

Abstract
Mia40 participates in oxidative protein folding within the mitochondrial intermembrane space (IMS) by mediating the transfer of reducing equivalents from client proteins to FAD-linked oxidoreductases of the Erv1 family (lfALR in mammals). Here we investigate the specificity of the human Mia40/lfALR system towards non-cognate unfolded protein substrates to assess whether the efficient introduction of disulfides requires a particular amino acid sequence context or the presence of an IMS targeting signal. Reduced pancreatic ribonuclease A (rRNase), avian lysozyme, and riboflavin binding protein are all competent substrates of the Mia40/lfALR system, although they lack those sequence features previously thought to direct disulfide bond formation in cognate IMS substrates. The oxidation of rRNase by Mia40 does not limit overall turnover of unfolded substrate by the Mia40/lfALR system. Mia40 is an ineffective protein disulfide isomerase when its ability to restore enzymatic activity from scrambled RNase is compared to that of protein disulfide isomerase. Mia40’s ability to bind amphipathic peptides is evident by avid binding to the isolated B-chain during the insulin reductase assay. In aggregate these data suggest that the Mia40/lfALR system has a broad sequence specificity and that potential substrates may be protected from adventitious oxidation by kinetic sequestration within the mitochondrial IMS.
Keywords: Augmenter of liver regeneration, Mia40, Mitochondrial intermembrane space targeting sequence, Protein disulfide isomerise, Oxidative protein folding, Disulfide exchange
INTRODUCTION
The finding that human Quiescin Q6 [1] was a multidomain FAD-dependent sulfhydryl oxidase [2– 4] led to the discovery that the yeast growth factor Erv1p (essential for respiration and viability) was also a disulfide-generating enzyme [5]. Both families of flavin-dependent sulfhydryl oxidases share fundamental commonalities in catalytic mechanism: a redox-active disulfide is positioned proximal to a FAD cofactor allowing the oxidation of remote thiol groups to be coupled to the reduction of molecular oxygen or alternate electron acceptors [4, 6–9]. Erv1p functions during oxidative folding in the yeast mitochondrial intermembrane space (IMS). Here, nascent cytosolic polypeptide chains, carrying a range of targeting sequence motifs, enter the mitochondrial IMS via the outer membrane translocase complex to be subsequently trapped via disulfide bond formation [10–16]. Each disulfide generated releases a pair of electrons which are passed to the oxidoreductases Mia40 and then to flavoprotein Erv1p [16–18].
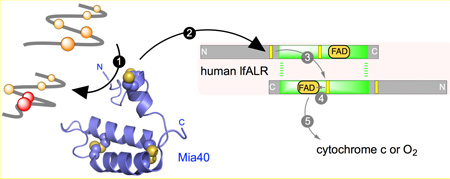
Our studies on the human counterpart of Erv1p (augmenter of liver regeneration, ALR; also abbreviated GFER, Growth Factor ERV-like) suggested that cytochrome c was an alternate physiological oxidant of the enzyme [19]. We further demonstrated that human Mia40 was obligatorily oxidized by the distal disulfide of lfALR with the flow of reducing equivalents depicted by arrows 2–5 in the schematic representation shown in Figure 1 [20]. These finding were corroborated with the yeast Erv1p/Mia40 system with the additional insight that redox equilibration between distal and proximal redox centers occurs across the dimer interface (Figure 1, step 3) [21–23]. Such inter-subunit redox communication was also proposed for lfALR [24]. The structural aspects of the communication between Mia40 and Erv1p or ALR have received considerable attention [22, 24–26].
Figure 1.

The flow of reducing equivalents during the oxidation of a protein dithiol catalyzed by Mia40 and lfALR. Mia40 (PDB 2K3J) is reduced by a reduced substrate (step 1) and then communicates with the distal CxxC disulfide in one subunit of homodimeric lfALR (step 2). Step 3 depicts intersubunit thiol-disulfide exchange, generating the reduced proximal disulfide, followed by transfer of reducing equivalents to the FAD prosthetic group (step 4). Oxidation is completed by reduction of cytochrome c or molecular oxygen (step 5).
Three-dimensional structures of both the yeast and mammalian Mia40 proteins [26–28] confirm the presence of a helix-turn-helix motif secured by 2 inter-strand disulfide bonds (Figure 1). Substrates bind orthogonally to the helical strands in a shallow hydrophobic depression that contributes to the recognition and retention of substrates, and to their subsequent efficient oxidation by the adjacent CPC redox motif of Mia40 (shown above the two main helices in Figure 1) [26–28]. Analyses of the amino acid sequences of IMS substrates of Mia40 have identified a cysteine-containing sequence (the mitochondrial IMS-targeting signal, MISS; or the IMS-targeting signal, ITS) that was believed to be necessary and sufficient for IMS targeting [11, 29, 30]. Tokatlidis and colleagues have suggested that a 9-residue amphipathic helix contained in client protein substrates within the CX9C and CX3C families forms a mixed disulfide with Mia40 facilitating the emergence of the native fold via a mechanism involving docking and sliding [11, 30]. However, a number of IMS proteins e.g. DRE2, ATP23, Erv1/lfALR, Ccs1, SOD1, and anamorsin contain neither the classical cysteine motifs nor ITS import sequences, although they have been shown to be Mia40 clients in vivo and/or in vitro [15, 31–34]. Schmid and coworkers further probed the substrate specificity of Mia40 by using cysteine mutants of IMS proteins [35, 36]. They demonstrated that rather than a precisely defined sequence, regions of hydrophobicity adjacent to cysteine residues are sufficient for binding and covalent capture by Mia40 [36]. In addition, Herrmann and Reimer and their colleagues have shown that Mia40 can bind a variety of non-cysteine-containing peptides and has chaperone-like activity towards unfolded proteins [31].
