values from the corresponding Bayesian analysis are provided below the branches;-: node not recognized).
3-cpDNA analysis
The 3-cpDNA tree of Melilotus based on 2284-bp of concatenated plastid sequences (rbcL, matK and trnL-F), with T. lupinaste and M. sativa as outgroups, is shown in Fig 2. Similar to the rbcL tree for the legume family (Fig 1), the 3-cpDNA tree showed that Melilotus species can also be divided into two clades, clade I and clade II, though with low bootstrap support. In clade II, M. spicatus, M. indicus and M. segetalis formed a subclade named clade 1, which was supported by a high bootstrap value of 85; M. infestus, M. siculus and M. sulcatus formed clade 2, with low bootstrap values of 54. Compared with the results of the rbcL tree for the legume family, subgroup IIb was found in clade 1.
Fig 2. Topology resulting from maximum parsimony analysis of the combined dataset of 3-cpDNA genes (rbcL, matK, trnL-F) using MEGA5.0.
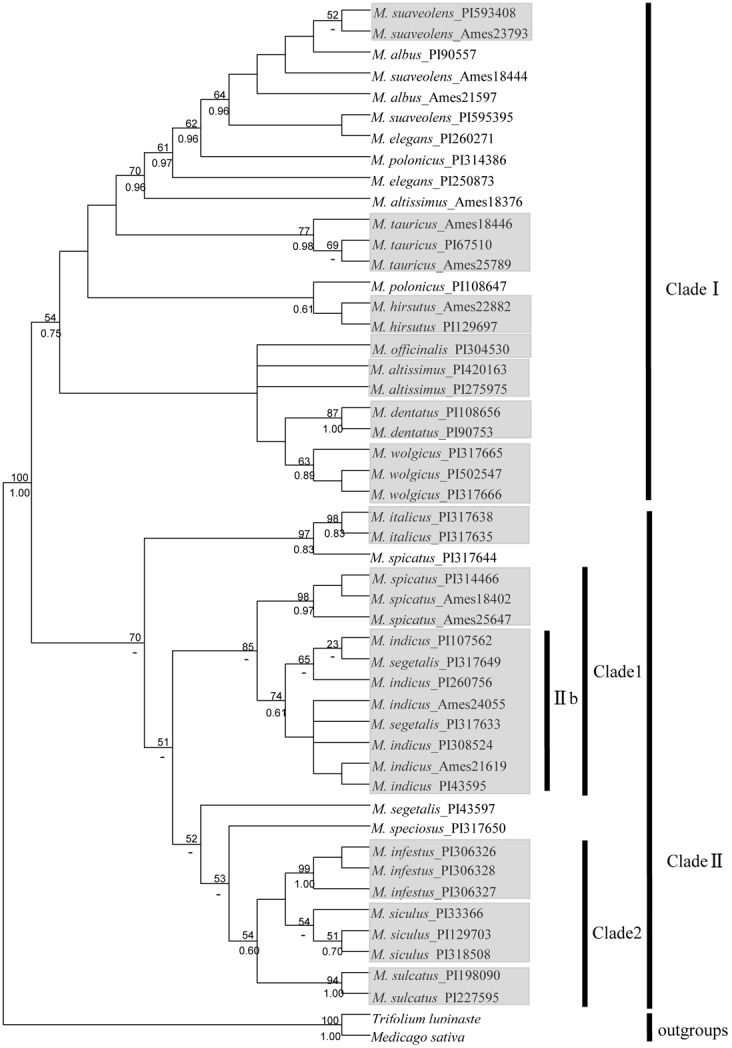
Bootstrap support values (> 50%) are indicated above the branches (posterior values from the corresponding Bayesian analysis are provided below the branches;-: node not recognized). The populations of the same species clustering together are indicated in a grey box. Except for clade 1 and clade 2, the definitions of clades follow those of Fig 1.
nrDNA analysis
The ITS-based phylogenetic tree of Melilotus based on the 714-bp alignment is shown in Fig 3, with T. lupinaste and M. sativa as outgroups. Two strongly divergent and highly supported clades with bootstrap values of 84 and 76, respectively, are shown in the ITS tree. In contrast to the results in Fig 2, clade A consists of two subclades, clade I and clade 1, with high bootstrap values of 91 and 88. Within clade 1, M. indicus and M. segetalis form a well-supported subgroup named IIb. As shown in Fig 3, subgroup IIa is evidently within clade 2, and subgroup IIa includes two species, M. sulcatus and M. siculus, with high bootstrap support.
