

Abstract
Background
Severe Combined Immunodeficiency (SCID) is a syndrome uniformly fatal during infancy unless recognized and treated successfully by bone marrow transplantation or gene therapy. Because SCID infants have no abnormal physical appearance, diagnosis is usually delayed unless newborn screening is performed.
Objective
In this study, we sought to evaluate the presenting features of all 172 SCID patients transplanted at this institution over the past 31 years.
Methods
We reviewed original charts from 172 consecutive classic SCID patients who received either T cell-depleted HLA-haploidentical (N=154) or HLA-identical (N=18) non-ablative related marrow transplants at Duke University Medical Center from 1982–2013.
Results
The mean age at presentation was 4.87 months. When there was a family history of early infant death or known SCID (63/172 or 37%), the mean presentation age was much earlier, 2.0 months compared to 6.6 months. Failure to thrive was common, with 84 patients (50%) having a weight less than the 5th percentile. The leading infections included oral moniliasis (43%), viral infections (61/172 35.5%) and Pneumocystis jiroveci (26%) pneumonia. The group mean ALC was 1454/cmm; 88% of the infants had an ALC less than 3000/cmm. Absent thymic shadow was seen in 92% of infants with electronic radiographic data available. An absence of T cell function was found in all patients.
Conclusions
SCID infants appear normal at birth but later present with failure to thrive and/or recurrent fungal, viral and bacterial infections. Low ALCs and absent thymic shadow on chest x-ray are key diagnostic clues. The absence of T cell function confirms the diagnosis.
Keywords: Absent Thymic Shadow, Failure to Thrive, Infections, Lymphopenia, Severe Combined Immunodeficiency
INTRODUCTION
Severe combined immunodeficiency (SCID) is a syndrome characterized by defects in both humoral and cellular immunity.1.2 Infants born with this disorder are healthy at birth, but are highly susceptible to infection as their passive immunity from transplacentally transferred maternal IgG wanes. Common symptoms in the first few months of life include failure to thrive, recurrent candidiasis and diarrhea. Despite these symptoms, the condition is often not immediately recognized and the historical mean age at presentation is 6 months, at which time there is usually a history of multiple infections.1,3
In addition to symptoms of recurrent infections and failure to thrive, other clues for diagnosis include absent tonsils and lack of palpable peripheral lymph nodes on clinical exam. A diagnosis of SCID should be suspected from the combination of clinical symptoms, physical examination and a low absolute lymphocyte count. However, low lymphocyte counts are often overlooked or their significance not appreciated. Ultimately, the SCID diagnosis is made based on an absence of T cells in flow cytometry and an absence of T cell function in lymphocyte proliferation studies.
Mutations in at least 13 different genes are known to cause the syndrome of SCID.2 The most common defect in the United States is X-linked SCID.4 Mutations in the gene encoding the common gamma chain (γc) shared by 6 different cytokine receptors: IL-2, IL-4, IL-7, IL-9, IL-15 and IL-21 account for these infants’ T cell developmental defect and the abnormal B cells and NK cells.5–7 The second most common cause of SCID in the United States is adenosine deaminase (ADA) deficiency.8 Other molecular defects in SCID include mutations in the genes encoding Janus kinase-3 (Jak-3),9 IL-7α receptor chain,10 recombinase activating genes 1 and 2 (RAG-1 and RAG-2),11 Artemis,12 DNA-PKcs,13 CD45,14 CD3δ,15 CD3ε and CD3ζ.16
Currently, the only curative treatment for SCID is bone marrow transplantation or gene therapy. However, a significant number of patients succumb to infections before they are able to be successfully immune reconstituted. Early recognition and treatment of this disorder saves the lives of these infants. The overall 31-year survival rate of SCID infants given non- ablative bone marrow transplants at Duke is 76%, with the rate for the 52 infants transplanted in the first 3.5 months of life being 94%, compared with 70% for the 113 transplanted after 3.5 months.17–19 In this study, we evaluated the presenting features of all 172 SCID patients transplanted at this institution over the past 31 years, only 18 of whom had an HLA-identical related donor.