These interesting studies leave uncertain the range of protein sequences that comprise substrates of the Mia40/lfALR disulfide generating system. Instead of bona-fide substrates of the IMS, with their generally conserved cysteine motifs and ITS sequences, we wanted to challenge the redox behavior of Mia40/lfALR with potential substrates lacking all of these features. For this work we chose three diverse proteins of the vertebrate secretory apparatus (ribonuclease A, egg white lysozyme, and riboflavin binding protein; RNase, lysozyme and RfBP respectively). We show that these reduced proteins are competent substrates of the Mia40/lfALR pathway reinforcing the idea that Mia40 dependent oxidative pathways have a broader oxidative scope than originally envisaged. To further investigate the catalytic potential of Mia40 to service structural disulfide bonds, we have benchmarked the suggested isomerase activity of Mia40 against the mammalian protein disulfide isomerase. In addition to providing new insights into Mia40 catalysis, our work identifies potential complications in the interpretation of the insulin reductase assays widely employed to explore the catalytic activities of disulfide oxidoreductases.
Experimental Procedures
Materials and General Methods
Commercial reagents were obtained as described previously [37–39]. Unless otherwise stated, 50 mM potassium phosphate buffer, pH 7.5, containing 1 mM EDTA was used throughout. Absorbance and fluorescence experiments were performed on HP8453 diode-array spectrophotometers and an SLM-Aminco Bowman 2 luminescence spectrometer respectively. Thiols were routinely quantitated by diluting samples into 180 µL of 0.2 mM DTNB in self-masking microcells and recording the increase in absorbance at 412 nm over a corresponding blank using a molar extinction coefficient for the 5-thio-2-nitobenzoate dianion of 14150 M−1 cm−1 at 412 nm [40]. Data were visualized using GraphPad Prism software.
Expression and Treatment of Proteins
lfALR was purified as described previously [20]. Wild type Mia40 was prepared in its short linker form as before [20]. The Mia40APA double-mutant (C53A, C55A) was treated similarly, except TCEP was excluded from the affinity chromatography step and β-mercaptoethanol was absent from the Ni- NTA elution buffers. Mia40 and Mia40APA were quantitated using a molar extinction coefficient of 13.3 mM−1 cm−1 at 280 nm (calculated using ProtParam [41]). Human PDI was purified and quantitated as described previously [42]. Bovine pancreatic RNase A (Sigma), hen egg white lysozyme (Sigma), and hen egg white riboflavin binding protein (prepared as in [43]) were reduced with a 20-fold molar excess of DTT per protein thiol for 1 h at 50 °C in 50 mM phosphate buffer, pH 8.0, containing 1 mM EDTA, 6 M guanidine hydrochloride and 100 mM NaCl. Excess reductant was subsequently removed by gel-filtration of 0.7 mL unfolded reduced protein on PD-10 gel filtration columns (GE Healthcare). Complete separation of reduced protein from excess reductant was verified by sampling small volumes of eluent using DTNB. Reduced RNase (rRNase) was eluted in 2 mM acetate buffer, pH 4.0, containing 1 mM EDTA. Reduced lysozyme and reduced RfBP were eluted in the same buffer supplemented with 3 M urea or 3 M guanidine HCl respectively. Reduced RNase, lysozyme and RfBP were quantitated using molar extinction coefficients of 9800 M−1 cm−1 at 278 nm; 34000 M−1 cm−1 at 280 nm; and 49000 M−1 cm−1 at 280 nm respectively. Scrambled RNase (sRNase) was prepared essentially as described previously [42]. In brief, RNase (2.9 mM, in 6 M guanidine HCl in 100 mM Tris buffer, pH 8.0, in the absence of EDTA) was incubated for 2 days under anaerobic conditions in the presence of one equivalent (2.9 mM) DTT. The solution was then stirred over an atmosphere of oxygen for 1 day yielding a solution with negligible free thiols. The oxidized protein was gel filtered using a PD10 column equilibrated with 3.5 mM potassium acetate buffer, pH 4, and stored before use at – 80 °C. The preparation used in this work exhibited 1.3 ± 0.7% activity (a value consistent with the random reformation of 4 disulfide bonds [44]).
RNase Activity Assay
Aliquots (20 µL) were withdrawn from incubation mixtures, as defined in the legend to Figure 5, and immediately mixed with 180 µL of 100 mM Tris-HCl buffer, pH 7.0, 25 °C, containing 1.11 mM cCMP and 1 mM EDTA. The increase in absorbance A296 – A310 reflects the hydrolysis of cCMP. Subtraction of the absorbance at 310 nm improved the signal to noise ratio as described previously [35]. Relative activities of the aliquots were determined by comparing the slope of the initial rates with those of an equivalent concentration of native RNase.
Figure 5. Assessing the isomerase activity of Mia40 using sRNase.