Fig 3. Topology resulting from maximum parsimony analysis of one ITS dataset using MEGA 5.0.
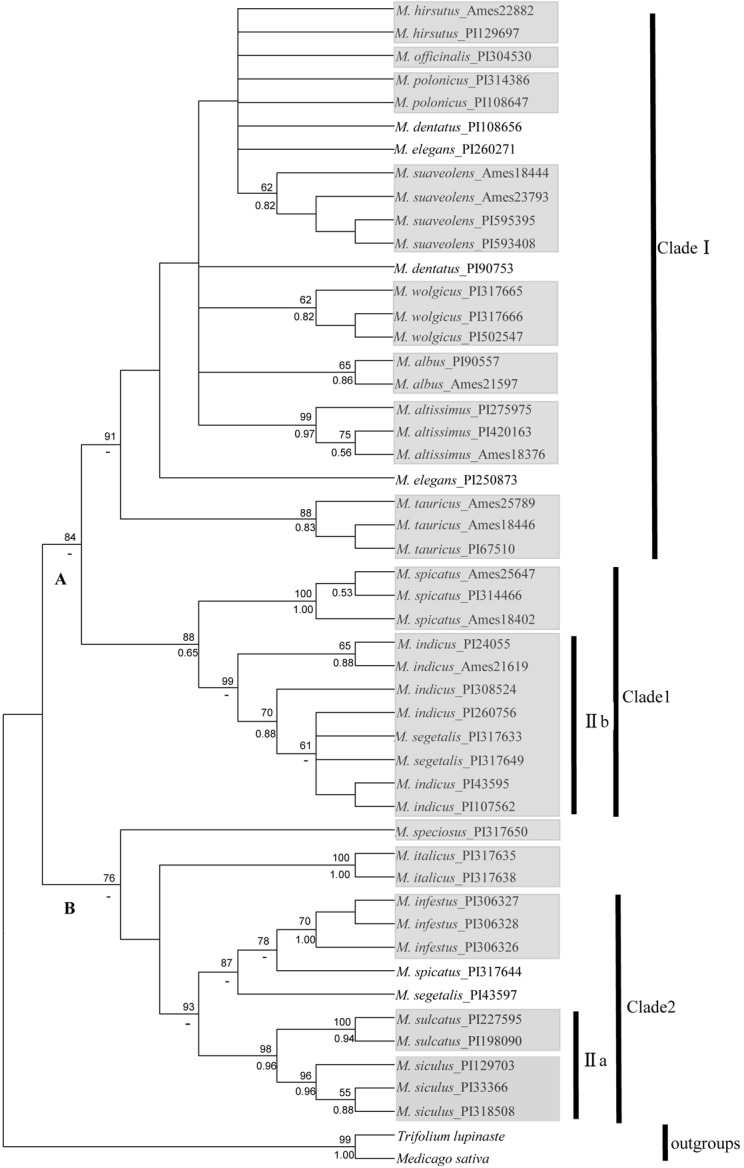
Bootstrap support values (> 50%) are indicated above the branches (posterior values from the corresponding Bayesian analysis are provided below the branches;-: node not recognized). The populations of the same species clustering together are indicated in a grey box. The definitions of clades follow those of Fig 1 and Fig 2.
4-gene analysis
The 4-gene tree of Melilotus yielded 2998 bp of four concatenated genes (rbcL, matK, trnL-F and ITS), with T. lupinaste and M. sativa as outgroups, is shown in Fig 4. The major clades recovered in the above tree were also successfully resolved by this analysis. Clade I was observed and contained 10 related species. Subgroup IIa and M. infestus formed a subclade, namely clade 2, and subgroup IIb with M. spicatus formed a highly supported subclade, clade 1. However, clade 2 clustered together with clade I, which is the major difference between this tree and the nrDNA tree. Except for populations of M. polonicus, M. spicatus and M. segetalis, other populations of the same species formed a subclade in the 4-gene tree.
