

Abstract
Certain human class I histocompatibility-linked leukocyte antigen (HLA)/killer cell immunoglobulin-like receptor (KIR) genotypic combinations confer more favourable prognoses upon exposure to human immunodeficiency virus (HIV). These combinations influence natural killer (NK) cell function, thereby implicating NK cells in protection from HIV infection or disease progression. Since CD8+ T cells restrict HIV replication, depend upon HLA class I antigen presentation and can also express KIR molecules, we investigated how these HLA/KIR combinations relate to the phenotype and function of CD8+ T cells from uninfected controls and individuals with chronic HIV infection. CD8+ T cells from KIR3DL1 and KIR3DS1 homozygous individuals, and expressing the corresponding KIR, were enumerated and phenotyped for CD127, CD57 and CD45RA expression. Ex vivo and in vitro responsiveness to antigen-specific and polyclonal stimulation was compared between KIR-expressing and non-expressing CD8+ T cells by interferon-γ production. There were higher numbers and fractions of KIR3DL1-expressing CD8+ T cells in HIV-infected individuals independent of HLA-Bw4 co-expression, whereas expansion of KIR3DS1-expressing CD8+ T cells reflected HLA-Bw4*80I co-expression. KIR3DL1+ and S1+ CD8+ T cells were predominantly CD127−CD57+CD45RA+. KIR3DL1-expressing CD8+ T cells were insensitive to ex vivo stimulation with peptides from HIV or common viruses, but responded to anti-CD3 and recovered responsiveness to common viruses in vitro. Ex vivo non-responsiveness of KIR3DL1-expressing CD8+ T cells was also independent of HLA-Bw4. KIR3DS1-expressing T cells responded normally to ex vivo antigenic stimulation, illustrating functional superiority over KIR3DL1+ CD8+ T cells.
Keywords: KIR3DL1, KIR3DS1, Bw4, CD8+ T cell, HIV
Introduction
Killer cell immunoglobulin-like receptors (KIR) are polymorphic transmembrane signaling proteins expressed on natural killer (NK) cells and some T cells. There are 15 different KIR proteins named for having either two (KIR2D) or three (KIR3D) extracellular domains with either long (L) or short (S) cytoplasmic tails1. Inhibitory KIR antagonize activation-related phosphorylation events by recruiting SHP-1 and SHP-2 phosphatases via intracellular tyrosine inhibitory motifs in their long cytoplasmic tails. Activating KIR, except KIR2DL4, have short cytoplasmic tails containing a basic amino that interacts with adaptor proteins such as DAP12 to recruit protein kinases and promote activation-related phosphorylation events. Specific extracellular ligands for KIR molecules are found within the highly diverse set of class one human histocompatibility linked leukocyte antigen (HLA) molecules that present peptides to CD8+ T cells. However, rather than directing their selective recognition on highly polymorphic regions of HLA-A, B or C molecules, individual KIR focus either on relatively non-polymorphic HLA-G molecules or amino acid (aa) sequences common to roughly half of the polymorphic HLA-B or C alleles2. From the perspective of KIR restriction, this collapses the vast polymorphism of class I HLA-B and C molecules into five groups (C1, C2, Bw4*80T, Bw4*80I and Bw6). In a process termed licensing, engagement of cognate class one HLA ligands by inhibitory KIR during NK cell maturation empowers subsequent NK cell activation by stressed, transformed, infected or otherwise altered autologous cells3. Regulation of mature NK cell activity is effected by integration of positive and negative signals received through KIR and other receptors1, 4–7. Multiple allelic variants of KIR and MHC molecules engender cognate interactions across a range of affinities and since KIR and MHC genes are inherited independently, identical KIR molecules function differently in MHC-disparate hosts. This idiosyncrasy is thought to underlie associations between outcomes of microbial infection or exposure and co-expression of particular allelic forms of KIR with their ligands8–15.
In HIV infection, delayed disease progression is associated with co-expression of KIR3DS1 and class 1 HLA-Bw4 molecules incorporating isoleucine at aa 80 (Bw4*80I)12. While this association as yet has no demonstrated physical basis, the role of KIR molecules and HLA in modulating NK cell function suggests the KIR3DS1/HLA-Bw4*80I combination delivers enhanced NK cell surveillance against HIV. Although subjects with a KIR genotype including either KIR3DS1 or the HLA Bw4 epitope have higher mean NK cell activity than subjects with neither, relative protection from HIV disease progression requires the combination12, 16. In vitro experiments suggest a specific interaction between KIR3DS1 on NK cells and Bw4*80I on HIV-infected CD4+ T cells enhances NK suppression of HIV replication17. During acute HIV infection, KIR3DS1+ NK cells selectively expand and a KIR3DS1-associated aa polymorphism in HIV Gag suggests immune selective pressure exerted on HIV by NK cells18, 19. However, no functional interaction between KIR3DS1 and HLA-Bw4*80I directly explaining the association with delayed disease progression has been delineated20. In another study, modest NK inhibition of HIV replication was observed irrespective of the donor’s HLA and KIR genotype21. Thus, the impact of co-ordinate KIR/HLA genotypes on NK surveillance against HIV and the contribution of NK cells in general to delayed disease progression in HIV infection remain controversial.
Another possible explanation for the relationship between slow disease progression and co-expression of HLA Bw4*80I and KIR3DS1 is that inhibitory KIR alleles negatively affect T cell behaviour compared to activating KIR. Activation of T cells is reduced by inhibitory KIR engaging their cognate class I ligands, suggesting that in certain settings, a lack of inhibitory signaling through HLA-Bw4 engagement favours KIR-negative and KIR3DS1-expressing T cell function22–26. Since the number of CD8+ T cells expressing KIR increases in HIV infection, the antiviral cellular immune responses of some KIR3DS1 versus KIR3DL1-expressing individuals might be differentially affected by their CD8+ T cell KIR expression27. If, as suggested by epidemiological associations and the specificity of closely related KIR3DL1 alleles, HIV infection transforms HLA-Bw4*80I into the KIR3DS1 ligand, any advantage to KIR3DS1 expression might be augmented by co-expression of HLA-Bw4*80I7, 12, 28, 29. A number of class I HLA molecules expressing Bw4*80I are associated with delayed disease progression in HIV infection independent of KIR genotype, presumably through better presentation of HIV peptides to CD8+ T cells. Ancillary stimulation of HIV-specific CD8+ T cells through KIR3DS1 and Bw4*80I engagement during HIV peptide recognition might further enhance CD8+ T cell surveillance against HIV30, 31.
To test the possibility that differential behaviour related to expression of either the inhibitory KIR3DL1 or activating KIR3DS1 molecule on CD8+ T cells could potentially contribute to the association between delayed HIV disease progression and co-expression of KIR3DS1 with HLA-Bw4*80I, we compared phenotypic and functional attributes of KIR3DL1-expressing CD8+ T cells to those of KIR3DS1-expressing CD8+ T cells in both HIV-infected individuals and uninfected controls, with or without co-ordinate expression of cognate class I HLA ligands. Our data are consistent with the functional superiority of KIR3DS1-expressing over KIR3DL1-expressing CD8+ T cells being a potential factor contributing to delayed disease progression in HIV infection.
