

Abstract
An umpolung approach to the synthesis of diaryl ketones has been developed based on in situ generation of acyl anion equivalents and their catalytic arylation. This method entails the base promoted palladium catalyzed direct C–H arylation of 2 The resulting 2,2-diaryl-1,3-dithianes with aryl bromides. Use of MN(SiMe3)2 (M=Li, Na) base results in reversible deprotonation of the weakly acidic dithiane. In the presence of a Pd(NiXantphos)-based catalyst and aryl bromide, cross-coupling of the metallated 2-aryl-1,3-dithiane takes place under mild conditions (2 h at rt) with yields as high as 96%. The resulting 2,2-diaryl-1,3-dithianes were converted into diaryl ketones by either molecular iodine, N-bromo succinimide (NBS) or Selectfluor in the presence of water. The dithiane arylation/hydrolysis can be performed in a one-pot procedure to yield a good to excellent yields. This method is suitable for rapid and large-scale synthesis of diaryl ketones. A one-pot preparation of anti-cholesterol drug fenofibrate (TriCor®) has been achieved on 10.0 mmol scale in 86% yield.
Keywords: C-C bond formation, cross-coupling, dithianes, ketones, palladium, umpolung
Introduction
Synthetic methods based on reversal of functional group polarity, or umpolung methods, constitute a valuable strategy in organic synthesis because they provide unconventional approaches to common structural motifs.[1] Nature also employs the umpolung reactivity to forge C–C bonds.[2] The most widely used umpolung methods involve the generation of acyl anion equivalents to elaborate carbonyl compounds.[3] In the early 1900s, the cyanide catalyzed homodimerization of aldehydes (benzoin condensation) led to the realization that the polarity of the carbonyl functionality could be reversed.[4] However, the great potential of the umpolung strategy was not broadly recognized until pioneering studies by Corey and Seebach on the use of metallated 1,3-dithianes as masked acyl anion equivalents.[5] To date, C–C bond formation reactions between metallated dithianes –almost always 2-lithio-1,3-dithianes– and various electrophilic reagents such as alkyl halides, epoxides and carbonyl compounds, have been extensively applied to the synthesis of intermediates in route to complex natural products.[6] 2-Lithio-1,3-dithianes can be generated by the action of strong bases (n-BuLi or t-BuLi) on dithianes at low temperatures.[7] Despite the widespread and elegant application of lithiated dithiane derivatives in organic synthesis, it is surprising that they have rarely been employed in catalytic reactions as acyl anion equivalents. To the best of our knowledge, there is only one publication claiming the use of dithianes in a catalytic reaction as acyl anion equivalents.[8] This manuscript involves the nickel-catalyzed reaction of preformed 2-lithio-1,3-dithianes with benzoyl halides to form 1,2-diketones derivatives (average yield 46%).[9] The conflicting description in this manuscript makes it unclear if benzoyl chlorides or bromides were actually employed.
Given the synthetic ease of accessing 2-aryl-1,3-dithianes, we wondered if it would be possible to use metallated 2-aryl-1,3-dithianes in palladium catalyzed direct arylation reactions as an umpolung strategy to prepare diaryl ketone derivatives. Rather than using highly reactive organolithium bases generally employed to deprotonate dithianes, we envisioned use of milder bases with the goal of reversibly metallating 2-aryl-1,3-dithianes. Generation of even low concentrations of metallated dithianes could be sufficient for efficient coupling reactions, if transmetallation to palladium is rapid. Herein we report the first direct arylation of dithianes in the presence of a transition metal catalyst and aryl bromides (Scheme 1). This method has been used to produce a diverse range of sterically and electronically differentiated 2,2-diaryl-1,3-dithianes in good to excellent yields. Moreover, we introduce a one-pot arylation of dithianes/hydrolysis to synthesize diaryl ketones with yields of up to 95% (Scheme 1). It is noteworthy that diaryl ketones comprise a common structural core of a large number of biologically relevant compounds and have gained considerable attention as active components of marketed medications, such as Ketoprofen (Oruvail®), Suprofen (Profenal®) and Fenofibrate (TriCor®).[10] We have applied our method to the one-pot synthesis the anti-cholesterol drug Fenofibrate on 10.0 mmol scale.
Scheme 1.