MATERIALS AND METHODS
We retrospectively reviewed original charts, electronic discharge summaries and lab results from 172 classic SCID patients who received either T cell-depleted HLA-haploidentical (N=154) or HLA-identical (N=18) related marrow transplants as treatment at Duke University Medical Center from 1982–2013. None of the patients had “leaky SCID” or Omenn’s syndrome. One Artemis SCID patient who also had DiGeorge syndrome and had a history of a thymus transplant prior to receiving an HLA-haploidentical bone marrow transplant was excluded from the analysis.20 Absolute lymphocyte counts were calculated from white blood cell counts and manual differentials performed at the time of presentation to this institution. Lymphocyte subpopulation analysis was performed via flow cytometry, and T cell function was assessed by in vitro lymphocyte stimulation studies, as previously described.3 Data regarding infections at the time of diagnosis were collected from laboratory reports from this institution and outside hospital records. Pneumocystis jiroveci pneumonia (PJP) was diagnosed via bronchoalveolar lavage (BAL) or presumptively based on clinical appearance, radiographic findings and improvement after therapy with trimethoprim and sulfamethoxazole. Pre-transplant electronic radiographs from a subset of recent patients (N=30) were reviewed with a Pediatric Radiologist to evaluate for the presence of a thymic shadow.
RESULTS
Demographics
A majority of the patients were male (80%), consistent with the large percentage of X linked SCID patients. Caucasian were the most common race (73%), followed by 15% black, 9% Hispanic, 2% Arab and 1% each Asian and American Indian.
Molecular type
The molecular type of all SCIDS in this report are shown in Fig 1, with the largest percentage being X linked SCIDs (n=78) followed by ADA deficient SCID (n=26) and IL7R deficient SCID (n=24). Other molecular defects were present at lower frequencies.
Fig 1. Percentage of SCIDs with each molecular defect.

The total number and percentage of the cohort are included for each molecular type of SCID.
Age at diagnosis
Of the 172 patients, mean age at presentation was 4.87 months (SD ±3.74). The age at diagnosis for all patients ranged from prenatal diagnosis to 18 months. Only 5/172 (2.9%) patients in our cohort presented after the age of 12 months, with 4 of those 5 patients ultimately dying. For the molecular subtypes of SCID, the age at diagnosis ranged from RAG deficient SCID patients, with a mean age of presentation at 3.57 months (SD ± 3.61), to JAK-3 SCID with a mean of 6.92 (SD ± 3.92). X-linked SCID had a mean diagnosis age of 4.42 months (SD ±3.92) (FIG 2).
Fig. 2. Age at diagnosis for the various molecular types of SCID.
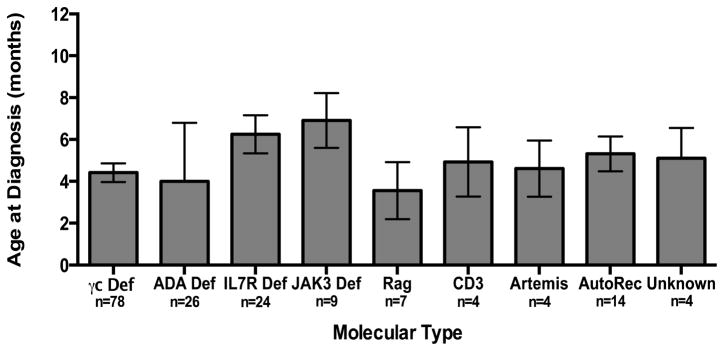
The mean age at which the infants in each group were diagnosed is shown with standard error bars. N indicates the number of patients within each group.
Family history
Family history of infant death due to infection or known SCID was present in 63/172 (36.6%) patients analyzed. Of those with a positive family history, the mean age at presentation was younger, 2.09 months (SD ± 2.92) months compared to 6.5 months (SD±3.2), p<0.0001, Mann Whitney test for those with no family history (Table 1).
Table 1.