Results
KIR3DL1/S1 expression on CD8+ T cells in HIV Infection
Since KIR expression is more common on CD8+ T cells than CD4+ T cells, we compared the percentage of CD8+ T cells expressing KIR3DL1 between KIR3DL1 homozygous HIV-infected subjects and uninfected controls. A higher median percentage of CD8+ T cells from HIV-infected individuals than uninfected controls expressed KIR3DL1 (0.6, IQR 0.25–1.15 versus 0.2, IQR 0.15–0.6, p = 0.0339, fig. 1a). To test whether this expansion of KIR3DL1-expressing CD8+ T cells was related to co-expression of the class I HLA Bw4 ligand, we compared the percentage of KIR3DL1-expressing CD8+ T cells between KIR3DL1 homozygous HIV-infected subjects who were either Bw6 homozygous, Bw4*80T or Bw4*80I. There was no significant difference in the percentage of CD8+ T cells expressing KIR3DL1 between these groups (p = 0.8274, fig. 1b). These data indicate that expansion of CD8+ T cells expressing inhibitory KIR3DL1 in HIV infection is independent of co-expression of the HLA-Bw4 ligand.
Figure 1.
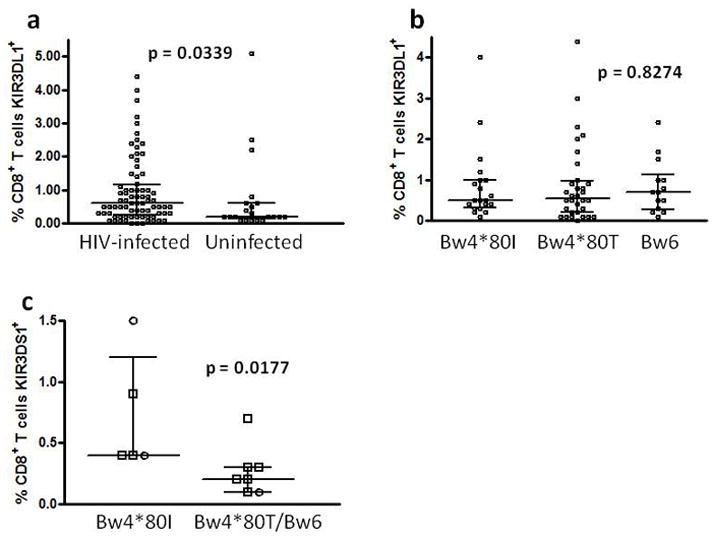
Distributions of percentage CD8+ T cells expressing KIR3DL1 or KIR3DS1 in different HIV-infected and uninfected subgroups. The percentage of CD8+ T cells expressing KIR3DL1 was compared between HIV-infected and uninfected KIR3DL1 homozygous subjects (a) and between subgroups of the HIV-infected individuals distinguished by HLA-Bw4*80I, Bw4*80T or Bw6 status (b). The percentage of CD8+ T cells expressing KIR3DS1 was compared between KIR3DS1 homozygous subjects expressing Bw4*80I or not with different symbols used to distinguish HIV-infected (□) from uninfected (○) individuals (c).
Multivariate regression analyses were performed to evaluate associations between the percentage of KIR3DL1+ T cells and plasma HIV viral load, β-2 microglobulin level, CD8+ T cell count, CD4+ T cell count, magnitude of CMV-specific CD8+ T cell response and relative CMV-specific humoral response. Univariate correlation of each potential variable with percentage of KIR3DL1+ T cells was performed and variables were included in the multivariate model if the two-sided p < 0.10. Univariate analysis demonstrated that only CD8+ T cell count (Spearman’s rho = 0.382, p = 0.001) and magnitude of CMV-specific CD8+ T cell response (Spearman’s rho = 0.348, p = 0.010) were significantly associated with the percentage of T cells expressing KIR3DL1 in the group of HIV-infected individuals. Model assumptions were tested and met and the final model containing CD8+ T cell count had a reasonable goodness of fit (R2 = 0.558). Magnitude of CMV-specific CD8+ T cell response did not explain a significant amount of the variation compared to CD8+ T cell count and was not included in the final model.
There was no significant difference in the percentage of CD8+ T cells expressing KIR3DS1 between the small number of HIV-infected and uninfected KIR3DS1 homozygotes identified. As KIR3DS1 homozygous individuals were rare, we compared the percentage of KIR3DS1-expressing CD8+ T cells between Bw4*80I expressers and others (Bw4*80T and Bw6) across HIV-infected and uninfected groups. Homozygous KIR3DS1 subjects co-expressing HLA-Bw4*80I had a significantly higher median percentage of CD8+ T cells expressing KIR3DS1 (0.4, IQR 0.4–1.2 versus 0.2, IQR 0.1–0.3, p = .0177, fig. 1c). This suggests that expansion of KIR3DS1+ CD8+ T cells in KIR3DS1 homozygous individuals relates to co-expression of the HLA-Bw4*80I epitope, which potentially serves as a ligand for KIR3DS1 under certain circumstances.
Phenotype of KIR3DL1/S1+ CD8+ T cells
Expression of KIRs on T cells is generally associated with previous activation22–26. Therefore, we evaluated expression of CD45RA, CD57 and CD127 on KIR3DL1+ and KIR3DS1+CD8+ T cells from homozygous HIV-infected and uninfected individuals to assess memory and effector cell status (fig. 2a, b). In the HIV-infected group, the median percentages (with IQR) of KIR3DL1+ CD8+ T cells that were CD45RA+, CD57+ and CD127+ respectively were 96.9 (91.7–100), 76.5 (71.5–92.6) and 25.6 (11.5–33.3) Values in the uninfected group were very similar 92.9 (68.9–98.9), 71.4 (50.0–95.5) and 31.0 (14.5–55.0) with no significant differences between them (two tailed Mann-Whitney test). This common predominant CD45RA+CD57+CD127− phenotype suggests that KIR3DL1 expression on CD8+ T cells from HIV-infected and uninfected individuals reflects similar histories of antigen exposure, activation, proliferation and differentiation. However, a small percentage of KIR3DL1+ CD8+ T cells do express CD127, a marker generally characteristic of naïve or central memory T cells. Although there were few KIR3DS1 homozygotes and a low percentage of CD8+ T cells expressing KIR3DS1 in most of them (fig. 1c), we assessed KIR3DS1+CD8+ T cell phenotypes in 7 HIV-infected individuals. The median percentages (with IQR) of KIR3DS1+ CD8+ T cells that were CD45RA+, CD57+ and CD127+ respectively were 43.5 (35.0–76.0), 93.5 (77.3–98.8) and 25.0 (19.5–50.) The same CD45RA+CD57+CD127− phenotype as expressed by the KIR3DL1+ CD8+ T cells from HIV-infected individuals was prominent, but there was a trend towards more KIR3DS1+CD8+ T cells expressing CD57 and significantly fewer KIR3DS1+ than KIR3DL1+ CD8+ T cells in HIV-infected individuals expressed CD45RA (p = 0.1164 and p = 0.0003) respectively, two-tailed Mann-Whitney test, fig. 3). Therefore, expression of inhibitory or stimulatory KIR3DL1 alleles on CD8+ T cells in HIV infected individuals is associated with a similar terminally differentiated, effector memory cell-like phenotype with some variation in CD57 and CD45RA expression.
Figure 2.
