

Abstract
An efficient route to substituted N-fused aromatic heterocycles, including indolizines, imidazo[1,2-a]pyridines, and imidazo[1,5-a]pyridines from azole aldehydes is reported. A three-component Wittig olefination of the aldehydes with fumaronitrile and triethylphosphine affords predominantly E-alkenes that undergo rapid cyclization upon in situ treatment with catalytic amounts of mild base. Substituent control of the 1-, 2-, and 3-positions of the resulting heteroaromatic bicycles is shown. Alternatively, the isolable E-alkene product from pyrrole-2-carboxaldehyde, under dianionic conditions, undergoes selective alkylation with various electrophiles, followed by in situ annulation to indolizines additionally substituted at the 6-position.
N-fused aromatic heterocycles, such as the indolizine and imidazopyridine classes, are attractive synthetic targets owing to their pharmacological potential and unique electronic properties. The wide array of biological effects elicited by members of theseheteroaromatic classes has been well documented, including antimicrobial,1 anti-viral,2 anti-inflammatory,3 anti-tubercular,4 anticancer,5 anti-nociceptive,6 anti-protozoal,7 and hypnoselective8 activities. In addition, these scaffolds, notably the imidazopyridines, have found utility as N-heterocyclic carbene (NHC) ligands in complexes with promising catalytic reactivity9 or as inks and dyes. As a consequence of their diverse uses, several synthetic methods have been developed for their preparation. Traditional syntheses employ substituted pyridines to annulate the 5-membered portion of the bicyclic system.10–11 For example, the most common indolizine syntheses involove the euphonious Tschitschibabin reaction or its conceptual variants, and proceed by reaction of 2-alkylpyridines with α-halo ketones.10a More recently, strategies have been developed to access these bicycles from their 5-membered heterocyclic precursors.12 Lee and Kim, for example, demonstrated the aldol-type cyclization of N-substituted 2-acetylpyrroles under alkaline conditions to furnish 6,8-disubstituted indolizines.12c
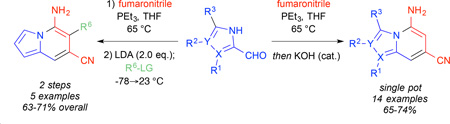
Despite recent developments to N-fused heteroaromatic bicycles, to our knowledge no routes exist that allow the direct synthesis of 5-amino-substituted indolizines. We recently reported a flexible, efficient route to highly-substituted 7-aminoindoles from pyrrole-3-carboxaldehydes.13 This route utilized a one-pot, three component Wittig olefination of the aldehydes with a trialkylphosphine and fumaronitrile to afford predominantly E-alkenes, followed by Lewis acid-mediated intramolecular Houben-Hoesch reaction to the corresponding indoles. We envisioned a mechanistically related route to obtain the 5-aminoindolizine, 5-aminoimidazo[1,2-a]pyridine, and 5-aminoimidazo[1,5-a]pyridine classes, as shown in Scheme 1. In this case, rather than nucleophilic attack of the pyrrolic α-carbon on a Lewis acid-activated nitrile, we reasoned that the relative acidity of the azolic N-H proton would allow for facile deprotonation to a nucleophilic azolide anion followed by cycloaromatization. Alternatively, a second deprotonation could furnish an allylic dianion that could react selectively with electrophiles prior to cyclization to allow for substituent control at the 6-position.
Scheme 1.

Proposed route to N-fused heterocycles.
Extending the conditions established in our laboratory,14 Wittig olefination of commercially-available pyrrole-2-carboxaldehyde afforded alkene 2a in excellent yield and good diastereoselectivity as shown in Table 1. To establish an optimal annulation procedure, the E-isomer, purified by recrystallization from the diastereomeric mixture, was then subjected to a variety of conditions. Under the Lewis acidic conditions employed previously for the Houben-Hoesch annulation to 7-aminoindoles (BF3•OEt2), no desired cyclization was observed. Similarly, treatment with LDA at −78 °C resulted in the isolation of only starting material. At 0 °C, however, treatment with stoichiometric amounts of the strong bases LDA or LiHMDS effected rapid and complete conversion to the indolizine 3a. The weaker bases KOH and K2CO3 also exhibited the desired cyclization although higher temperatures were required. For KOH, complete conversion to the indolizine was observed at room temperature after 15 minutes. K2CO3 was far less efficient and required reflux temperature, longer reaction time, and afforded lower yields. We hypothesized that the initial cyclized product, an anilide anion, could also act as a base itself thereby necessitating only a catalytic amount of base to initiate the cycle. Indeed, treatment with catalytic amounts of LDA at 0 °C or KOH at room temperature effected complete conversion to the cyclized product. Due to the milder conditions, short reaction time, low cost, and ease of workup, we proceeded with catalytic KOH as the optimal method for annulation.