Fig 4. Topology resulting from maximum parsimony analysis of one 4-gene dataset using MEGA5.0.
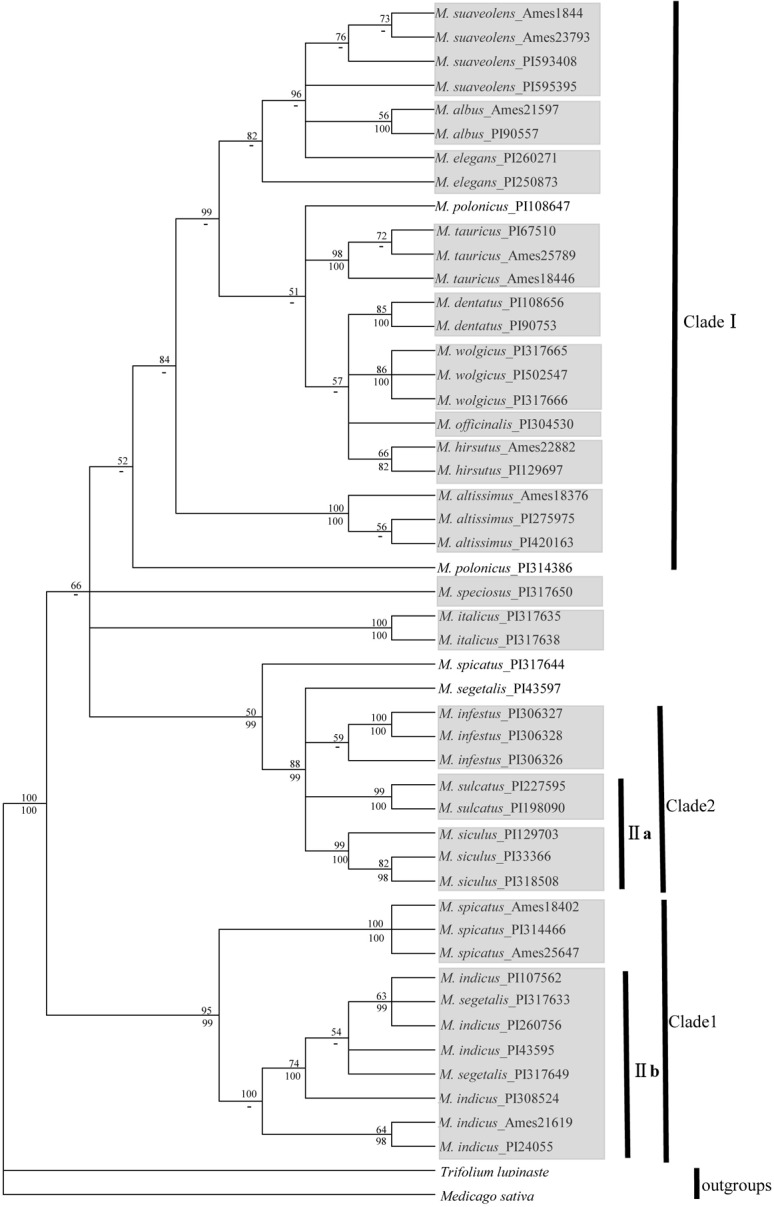
Bootstrap support values (> 50%) are indicated above the branches (posterior values from the corresponding Bayesian analysis are provided below the branches;-: node not recognized). The populations of the same species clustering together are indicated in a grey box. The definitions of clades follow those of Fig 1 and Fig 2.