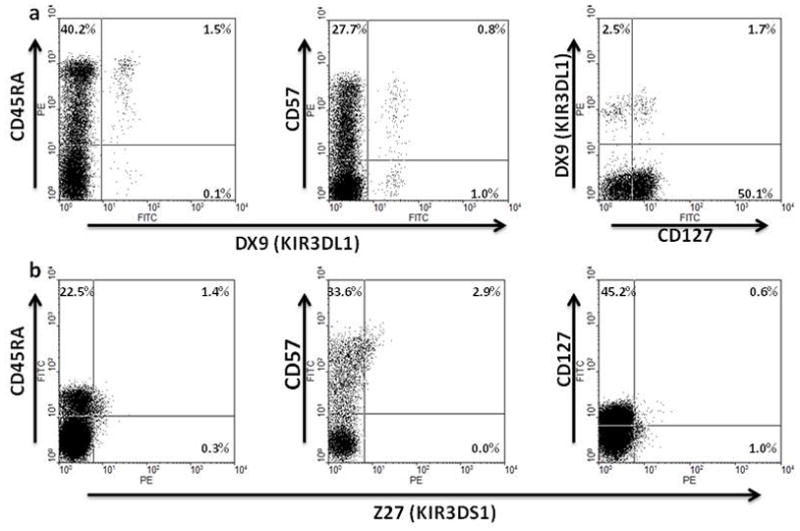
Representative examples of analysis of CD45RA, CD57 and CD127 expression on KIR3DL1+ (a) or KIR3DS1+ (b) CD8+ T cells from KIR3DL1 or KIR3DS1 homozygous subjects by flow cytometry. Gating for analysis was on CD3+CD8+ lymphocytes with DX9-FITC or DX9-PE identifying KIR3DL1+ cells and Z27-PE identifying KIR3DS1+ cells.
Figure 3.
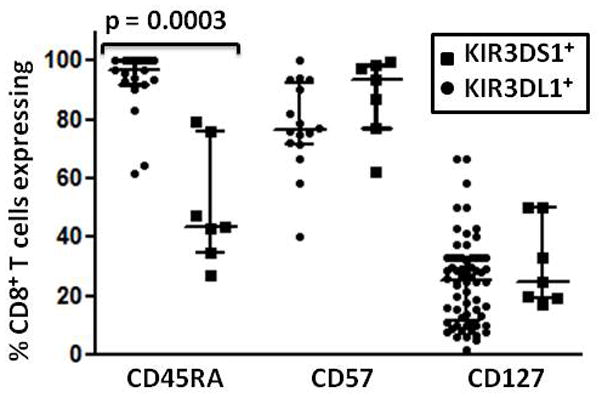
Distribution of KIR3DL1/S1+CD8+ T cell phenotypes. The percentage of KIR3DL1+ and KIR3DS1+ CD8+ T cells expressing CD45RA, CD57 and CD127 in groups of HIV-infected KIR3DL1 or KIR3DS1 homozygous individuals was compared by flow cytometry.
Ex vivo responsiveness of KIR3DL1/S1+CD8+ T cells
The responsiveness of KIR3DL1+ and KIR3DS1+ CD8+ T cells to antigen specific and anti-CD3 stimulation ex vivo was tested in the setting of different genotypic combinations. When CD8+ T cells from KIR3DL1 homozygous individuals co-expressing HLA-Bw4 were stimulated with single antigenic peptides or peptide pools from CMV or HIV, KIR3DL1+CD8+ T cells did not produce interferon-gamma (IFN-γ), despite specific IFN-γ responses in the homologous KIR3DL1−CD8+ T cell population. Data shown in figure 4 is representative of experiments carried out with 12 HIV-infected individuals and 6 uninfected controls. This suggests that in the setting of HLA-Bw4 co-expression, KIR3DL1+CD8+ T cells are anergic to antigen-specific stimulation. Anergy to antigen-specific T cell stimulation was largely independent of in vivo exposure to the HLA-Bw4 ligand of KIR3DL1 as KIR3DL1+ CD8+ T cells from 9 of 10 HLA-Bw6 homozygous individuals were also anergic to antigen-specific stimulation with peptides from common viruses or HIV (fig. 5a, b). Only 1 individual with a robust CMV-specific response had KIR3DL1+CD8+ T cell responses against CMV and the fraction of KIR3DL1+CD8+ T cells responding was less than that of the KIR3DL1−CD8+ T cell population (fig. 5c). The unresponsiveness of KIR3DL1+CD8+ T cells from HLA-Bw4 expressing individuals to stimulation through the T cell receptor complex was overcome by anti-CD3 in 10/10 individuals tested (fig. 5d), showing that even in this setting, KIR3DL1+CD8+ T cells were not completely anergic. However, ex-vivo antigen-specific responsiveness of KIR3DL1+CD8+ T cells is absent or greatly reduced greatly reduced in both HLA-Bw4-expressing and HLA-Bw6 homozygous individuals. In contrast to KIR3DL1+CD8+ T cells, KIR3DS1+CD8+ T cells from 3/3 HIV-infected individuals tested responded to antigen-specific stimulation with common viral peptides and HIV peptides by producing IFN-γ (fig. 6). Despite their similar phenotype, KIR3DS1+ CD8+ T cells are functionally superior to KIR3DL1+ CD8+ T cells in terms of ex vivo IFN-γ production following antigen-specific stimulation with HIV or other common viral peptides.
Figure 4.
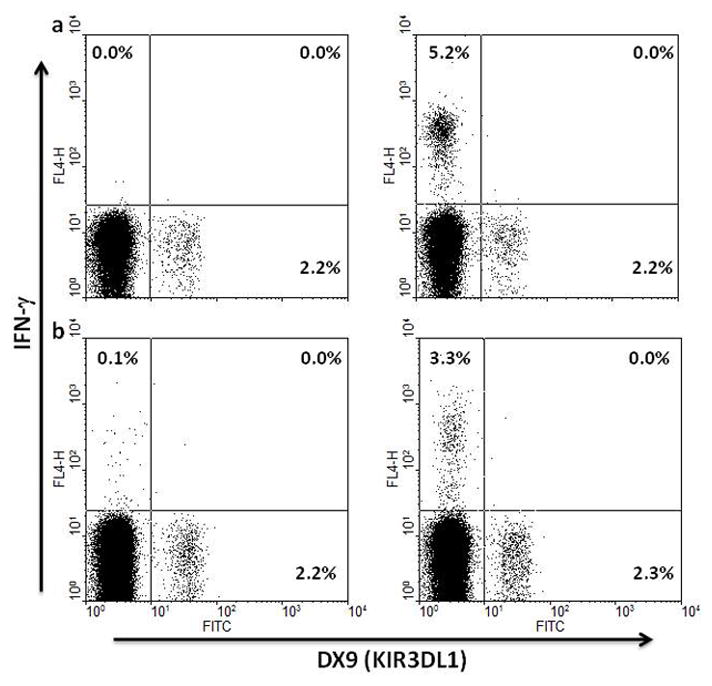
Representative ex vivo antigen-specific responses of KIR3DL1+CD8+ T cells from KIR3DL1 homozygous individuals co-expressing HLA-Bw4. Freshly-isolated PBMC from an uninfected (a) and HIV-infected individual (b) were incubated with overlapping peptides from CMV pp65 (a) or HIV Gag (b) as described in the methods section and production of intracellular IFN-γ was assessed by flow cytometry with gating on CD3+CD8+ lymphocytes. Unstimulated controls are shown in the left hand panel of each set.
Figure 5.