Table 1.
Optimization of Annulation of Alkene 2a to Indolizine 3a.
 | ||||||
|---|---|---|---|---|---|---|
| entry | Reagent | X (eq.) |
temp. (°C) |
time (h) |
yield (%)b |
3a |
| 1 | BF3•OEt2a | 2.5 | 90 | 12 | 0 | |
| 2 | LDA | 1.0 | −78 | 1 | 0 | |
| 3 | LDA | 1.0 | 0 | 0.5 | 98 | |
| 4 | LDA | 0.4 | 0 | 0.5 | 97 | |
| 5 | LiHMDS | 1.0 | 0 | 0.5 | 97 | |
| 6 | KOH | 4.0 | 23 | 0.25 | 97 | |
| 7 | KOH | 0.4 | 23 | 0.25 | 96 | |
| 8 | K2CO3 | 4.0 | 70 | 1 | 86 | |
Reaction run in 1,2-dichloroethane rather than THF.
Isolated yield
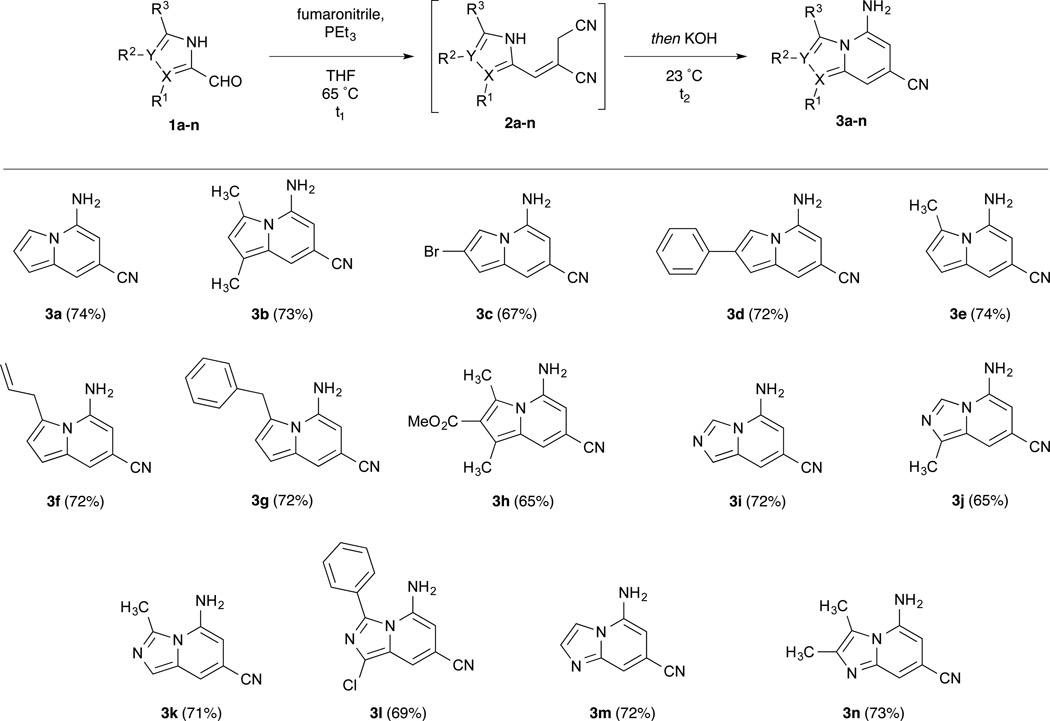
The mild and efficient cyclization conditions encouraged us to combine the Wittig olefination and cyclization steps into a tandem, one-pot synthesis of highly-substituted N-fused heteroaromatic bicycles from azole aldehydes as shown in Table 2. The unadorned pyrrole-2-carboxaldehyde 1a was reacted as before with PEt3 and fumaronitrile at 65 °C, but for the one-pot procedure, the E/Z mixture was cooled to room temperature and catalytic KOH was added. The E-isomer was completely converted to 5-amino-7-cyanoindolizine within 30 minutes while the Z-isomer was left unreacted. The 74% yield for this reaction approaches the stoichiometric conversion of the 3:1 E/Z mixture. To demonstrate the scope of the olefination/cyclization sequence, a variety of substituted pyrrole and imidazole aldehydes were employed.14 Under the same conditions as before, aldehydes 1b–n underwent alkene formation and cyclization to indolizines, imidazo[1,2-a]pyridines, and imidazo[1,5-a]pyridines in moderate to good yields. Like the unsubstituted pyrrole-2-aldehyde, yields approached the stoichiometric limit for the E-isomer present in the diastereomeric mixture of the crude Wittig reaction. The imidazole aldehydes exhibited much faster reaction rates for the Wittig reaction but much slower rates of annulation (see Supporting Information for reaction times). The additional nitrogen on the imidazole activates the aldehyde for attack by the phosphonium ylide but the increased acidity of the imidazole stabilizes the azolide anion, thereby decreasing its nucleophilicity and slowing cyclization.