Panel A: mixtures containing 50 µM scrambled RNase in 50 mM phosphate buffer, pH 7.5 containing 3 mM GSH and 0.3 mM GSSG, were incubated at 25 °C with 1 µM PDI (black circles), without (grey), with 2 µM Mia40 (red), 10 µM Mia40 (blue), 2 µM Mia40APA(purple) or 10 µM Mia40APA (green circles). Aliquots were withdrawn and measured for RNase activity (see Methods). The average activity of 3 independent reactions is plotted in Panel A and the values recorded after 2 h are summarized in Panel B.
Insulin Reductase Assay
Stock solutions of bovine insulin were prepared in water by bringing a suspension of the solid to a pH of 3 with HCl and then returning the clear solution to a pH of 6 with KOH. Reaction mixtures contained 50 µM insulin in 50 mM phosphate buffer, pH 7.5, 25 °C, containing 1 mM EDTA in the presence or absence of wild-type Mia40 or the Mia40APA double mutant. Reactions were started by the addition of 5 mM DTT and the subsequent increase in light scattering monitored at 600 nm.
Stopped Flow Methods
An SF-61 DX2 stopped-flow spectrometer (Hi-Tech Scientific) was used for rapid mixing kinetics experiments in fluorescence mode exciting at 290 nm with a 300 nm cutoff filter in the emission beam. Oxidized Mia40 was mixed at 25 °C with rRNAse to give final concentrations of 0.5 µM and 10–50 µM respectively in 50 mM phosphate buffer, pH 7.5. Fluorescence traces were visualized and analyzed using the instrument software, KinetAsyst 3, and with KinTek Explorer [45]. The apparent rate constants from biphasic fits to the first 5 s of fluorescence increase were plotted as a function of rRNase concentration. The fast phase was fit to saturation kinetics using GraphPad Prism.
Sequence Analysis
The primary amino acid sequences of RNase, chicken lysozyme and RfBP were analyzed using the ScanProsite tool (http://prosite.expasy.org/scanprosite/) for potential ITS motifs using: [YWF] -x(2)- [AILVFYWGMP]-[AILVFYWGMP]-x(2)-C, C-x(2)-[AILVFYWGMP]-[AILVFYWGMP]-x(2)-[YWF]. ScanProsite analysis identified no ITS motifs using these sequences. ScanProsite, in conjunction with PDB structures visualized with PyMol (Schrödinger, LLC), showed that neither paired CX3C nor CX9C motifs are found in the context of helix-coil-helix domains in RNase, lysozyme or RfBP.
RESULTS AND DISCUSSION
Mia40/lfALR is a general catalyst of protein oxidation
Three diverse disulfide-containing products of the vertebrate secretory apparatus were chosen for this study exploring the ability of Mia40/lfALR to oxidize non-cognate substrates using oxygen as the terminal electron acceptor (Figure 1). The proteins show a range of secondary structures, disulfide connectivities, and pI values (RNase, 9.6; lysozyme, 11.35, RfBP, ~ 4.0). ITS sequences are absent in all three proteins (Figure 2; see Methods; [29, 30]), as are paired CX3C or CX9C motifs frequently encountered in the context of helix-coil-helix super secondary structures in IMS-resident proteins (see Methods). RNase, lysozyme and RfBP were reduced under denaturing conditions and freed from excess reductant by size-exclusion chromatography (see Methods). In all three cases efficient oxidation of these proteins requires the presence of both Mia40 and lfALR (Figure 3). Panel A shows the disappearance of rRNase thiols mediated by the Mia40/lfALR system. The conditions chosen (500 µM RNase thiols, 20 µM oxidized Mia40 and 1 µM lfALR) require 250 turnovers of lfALR to completely oxidize substrate thiols to disulfides. Under these conditions the initial rate corresponds to an apparent turnover of 8.9 ± 0.1 RNase disulfide bonds generated/min per lfALR active site. In a previous study, Daithankar et al. examined the concentration dependence for the turnover number of reduced Mia40 by lfALR; at 20 µM Mia40 a value of 7/min was found under otherwise identical conditions to those in Figure 3 [20]. The similarity between these two turnover numbers for lfALR (encompassing steps 1–5 or 2–5 in Figure 1) suggests that the reduction of Mia40 by rRNase thiols (step 1) is not rate-limiting in these experiments. Figure 3A also shows that Mia40 is an essential mediator between rRNase and lfALR; reaction mixtures lacking either enzyme give background levels of rRNase autoxidation. Further, when the redox-active CPC motif of Mia40 is replaced with an APA sequence (Mia40APA) the oxidation of rRNase is again ablated (Figure 3A). The time-dependent slowing of the rate of oxidation observed in Figure 3A likely reflects the decreasing access to substrate thiols as the number of disulfides accumulates, as is observed with the oxidation of rRNase catalyzed by QSOX [46–48].
Figure 2. RNase, lysozyme and RfBP lack ITS sequences.

RNase, lysozyme and riboflavin binding protein, following their signal sequences, show neither ITS sequences nor paired CX3C or CX9C motifs (see the Text). Cysteine residues are shown in inverse font. RNase, lysozyme and RfBP contain 4, 4 and 9 disulfides respectively.
Figure 3. Oxidation of reduced protein non-cognate substrates of the Mia40/lfALR system.