Clinical symptoms at diagnosis
| Clinical Symptoms at diagnosis | # | % |
|---|---|---|
| Failure to thrive (weight at or less than 5th percentile) | 84 | 50.6 |
| Diarrhea/Frequent loose stools | 38 | 22.1 |
| Pneumonia | 34 | 19.8 |
| Recurrent otitis media | 30 | 17.4 |
| Family History of immunodeficiency or unexplained deaths in infancy | # | Mean Age (months) |
|---|---|---|
| Family History present | 63 | 2.09 |
| Family History absent | 109 | 6.5 |
| Radiographic Evidence of SCID | # | % |
|---|---|---|
| Absent thymic shadow | 28 | 93.3 |
| Indeterminate | 2 | 6.7 |
Results indicate numbers of patients and percentage of total patients in which clinical symptoms were reported at diagnosis.
Clinical symptoms at presentation
Patients commonly presented with failure to thrive, with 84 patients (50%) presenting with weight less than or equal to the 5th percentile (Table 1). In the 109 infants without a family history, 69 presented with failure to thrive (65%). Other relevant history at time of presentation included complaints of diarrhea or of frequent loose stools in 22.1%, pneumonia in 19.8% and recurrent otitis media in 17.4% of patients (Table 1).
Considering the 4 major clinical findings: failure to thrive, diarrhea, pneumonia and recurrent otitis media, 37% presented with 0/4, 27% presented with 1/4, 26% presented with 2/4, 9% presented with 3/4 and 1% presented with 4/4 findings.
Infections at the time of presentation
The most common infections (Table 2) at the time of presentation were oral moniliasis (74/172, 43%), viral infections (61/172, 35.5 %) and Pneumocystis jiroveci (45/172, 26.2 %) of patients. Over half of the patients in this cohort presented with a history of a fungal infection. Looking specifically at the 109 patients who presented without a family history of SCID, 58 (53.2%) presented with thrush, and 40 (36.7%) patients presented with PJP. The most common viral infections at diagnosis included: Parainfluenza (18/172), RSV (17/172), rotavirus (16/172), and adenovirus (11/172). Bacterial infections with identified organisms were found at a lower frequency than viral or fungal infections with Pseudomonas aeruginosa (8/172) and Staphylococcus sp. (5/172) being the most commonly isolated. Mycobacterial sp. infections were present in 4/172 patients (2.3%). In two of these patients, they were known to have received BCG vaccination.
Table 2.
Infections at Diagnosis
| # | % | |
|---|---|---|
| VIRAL | 61 | 35.5 |
|
| ||
| Parainfluenza | 18 | 10.5 |
| RSV | 17 | 9.9 |
| Rotavirus | 16 | 9.3 |
| Adenovirus | 11 | 6.4 |
| Influenza | 4 | 2.3 |
| Enterovirus | 4 | 2.3 |
| Varicella | 3 | 1.7 |
| CMV | 2 | 1.2 |
| Coxsackie | 1 | 0.6 |
| Rhinovirus | 1 | 0.6 |
| HSV | 1 | 0.6 |
|
| ||
| FUNGAL | 96 | 55.8 |
|
| ||
| Thrush or candida skin infection | 74 | 43.0 |
| Pneumocystis jiroveci* | 45 | 26.2 |
| Aspergillus sp. | 2 | 1.2 |
|
| ||
| BACTERIAL | 24 | 14.0 |
|
| ||
| Pseudomonas aeruginosa | 8 | 4.7 |
| Staphylococcus sp. | 5 | 2.9 |
| Mycobacterial sp. | 4 | 2.3 |
| Klebsiella sp. | 3 | 1.7 |
| Haemophilus influenzae | 3 | 1.2 |
| Streptococcus pneumoniae | 2 | 1.2 |
| Escherichia coli | 2 | 1.2 |
| Enterococcus sp. | 2 | 1.2 |
| Listeria sp. | 1 | 0.6 |
| Salmonella sp. | 1 | 0.6 |
| Streptococcus viridans | 1 | 0.6 |
| Moraxella sp. | 1 | 0.6 |
| Chlamydia sp. | 1 | 0.6 |
Results indicate numbers of patients and percentage of total patients in which viral, fungal and bacterial agents were identified by historical report and/or laboratory reports.
In some cases, the diagnosis of Pneumocystis jiroveci was presumed by the clinical symptoms without confirmatory testing.