Table 2.
Scope of one-pot annulation of azole aldehydes.
 |
See Supporting Information for reaction times.
The conditions were tolerant of a range of substituents including halide, ester, and aryl functionalities, as well as alkyl substituents at each of the possible positions on the azole aldehydes. Deactivated azole aldehydes, for example those containing halogen and ester functionalities, showed a modest decline in yield. Aldehydes with substituents at the α-position (e.g. 1b,h,l) showed a moderate decrease in cyclization rate, potentially due to the A1,3 strain present between the C-3 and C-5 substituents in the cyclized products.
With no cyclization occurring at −78 °C, we hypothesized that the dianion of alkene 2a formed at low temperatures could react with electrophiles regioselectively at the allylic position. The resulting alkylated monoanions would then cyclize in situ upon warming. This method would allow for selective installation of C6 substituents in the indolizine products. A set of electrophiles was chosen and the results are summarized in Table 3. Selective addition of alkyl-, allyl-, propargyl-, benzyl-, and α-keto halides was achieved at −78 °C. After 1 h of alkylation, the acetone/CO2 bath was removed effecting rapid cyclization to the 5,6,7-trisubstituted indolizines 4a–e as the reaction mixture warmed to room temperature. These tandem alkylation/cyclization reactions exhibited excellent yields. The activated electrophiles afforded slightly higher yields than the non-activated MeI. Notably, the reaction with ethyl bromoacetate effected selective alkylation of the dianion, annulation to the indolizine, and further condensation to the ɣ-lactam 4e in a single pot, highlighting the efficiency and synthetic potential of this method.
Table 3.
Tandem, One-Pot Alkylation/Cyclization Sequence to 5,6,7-Trisubstituted Indolizines 4a–e.
 | ||||
|---|---|---|---|---|
| entry | Electrophile | Product | R6 | yield (%)a |
| 1 | MeI | 4a | -CH3 | 87 |
| 2 | H2C=CHCH2Br | 4b | -CH2CH=CH2 | 98 |
| 3 | H2C≡CHCH2Br | 4c | -CH2CH≡CH2 | 94 |
| 4 | PhCH2Br | 4d | -CH2Ph | 98 |
| 5 | BrCH2CO2Et | 4e | 90 | |
Isolated yield.
In conclusion, we have developed a one-pot, olefination/cyclization sequence for the synthesis of highly-substituted indolizines, imidazo[1,2-a]pyridines, and imidazo[1,5-a]pyridines from pyrrole-2-, imidazole-2-, and imidazole-4-carboxaldehydes, respectively. Wittig olefination of the aldehydes with an ylide formed in situ from fumaronitrile and triethylphosphine affords E-enriched alkenes which undergo cycloaromatization when treated with catalytic and mild bases, allowing for control of the C-1, C-2, and C-3 substituents. Alternatively, the isolable E-isomers undergo selective dianionic alkylation at −78 °C followed by spontaneous cyclization upon warming to room temperature, allowing for C-6 substituent control. The efficiency and tailorability of this method simplifies the generation of large arrays of functionalized heteroaromatic compounds. Isosteres and nitrogen-rich homologs of indole present geometrically definable hydrogen bond donors and acceptors that greatly expand the usefulness of this privileged nucleus in medicinal chemistry. The substituents and substitution patterns readily achievable by this approach enrich and complement those available by current methods.15
Supplementary Material
ACKNOWLEDGMENT
We thank Dr. I. P. Mortimer for mass spectral analysis.