Discussion
In this study, clade I, which contains 10 species, was found in all four trees of ITS and cpDNA genes. However, in the two cpDNA trees (Fig 1 and Fig 2), the 8 species formed another large clade, clade II. In the nrDNA tree (Fig 3) and 4-gene tree (Fig 4), clade 1, which was clustered into clade II in other two trees, was clustered into clade I; therefore, no clade II was shown. Several species are closely related in 3-cpDNA tree, nrDNA tree and 4-gene tree, e.g., M. spicatus, M. segetalis and M. indicus in clade 1, and M. siculus, M. sulcatus and M. infestus in clade 2. As shown in Table 3, the ITS region provided more informative characteristics (9.8%) than cpDNA (3.0%). Liu et al. [38] reported a similar result for Ligularia–Cremanthodium–Parasenecio in a study showing that ITS (39.6%) had more parsimony-informative characters than cpDNA (2.5%) using an NdhF and trnL-trnF combination. The higher sequence variability in the ITS region compared with cpDNA, which has also been demonstrated in many other taxa [39, 40, 41, 42] may lead to incongruence in phylogenetic tree. As nrDNA is biparentally inherited and has high rates of intraspecific gene flow which can enhance species delimitation. Howevre, the maternally inherited chloroplast DNA is more frequently introgressed and more limited use in species delimitation than nuclear DNA [43, 44]. In addition, incomplete lineage sorting [45] and hybridization between and within species [20] may also cause phylogeny incongruent.
According to Steven [16], plant morphology may show great variation within a single plant, which was not used for species classification. Steven studied agronomic and taxonomic reviews of the genus Melilotus and divided Melilotus into two groups according to flower color, namely, white and yellow. The white group contains four species, M. albus, M. tauricus, M. wolgicus and M. speciosus, and the other species compose the yellow group (Table 4). Our results showed that flower color has no obvious link with the phylogenetic classification in our study.
Table 4. The four classifications of flower color, seed morphology, karyotype and molecular phylogeny in Melilotus.
| Categories | Classification | Subclassification | Species | |
|---|---|---|---|---|
| Flower color[11] | white | M. albus, M. tauricus, M. wolgicus, M. speciosus | ||
| yellow | M. altissimus, M. dentatus, M. hirsatus, M. officinalis, M. polonicus, M. suaveolens, M. elegans, M. spicatus ※ , M. indicus, M. segetalis, M. infestus, M. siculus ※ , M. sulcatus, M. italicus | |||
| Karyotype[39, 40] | Type A | M. albus, M. altissimus, M. dentatus, M. hirsutus, M. officinalis, M. polonicus, M. suaveolens, M. tauricus, M. wolgicus | ||
| Type B | B-1 | M. elegans, M. indicus, M. neapolitana ※ | ||
| B-2 | M. infestus, M. macrocarpus, M. messanensis ※ , M. segetalis, M. speciosus, M. sulcatus | |||
| Type C | M. italicus | |||
| Molecular phylogeny | Clade Ⅰ | M. albus, M. altissimus, M. dentatus, M. hirsatus, M. officinalis, M. polonicus, M. suaveolens, M. tauricus, M. wolgicus, M. elegans | ||
| Clade Ⅱ | Clade 1 | M. spicatus ※ | ||
| Ⅱb | M. indicus, M. segetalis | |||
| Clade 2 | M. infestus | |||
| Ⅱa | M. siculus ※ , M. sulcatus | |||
| M. speciosus, M. italicus | ||||
※: Species incongruence in flower color, karyotype and molecular phylogeny
Clarke studied the number and morphology of chromosomes in the genus Melilotus [46], reporting a chromosome number of 2n = 16. Karyotype analyses of all Melilotus species were conducted by Kita [47]. The 19 species examined are grouped into three types: A, B and C. Type B is further divided into Type B-1 and Type B-2 (Table 4). The grouping information based on karyotype analyses indicates that the species within each type are closely related [47]. Except for M. elegans, clade 1 of the phylogenetic trees is consistent with type A, and clade 2 comprises all Type B and Type C species. The molecular phylogeny classification in our study well support the karyotype classification. The better consistency between molecular phylogenetic and karyotype indicate that karyotype may be the significant phylogenetic signal in the Melilotus genus.
However, the phylogeography of Melilotus species and populations which rely on their distributions around the world remains largely unknown. Genetic diversity analysis within Melilotus genus is on going in our group with SSR markers, which will also provide a supplement conclusion of the interspecific relationship.