Lymphocyte counts
The 95% confidence interval for normal absolute lymphocyte counts at 6 months of age ranged from 4000 to 11000/mm3.21 The overall mean ALC for all SCID patients transplanted at this institution at the time of diagnosis was 1454/cmm (SD±1279); 152/172 (88.4%) of the patients transplanted at Duke presented with an ALC less than 3000/cmm. The lowest mean ALC (367/cmm, SD± 504) was found in the ADA deficient SCID group (FIG. 3), p<0.0001, Kruskal-Wallis. The highest mean ALC was found in the JAK3 deficient SCID group at 3077 ± 2869/cmm, likely attributed to a single JAK3 patient who had a very large number of transplacentally transferred maternal T lymphocytes resulting in an ALC of 10,207/cmm.
Fig. 3. Absolute lymphocyte counts for molecular types of SCID.
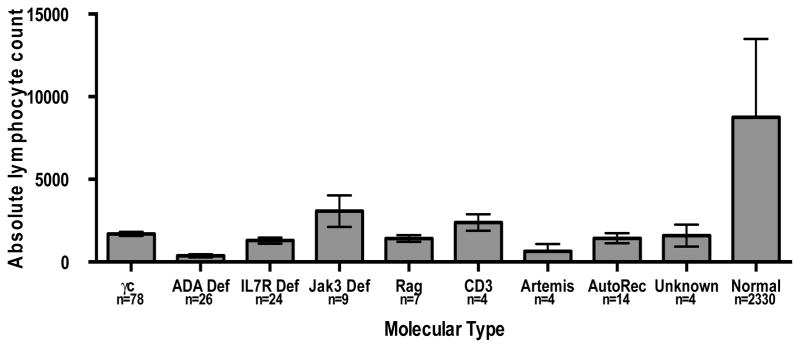
The mean absolute lymphocyte counts in each group at the time of diagnosis are shown with standard error bars. The normal lymphocyte counts are shown for healthy individuals (6 months of age).
Radiographic findings
Review of radiographs from recent patients with electronic radiograph data (N=30) revealed an obvious absence of a thymic shadow in 92.3% (Table 1). Of the two patients with indeterminate films, one had a poor quality film due to low lung volumes and one had a pneumonia obscuring the mediastinal contour. An example of a radiograph from one of the SCID patients is shown in Figure 4b, revealing an absent thymic shadow. A normal radiograph from a healthy infant is shown in Figure 4b. There is a clear “sail sign” in the right cardiomediastinal contour indicating the presence of thymic tissue.
Fig. 4.


Fig. 4a. Absent thymic shadow for a patient with SCID. No thymic shadow in the cardiomediastinal contour is evident on the radiographic image
Fig 4b. Present thymic shadow for a normal infant. A thymic shadow or “sail sign” is present in the right cardiomediastinal contour.
Immunologic findings
Maternal IgG is transplacentally transferred to the fetus in utero and persists until approximately 6 months of age, negating the ability to use serum IgG concentrations as a useful diagnostic finding. Immunoglobulins A, M and E are not transferred from the mother to the fetus, so the presence of these molecules indicates that the infant produced them. IgA, IgM and IgE concentrations at the time of presentation are shown in FIG 5a, with some molecular types of SCID having almost normal levels of some isotypes, while others had very low levels; IgA was usually consistently very low or absent in all types. T cells were absent or markedly reduced in all molecular types of SCID (FIG 5b). The highest mean T cell count was found in the JAK3 deficient group as a result of the above-mentioned very high number of transplacentally transferred maternal T cells in one patient. The numbers of B and NK cells varied among the molecular types, reflective of the phenotype of that cause of SCID. No matter the molecular type of SCID, there was nearly absent T cell proliferation in response to mitogen stimulation (FIG 5c).
Fig. 5.



Fig. 5a. Levels of Immunoglobulins at diagnosis for each molecular type of SCID. Open bars indicate IgA (mg/dL), black filled bars indicate IgM (mg/dL) and gray filled bars indicated IgE (I.U./mL). Normal 6 month old infants’ immunoglobulin levels are shown for comparison.
Fig. 5b. Absolute numbers of B, T and NK cells for each molecular type of SCID. Open bars indicate B cells, black filled bars indicate T cells and gray filled bars indicated NK cells. All values are lymphocytes/cmm. Normal 6 month old infants’ levels are shown for comparison.