Funding Sources
This work was supported by the National Institutes of Health research grant ES001670.
Footnotes
ASSOCIATED CONTENT
Detailed synthetic procedures and characterization data can be found in the Supplemental Information. This material is available free of charge via the Internet at http://pubs.acs.org.
Notes
The authors declare no competing financial interest.
REFERENCES
- 1.a) Hazra A, Mondal S, Maity A, Naskar S, Saha P, Paira R, Sahu KB, Paira P, Ghosh S, Sinha C, Samanta A, Banerjee S, Mondal NB. Eur. J. Med. Chem. 2011;46:2132–2140. doi: 10.1016/j.ejmech.2011.02.066. [DOI] [PubMed] [Google Scholar]; b) Darwish ES. Molecules. 2008;13:1066–1078. doi: 10.3390/molecules13051066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.a) Reddy MV, Rao MR, Rhodes D, Hansen MS, Rubins K, Bushman FD, Venkateswarlu Y, Faulkner DJ. J. Med. Chem. 1999;42:1901. doi: 10.1021/jm9806650. [DOI] [PubMed] [Google Scholar]; b) Facompré M, Tardy C, Bal-Mahieu C, Colson P, Perez C, Manzanares I, Cuevas C, Bailly C. Cancer research. 2003;63:7392–7399. [PubMed] [Google Scholar]
- 3.a) Hagishita S, Yamada M, Shirahase K, Okada T, Murakami Y, Ito Y, Matsuura T, Wada M, Kato T, Ueno M, Chikazawa Y, Yamada K, Ono T, Teshirogi I, Ohtani M. J. Med. Chem. 1996;39:3636–3658. doi: 10.1021/jm960395q. [DOI] [PubMed] [Google Scholar]; b) Yokota Y, Hanasaki K, Ono T, Nakazato H, Kobayashi T, Arita H. Biochim. Biophys. Acta. 1999;1438:213–222. doi: 10.1016/s1388-1981(99)00053-0. [DOI] [PubMed] [Google Scholar]
- 4.a) Dannhardt G, Meindl W, Gussmann S, Ajili S, Kappe T. Eur. J. Med. Chem. 1987;22:505–510. [Google Scholar]; b) Gundersen LL, Negussie AH, Rise F, Ostby OB. Arch. Pharm. Pharm. Med. Chem. 2003;336:191–195. doi: 10.1002/ardp.200390019. [DOI] [PubMed] [Google Scholar]
- 5.a) Cheng Y, An LK, Wu N, Wang XD, Bu XZ, Huang ZS, Gu LQ. Bioorg. Med. Chem. 2008;16:4617–4625. doi: 10.1016/j.bmc.2008.02.036. [DOI] [PubMed] [Google Scholar]; b) James DA, Koya K, Li H, Liang G, Xia Z, Ying W, Wu Y, Sun L. Bioorg. Med. Chem. Lett. 2008;18:1784–1787. doi: 10.1016/j.bmcl.2008.02.029. [DOI] [PubMed] [Google Scholar]; c) Shen YM, Lv PC, Chen W, Liu PG, Zhang MZ, Zhu HL. Eur. J. Med. Chem. 2010;45:3184–3190. doi: 10.1016/j.ejmech.2010.02.056. [DOI] [PubMed] [Google Scholar]; d) Bloch WM, Derwent-Smith SM, Issa F, Morris JC, Rendina LM, Sumby CJ. Tetrahedron. 2011;67:9368–9375. [Google Scholar]
- 6.Vaught JL, Carson JR, Carmosin RJ, Blum PS, Persico FJ, Hageman WE, Shank RP, Raffa RB. J. Pharmacol. Exp. Ther. 1990;255:1–10. [PubMed] [Google Scholar]
- 7.Ismail MA, Arafa RK, Wenzler T, Brun R, Tanious FA, Wilson WD, Boykin DW. Bioorg. Med. Chem. 2008;16:683–691. doi: 10.1016/j.bmc.2007.10.042. [DOI] [PubMed] [Google Scholar]
- 8.Depoortere H, Zivkovic B, Lloyd KG, Sanger DJ, Perrault G, Langer SZ, Bartholini G. J. Pharmacol. Exp. Ther. 1986;237:649–658. [PubMed] [Google Scholar]