Acknowledgments
This research was supported by grants from Ministry of Agriculture, China (20120304205), Ministry of S&T, China (31101759), and the Department of Agriculture and Animal Husbandry, Gansu Province (GNSW-2011-16).
The authors thank NPGS for providing Melilotus seeds, Xue Gao and Peiling Chen for support in cultivating and sampling Melilotus species, and Dong Luo and Qiang Zhou for help in data processing. We also thank many other students for their assistance with our study.
Data Availability
All relevant data are within the paper.
Funding Statement
This research was supported by grants from Ministry of Agriculture, China (20120304205), Ministry of S&T, China (31101759), the Department of Agriculture and Animal Husbandry, Gansu province (GNSW-2011-16).
References
- 1. Bowman G, Shirley C, Cramer C (1998) Managing cover crops profitably. Sustainable Agriculture Network Handbook Series 15:100–104. [Google Scholar]
- 2. Aboel-Atta AI (2009) Isozymes, RAPD and ISSR variation in Melilotus indica (L.) All. and M. siculus(Turra) BG Jacks.(Leguminosae). Int J Plant Sci 2: 113–118. [Google Scholar]
- 3. Brenner DM (1970) Sweetclover descriptors for GRIN. Newsletter 141: 51–55. [Google Scholar]
- 4. Stevenson TM, Kirk LE (1935) Studies in interspecific crossing with Melilotus, and intergeneric crossing with Melilotus, Medicago and Trigonella . Sci. Agric. 15: 580–589. [Google Scholar]
- 5. Clarke AE (1935) Inheritance of annual habit and mode of pollination in An annual white sweet clover. J. Amer. Soc. Agron 27: 492–496. [Google Scholar]
- 6. Conn JS, Werdin-Pfisterer NR, Beattie KL, Densmore RV (2011) Ecology of Invasive Melilotus albus on Alaskan Glacial River Floodplains. Arct Antarct Alpres 43: 343–354. [Google Scholar]
- 7. Van Ripre LC, Larson DL (2009) Role of invasive Melilotus officinalis in two native plant communities. Plant Ecol 200:129–139. [Google Scholar]
- 8. Reinhardt AC, Galatowitsch SM (2008). The transition from invasive species control to native species promotion and its dependence on seed density thresholds. Appl Veg Sci 11:131–138. [Google Scholar]
- 9. Rogers ME, Colmer TD, Frost K, Henry D, Cornwall D, Hulm, et al. (2008) Diversity in the genus Melilotus for tolerance to salinity and water logging. Plant Soil 304: 89–101. [Google Scholar]
- 10. Sherif EA (2009) Melilotus indicus (L.) All., a salt-tolerant wild leguminous herb with high potential for use as a forage crop in salt-affected soils. Flora-Journal-Elsevier 204: 737–746. [Google Scholar]
- 11. Cong JM, Chen FQ, Sun CL (2012) Study on comprehensive development of Metlilotus suaverolens L. Journal of Anhui Agricultural Sciences 5: 2962–2963. [Google Scholar]
- 12. Stickler F, Johnson I (1959) Dry matter and nitrogen production of legumes and legume associations in the fall of the seeding year. Agron J 51: 135–137. [Google Scholar]
- 13. Moussavi SM (2001) Species of Melilotus in Iran (key to the species, descriptions and their distributions). Rostaniha 2: 57–73. [Google Scholar]
- 14. Klebesadel LJ (1954) Morphological, physiological, and winter hardiness comparisons among latitudinal ecotypes of biennial sweetclover (Melilotus species) in subarctic Alaska School of Agriculture and Land Resources Management, Agricultural and Forestry Experiment Station. [Google Scholar]
- 15. Isely D (1969) Keys to sweetclovers (Melilotus). Proc Iowa Acad Sci 61: 119–131. [Google Scholar]
- 16. Stevenson G. A. An agronomic and taxonomic review of the genus Melilotus Mill. Can J of Plant Sci 49: 1–20. [Google Scholar]
- 17. Fordyce JA, Forister ML, Nice CC, Burns JM, Shapiro AM (2008) Patterns of genetic variation between the checkered skippers Pyrgus communis and Pyrgus albescens (Lepidoptera: Hesperiidae). Ann Entomol Soc Am 101: 794–800. [Google Scholar]