Fig. 5c. Lymphocyte proliferation at diagnosis for each Molecular Type of SCID. Patterned bars indicate media only controls. Black filled bars indicate Phytohemagglutinin (PHA) stimulated cells, open bars indicated Concanavalin (conA) stimulated cells and gray filled bars indicate Pokeweed mitogen (PWM) stimulated cells. Responses of normal 6 month old infants’ controls are shown for comparison.
Time between diagnosis and transplantation
Patients were transplanted shortly after arrival at Duke. The time between formal diagnosis of SCID and transplantation was an average of 3 weeks, with a range of 1 week to 7 months. In most cases, the patients who were transplanted a week after diagnosis reflected those patients with a family history who were diagnosed prenatally or at birth. For those patients with a delay between diagnosis and transplantation, in most cases this reflected delays in arranging transfer from the referring hospital or physician to our institution.
DISCUSSION
We have presented the clinical and immunologic features of the largest single institution cohort of classic SCID patients transplanted to date. Our previous report of the diagnostic features of this cohort was nearly a decade ago and included the findings from 89 infants transplanted at Duke.3 Other cohorts have reported as many as 117 SCIDs, however, the molecular etiology of SCID was not known for a majority of the latter patients.22
The initial clinical symptoms in patients ultimately diagnosed with SCID included failure to thrive and common infections such as recurrent otitis media and pneumonias. Later they present with recurrent viral, fungal and bacterial infections. Our cohort’s infection history at the time of presentation revealed a high rate of fungal infections due to candida and PJP, as well as viral infections. Bacterial infections were less commonly identified; however, because of the frequent history of otitis and pneumonia, we know that bacterial infections are also common. It is likely specific bacterial infections are underrepresented in our analysis due to the prior use of antibiotics.
Unless diagnosed early, which is difficult because SCID patients look outwardly normal until they become ill, they are at risk from live vaccines and graft-versus-host disease from non-irradiated blood products. The only significant past medical history leading to a significantly earlier diagnosis was a positive family history of SCID.
However, blood counts are often done when infants become ill and they can provide very important information that could lead to the diagnosis of SCID. Lymphopenia represents a simple and rapid test for SCID or other T cell defects, since T cells normally represent 70% of circulating lymphocytes and are missing in all molecular types of SCID. In a young infant with recurrent infections, an ALC less than 3000/cmm should warrant immediate referral and further testing. In our cohort, 88% of the 172 classic SCID patients had an ALC less than 3000/cmm. Rarely transplacentally-transferred maternal T cells can confound the diagnosis of SCID, as demonstrated by the few patients in our cohort with higher lymphocyte counts due to this phenomenon.
Often radiologists do not comment on the presence or absence of a thymic shadow on an infant chest radiograph. A thymic shadow is clearly visible as part of the mediastinal contour on chest radiograph of an infant without SCID.23 In our study, 92% of our SCID patients’ chest radiographs clearly had absent thymic shadows. In the two patients whose films were indeterminant, quality and technique did not allow for accurate assessment. While this finding should not be relied upon as the sole diagnostic test for SCID, it can provide an early and rapid clue to this diagnosis during an initial evaluation of an infant with recurrent infections.
Further immunologic testing should include flow cytometry for enumeration of T, B and NK cells. The presence of low absolute numbers and percentages of T cells is strongly suggestive of SCID, with B and NK cell numbers defining the phenotype and, therefore, hinting at the type of molecular defect that might be involved. However, low T cell numbers must be supported by the absence of T cell function in order to confirm a diagnosis of SCID. This requires lymphocyte culture and stimulation with mitogens such as PHA, Con A and PWM. When patients have transplacentally-transferred maternal T cells, flow cytometry can reveal near normal levels of total T cells, but these T cells are usually anergic so T cell function is still very low. Conversely, patients with intestinal lymphangiectasia or cartilage hair hypoplasia can have profound lymphopenia with low absolute numbers of T cells, but normal T cell function on proliferation studies, effectively ruling out SCID. In summary, there have not been any significant changes over the past decade in the clinical or laboratory presentation of SCID, including lymphopenia and the use of chest x-rays to assess thymus size. However, there has been a change in vaccine recommendations: the American Academy of Pediatrics recommends giving live rotaviral vaccine to all infants by age 2 months, emphasizing the need for very early diagnosis.