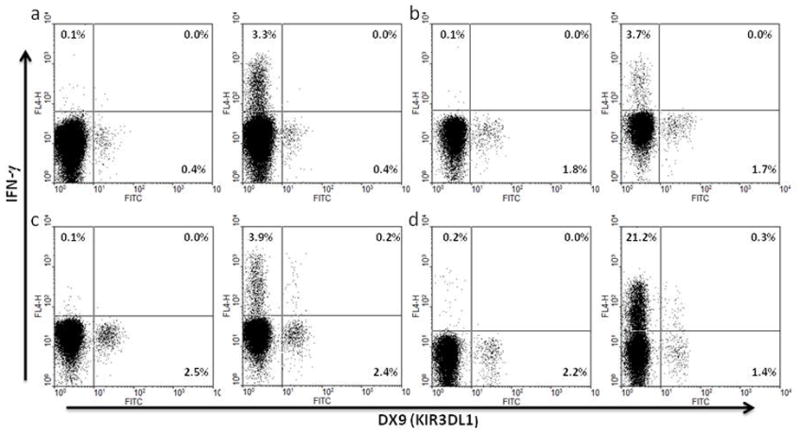
Representative ex vivo antigen-specific responses of KIR3DL1+CD8+ T cells from HLA-Bw6 subjects and representative ex vivo response to P815/anti-CD3 stimulation. Freshly-isolated PBMC from three KIR3DL1 homozygous HIV-infected individuals defined as HLA-Bw6 (a, b, c) and one uninfected KIR3DL1 homozygous individual co-expressing HLA-Bw4 were incubated with overlapping peptides from CMV pp65 (a, c) or HIV Gag (b) as described in the methods. Freshly-isolated PBMC from an uninfected KIR3DL1 homozygous individual who co-expressed HLA-Bw4 were incubated with P815 cells and anti-CD3 as described in the methods section (d). Intracellular IFN-γ production was assessed by flow cytometry with gating on CD3+CD8+ (a, b, c) or CD8+ lymphocytes (d). Unstimulated controls, including PBMC incubated with P815 cells without anti-CD3 (d) are shown in the left hand panel of each set.
Figure 6.
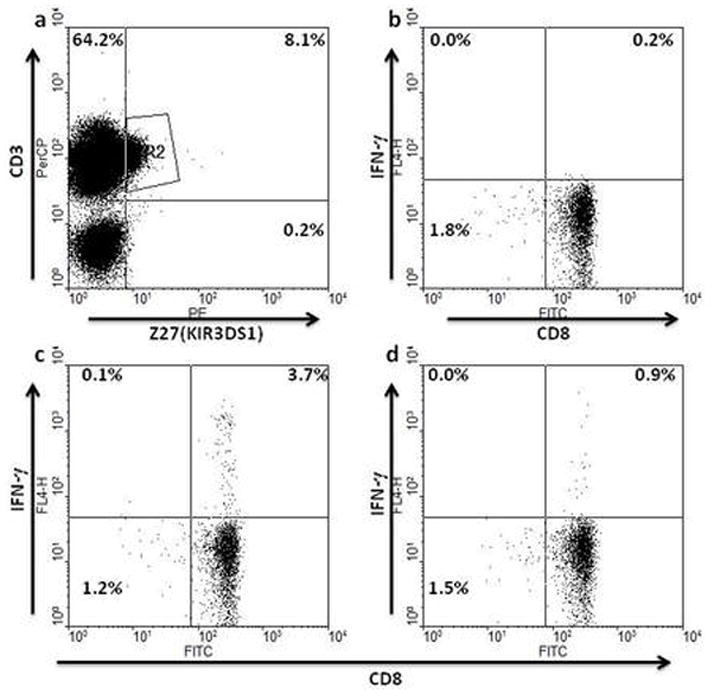
Representative ex vivo antigen-specific responses of KIR3DS1+CD8+ T cells. Gating was on CD3+KIR3DS1+ cells as in (a) with CD8+ cells producing IFN-γ shown in the upper right hand quadrants of the subsequent plots. Freshly-isolated PBMC from a KIR3DS1 homozygous individual were unstimulated (b) or incubated with overlapping peptides from CMV pp65 (c) or HIV Gag (d).
In vitro responsiveness of KIR3DL1+ CD8+ T cells
To test whether KIR3DL1+ CD8+ T cells from HLA-Bw4+ KIR3DL1 homozygous individuals could recover antigen-specific responsiveness after in vitro culture, we stimulated PBMC with specific peptides, intereleukin-7 (IL-7) and IL-2 and reassessed peptide-specific IFN-γ responses of KIR3DL1+ CD8+ T cells by secondary stimulation with peptide-pulsed autologous BLCL. Under these conditions, KIR3DL1+ CD8+ T cells responded to antigen-specific stimulation with common viral peptides (fig. 7a), but not HIV peptides (fig. 7b). Therefore, KIR3DL1+ CD8+ T cells from HLA-Bw4+ individuals are not uniformly unresponsive to antigen-specific stimulation. However, with this protocol, we could not determine whether the KIR3DL1+ CD8+T cells producing IFN-γ in response to the common viral peptides after in vitro stimulation were originally KIR3DL1+ or acquired KIR3DL1 in vitro. To address this issue, we purified KIR3DL1+ cells from freshly-isolated PBMC and expanded these cells by mitogenic stimulation with concanavalin A (Con A)/IL-2 or peptide-specific stimulation together with allogeneic feeder cells as described in the methods. Under these conditions, KIR3DL1+ cells also responded to antigen-specific stimulation with other viral peptides (fig. 7c). Results of ex vivo and in vitro stimulation experiments are summarized in Table 1.
Figure 7.
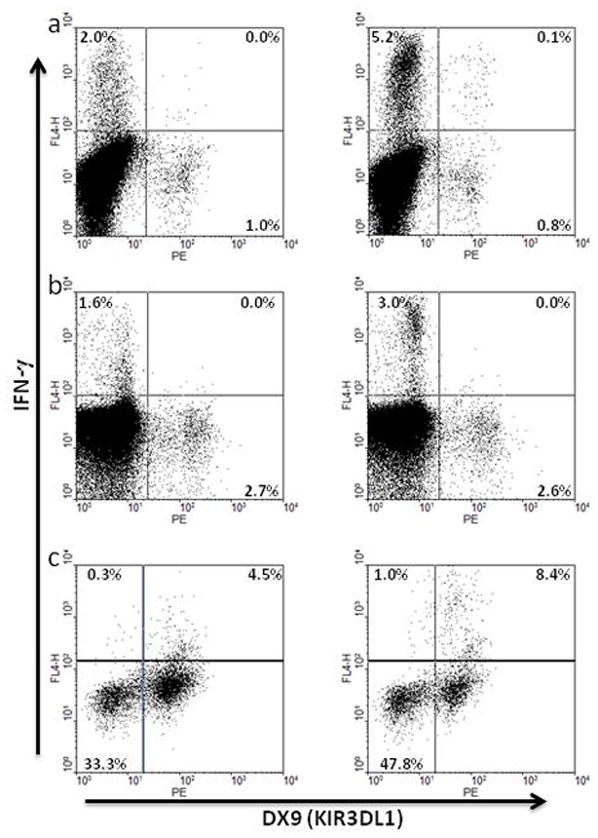
Representative secondary antigen-specific responses of KIR3DL1+CD8+ T cells following in vitro stimulation and expansion. Freshly-isolated PBMC from 2 HLA-Bw4+ HIV-infected individuals were cultured for 7 days with specific HLA-A2-restricted flu (a) or HIV-Gag peptides (b), restimulated with peptide-pulsed autologous BLCL as described in the methods section, and tested for antigen-specific IFN-γ production by intracellular flow cytometry with gating on CD3+CD8+ lymphocytes. Cells expressing KIR3DL1 were positively selected from freshly isolated PBMC of an uninfected HLA-Bw4 individual, incubated with a specific HLA-A2-restricted CMV peptide for 7 days, restimulated with peptide-pulsed autologous BLCL as described in the methods section, and tested for antigen-specific IFN-γ production by intracellular flow cytometry with gating on CD3+CD8+ lymphocytes (c).
Table 1.