- 18.Hao JS, Sun QQ, Zhao HB, Sun XY, Gai YH, Yang Q (2012) The complete mitochondrial genome of Ctenoptilum vasava (Lepidoptera: Hesperiidae: Pyrginae) and its phylogenetic implication. Comp Funct Genom [DOI] [PMC free article] [PubMed]
- 19. Doyle JJ (1992) Gene trees and species trees: Molecular systematics as one-character taxonomy. Syst Bot 17: 144–163. [Google Scholar]
- 20. Käss E, Wink M (1997) Phylogenetic relationships in the Papilionoideae (family Leguminosae) based on nucleotide sequences of cpDNA (rbcL) and ncDNA (ITS 1 and 2). Mol Phylogenet Evol 8: 65–88. [DOI] [PubMed] [Google Scholar]
- 21. Arnheim N, Krystal M, Schmickel R, Wilson G, Ryder O, Zimmer E (1980) Molecular evidence for genetic exchanges among ribosomal genes on non-homologous chromosomes in man and apes. Proc Natl Acad Sci 77: 7323–7327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Zimmer EA, Martin SL, Beverley SM, Kan YW, Wilson AC (1991) Rapid duplication and loss of genes coding for the a chains of hemoglobin. Proc Natl Acad Sci 77: 2158–2162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hillis DM, Dixon MT (1991) Ribosomal DNA: Molecular evolution and phylogenetic inference. Quart Rev Biol 66: 411–453. [DOI] [PubMed] [Google Scholar]
- 24. Lindner DL, Banik MT (2011) Intragenomic variation in the ITS rDNA region obscures phylogenetic relationships and inflates estimates of operational taxonomic units in genus Laetiporus . Mycologia 103: 731–740. 10.3852/10-331 [DOI] [PubMed] [Google Scholar]
- 25. Scarcelli N, Barnaud A, Eiserhardt W, Treier UA, Seveno M, Anfray A, et al. (2011) A set of 100 chloroplast DNA primer pairs to study population genetics and phylogeny in monocotyledons. Plos One 6, e19954 10.1371/journal.pone.0019954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hamby RK, Zimmer EA (1992) Ribosomal RNA as a phylogenetic tool in plant systematics. Molecular Systematics of Plants 12: 50–91. [Google Scholar]
- 27. Zhang W, Kan SL, Zhao H, Li ZY, Wang XQ (2014) Molecular phylogeny of Tribe Theeae (Theaceae s.s.) and its implications for generic delimitation. Plos One 9: e98133 10.1371/journal.pone.0098133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Cardoso D, Queiroz LP, Lima HC, Suganuma E, Berg CV, Lavin M (2013) A molecular phylogeny of the vataireoid legumes underscores floral evolvability that is general to many early branching papilionoid lineages. Am J Bot 100: 403–421. 10.3732/ajb.1200276 [DOI] [PubMed] [Google Scholar]
- 29. Lee CS, Yeau SH, Lee NS (2012) Taxonomic status and genetic variation of Korean endemic plants, Eranthis byunsanensis and Eranthis pungdoensis (Ranunculaceae) based on nrDNA ITS and cpDNA sequences. J Plant Biol 55: 165–177. [Google Scholar]
- 30. Jenks AA, Walker JB, Kim SC (2013) Phylogeny of new world Salvia subgenus Calosphace (Lamiaceae) based on cpDNA (psbA-trnH) and nrDNA (ITS) sequence data. J Plant Res 126: 483–496. 10.1007/s10265-012-0543-1 [DOI] [PubMed] [Google Scholar]
- 31. Shan Z (2011) Improved SDS method for general plant genomic DNA extraction. Guangdong Agricultural Sciences 8: 113–115. [Google Scholar]
- 32. Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA (2007) Clustal W and Clustal X version 2.0. Bioinformatics 23: 2947–2948. [DOI] [PubMed] [Google Scholar]
- 33. Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S (2011) MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol and Evol 28: 2731–2739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Huelsenbeck JP, Ronquist F (2001) MRBAYES: Bayesian inference of phylogenetic trees. Bioinformatics 17: 754–755. [DOI] [PubMed] [Google Scholar]
- 35. Posada D, Crandall KA (1998) Modeltest: testing the model of DNA substitution. Bioinformatics 14: 817–818. [DOI] [PubMed] [Google Scholar]
- 36.Nuin P (2005) MrMTgui 1.0 (version 1.6). Program distributed by the author.