It is clear that patients transplanted within the first three and one half months of life have a superior outcome, both in terms of survival and level of immune reconstitution. 3,17,18 In 2010, the US Department of Health and Human Services recommended adding SCID to the uniform panel of newborn screening.24 The screening test employed is for T cell lymphopenia by quantifying T cell receptor excision circles (TRECs) by real time PCR performed on punches from the dried blood spots. Implementation of this testing in all states will allow patients to be diagnosed shortly after birth and improve outcomes. Twenty-one states have adopted newborn screening with more planning to move forward with this in the future. Early diagnosis of SCID is imperative, given the recommendation by the American Academy of Pediatrics for routine immunization of infants with rotavirus vaccine at 2 months of age.25 Infants with unrecognized SCID are susceptible to vaccine-acquired disease from live vaccines, as demonstrated by multiple SCID infants with rotavirus infection who became infected with vaccine strain virus.26–28 Some of the SCID infants reported to have acquired rotavirus from vaccine had persistence of the acquired infection until after T cell function was restored by stem cell transplantation.26, 28. Routine immunization with rotavirus vaccine in the first few months of life, before the typical age of presentation of SCID, will likely result in more infants presenting with vaccine-acquired disease in the future until there is universal newborn screening. When SCID is diagnosed in the first few weeks of life by newborn screening, it will be possible to avoid live virus immunizations and minimize exposure to infections that can lead to death in these infants. The success of identifying infants with SCID and other T-cell numerical deficiencies has recently been reported in New York29, in California30, and additional screening programs 31 When TREC screening becomes universal in the US, all SCID babies will be identified without the need of obtaining a complete blood count and differential.
Until newborn screening is performed in all states, it is important for physicians to be aware of clinical symptoms and signs of SCID in order to make as early a diagnosis as possible. In addition to the risks of rotavirus vaccine in those with delayed diagnosis, physicians in certain countries should be aware of the risks of Bacille Calmette Guérin (BCG) vaccination, a live bovine mycobacterial vaccine administered shortly after birth in Middle Eastern, South American, African and other countries to protect against tuberculosis. Later in infancy, the live vaccine given to immunize against varicella in all countries poses a risk for disseminated vaccine-acquired varicella if the infant has SCID. Two of the infants in our cohort presented with vaccine-acquired varicella. Additionally, any blood products a SCID patient receives should be irradiated due to the risk of graft-vs-host disease from alloreactive donor lymphocytes. Without recognition that an infant has SCID, any one of these interventions could be fatal.
Highlights section.
1. What is already known about this topic?
Classic SCID is a fatal syndrome caused by mutations in at least 13 different genes.
2. What does this article add to our knowledge?
This article highlights the presenting symptoms, exam findings along with laboratory values for the largest single center cohort of SCID patients.
3. How does this study impact current management guidelines?
This study may lead to increased recognition of SCID until newborn screening is implemented in all states and before live vaccines are administered.
Acknowledgments
Funding: The authors thank Roberta E. Parrott, B.S. for her performance of the initial immune studies and transplants in these patients. They would also like to thank Dr. Donald Frush for his assistance in the review of the patients’ chest x-rays.
This research was supported by Grants AI042951 and AI47605 from the National Institute of Allergy and Infectious Diseases and by Grant # 1 UL1 RR024128-01 from the National Center for Research Resources (NCRR), a component of the National Institutes of Health (NIH), and NIH Roadmap for Medical Research.