Reactions were started by the addition of the enzymes indicated below and aliquots were withdrawn at the indicated times for measurement of thiol titer using DTNB. Panel A represents 62.5 µM of rRNase (8 cysteines; 500 µM thiols in phosphate buffer, pH 7.5) in the additional presence of 1 µM lfALR and 20 µM Mia40 (filled black circles), 1 µM lfALR and 20 µM Mia40APA (open black circles), 20 µM Mia40 (red filled squares), 20 µM Mia40APA (red open squares), or 1 µM lfALR (blue filled triangles). Panel B shows the incubation of 62.5 µM of reduced lysozyme (8 cysteines; 500 µM thiols in phosphate buffer with carry over of 0.15 M urea; see Methods) with 1 µM lfALR and 20 µM Mia40 (filled black circles), 20 µM Mia40 (filled red squares), or 1 µM lfALR (filled blue triangles). Panel C shows the comparable treatment of 27.8 µM reduced RfBP (18 cysteines; 500 µM thiols in phosphate buffer with carry over of 0.2 M guanidine hydrochloride; see Methods) using 1 µM lfALR and 20 µM Mia40 (filled black circles), 20 µM Mia40 (filled red squares), or 1 µM lfALR (filled blue triangles). All experiments were performed in triplicate at 25 °C in 50 mM phosphate buffer, pH 7.5, containing 1mM EDTA.
Figure 3B shows comparable experiments performed with reduced lysozyme. As observed previously the inclusion of 3 M urea was necessary to avoid precipitation of the unfolded reduced protein. Clearly, the presence of both Mia40 and lfALR were required for significant thiol oxidation in aerobic solution. The turnover number calculated from the initial rate of disulfide bond generation was 10 ± 0.4 /min/per lfALR active site comparable to that obtained for rRNase. Finally, RfBP is a secreted protein with a much more complicated disulfide connectivity than that of RNase or lysozyme. However upon reduction of this 9-disulfide protein it also becomes a competent substrate in the Mia40/lfALR system with a turnover number of 11.4 ± 1.1 disulfides/min/lfALR active site (Figure 3C). In summary, Figure 3 shows that Mia40 can efficiently mediate the transfer of reducing equivalents from diverse non-cognate substrates directly to lfALR.
Reduced RNase interacts with oxidized Mia40
Schmidt and coworkers have used rapid reaction approaches to study the interaction between Mia40 and wild-type and mutant forms of its cognate partner reduced COX17. Utilizing single cysteine mutants of Cox17 they reported apparent rate constants for formation of a mixed disulfide with Mia40 of between 0.12 and 55/s following a rapid pre-equilibrium binding step (with Kd values from ~ 1 to ~ 50 µM; [36]). It was not the purpose of this work to conduct a detailed characterization of the interaction between a reduced unfolded non-cognate substrate and Mia40. Here we are only interested in evaluating whether the interaction between Mia40 and reduced substrate is sufficiently rapid to support the turnover numbers obtained for the experiments in Figure 3. We chose rRNase because it does not contain tryptophan residues thus allowing the reaction to be followed more readily. In addition to mixed disulfide bond formation the release of free reduced Mia40 would result in a further increase in the fluorescence of the TRP residue adjacent to the CPC redox active motif [27, 36]. Under pseudo first-order conditions the reaction between 0.5 µM oxidized Mia40 and a 50-fold molar excess of rRNase can be fit to 3 exponentials (inset). The slowest phase (0.02/s) is too slow to be catalytically significant and was not considered further. The concentration dependence of the first and second phases are shown in the main panel of Figure 4. The rate constants for the fastest phase can be fit to a Kd of 10.4 ± 0.2 µM rRNase with a limiting constant of 7.0 ± 0.4/s. In contrast the second phase appears concentration independent over this range (~ 0.36/s) consistent with resolution of a mixed disulfide between Mia40 and rRNase. Importantly, both processes are fast enough to ensure that the reduction of Mia40 by rRNase is not a serious limitation to the overall oxidation depicted in Figure 1 (the overall turnover in Figure 3A corresponds to 0.15/s). In sum Mia40 is effectively reduced by a reduced protein lacking ITS sequences [29, 30] at a rate that can support the oxidase activity of lfALR. Further, the Kd values and rate of disulfide exchange reactions for the non-cognate substrate, RNase, are comparable to those reported previously for the interaction between reduced Cox17 and Mia40 [36].
Figure 4. Reduction of Mia40 by reduced RNase.

Oxidized Mia40 was mixed with rRNase at 25 °C in a stopped flow spectrofluorimeter to give final concentrations of 0.25 µM Mia40 and 10, 20, 25 and 50 µM rRNase (see Methods). The main panel shows apparent first-order rate constants for first and second phases of a reaction time course that can be fit using 3 exponentials as shown in the inset for 25 µM rRNAse. The rate constant for the fast phase saturates at 7.0 ± 0.4/s with an apparent Kd of 10.4 ± 0.2 µM. The rate constant for the second phase shows insignificant concentration dependence (averaging ~ 0.36/s). The third phase of ~ 0.02/s is significantly slower than the overall turnover number for RNase oxidation (~ 0.15/s) and was not considered further here.
Does Mia40 show significant protein disulfide isomerase activity?