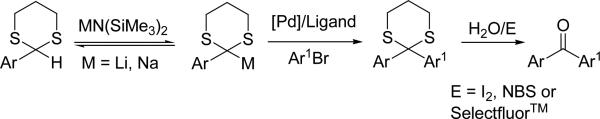
One-pot synthesis of diaryl ketones by tandem arylation/hydrolysis of dithianes
Results and Discussion
Background and Approach
Various methods have been introduced for the synthesis of diaryl ketones. Friedel-Crafts acylations and the addition of organometallic reagents to carbonyl compounds followed by oxidation are classic routes for aryl ketone synthesis.[11] These methods typically employ stoichiometric amounts of metal-containing reagents and often perform poorly with heteroaryls or sterically crowded substrates. Transition metal catalyzed carbonylative reactions have also received significant attention and emerged as useful routes to diaryl ketones.[12]
More recent developments in the synthesis of diaryl ketones have focused on acyl anion equivalents. Among them, organometallic acyl reagents such as acylzirconocenes,[13] acylstannanes,[14] and acylindiums[15] have been applied in palladium and copper catalyzed reactions to prepare various carbonyl compounds. These acyl anion reagents can be challenging to synthesize and to handle.[16] An elegant use of N-tert-butylhydrazones as acyl anion equivalents in the palladium catalysed cross-coupling with aryl bromides has been communicated by Takemiya and Hartwig. The arylated products are easily converted to various ketones via acidic hydrolysis.[17] An innovative example of the umpolung approach to diaryl ketones was reported in 2011 by Schmink and Krska and is illustrated in Scheme 2.[18] After a palladium-catalyzed cross-coupling of acyl chlorides and hexamethyldisilane, the resulting acyl silanes were coupled with aryl bromides to furnish the desired diaryl ketones in 21–85% yield (from the acyl silane).
Scheme 2.
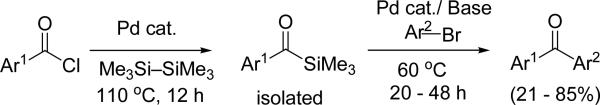
Two step umpolung synthesis of diaryl ketones.
Our approach to the synthesis of diaryl ketones stems from a broader program involving C–H functionalization of weakly acidic substrates (pKa values 28–35 in DMSO). This approach relies on a reversible in situ deprotonation of these substrates to generate reactive organolithium, –sodium, or –potassium intermediates that then undergo transmetallation with a palladium catalyst followed by C–C bond formation. We have found that different phosphine ligands are required (Figure 1, ligands L1–L5) for the diverse substrate classes and bases we have employed. This method has proven successful for the direct C–H arylation of diarylmethanes (L1),[19] benzylic ketimines (L1),[20] benzyl thioethers,[21] benzylic phosphine oxides (L2),[22] methyl sulfoxides and sulfones (L3),[23] amides (L3),[24] benzylic phosphonates (L4),[25] and allyl benzenes (L5).[26]
Figure 1.

Ligands employed in the deprotonative cross-coupling processes of weakly acidic substrates.
Development of the direct arylation of 1,3-dithianes
The steps in the deprotonative cross-coupling process (DCCP) in Scheme 1 involve oxidative addition of the aryl bromide to Pd(0), deprotonation of the substrate and transmetallation to palladium followed by reductive elimination. Given that the pKa of 2-aryl-1,3-dithianes is ~31 in DMSO, the deprotonation was not viewed as the most challenging step. We were more concerned with the transmetallation, because the unfavorable steric interactions necessary to form a fully substituted carbon bound to palladium.
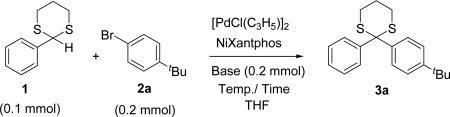
Based on the ability of palladium complexes of van Leeuwen's NiXantphos (L1)[27] to effectively cross-couple organolithium, -sodium, and -potassium organometallics, we examined the reaction of 2-phenyl-1,3-dithiane (1) with 4-tert-butyl bromobenzene (2a) in the presence of dimeric [PdCl(allyl)]2 (20 mol % Pd), NiXantphos (20 mol %) and LiN(SiMe3)2 (2 equiv) and achieved 71% assay yield of 2,2-diaryl-1,3-dithiane 3a after 2 h at room temperature (Table 1, entry 1). We next examined three additional bases [NaN(SiMe3)2, KN(SiMe3)2, NaOt-Bu] to increase the yield (entries 2–4). While NaN(SiMe3)2 and KN(SiMe3)2 each furnished the product in 83% assay yield, NaOt-Bu was totally ineffective. At this point, we chose NaN(SiMe3)2 for further optimization of the reaction conditions.