Abbreviations
- 70 ADA
adenosine deaminase
- ALC
absolute lymphocyte count
- BAL
bronchoalveolar lavage
- JAK3
Janus kinase 3
- PJP
Pneumocystis jiroveci pneumonia
- RAG
recombinase activating genes
- RSV
respiratory syncytial virus
- SCID
Severe Combined Immunodeficiency
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Buckley RH, Schiff RI, Schiff SE, et al. Human severe combined immunodeficiency: Genetic, phenotypic, and functional diversity in one hundred eight infants. Journal of Pediatrics. 1997;130:378–87. doi: 10.1016/s0022-3476(97)70199-9. [DOI] [PubMed] [Google Scholar]
- 2.Buckley RH. Molecular defects in human severe combined immunodeficiency and approaches to immune reconstitution. Annual Review of Immunology. 2004;22:625–55. doi: 10.1146/annurev.immunol.22.012703.104614. [DOI] [PubMed] [Google Scholar]
- 3.Buckley RH, Schiff SE, Schiff RI, et al. Hematopoietic stem-cell transplantation for the treatment of severe combined immunodeficiency. New Engl J Med. 1999;340:508–16. doi: 10.1056/NEJM199902183400703. [DOI] [PubMed] [Google Scholar]
- 4.Noguchi M, Yi HF, Rosenblatt HM, et al. Interleukin-2 Receptor Gamma Chain Mutation Results in X-Linked Severe Combined Immunodeficiency in Humans. Cell. 1993;73:147–57. doi: 10.1016/0092-8674(93)90167-o. [DOI] [PubMed] [Google Scholar]
- 5.Russell SM, Keegan AD, Harada N, et al. Interleukin-2 Receptor Gamma- Chain - a Functional Component of the Interleukin-4 Receptor. Science. 1993;262:1880–83. doi: 10.1126/science.8266078. [DOI] [PubMed] [Google Scholar]
- 6.Noguchi M, Nakamura Y, Russell SM, et al. Interleukin-2 Receptor Gamma-Chain - a Functional Component of the Interleukin-7 Receptor. Science. 1993;262:1877–80. doi: 10.1126/science.8266077. [DOI] [PubMed] [Google Scholar]
- 7.Asao H, Okuyama C, Kumaki S, et al. Cutting edge: The common gamma- chain is an indispensable subunit of the IL-21 receptor complex. Journal of Immunology. 2001;167:1–5. doi: 10.4049/jimmunol.167.1.1. [DOI] [PubMed] [Google Scholar]
- 8.Hirschhorn RCF, editor. Immunodeficiency due to defects of purine metabolism. New York: Oxford University Press; 2007. [Google Scholar]
- 9.Roberts JL, Lengi A, Brown SM, et al. Janus kinase 3 (JAK3) deficiency: clinical, immunologic, and molecular analyses of 10 patients and outcomes of stem cell transplantation. Blood. 2004;103:2009–18. doi: 10.1182/blood-2003-06-2104. [DOI] [PubMed] [Google Scholar]
- 10.Puel A, Ziegler SF, Buckley RH, Leonard WJ. Defective IL7R expression in T-B+NK+ severe combined immunodeficiency. Nat Genet. 1998;20:394–97. doi: 10.1038/3877. [DOI] [PubMed] [Google Scholar]
- 11.Schwarz K, Gauss GH, Ludwig L, et al. RAG mutations in human B cell-negative SCID. Science. 1996;274:97–99. doi: 10.1126/science.274.5284.97. [DOI] [PubMed] [Google Scholar]
- 12.Moshous D, Callebaut I, de Chasseval R, et al. Artemis, a novel DNA double-strand break repair/V(D)J recombination protein, is mutated in human severe combined immune deficiency. Cell. 2001;105:177–86. doi: 10.1016/s0092-8674(01)00309-9. [DOI] [PubMed] [Google Scholar]
- 13.van der Burg M, Ijspeert H, Verkaik NS, et al. A DNA-PKcs mutation in a radiosensitive T-B- SCID patient inhibits Artemis activation and nonhomologous end-joining. J Clin Invest. 2009;119:91–8. doi: 10.1172/JCI37141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kung C, Pingel JT, Heikinheimo M, et al. Mutations in the tyrosine phosphatase CD45 gene in a child with severe combined immunodeficiency disease. Nature Medicine. 2000;6:343–45. doi: 10.1038/73208. [DOI] [PubMed] [Google Scholar]
- 15.Dadi HK, Simon AJ, Roifman CM. Effect of CD3 delta deficiency on maturation of alpha/beta and gamma/delta T-cell lineages in severe combined immunodeficiency. New Engl J Med. 2003;349:1821–28. doi: 10.1056/NEJMoa031178. [DOI] [PubMed] [Google Scholar]