There has been considerable discussion regarding the need for a protein disulfide isomerase activity in the context of oxidative protein folding within the IMS [16, 31, 32, 35]. While a number of IMS client proteins have simple patterns of disulfide connectivity, and might therefore not require a dedicated isomerase, others proteins show more complicated folds and contain multiple disulfide bonds. In an interesting prior study, Koch and Schmid [35] followed the oxidative refolding of 5 µM rRNase in the presence of 2 µM Mia40 and a glutathione redox buffer (3 mM GSH/ 0.3 mM GSSG). The regain of enzymatic activity observed over corresponding controls (amounting to ~25% over 5 h) was very modest considering that the substrate RNase was only present at a 2.5-fold higher concentration over Mia40.
A more stringent assessment of isomerase activity of thiol/disulfide oxidoreductases follows their ability to correct disulfide pairings starting with a fully oxidized RNase populated by random pairings. Scrambled RNase (sRNase; 105 possible pairings in a protein with 4 disulfide bonds; see Methods) shows the expected ~1% residual activity [44]. Figure 5 uses sRNase in the presence of a GSH/GSSG redox buffer to compare the isomerase activity of protein disulfide isomerase (PDI) to that of Mia40. Using 1 µM PDI, half-maximal RNase activity was recovered in ~35 min (Figure 5A) with 83 ± 5 % activity regained in 2h (Figure 5B). These activities need to be compared to controls without PDI because the GSH/GSSG redox buffer can directly stimulate non-enzymatic disulfide exchange reactions [49]; in Figure 5B, this non-enzymatic component amounts to 33 ± 3 % activity over 2 h. Importantly, Mia40 proved a negligible isomerase towards sRNase at both 2 and 10 µM (Figure 5A and B). Figure 5 also shows, as expected, that the redox-inactive Mia40APA construct was similarly ineffective at promoting disulfide exchange.
It could be argued that Mia40 would be a better catalyst for the isomerization of bona-fide IMS substrates. However Mia40 has been shown to be a poor isomerase towards scrambled Cox17 in the presence of the same redox buffer used in Figure 5 [35]. The Cox17 construct employed for those experiments contained two structural disulfides and no free cysteine residues. Given the permissive conditions used for that experiment (10 µM Mia40, 30 µM scrambled Cox17 in the presence of 3 mM GSH and 0.3 mM GSSG over 6 h), and the fact that there are only 3 ways to pair 4 cysteine residues, the comparative ineffectiveness of Mia40 in correcting scrambled Cox17 is striking. In summary, Mia40 is ineffective as an isomerase under these conditions towards either cognate proteins with simple disulfide patterns or against non-cognate proteins with more complicated disulfide connectivities.
Finally, a range of thiol/disulfide oxidoreductases, including PDI itself, have been characterized by their ability to catalyze the reduction of insulin driven by low concentrations of glutathione, DTT or other disulfide reductants [50–52]. Reduction of the two interchain disulfide bonds between A and B chains of insulin leads to aggregation of the poorly-soluble B chain [53]. The time required for the onset of turbidity has been widely used for assays of PDI activity [39, 54–58]. However, implementation of the insulin reductase assay with Mia40 provided a strikingly different behavior from that found with PDI. Rather than accelerate the onset of turbidity, Figure 6A shows that increasing concentrations of Mia40 impede insulin B-chain aggregation; when Mia40 and insulin are equimolar the reaction becomes very slow. The redox-inactive Mia40APA form has a less pronounced effect (Panel B) but still significantly retards the onset of turbidity in a concentration-dependent way (Panel C). These data strongly suggest that the isolated reduced insulin B chain can be sequestered by binding to the hydrophobic cleft on Mia40 even in the absence of mixed-disulfide bond formation. It is notable that a previous study identified the ability of Mia40 to bind non-cysteine containing peptides in a form of Atp23 [31]. Further Mia40 has been recognized for its holdase /chaperone activities [27, 31, 32]. On a more practical level, our data shows that the widely-used insulin reductase activity can be undermined by sequestration of B-chains via a combination of hydrophobic binding and covalent mixed disulfide bond formation. Hence the ability of an oxidoreductase to accelerate the reduction of insulin disulfides assessed by this standard turbidometric method might be masked by the tendency of the hydrophobic B-chain to bind to the enzyme under examination.
Figure 6. Mia40 suppresses the aggregation of the insulin B chain.

A solution of 50 µM insulin in 50 mM phosphate buffer, pH 7.5, containing 1 mM EDTA was incubated at 25 °C with 5 mM DTT. Reduction of the two interchain disulfide bonds in insulin leads to the development of turbidity (followed at 600 nm) following a lag phase of ~ 600 s under these conditions (Figure 6A). Increasing concentrations of Mia40 (5–50 µM) markedly lengthen the lag phase as shown in panel A. Panel B shows the corresponding behavior of the redox-inactive Mia40APA construct. Reactions including 0, 5, 10, 20, and 50 µM Mia40, or Mia40APA, are shown as black, purple, green, blue, and red traces respectively in panels A and B. In both panels the traces represent the average between two consecutive runs. Panel C: For each indicated Mia40 or Mia40APA concentration, the bars reflect the additional lag time required for incubations to reach an absorbance of 0.05 at 600 nm over the corresponding control (black lines panel A and B).