KIR3DL1+ and KIR3DS1+ CD8+ T cell response rates to different stimulations.
| HIV peptides | CMV peptides | P815/anti-CD3 | |
|---|---|---|---|
| PBMC Uninfected controls | |||
| KIR3DL1++ HLA-Bw41 | 0/6 | 6/6 | |
| KIR3DL1++ HLA-Bw6 | 0/3 | 3/3 | |
| KIR3DS1++ | 1/1 | 1/1 | |
| PBMC HIV-infected subjects | |||
| KIR3DL1++ HLA-Bw4 | 0/10 | 0/6 | 4/4 |
| KIR3DL1++ HLA-Bw6 | 0/6 | 1/8 | 2/2 |
| KIR3DS1++ | 3/3 | 3/3 | 3/3 |
| In vitro restimulation2 purified KIR3DL1++ | Flu peptide | ||
| peptide + IL-2 + IL-7 | 0/4 | 2/2 | 1/1 |
| Con A + IL-2 | 0/2 | 2/2 |
CD8+ KIR3DL1+ T cell responses against HIV or CMV peptides in PBMC from control and HIV-infected individuals with different genetic backgrounds and strong overall T cell responses against the corresponding peptides were assessed by surface and intracellular flow cytometry.
KIR3DL1+ cells purified from PBMC of KIR3DL1 homozygous individuals were restimulated with either specific peptides or mitogen (Con A) plus cytokines and the reactivity of expanded cells was tested against HIV, CMV or flu peptides by surface and intracellular flow cytometry.
Discussion
Several studies suggest that the epidemiological association between certain HLA/KIR genotypic combinations and slow HIV disease progression relates to differential NK cell behaviour within that genetic background12, 17, 19, 29. Stochastic expression of some subset of available inhibitory and activating receptors on NK cells, together with allele-dependent variation in the potency of signaling through those receptors and independent inheritance of HLA ligands mediating licensing, inhibition or activation, produces an idiosyncratic NK repertoire from which certain clones can undergo selective expansion. Since a subset of T cells, primarily CD8+, also express inhibitory and activating receptors that modulate NK cell behaviour, differential T cell behavior may also occur in relation to the expression pattern of these receptors in various genetic backgrounds22, 24–27, 32. We investigated this possibility and found that the behaviour of CD8+ T cells in HIV-infected individuals did vary with their expression of either inhibitory KIR3DL1 or activating KIR3DS1 receptors. Ex vivo antigen-specific activation of CD8+ T cells expressing KIR3DL1 was severely limited relative to that of the CD8+ T cell population not expressing KIR3DL1, irrespective of HLA Bw4/Bw6 background. This activation deficit or anergy was not absolute as it was overcome in all cases tested by stimulation through the TCR complex with anti-CD3 antibodies. Identical results were obtained in a recent study with CD8+KIR+ T cells from non-HIV-infected individuals using the same P815/anti-CD3 stimulation protocol33. After in vitro stimulation with IL-7, IL-2 and peptides, KIR3DL1+ CD8+ T cells did respond to secondary antigen-specific stimulation with peptide-pulsed autologous B lymphoblastoid cells, reiterating our conclusion, and that of several other studies, that the ex vivo anergy demonstrated by KIR3DL1+ CD8+ T cells is neither absolute, nor intransigent22, 24, 32–34. Although many KIR3DS1 and L1-expressing CD8+ T cells share a similar CD127−CD45RA+CD57+ phenotype indicative of effector memory cells, the KIR3DS1-expressing CD8+ T cells of homozygous individuals exhibited no ex vivo deficit in antigen-specific activation compared to KIR3DS1-negative CD8+ T cells from the same individual. Thus, the behaviour of a subset of the CD8+ T cell population is related to the KIR phenotype of individual cells and hence, to the KIR genotype of the host. Although we and several other groups found that co-expression of the HLA-encoded Bw4 ligand does not appear to dictate the effect of KIR3DL1 expression on CD8+ T cell behaviour, co-expression of HLA-Bw4*80I, which represents a putative ligand of KIR3DS1, does appear to influence in vivo expansion of KIR3DS1+ CD8+ T cells32, 33.
Although we found that the inability of KIR3DL1+ CD8+ T cells to respond to conventional antigen-specific stimulation ex vivo was independent of engagement of HLA-Bw4 ligands by KIR3DL1, several previous studies, measuring longer term proliferation as opposed to ex vivo antigen-specific IFN-γ production, reported some dependency on specific KIR ligation for reduced responsiveness24, 32. More recent studies suggest that even in the absence of ligand binding, KIR3DL1 may co-localize with the T cell receptor at the immunological synapse and recruit phosphatases that inhibit T cell activation35. While this is generally consistent with our observations, CD8+ T cells expressing KIR3DL1 after in vitro culture no longer exhibited an antigen-specific activation deficit, and within a licensing environment, a greater fraction of NK cells expressing KIR3DL1 than not expressing KIR3DL1 respond to ex vivo stimulation36. At least in this situation, expression of KIR3DL1 is not constitutively inhibitory, possibly through altered programming imprinted during the NK licensing process. Another possibility is that CD8+ T cell expression of KIR3DL1 reflects recent or repeated previous antigenic exposure in vivo that leaves the T cells refractory to ex vivo stimulation37. However, in that case, we would also expect KIR3DS1-expressing T cells to be refractory to ex vivo antigen-specific stimulation, which was not observed. Whatever the mechanism, a subset of the CD8+ T cells of a KIR3DS1 homozygous individual is functionally superior to the homologous subset of a KIR3DL1 homozygous individual. Unlike KIR3DL1+ CD8+ T cells, expansion of KIR3DS1+ T cells appears to depend on co-expression of the putative HLA-Bw4*80I ligand. If this expansion is predicated on positive signaling that is at least partially collateral to TCR signaling, this might produce cells less refractory to antigen-specific signaling ex vivo. Our phenotypic analysis of KIR3DL1/S1+ CD8+ T cells indicates that the KIR+CD8+ T cell population of HIV-infected individuals generally expresses the same markers as in non-HIV-infected individuals, and thus, is mostly comprised of CD8+ T cells with similar effector memory status24, 33. However, we did observe one potentially meaningful phenotypic difference based on expression of the activating KIR3DS1 versus inhibitory KIR3DL1 receptor. While KIR3DL1 and KIR3DS1 CD8+ T cells were similar in terms of CD127 expression, a significantly lesser fraction of KIR3DS1+ CD8+ T cells expressed CD45RA. This could reflect a stage of differentiation or activation less proximal to terminal differentiation and more sensitive to activation. Antigen-specific responses against HIV were not detected even following culture of KIR3DL1+ CD8+ T cells, which raises several possibilities. Firstly, there could be no entry of HIV-specific CD8+ T cells into the KIR3DL1+ population. This seems unlikely given the prominence of KIR3DL1+ CD8+ T cells specific for other viruses and previous evidence for clonal overlap between the KIR-expressing and KIR-negative CD8+ T cell populations35. Secondly, the chronicity of HIV infection relative to CMV, Flu and EBV might differentially affect the fate of HIV-specific KIR3DL1+ CD8+ T cells in regard to clonal deletion or development of a more intransigent anergy.
The functional superiority of KIR3DS1+ over KIR3DL1+ CD8+ T cells and their selective expansion in the setting of HLA-Bw4*80I co-expression that we describe herein is consistent with epidemiological data indicating a protective effect of this genotypic combination against HIV disease progression. Co-expression of KIR3DL1 with certain HLA-Bw4 ligands is associated with resistance to HIV infection, but not always with protection against disease progression8, 12, 29. Since there was no evidence of superior function for CD8+ T cells expressing KIR3DL1, with or without co-expression of HLA-Bw4, the association with protection from infection, in this case, is more likely to reflect effective licensing of NK cells and their potential role in the early stages of viral infection than to reflect a functional advantage for CD8+ T cells expressing KIR3DL1.