- 16.Roberts JL, Lauritsen JPH, Cooney M, et al. T-B+NK+ severe combined immunodeficiency caused by complete deficiency of the CD3 xi subunit of the T-cell antigen receptor complex. Blood. 2007;109:3198–206. doi: 10.1182/blood-2006-08-043166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Myers LA, Patel DD, Puck JM, Buckley RH. Hematopoietic stem cell transplantation for severe combined immunodeficiency in the neonatal period leads to superior thymic output and improved survival. Blood. 2002;99:872–8. doi: 10.1182/blood.v99.3.872. [DOI] [PubMed] [Google Scholar]
- 18.Railey MD, Lokhnygina Y, Buckley RH. Long-term clinical outcome of patients with severe combined immunodeficiency who received related donor bone marrow transplants without pretransplant chemotherapy or post-transplant GVHD prophylaxis. J Pediatr. 2009;155:834–40. e1. doi: 10.1016/j.jpeds.2009.07.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Buckley RH. Transplantation of hematopoietic stem cells in human severe combined immunodeficiency: longterm outcomes. Immunol Res. 2011;49:25–43. doi: 10.1007/s12026-010-8191-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Heimall J, Keller M, Saltzman R, et al. Diagnosis of 22q11.2 deletion syndrome and artemis deficiency in two children with T-B-NK+ immunodeficiency. J Clin Immunol. 2012;32:1141–4. doi: 10.1007/s10875-012-9741-9. [DOI] [PubMed] [Google Scholar]
- 21.Altman P. Blood leukocyte values: man. Washington, D.C: 1961. [Google Scholar]
- 22.Stephan JL. Severe combined immunodeficiency:retrospective single-center study of clinical presentation and outcome in 117 patients. J Pediatr. 1993;123:564–72. doi: 10.1016/s0022-3476(05)80951-5. [DOI] [PubMed] [Google Scholar]
- 23.Buckley RH. Primary immunodeficiency or not? Making the correct diagnosis. J Allergy Clin Immun. 2006;117:756–58. doi: 10.1016/j.jaci.2006.01.008. [DOI] [PubMed] [Google Scholar]
- 24.Buckley RH. The long quest for neonatal screening for SCID. J Allergy Clin Immunol. 2012;129:597–604. doi: 10.1016/j.jaci.2011.12.964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Prevention of rotavirus disease: updated guidelines for use of rotavirus vaccine. Pediatrics. 2009;123:1412–20. doi: 10.1542/peds.2009-0466. [DOI] [PubMed] [Google Scholar]
- 26.Patel NC, Hertel PM, Estes MK, et al. Vaccine-acquired rotavirus in infants with severe combined immunodeficiency. N Engl J Med. 2010;362:314–9. doi: 10.1056/NEJMoa0904485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Uygungil B, Bleesing JJ, Risma KA, McNeal MM, Rothenberg ME. Persistent rotavirus vaccine shedding in a new case of severe combined immunodeficiency: A reason to screen. J Allergy Clin Immunol. 2010;125:270–1. doi: 10.1016/j.jaci.2009.10.029. [DOI] [PubMed] [Google Scholar]
- 28.Werther RL, Crawford NW, Boniface K, Kirkwood CD, Smart JM. Rotavirus vaccine induced diarrhea in a child with severe combined immune deficiency. J Allergy Clin Immunol. 2009;124:600. doi: 10.1016/j.jaci.2009.07.005. [DOI] [PubMed] [Google Scholar]
- 29.Vogel BH, Bonagura V, Weinburg GA, et al. Newborn Screening for SCID in New York State: Experience from the First Two Years. J Clin Immunol. 2014;34:289–303. doi: 10.1007/s10875-014-0006-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kwan A, Church JA, Cowan MJ, et al. Newborn screening for severe combined immunodeficiency and T-cell lymphopenia in California: Results of the first 2 years. J Allergy Clin Immunol. 2013;132:140–150. doi: 10.1016/j.jaci.2013.04.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kwan A, Abrahams RS, Currier R, et al. Newborn screening for severe combined immunodeficiency in 11 screening programs in the United States. JAMA. 2014;312:729–738. doi: 10.1001/jama.2014.9132. [DOI] [PMC free article] [PubMed] [Google Scholar]