Conclusions
While the ITS plays an important role in the import of a range of proteins destined for the IMS, our studies reinforce earlier suggestions that it is not an obligatory requirement for oxidative folding at that locale. We report turnover numbers for the insertion of disulfides into non-cognate substrates by the Mia40/lfALR system, and show that overall oxidation is not limited by the transfer of reducing equivalents from unfolded substrates to Mia40. In view of the general lack of substrate specificity observed in this work, all proteins transiting the IMS en route to their final location are potential substrates of the Mia40/lfALR system. However oxidation might be prevented by efficient sequestration during interactions with translocases [12, 13, 59] or chaperones [59, 60], or might be reversed via IMS-resident reductase sytems [23, 61–63]. In terms of an expanding role for the Mia40/lfALR system, it is interesting to note that a mitochondrial matrix protein Mrp10 contains two disulfide bonds that are inserted during transit through the IMS [34].
Finally, the minimal isomerase activity observed for a simple potential substrate, scrambled Cox17, and the more challenging example studied here, sRNase, shows that IMS proteins with complex disulfide connectivities would likely need a more proficient isomerase activity to fold correctly. An interesting question for future investigation is the molecular basis for the negligible activity of Mia40 as an isomerase when benchmarked against PDI itself. Both proteins have redox active disulfide motifs that are solvent accessible; both provide areas of hydrophobic surface for substrate binding, both can be readily reduced by unfolded reduced proteins and both form mixed disulfides with their redox clients. Exploring this question should lead to a more nuanced understanding of both these thiol/disulfide oxidoreductases catalysts of oxidative protein folding.
HIGHLIGHTS.
Mia40 is a facile oxidant of a range of non-cognate unfolded reduced proteins
This broad specificity impacts consideration of protein trafficking within the IMS
Mia40 shows minimal disulfide isomerase activity with scrambled proteins
The need for additional disulfide isomerase activity in the IMS is considered
ACKNOWLEDGMENTS
This work was supported by NIH Grant GM26643 (CT) and USPHS Training Grant 1-T32- GM008550 (DH). We thank Dr. Vidyadhar Daithankar and Mr. Jason Cargill for helpful discussions.
Abbreviations
- cCMP
cytidine 2′-3′-cyclic monophosphate
- DTNB
5-5'-dithiobis(2-nitrobenzoate)
- GSH
reduced glutathione
- GSSG
oxidized glutathione
- Mia40APA
redox-inactive mutant of CPC active site sequence of wild type human Mia40
- RfBP
chicken egg white riboflavin binding protein
- RNase
bovine pancreatic ribonuclease A
- rRNase
reduced RNase
- sRNase
scrambled RNase
- TCEP
tris(2-carboxyethyl)phosphine hydrochloride
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.Coppock DL, Cina-Poppe D, Gilleran S. Genomics. 1998;54:460–468. doi: 10.1006/geno.1998.5605. [DOI] [PubMed] [Google Scholar]
- 2.Hoober KL, Glynn NM, Burnside J, Coppock DL, Thorpe C. J. Biol. Chem. 1999;274:31759–31762. doi: 10.1074/jbc.274.45.31759. [DOI] [PubMed] [Google Scholar]
- 3.Thorpe C, Hoober K, Raje S, Glynn N, Burnside J, Turi G, Coppock D. Arch. Biochem. Biophys. 2002;405:1–12. doi: 10.1016/s0003-9861(02)00337-5. [DOI] [PubMed] [Google Scholar]
- 4.Kodali VK, Thorpe C. Antioxid. Redox Signal. 2010;13:1217–1230. doi: 10.1089/ars.2010.3098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lee J, Hofhaus G, Lisowsky T. FEBS Lett. 2000;477(1–2):62–66. doi: 10.1016/s0014-5793(00)01767-1. [DOI] [PubMed] [Google Scholar]
- 6.Thorpe C, Coppock DL. J. Biol Chem. 2007;282:13929–13933. doi: 10.1074/jbc.R600037200. [DOI] [PubMed] [Google Scholar]
- 7.Heckler EJ, Alon A, Fass D, Thorpe C. Biochemistry. 2008;47:4955–4963. doi: 10.1021/bi702522q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fass D. Biochim. Biophys. Acta. 2008;1783:557–566. doi: 10.1016/j.bbamcr.2007.11.009. [DOI] [PubMed] [Google Scholar]