Table 1.
Optimization of the direct arylation of 2-phenyl-1,3-dithiane.
| |||||
|---|---|---|---|---|---|
| Entry | Rase | [PdCl(C3H5)]2 mol % | NiXantphos mol % | Temp./Time °C/ h | Yield (%)[a] 3a |
| 1 | LiN(SiMe3)2 | 10 | 20 | 24 /2 | 71 |
| 2 | NaN(SiMe3)2 | 10 | 20 | 24 /2 | 83 |
| 3 | KN(SiMe3)2 | 10 | 20 | 24 /2 | 83 |
| 4 | NaOt-Ru | 10 | 20 | 24 /2 | - |
| 5 | NaN(SiMe3)2 | 5 | 10 | 24 /2 | 89 |
| 6 | NaN(SiMe3)2 | 5 | 10 | 24 /1 | 79 |
| 7 | NaN(SiMe3)2 | 2.5 | 5 | 24 /2 | 92 |
| 8[b] | NaN(SiMe3)2 | 2.5 | 5 | 24 /2 | 93 |
| 9 | NaN(SiMe3)2 | 2.5 | 5 | 60 /0.5 | 95 |
| 10 | NaN(SiMe3)2 | 1.25 | 2.5 | 24 /2 | 35 |
| 11 | NaN(SiMe3)2 | 1.25 | 2.5 | 24 /3 | 37 |
Yield determined by 1H NMR integration of the crude reaction mixture using 0.1 mmol CH2Br2 as the internal standard.
Reaction concentration is 0.07 M (entry 8). Reaction concentration for other entries is 0.10 M.
We next explored the possibility of decreasing the catalyst and ligand loading. Using a concentration of 0.1 M and decreasing the palladium loading from 20 mol % to 10 mol % provided the arylated product in 89% assay yield after 2 h at rt (entry 5). Under identical reaction conditions, however, the reaction did not reach completion in 1 h (entry 6). A further decrease of catalyst loading to 5 mol% furnished the desired dithiane in 92% yield in 2 h (entry 7). When the same reaction was carried out in lower concentration (0.07 M) or at higher temperature (60 °C), 93–95% assay yield was observed (entries 8 and 9). Lowering the catalyst loading to 2.5 mol % (entries 10 and 11) resulted in a significant decrease in the assay yield after 2 or 3 h (~37%). Based on these results, we investigated the scope and limitations of this DCCP employing 2.5 mol % [PdCl(allyl)]2 and 5.0 mol % NiXanthphos in THF at room temperature.
Scope of the aryl bromide in the direct arylation of 2-phenyl-1,3-dithianes
Based on the results in Table 1, and the value of the dithiane component, we decided to utilize dithiane derivatives as the limiting reagent with 2 equiv each of aryl bromide and NaN(SiMe3)2 or LiN(SiMe3)2. The results of our study are summarized in Tables 2 and 3.
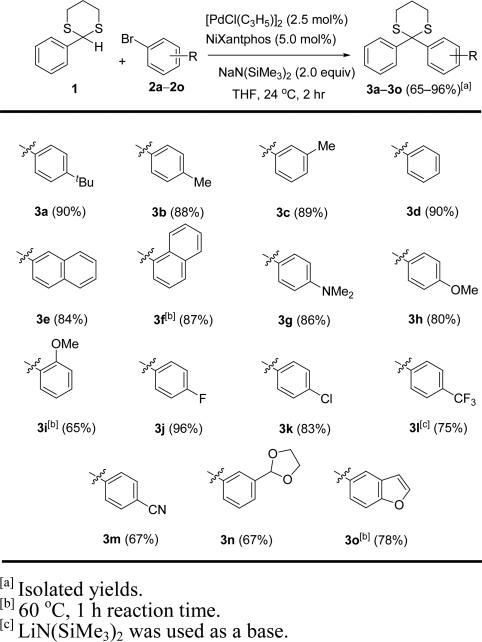
Table 2.
Substrate scope of the aryl bromide with 2-phenyl-1,3 -dithiane.a
|
Table 3.