- 37. Posada D, Buckley T (2004) Model selection and model averaging in phylogenetics: advantages of Akaike Information Criterion and Bayesian approaches over likelihood ratio tests. Syst Biol 53: 793–808. [DOI] [PubMed] [Google Scholar]
- 38. Liu JQ, Wang TJ, Wang AL, Ohba H, Abbott RJ (2006) Radiation and diversification within the Ligularia–Cremanthodium–Parasenecio complex (Asteraceae) triggered by uplift of the Qinghai-Tibetan Plateau. Mol Phylogenet Evol 38: 31–49. [DOI] [PubMed] [Google Scholar]
- 39. Clevinger JA, Panero JL (2000) Phylogenetic analysis of Silphium and subtribe Engelmanniinae (Asteraceae: Heliantheae) based on ITS and ETS sequence data. Am J Bot 87: 565–572. [PubMed] [Google Scholar]
- 40. Linder CR, Goertzen LR, Heuval BV, Francisco-Ortega J, Jansen RK (2000) The complete external transcribed spacer of 18S-26S rDNA: amplification and phylogenetic utility at low taxonomic levels in Asteraceae and closely allied families. Mol Phylogenet Evo 14: 285–303. [DOI] [PubMed] [Google Scholar]
- 41. Fior S, Karis PO, Casazza G, Minuto L, Sala F (2006) Molecular phylogeny of the Caryophyllaceae (Caryophyllales) inferred from chloroplast matK and nuclear rDNA ITS sequences. Am J Bo. 93: 399–411. [DOI] [PubMed] [Google Scholar]
- 42. Mitsui Y, Chen ST, Zhou ZK, Peng CI, Deng YF, Setoguchi H (2008) Phylogeny and biogeography of the genus Ainsliaea (Asteraceae) in the Sino-Japanese Region based on nuclear rDNA and cpDNA sequence data. Ann Bot-london 101: 111–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Degnan JH, Rosenberg NA (2009) Gene tree discordance, phylogenetic inference and the multispecies coalescent. Trends Ecol Evol 24: 332–340. 10.1016/j.tree.2009.01.009 [DOI] [PubMed] [Google Scholar]
- 44. Maddison WP (1997) Gene trees in species trees. Syst Biol 46: 523–536. [Google Scholar]
- 45. Maddison WP, Knowles LL (2006) Inferring phylogeny despite incomplete lineage sorting. Syst Biol 55: 21–30. [DOI] [PubMed] [Google Scholar]
- 46. Clarke AE (1934) The number and morphology of chromosomes in the genus Melilotus University of California Press. [Google Scholar]
- 47. Kita F (1965) Studies on the genus Melilotus (sweetclover) with special reference to interrelationships among species from a cytological point of view. J Fac Agr 54: 23–122. [Google Scholar]
- 48. Dong WP, Cheng T, Li CH, Xu C, Long P, Chen CM, et al. (2013) Discriminating plants using the DNA barcode rbcLb: an appraisal based on a large data set. Mol Ecol Resour 14: 336–343. 10.1111/1755-0998.12185 [DOI] [PubMed] [Google Scholar]
- 49. Yu J, Xue JH, Zhou SL (2011) New universal matK primers for DNA barcoding angiosperms. J Syst Evol 49: 176–181. [Google Scholar]
- 50. Taberlet P, Gielly L, Pautou G, Bouvet J (1991) Universal primers for amplification of three noncoding regions of chloroplast DNA. Plant Mol Biol 17: 1105–1109. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All relevant data are within the paper.