- 9.Heckler EJ, Rancy PC, Kodali VK, Thorpe C. Biochim. Biophys. Acta. 2008;1783:567–577. doi: 10.1016/j.bbamcr.2007.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chacinska A, Koehler CM, Milenkovic D, Lithgow T, Pfanner N. Cell. 2009;138:628–644. doi: 10.1016/j.cell.2009.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sideris DP, Tokatlidis K. Antioxid. Redox Sign. 2010;13:1189–1204. doi: 10.1089/ars.2010.3157. [DOI] [PubMed] [Google Scholar]
- 12.Fischer M, Riemer J. Int. J. Cell Biol. 2013;2013:742923. doi: 10.1155/2013/742923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wenz LS, Opalinski L, Wiedemann N, Becker T. Biochim. Biophys. Acta. 2015;1853:1119–1129. doi: 10.1016/j.bbamcr.2015.01.012. [DOI] [PubMed] [Google Scholar]
- 14.Riemer J, Bulleid N, Herrmann JM. Science. 2009;324:1284–1287. doi: 10.1126/science.1170653. [DOI] [PubMed] [Google Scholar]
- 15.Herrmann JM, Kohl R. J. Cell Biol. 2007;176:559–563. doi: 10.1083/jcb.200611060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Deponte M, Hell K. J. Biochem. 2009;146:599–608. doi: 10.1093/jb/mvp133. [DOI] [PubMed] [Google Scholar]
- 17.Chacinska A, Pfannschmidt S, Wiedemann N, Kozjak V, Sanjuan Szklarz LK, Schulze- Specking A, Truscott KN, Guiard B, Meisinger C, Pfanner N. EMBO J. 2004;23:3735–3746. doi: 10.1038/sj.emboj.7600389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mesecke N, Terziyska N, Kozany C, Baumann F, Neupert W, Hell K, Herrmann JM. Cell. 2005;121:1059–1069. doi: 10.1016/j.cell.2005.04.011. [DOI] [PubMed] [Google Scholar]
- 19.Farrell SR, Thorpe C. Biochemistry. 2005;44:1532–1541. doi: 10.1021/bi0479555. [DOI] [PubMed] [Google Scholar]
- 20.Daithankar VN, Farrell SR, Thorpe C. Biochemistry. 2009;48:4828–4837. doi: 10.1021/bi900347v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ang SK, Lu H. J. Biol. Chem. 2009;284:28754–28761. doi: 10.1074/jbc.M109.021113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Guo PC, Ma JD, Jiang YL, Wang SJ, Bao ZZ, Yu XJ, Chen Y, Zhou CZ. J. Biol. Chem. 2012;287:34961–34969. doi: 10.1074/jbc.M112.394759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bien M, Longen S, Wagener N, Chwalla I, Herrmann JM, Riemer J. Mol. Cell. 2010;37:516–528. doi: 10.1016/j.molcel.2010.01.017. [DOI] [PubMed] [Google Scholar]
- 24.Banci L, Bertini I, Calderone V, Cefaro C, Ciofi-Baffoni S, Gallo A, Tokatlidis K. J. Am. Chem. Soc. 2012;134:1442–1445. doi: 10.1021/ja209881f. [DOI] [PubMed] [Google Scholar]
- 25.Banci L, Bertini I, Calderone V, Cefaro C, Ciofi-Baffoni S, Gallo A, Kallergi E, Lionaki E, Pozidis C, Tokatlidis K. Proc Natl. Acad. Sci. USA. 2011;108:4811–4816. doi: 10.1073/pnas.1014542108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Banci L, Bertini I, Cefaro C, Cenacchi L, Ciofi-Baffoni S, Felli IC, Gallo A, Gonnelli L, Luchinat E, Sideris D, Tokatlidis K. Proc. Natl. Acad. Sci. USA. 2010;107:20190–20195. doi: 10.1073/pnas.1010095107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Banci L, Bertini I, Cefaro C, Ciofi-Baffoni S, Gallo A, Martinelli M, Sideris DP, Katrakili N, Tokatlidis K. Nat. Struct. Mol. Biol. 2009;16:198–206. doi: 10.1038/nsmb.1553. [DOI] [PubMed] [Google Scholar]
- 28.Endo T, Yamano K, Kawano S. Antioxid. Redox Sign. 2010;13:1359–1373. doi: 10.1089/ars.2010.3099. [DOI] [PubMed] [Google Scholar]
- 29.Milenkovic D, Ramming T, Muller JM, Wenz LS, Gebert N, Schulze-Specking A, Stojanovski D, Rospert S, Chacinska A. Mol. Biol. Cell. 2009;20:2530–2539. doi: 10.1091/mbc.E08-11-1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sideris DP, Petrakis N, Katrakili N, Mikropoulou D, Gallo A, Ciofi-Baffoni S, Banci L, Bertini I, Tokatlidis K. J. Cell Biol. 2009;187:1007–1022. doi: 10.1083/jcb.200905134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Weckbecker D, Longen S, Riemer J, Herrmann JM. Embo Journal. 2012;31:4348–4358. doi: 10.1038/emboj.2012.263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chatzi A, Tokatlidis K. Antioxid. Redox Sign. 2013;19:54–62. doi: 10.1089/ars.2012.4855. [DOI] [PubMed] [Google Scholar]
- 33.Banci L, Bertini I, Ciofi-Baffoni S, Boscaro F, Chatzi A, Mikolajczyk M, Tokatlidis K, Winkelmann J. Chem. Biol. 2011;18:794–804. doi: 10.1016/j.chembiol.2011.03.015. [DOI] [PubMed] [Google Scholar]
- 34.Longen S, Woellhaf MW, Petrungaro C, Riemer J, Herrmann JM. Devel. Cell. 2014;28:30–42. doi: 10.1016/j.devcel.2013.11.007. [DOI] [PubMed] [Google Scholar]