Substrate scope of 2-aryl-1,3-dithiane and aryl bromides.
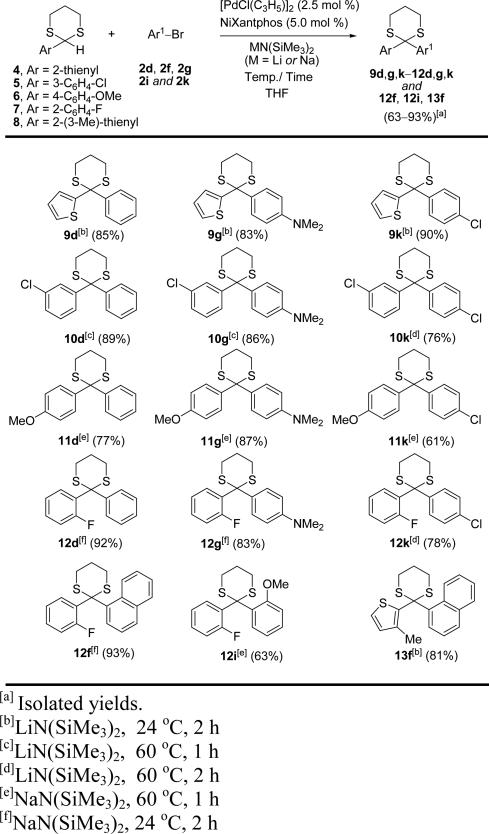
|
Commercially available 2-phenyl-1,3-dithiane (1) underwent DCCP with a wide range of aryl bromides to give the desired 2,2-diaryl-1,3-dithianes (3a–3o). Alkyl substituted aryl bromides (2a–c), bromobenzene (2d) and 2-bromonaphthalene (2e) furnished over 80% yield at room temperature in 2 h. The reaction with sterically encumbered 1-bromonaphthalene (2f) required heating to 60 °C for 1 h for full conversion and provided 3f in 87% yield. Reactions with aryl bromides bearing electron donating 4-N,N-dimethylamino (2g) and 4-methoxy (2h) groups were good substrates, producing 3g and 3h in 86 and 80% yield, respectively. The reaction with sterically demanding 2-bromoanisole (2i) required heating to 60 °C for 1 h to drive the reaction to completion and afforded diaryl dithiane 3i in 65% yield. With 4-fluorobromobenzene (2j), DCCP was particularly successful, providing desired product 3j in 96% yield at room temperature.
To test the scalability of the arylation, we performed this reaction with 6.0 mmol 2-phenyl-1,3-dithiane (1), 12.0 mmol 4-fluorobromobenzene (2j) and 12.0 mmol NaN(SiMe3)2 in the presence of 2.5 mol % [PdCl(allyl)]2 and 5.0 mol % NiXanthphos at room temperature. The starting dithiane 1 was consumed after 3 h and the product 3j was isolated in 85% yield (1.5 g) after purification by column chromatography.
With 4-chlorobromobenzene (2k), the reaction afforded the product 3k in a good yield (83%) and high chemoselectivity, despite the known ability of Pd°(NiXantphos)-based intermediates to oxidatively add aryl chlorides at room temperature.[18a] On the other hand, the reaction with 4-bromobenzotrifluoride (2l), having a strongly electron with drawing –CF3 group, proceeded more efficiently with LiN(SiMe3)2 and gave the product 3l in 75% yield after 2 h at room temperature. Moreover, the sensitive 4-cyano (2m) and 3-cyclic acetal (2n) functional groups were tolerated reasonably well under our reaction conditions, furnishing arylation products 3m and 3n each in 67% yield. The reaction with 5-bromobenzofuran (2o) required heating to 60 °C to afford the diaryl dithiane 3o in 78% yield. The successful coupling of a variety of aryl bromides to 2-phenyl-1,3-dithiane inspired us to next examine the scope of the dithiane coupling partner.