- 9.a) Alcarazo M, Roseblade SJ, Cowley AR, Fernández R, Brown JM, Lassaletta JM. J. Am. Chem. Soc. 2005;127:3290–3291. doi: 10.1021/ja0423769. [DOI] [PubMed] [Google Scholar]; Klingele J, Kaase D, Hilgert J, Steinfeld G, Klingele MH, Lach J. Dalton Trans. 2010;39:4495. doi: 10.1039/b925107c. [DOI] [PubMed] [Google Scholar]
- 10.For pyridine-based indolizine syntheses see: Tschitschibabin AE. Ber. Dtsch. Chem. Ges. 1927;60:1607. Behnisch R. Houben-Weyl, Methoden der Organischen Chemie. E6b1. Thieme: Stuttgart: 1994. pp. 323–450. Uchida T, Matsumoto K. Synthesis. 1976:209. Katritzky AR, Qui G, Yang B, He H-Y. J. Org. Chem. 1999;64:7618. Kostik EI, Abiko A, Oku A. J. Org. Chem. 2001;66:2618–2623. doi: 10.1021/jo0011639. Smith CR, Bunnelle EM, Rhodes AJ, Sarpong R. Org. Lett. 2007;9:1169–1171. doi: 10.1021/ol0701971. Basavaiah D, Devendar B, Lenin D, Satyanarayana T. Synlett. 2009:411–416.
- 11.For pyridine-based imidazopyridine syntheses see: Barun O, Ila H, Junjappa H, Singh OM. J. Org. Chem. 2000;65:1583–1587. doi: 10.1021/jo9916129. Loones KTJ, Maes BUW, Meyers C, Deruytter J. J. Org. Chem. 2006;71:260–264. doi: 10.1021/jo052091u. Chernyak N, Gevorgyan V. Angew. Chem. Int. Ed. 2010;49:2743–2746. doi: 10.1002/anie.200907291. Seregin IV, Schammel AW, Gevorgyan V. Org. Lett. 2007;9:3433–3436. doi: 10.1021/ol701464j. Katritzky AR, Xu Y-J, Tu H. J. Org. Chem. 2003;68:4935–4937. doi: 10.1021/jo026797p. Masquelin T, Bui H, Brickley B, Stephenson G, Schwerkoske J, Hulme C. Tetrahedron Letters. 2006;47:2989–2991.
- 12.For syntheses originating from 5-membered ring see: Kim M, Vedejs E. J. Org. Chem. 2004;69:6945–6948. doi: 10.1021/jo040191e. Zhu H, Stöckigt J, Yu Y, Zou H. Org. Lett. 2011;13:2792–2794. doi: 10.1021/ol200883w. Lee JH, Kim I. J. Org. Chem. 2013;78:1283–1288. doi: 10.1021/jo302590a. Kucukdisli M, Opatz T. J. Org. Chem. 2013;78:6670–6676. doi: 10.1021/jo400992n. Galons H, Bergerat I, Combet Farnoux C, Miocque M. Synthesis. 1982:1103. Knölker HJ, Hitzemann R, Boese R. Chem. Ber. 1990;123:327–339. Virieux D, Guillouzic A-F, Cristau H-J. Tetrahedron. 2006;62:3710–3720.
- 13.Outlaw VK, Townsend CA. Org. Lett. 2014;16:6334–6337. doi: 10.1021/ol503078h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.For preparation of known substituted pyrrole-2-aldehydes shown in SI see: Krayer M, Balasubramanian T, Ruzié C, Ptaszek M, Cramer DL, Taniguchi M, Lindsey JS. J Porphyr Phthalocyanines. 2009;13:1098–1110. Bergauer M, Gmeiner P. Synthesis. 2002;2:0274–0278. doi: 10.1016/s0960-894x(02)00316-5. Muchowski JM, Hess P. Tetrahedron Letters. 1988;29:777–780.
- 15.Singh GS, Mmatli EE. European Journal of Medicinal Chemistry. 2011;46:5237–5257. doi: 10.1016/j.ejmech.2011.08.042. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.