- 35.Koch JR, Schmid FX. J. Mol. Biol. 2014;426:4087–4098. doi: 10.1016/j.jmb.2014.10.022. [DOI] [PubMed] [Google Scholar]
- 36.Koch JR, Schmid FX. Nat. Comm. 2014;5:3041. doi: 10.1038/ncomms4041. [DOI] [PubMed] [Google Scholar]
- 37.Schaefer-Ramadan SA, Gannon SA, Thorpe C. Biochemistry. 2013;52:8323–8332. doi: 10.1021/bi401305w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Israel BA, Kodali VK, Thorpe C. J. Biol. Chem. 2014;289:5274–5284. doi: 10.1074/jbc.M113.536219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sapra A, Ramadan D, Thorpe C. Biochemistry. 2015;54:612–621. doi: 10.1021/bi501360e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Eyer P, Worek F, Kiderlen D, Sinko G, Stuglin A, Simeon-Rudolf V, Reiner E. Anal. Biochem. 2003;312:224–227. doi: 10.1016/s0003-2697(02)00506-7. [DOI] [PubMed] [Google Scholar]
- 41.Gasteiger E, Gattiker A, Hoogland C, Ivanyi I, Appel RD, Bairoch A. Nucleic Acids Res. 2003;31:3784–3788. doi: 10.1093/nar/gkg563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rancy PC, Thorpe C. Biochemistry. 2008;47:12047–12056. doi: 10.1021/bi801604x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Miller MS, White HB., 3rd Methods Enzymol. 1986;122:227–234. doi: 10.1016/0076-6879(86)22174-6. [DOI] [PubMed] [Google Scholar]
- 44.Haber E, Anfinsen CB. J. Biol. Chem. 1962;237:1839–1844. [PubMed] [Google Scholar]
- 45.Johnson KA, Simpson ZB, Blom T. Anal. Biochem. 2009;387:20–29. doi: 10.1016/j.ab.2008.12.024. [DOI] [PubMed] [Google Scholar]
- 46.Hoober KL, Joneja B, White HB, III, Thorpe C. J. Biol. Chem. 1996;271:30510–30516. doi: 10.1074/jbc.271.48.30510. [DOI] [PubMed] [Google Scholar]
- 47.Hoober KL, Sheasley SS, Gilbert HF, Thorpe C. J. Biol. Chem. 1999;274:22147–22150. doi: 10.1074/jbc.274.32.22147. [DOI] [PubMed] [Google Scholar]
- 48.Hoober KL, Thorpe C. Biochemistry. 1999;38:3211–3217. doi: 10.1021/bi9820816. [DOI] [PubMed] [Google Scholar]
- 49.Lyles MM, Gilbert HF. Biochemistry. 1991;30:613–619. doi: 10.1021/bi00217a004. [DOI] [PubMed] [Google Scholar]
- 50.Holmgren A. J. Biol. Chem. 1979;254:9627–9632. [PubMed] [Google Scholar]
- 51.Luthman M, Holmgren A. J. Biol. Chem. 1982;257:6686–6690. [PubMed] [Google Scholar]
- 52.Lundstrom J, Holmgren A. J. Biol. Chem. 1990;265:9114–9120. [PubMed] [Google Scholar]
- 53.Sanger F. Biochem. J. 1949;44:126–128. doi: 10.1042/bj0440126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Smith AM, Chan J, Oksenberg D, Urfer R, Wexler DS, Ow A, Gao L, McAlorum A, Huang SG. J. Biomol. Screen. 2004;9:614–620. doi: 10.1177/1087057104265292. [DOI] [PubMed] [Google Scholar]
- 55.Jasuja R, Passam FH, Kennedy DR, Kim SH, van Hessem L, Lin L, Bowley SR, Joshi SS, Dilks JR, Furie B, Furie BC, Flaumenhaft R, Clin J. Invest. 2012;122:2104–2113. doi: 10.1172/JCI61228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ge J, Zhang CJ, Li L, Chong LM, Wu X, Hao P, Sze SK, Yao SQ. ACS Chem. Biol. 2013;8:2577–2585. doi: 10.1021/cb4002602. [DOI] [PubMed] [Google Scholar]
- 57.Horibe T, Torisawa A, Okuno Y, Kawakami K. Chem bio chem. 2014;15:1599–1606. doi: 10.1002/cbic.201400050. [DOI] [PubMed] [Google Scholar]
- 58.Xu S, Butkevich AN, Yamada R, Zhou Y, Debnath B, Duncan R, Zandi E, Petasis NA, Neamati N. Proc. Natl. Acad. Sci. USA. 2012;109:16348–16353. doi: 10.1073/pnas.1205226109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Dudek J, Rehling P, van der Laan M. Biochim. Biophys. Acta. 2013;1833:274–285. doi: 10.1016/j.bbamcr.2012.05.028. [DOI] [PubMed] [Google Scholar]
- 60.Stojanovski D, Bohnert M, Pfanner N, van der Laan M. Cold Spring Harbor Perspect Biol. 2012;4 doi: 10.1101/cshperspect.a011320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kojer K, Peleh V, Calabrese G, Herrmann JM, Riemer J. Mol. Biol. Cell. 2015;26:195–204. doi: 10.1091/mbc.E14-10-1422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kojer K, Riemer J. Biochim. Biophys. Acta. 2014 doi: 10.1016/j.bbapap.2014.02.004. [DOI] [PubMed] [Google Scholar]
- 63.Vogtle FN, Burkhart JM, Rao S, Gerbeth C, Hinrichs J, Martinou JC, Chacinska A, Sickmann A, Zahedi RP, Meisinger C. Mol. Cell. Proteom. 2012;11:1840–1852. doi: 10.1074/mcp.M112.021105. [DOI] [PMC free article] [PubMed] [Google Scholar]