Scope of 2-aryl-1,3-dithianes in the direct arylation
We next explored the DCCPs of sterically and electronically diverse 2-aryl-1,3-dithianes (4–8) with various aryl bromides (Table 3). Straightforward procedures for the synthesis of a variety of 2-aryl-1,3-dithiane derivatives have been reported.[28] For example, Chakraborti and coworkers reported that 2-thienyl-1,3-dithiane (4) and 2-(4-methoxyphenyl)-1,3-dithiane (6) could be prepared in quantitative yields from their respective aldehydes and 1,3-dithiopropane in a one-minute reaction time under solvent free copper catalyzed reaction conditions.[28a]
For the reactions of 2-aryl-1,3-dithianes with aryl bromides, we initially employed NaN(SiMe3)2 in the presence of 2.5 mol % [PdCl(allyl)]2 and 5.0 mol % NiXanthphos in THF at room temperature for 2 h. If necessary, we modified the conditions to increase the yield. For instance, the reaction of 2-thienyl-1,3-dithiane (4) with bromobenzene (2d) gave dithiane 9d in only 49% yield when NaN(SiMe3)2 was used. The yield could be increased to 85% with LiN(SiMe3)2 under otherwise the identical conditions (Table 3). 2-Thienyl-1,3-dithiane (4) afforded good yield with 4-N,N-dimethylamino bromobenzene (83%) and 4-chloro bromobenzene (90%) with LiN(SiMe3)2. Similarly, 2-(3-chlorophenyl)-1,3-dithiane (5) underwent DCCPs with bromobenzene (2d), 4-N,N-dimethylamino bromobenzene (2g) and 4-chloro bromobenzene (2k) more effectively in the presence of LiN(SiMe3)2, producing dithianes 10d, 10g and 10k in 89, 86 and 76% yields, respectively. It is noteworthy that use of LiN(SiMe3)2 in these reactions required heating to 60 °C. On the other hand, for reactions of less acidic 2-(4-methoxyphenyl)-1,3-dithiane (6), use of NaN(SiMe3)2 at 60 °C for 1 h was necessary to obtain higher yields of dithianes 11d, 11g and 11k (77, 87, and 61% yields, respectively).
Reactions of 2-fluorophenyl substituted dithiane 7 with aryl bromides were particularly efficient. Dithianes 12d and 12g were obtained by coupling bromobenzene (2d) and 4-N,N-dimethylamino bromobenzene (2g) with 2-(2-fluorophenyl)-1,3-dithiane (7) in 92 and 83% yields, respectively, using NaN(SiMe3)2 at room temperature in 2 h. Reaction of dithiane 7 with 4-chloro bromobenzene (2k) was rather sluggish, however, and reaction had to be performed in the presence of LiN(SiMe3)2 at 60 °C to furnish 12k in 78% yield in 2 h. Recently, the synthesis of diaryl ketones bearing ortho-substituents on both aryl groups under mild conditions has gained importance,[29] because many biologically active diaryl ketones are sterically congested.[10c-e] To explore the potential of our method for the construction of more hindered ketone precursors, 2-fluorophenyl substituted dithiane 7 was coupled with 1-bromonaphthalene (2f) and 2-bromoanisole (2i) to achieve dithianes 12f (93%) and 12i (63%), respectively. Furthermore, sterically more crowded 3-methylthiophene containing dithiane 8 was coupled with 1-bromo naphthalene to afford 13f in 81% yield. Taken together, the results in Tables 2 and 3 indicate that the DCCP of 2-aryl-1,3-dithianes with aryl bromides is a general and mild method for the synthesis of diaryl dithianes.
Development of a one-pot umpolung synthesis of diaryl ketones
Having demonstrated the direct arylation of 2-aryl-1,3-dithiane derivatives, we set out to develop a one-pot arylation/hydrolysis procedure. There are numerous protocols for the hydrolysis of dithianes. In the tandem arylation/ketone release, we explored three oxidative methods for the hydrolysis (Table 4). Thus, upon complete consumption of the 2-aryl-1,3-dithiane starting material in the DCCP, water (0.5 mL) and electrophile (I2, NBS or Selectfluor™) were added in procedures A, B, and C, respectively (Table 4). The reaction mixtures were stirred for 1 h at rt before workup. In general, all three procedures gave diaryl ketone derivatives in good yields. Furthermore, in most cases, we observed that the scope of aryl bromides and 2-aryl-1,3-dithianes described in Tables 2 and 3 readily translated to the one-pot procedure. Simple alkyl (16a), amino (16g) and halogen (18j, k) substituted diaryl ketones were obtained in moderate to excellent yields (66–95%). Likewise, sterically encumbered diaryl ketones (19i and 20f, 20i) formed in 75–95%. This sequential procedure tolerated different functionalities, although 4-N,N-dimethylamino (16g, 66% and 17g, 57%) and 4-cyano (16m, 50%) substituted diaryl ketones exhibited noticeably lower yields than their corresponding dithianes 3g (86%), 9g (83%) and 3m (67%).