The list of cell surface molecules that can modulate T cell function during secondary antigenic exposure continues to grow, mostly with members that reduce the likelihood of T cell activation and limit acquisition or expression of a full range of effector functions. However, just as inhibitory molecules may play a negative role in chronic conditions such as cancer and HIV infection, acquired cell surface molecules that can increase the likelihood of T cell activation, such as KIR2DL4, KIR2DS1-5, KIR3DS1, NKG2C-E, and CD16, might actually enhance T cell activation and their retention of a full range of effector functions in certain settings. Exploitation of this possibility offers a rational complement to ongoing strategies aimed at reducing delivery of negative signals to T cells in chronic infection and cancer.
Methods
Subjects and peripheral blood mononuclear cell isolation
Study subjects infected with HIV were recruited through the Newfoundland and Labrador Provincial HIV Clinic. Uninfected subjects were recruited from laboratory and hospital personnel. All provided informed consent for whole blood collection, immunological studies and researcher access to medical laboratory records. The Newfoundland and Labrador Health Research Ethics Authority gave ethical approval for this study. Acid-citrate-dextrose treated whole blood was collected by forearm venipuncture, diluted 1:2 with phosphate buffered saline (PBS) and peripheral blood mononuclear cells (PBMC) isolated by Ficoll-Hypaque (GE Healthcare Bio-Science) density gradient centrifugation. Interface cells were washed once in PBS with 1% fetal calf serum (FCS) and resuspended in lymphocyte medium comprised of RPMI 1640 with 10% FCS, 100 IU/ml penicillin, 100 μg/ml streptomycin, 2 mM L-glutamine, 10 mM HEPES buffer solution and 2 × 10−5 M 2-mercaptoethanol (all from Invitrogen).
Analysis of KIR3DL/S1 and class I HLA-A and B genotype
Subject DNA was isolated from B lymphoblastoid cells (BLCL) transformed as previously described or from fresh PBMC using the illustra genomicPrep Mini Spin kit (GE Healthcare)38. To determine KIR3DL1 genotype, 200 ng genomic DNA was used in separate KIR3DL1 and KIR3DS1 gene-specific polymerase chain reactions (PCR) as previously described39. The KIR3DL1 primers (forward 5′ CCA TCG GTC CCA TGA TGC T 3′ and reverse 5′ AGA GAG AAG GTT TCT CAT ATG 3′) and KIR3DS1 primers (forward 5′ GGC AGA ATA TTC CAG GAG G 3′ and reverse 5′ AGG GGT CCT TAG AGA TCC A 3′) were used at 0.5 μM each in 50 μl reaction volume with 0.2 mM dNTPs, 1.5 mM magnesium chloride, 1X Taq buffer and 2.5 U Taq polymerase (all from Invitrogen). The PCR was run on a PTC-100 thermal cycler (MJ Research) under the following conditions: initial denaturation for 5 min at 95° C, then 20 s at 97° C, 45 s at 62° C and 90 s at 72° C for 5 cycles followed by 25 cycles of 20 s at 95° C, 45 s at 60° C and 90 s at 72° C. Products were visualized by ethidium bromide staining after separation on 1.5% agarose gels. For 2 digit resolution HLA-A/B typing, subject DNA was isolated from BLCL as above and PCR with allele-specific primers carried out using commercial kits from One Lambda. Reaction conditions, product separation and visualization on agarose gels and interpretation of electrophoresis banding pattern were as per the manufacturer’s instructions.
Phenotypic analysis of KIR3DL/S1 expressing CD8+ T cells
Aliquots of 1 × 106 PBMC were stained with isotype controls or allophycocyanin (APC)-conjugated anti-CD3 (clone HIT3a, Biolegend), peridinyll chlorophyll protein (PerCP)-conjugated anti-CD8 (clone HIT8a, Biolegend) fluorescein isothiocyanate (FITC) or phycoerythrin (PE)-conjugated anti-KIR3DL1 (clone DX9, Miltenyi Biotech), or anti-CD3•APC, anti-CD8•PerCP and phycoerythrin (PE) conjugated anti-KIR3DL1/S1 (clone Z27, Beckman Coulter). This allowed confirmation of KIR3DL1/S1 genotypic analysis, measurement of the percentage of CD8+ T cells expressing KIR3DL1 and measurement of the percentage of CD8+ T cells from KIR3DS1 homozygotes expressing KIR3DS1. Further characterization of KIR3DL1++ or KIR3DL1S1++ CD8+ T cells for expression of CD45RA, CD57 and CD127 using anti-CD45RA•PE or FITC (clone JS.83, eBioscience), anti-CD57•FITC (clone TB03, Miltenyi Biotech) and anti-CD127•FITC (clone BioRDR5 eBioscience) as appropriate was carried out using PBMC from homozygous individuals with discernable KIR3DLI/S1++ CD8+ T cell populations.
Functional analysis of KIR3DL1/S1 expressing CD8+ T cells
Intracellular detection of IFN-γ production following different forms of stimulation was carried out to assess the functional status of KIR3DL1/S1-expressing CD8+ T cells. Antigen-specific responsiveness ex vivo was assessed by stimulation with individual peptides from HIV, cytomegalovirus (CMV), EBV or Flu, (synthesized by EZBioLab, 95% pure, 4.0 μg/mL final concentration), overlapping peptide pools from HIV Gag, Pol, Env or Nef proteins (National Institute of AIDS and Infectious Diseases AIDS Reagent Repository, 0.15 μg/mL final individual peptide concentration), or overlapping peptide pools from CMV pp65 and IE-1 proteins (Miltenyi Biotech, 0.5 μg/mL final individual peptide concentration). Cells were cultured with peptides at 1 × 106/ml at 37°C in a 5% CO2 humidity controlled incubator for 1 hr after which brefeldin A (Sigma) was added to 10 μg/ml and incubation continued for another 4 hours. Cells were then washed and surface stained with anti-CD3•FITC, anti-CD8•PerCP, DX9•PE or Z27•PE as appropriate, fixed, permeabilized (Dako intrastain kits) and stained with anti-IFN-γ•APC (clone 4S.B3, eBioscience). For non-specific stimulation through the T cell receptor (TCR) complex, 1 × 106 PBMC were incubated 5 hours with 1 μg/ml anti-CD3 (clone OKT3, ATCC CRL 8001) and 1 × 105 Fc receptor-expressing P815 cells (ATCC TIB-64). Brefeldin A was added after the first hour with staining as above, with exclusion of anti-CD3.
Selection and expansion of KIR3DL1-expressing T cells
KIR3DL1-expressing cells were isolated from PBMC of individuals homozygous for high expressing KIR3DL1 alleles, and expanded in vitro by either mitogenic or peptide-specific stimulation together with exogenous cytokines as described below. To select KIR3DL1+ cells, freshly-isolated PBMC were washed with separation buffer (PBS with 5 mM ethylene diamine tetraacetic acid and 0.1 % bovine serum albumin, Sigma), resuspended in a small volume and incubated with 1 μg purified DX9/106 cells for 30 min at 4°C. Cells were washed and resuspended in separation buffer at 3 × 106/ml with goat anti-mouse IgG conjugated magnetic beads (Invitrogen) at a bead-to-target cell ratio of 10:1 and incubated at 4°C on a rotator for 45 min. Bead-bound cells were collected by magnetic attraction and unbound KIR3DL1-negative cells passaged twice with remaining bead-bound cells removed by magnetic attraction each time. Bead-bound cells were expanded with 10 μg/ml Con A, 10 U/ml IL-2 and irradiated (3000 Rad) allogeneic PBMC. In several cases, bead-bound cells were expanded by peptide specific stimulation with heterologous feeder cells and 25 ng/ml IL-7 added at the time of initial stimulation and IL-2 added to 10 U/ml after day 3 of culture38, 40. Beads were removed after 9 days of culture by vigorous pipetting and magnetic collection 24 hours before secondary stimulation with peptide-pulsed autologous BLCL. Autologous BLCL were pulsed with the peptide or peptide pool of interest for 1 hour prior to addition of responder cells at a 10:1 responder to stimulator ratio such that the final peptide concentration was as stated above for secondary stimulations. Brefeldin A as added to 10 μg/mL after 1 hour and stimulation continued for a further 4 hours before surface staining and detection of intracellular IFN-γ by flow cytometry was carried out as previously described.
Statistical Analysis
Data sets were assessed for normal distribution by the Kolmogorov-Smirnov, D’Agostini and Pearson and Shapiro-Wilk tests. If all tests indicated normal distribution, data were represented with mean ± standard deviation (SD) shown for the different groups and Student’s t test used to compare groups. If any data sets being compared did not meet test criteria for normal distribution, data were represented with median and interquartile range (IQR) shown and groups compared by Mann-Whitney test. Anova was used if more than two groups were compared. A two-tailed probability value of less than 0.05 was considered to indicate a significant difference. For multivariable regression analyses, univariate correlation was first assessed by Spearman’s rho correlation test and variables included in the multivariate linear regression model if p < 0.10. A two sided 0.05 level test determined statistical significance. Data analyses were conducted with IBM SPSS version 21 for Macintosh.
Acknowledgments
The authors would like to thank all individuals who provided blood samples for this study. This research was supported by a Canadian Institutes of Health Research (CIHR) operating grant HOP 93428 awarded to MGr. SS was supported in part by the Newfoundland and Labrador graduate transition to employment program.
References
- 1.Lanier LL. NK cell receptors. Annu Rev Immunol. 1998;16:359–93. doi: 10.1146/annurev.immunol.16.1.359. [DOI] [PubMed] [Google Scholar]
- 2.Williams AP, Bateman AR, Khakoo SI. Hanging in the balance. KIR and their role in disease. Mol Interv. 2005;5:226–40. doi: 10.1124/mi.5.4.6. [DOI] [PubMed] [Google Scholar]
- 3.Kim S, Poursine-Laurent J, Truscott SM, Lybarger L, Song YJ, Yang L, et al. Licensing of natural killer cells by host major histocompatibility complex class I molecules. Nature. 2005;436:709–13. doi: 10.1038/nature03847. [DOI] [PubMed] [Google Scholar]
- 4.Gumperz JE, Litwin V, Phillips JH, Lanier LL, Parham P. The Bw4 public epitope of HLA-B molecules confers reactivity with natural killer cell clones that express NKB1, a putative HLA receptor. J Exp Med. 1995;181:1133–44. doi: 10.1084/jem.181.3.1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Malnati MS, Lusso P, Ciccone E, Moretta A, Moretta L, Long EO. Recognition of virus-infected cells by natural killer cell clones is controlled by polymorphic target cell elements. J Exp Med. 1993;178:961–9. doi: 10.1084/jem.178.3.961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Moretta A, Vitale M, Bottino C, Orengo AM, Morelli L, Augugliaro R, et al. P58 molecules as putative receptors for major histocompatibility complex (MHC) class I molecules in human natural killer (NK) cells. Anti-p58 antibodies reconstitute lysis of MHC class I-protected cells in NK clones displaying different specificities. J Exp Med. 1993;178:597–604. doi: 10.1084/jem.178.2.597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cella M, Longo A, Ferrara GB, Strominger JL, Colonna M. NK3-specific natural killer cells are selectively inhibited by Bw4-positive HLA alleles with isoleucine 80. J Exp Med. 1994;180:1235–42. doi: 10.1084/jem.180.4.1235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Boulet S, Kleyman M, Kim JY, Kamya P, Sharafi S, Simic N, et al. A combined genotype of KIR3DL1 high expressing alleles and HLA-B*57 is associated with a reduced risk of HIV infection. AIDS. 2008;22:1487–91. doi: 10.1097/QAD.0b013e3282ffde7e. [DOI] [PubMed] [Google Scholar]
- 9.Boulet S, Sharafi S, Simic N, Bruneau J, Routy JP, Tsoukas CM, et al. Increased proportion of KIR3DS1 homozygotes in HIV-exposed uninfected individuals. AIDS. 2008;22:595–9. doi: 10.1097/QAD.0b013e3282f56b23. [DOI] [PubMed] [Google Scholar]
- 10.Giebel S, Locatelli F, Lamparelli T, Velardi A, Davies S, Frumento G, et al. Survival advantage with KIR ligand incompatibility in hematopoietic stem cell transplantation from unrelated donors. Blood. 2003;102:814–9. doi: 10.1182/blood-2003-01-0091. [DOI] [PubMed] [Google Scholar]
- 11.Kim S, Sunwoo JB, Yang L, Choi T, Song YJ, French AR, et al. HLA alleles determine differences in human natural killer cell responsiveness and potency. Proc Natl Acad Sci U S A. 2008;105:3053–8. doi: 10.1073/pnas.0712229105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Martin MP, Gao X, Lee JH, Nelson GW, Detels R, Goedert JJ, et al. Epistatic interaction between KIR3DS1 and HLA-B delays the progression to AIDS. Nat Genet. 2002;31:429–34. doi: 10.1038/ng934. [DOI] [PubMed] [Google Scholar]
- 13.Miller JS, Cooley S, Parham P, Farag SS, Verneris MR, McQueen KL, et al. Missing KIR ligands are associated with less relapse and increased graft-versus-host disease (GVHD) following unrelated donor allogeneic HCT. Blood. 2007;109:5058–61. doi: 10.1182/blood-2007-01-065383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rauch A, Laird R, McKinnon E, Telenti A, Furrer H, Weber R, et al. Influence of inhibitory killer immunoglobulin-like receptors and their HLA-C ligands on resolving hepatitis C virus infection. Tissue Antigens. 2007;69 (Suppl 1):237–40. doi: 10.1111/j.1399-0039.2006.773_4.x. [DOI] [PubMed] [Google Scholar]
- 15.Ruggeri L, Capanni M, Urbani E, Perruccio K, Shlomchik WD, Tosti A, et al. Effectiveness of donor natural killer cell alloreactivity in mismatched hematopoietic transplants. Science. 2002;295:2097–100. doi: 10.1126/science.1068440. [DOI] [PubMed] [Google Scholar]
- 16.Long BR, Ndhlovu LC, Oksenberg JR, Lanier LL, Hecht FM, Nixon DF, et al. Conferral of enhanced natural killer cell function by KIR3DS1 in early human immunodeficiency virus type 1 infection. J Virol. 2008;82:4785–92. doi: 10.1128/JVI.02449-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Alter G, Martin MP, Teigen N, Carr WH, Suscovich TJ, Schneidewind A, et al. Differential natural killer cell-mediated inhibition of HIV-1 replication based on distinct KIR/HLA subtypes. J Exp Med. 2007;204:3027–36. doi: 10.1084/jem.20070695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Alter G, Heckerman D, Schneidewind A, Fadda L, Kadie CM, Carlson JM, et al. HIV-1 adaptation to NK-cell-mediated immune pressure. Nature. 476:96–100. doi: 10.1038/nature10237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Alter G, Rihn S, Walter K, Nolting A, Martin M, Rosenberg ES, et al. HLA class I subtype-dependent expansion of KIR3DS1+ and KIR3DL1+ NK cells during acute human immunodeficiency virus type 1 infection. J Virol. 2009;83:6798–805. doi: 10.1128/JVI.00256-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gillespie GM, Bashirova A, Dong T, McVicar DW, Rowland-Jones SL, Carrington M. Lack of KIR3DS1 binding to MHC class I Bw4 tetramers in complex with CD8+ T cell epitopes. AIDS Res Hum Retroviruses. 2007;23:451–5. doi: 10.1089/aid.2006.0165. [DOI] [PubMed] [Google Scholar]
- 21.O’Connell KA, Han Y, Williams TM, Siliciano RF, Blankson JN. Role of natural killer cells in a cohort of elite suppressors: low frequency of the protective KIR3DS1 allele and limited inhibition of human immunodeficiency virus type 1 replication in vitro. J Virol. 2009;83:5028–34. doi: 10.1128/JVI.02551-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Anfossi N, Doisne JM, Peyrat MA, Ugolini S, Bonnaud O, Bossy D, et al. Coordinated expression of Ig-like inhibitory MHC class I receptors and acquisition of cytotoxic function in human CD8+ T cells. J Immunol. 2004;173:7223–9. doi: 10.4049/jimmunol.173.12.7223. [DOI] [PubMed] [Google Scholar]
- 23.Anfossi N, Robbins SH, Ugolini S, Georgel P, Hoebe K, Bouneaud C, et al. Expansion and function of CD8+ T cells expressing Ly49 inhibitory receptors specific for MHC class I molecules. J Immunol. 2004;173:3773–82. doi: 10.4049/jimmunol.173.6.3773. [DOI] [PubMed] [Google Scholar]
- 24.Arlettaz L, Degermann S, De Rham C, Roosnek E, Huard B. Expression of inhibitory KIR is confined to CD8+ effector T cells and limits their proliferative capacity. Eur J Immunol. 2004;34:3413–22. doi: 10.1002/eji.200324756. [DOI] [PubMed] [Google Scholar]
- 25.Arlettaz L, Villard J, de Rham C, Degermann S, Chapuis B, Huard B, et al. Activating CD94:NKG2C and inhibitory CD94:NKG2A receptors are expressed by distinct subsets of committed CD8+ TCR alphabeta lymphocytes. Eur J Immunol. 2004;34:3456–64. doi: 10.1002/eji.200425210. [DOI] [PubMed] [Google Scholar]
- 26.Mingari MC, Ponte M, Vitale C, Bellomo R, Moretta L. Expression of HLA class I-specific inhibitory receptors in human cytolytic T lymphocytes: a regulated mechanism that controls T-cell activation and function. Hum Immunol. 2000;61:44–50. doi: 10.1016/s0198-8859(99)00158-5. [DOI] [PubMed] [Google Scholar]
- 27.Galiani MD, Aguado E, Tarazona R, Romero P, Molina I, Santamaria M, et al. Expression of killer inhibitory receptors on cytotoxic cells from HIV-1-infected individuals. Clin Exp Immunol. 1999;115:472–6. doi: 10.1046/j.1365-2249.1999.00833.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Luque I, Solana R, Galiani MD, Gonzalez R, Garcia F, Lopez de Castro JA, et al. Threonine 80 on HLA-B27 confers protection against lysis by a group of natural killer clones. Eur J Immunol. 1996;26:1974–7. doi: 10.1002/eji.1830260845. [DOI] [PubMed] [Google Scholar]
- 29.Martin MP, Qi Y, Gao X, Yamada E, Martin JN, Pereyra F, et al. Innate partnership of HLA-B and KIR3DL1 subtypes against HIV-1. Nat Genet. 2007;39:733–40. doi: 10.1038/ng2035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Migueles SA, Sabbaghian MS, Shupert WL, Bettinotti MP, Marincola FM, Martino L, et al. HLA B*5701 is highly associated with restriction of virus replication in a subgroup of HIV-infected long term nonprogressors. Proc Natl Acad Sci U S A. 2000;97:2709–14. doi: 10.1073/pnas.050567397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Carrington M, Nelson GW, Martin MP, Kissner T, Vlahov D, Goedert JJ, et al. HLA and HIV-1: heterozygote advantage and B*35-Cw*04 disadvantage. Science. 1999;283:1748–52. doi: 10.1126/science.283.5408.1748. [DOI] [PubMed] [Google Scholar]
- 32.van der Veken LT, Campelo MD, van der Hoorn MA, Hagedoorn RS, van Egmond HM, van Bergen J, et al. Functional analysis of killer Ig-like receptor-expressing cytomegalovirus-specific CD8+ T cells. J Immunol. 2009;182:92–101. doi: 10.4049/jimmunol.182.1.92. [DOI] [PubMed] [Google Scholar]
- 33.Bjorkstrom NK, Beziat V, Cichocki F, Liu LL, Levine J, Larsson S, et al. CD8 T cells express randomly selected KIRs with distinct specificities compared with NK cells. Blood. 120:3455–65. doi: 10.1182/blood-2012-03-416867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Young NT, Uhrberg M, Phillips JH, Lanier LL, Parham P. Differential expression of leukocyte receptor complex-encoded Ig-like receptors correlates with the transition from effector to memory CTL. J Immunol. 2001;166:3933–41. doi: 10.4049/jimmunol.166.6.3933. [DOI] [PubMed] [Google Scholar]
- 35.Alter G, Rihn S, Streeck H, Teigen N, Piechocka-Trocha A, Moss K, et al. Ligand-independent exhaustion of killer immunoglobulin-like receptor-positive CD8+ T cells in human immunodeficiency virus type 1 infection. J Virol. 2008;82:9668–77. doi: 10.1128/JVI.00341-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Parsons MS, Zipperlen K, Gallant M, Grant M. Killer cell immunoglobulin-like receptor 3DL1 licenses CD16-mediated effector functions of natural killer cells. J Leukoc Biol. 88:905–12. doi: 10.1189/jlb.1009687. [DOI] [PubMed] [Google Scholar]
- 37.Huard B, Karlsson L. KIR expression on self-reactive CD8+ T cells is controlled by T-cell receptor engagement. Nature. 2000;403:325–8. doi: 10.1038/35002105. [DOI] [PubMed] [Google Scholar]
- 38.Mason RD, Bowmer MI, Howley CM, Gallant M, Myers JC, Grant MD. Antiretroviral drug resistance mutations sustain or enhance CTL recognition of common HIV-1 Pol epitopes. J Immunol. 2004;172:7212–9. doi: 10.4049/jimmunol.172.11.7212. [DOI] [PubMed] [Google Scholar]
- 39.Uhrberg M, Valiante NM, Shum BP, Shilling HG, Lienert-Weidenbach K, Corliss B, et al. Human diversity in killer cell inhibitory receptor genes. Immunity. 1997;7:753–63. doi: 10.1016/s1074-7613(00)80394-5. [DOI] [PubMed] [Google Scholar]
- 40.Lalvani A, Dong T, Ogg G, Patham AA, Newell H, Hill AV, et al. Optimization of a peptide-based protocol employing IL-7 for in vitro restimulation of human cytotoxic T lymphocyte precursors. J Immunol Methods. 1997;210:65–77. doi: 10.1016/s0022-1759(97)00177-4. [DOI] [PubMed] [Google Scholar]