Table 4.
Substrate scope of the one-pot umpolung synthesis of diaryl ketones
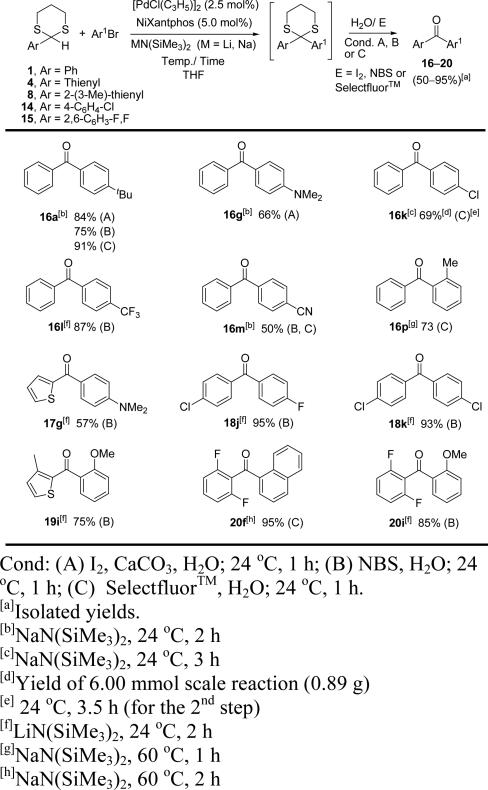
|
On the other hand, trifluoromethyl substituted diaryl ketone 16l was obtained in higher yield (87%) compared to its corresponding dithiane 3l (75%). Moreover, o-tolyl substituted ketone 16p was synthesized in 73% yield in this one-pot procedure. In contrast, its corresponding dithiane 3p was rather unstable to column chromatography and it could not be isolated in pure form. We have also tested the scalability of this process with the 6.0 mmol scale synthesis of chloro substituted benzophenone 16k in 69% isolated yield (0.89 g). The reaction of dithiane 14 with carbonyl containing substrate methyl 4-bromobenzoate in the presence of LiN(SiMe3)2 at room temperature failed in the first step. Dithiane 14 was partially recovered from the reaction mixture.
Synthesis of fenofibrate
To demonstrate the utility of the method presented herein, we synthesized marketed drug fenofibrate (TriCor by Abbott) used for the treatment of hypercholesterolemia and hyper-triglyceridemia (Scheme 3).[30] Initial optimization involved 0.2 mmol scale reactions with 2.0, 1.2, 1.1 and 1.0 equiv. of aryl bromide having hindered carbonyl functionality 2r, 1.0 equiv. of 2-(4-chlorophenyl)-1,3-dithiane (14), and 2.0 equiv. of LiN(SiMe3)2 at room temperature with 2.5 mol % catalyst. Selectfluor™ (4.0 equiv.) was used in the hydrolysis step. Reactions with 2.0 and 1.2 equiv. of 2r gave fenofibrate in 86% and 90% yields, respectively. However, the yield of fenofibrate decreased gradually with the use of 1.1 (82%) and 1.0 equiv. (77%) of aryl bromide 2r. The efficiency of hydrolysis step was examined employing more economical NBS in the tandem arylation/hydrolysis reaction using 2.0 equiv. of 2r. This reaction produced fenofibrate in very good yield (92%).
Scheme 3.
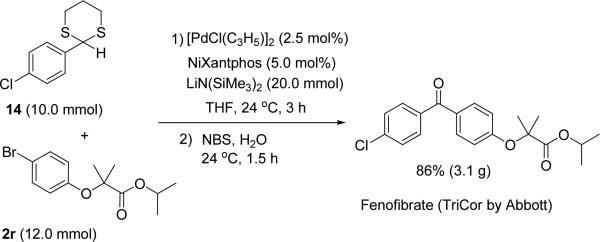
One-pot umpolung synthesis of fenofibrate
With the successful optimization of the small scale reactions, we tested the scalability of the DCCP for the one-pot synthesis of fenofibrate. We conducted the large scale reaction without using a drybox and employed commercially available 1.0 M solution of LiN(SiMe3)2 in THF to simplify the method. The arylation was performed with 10.0 mmol 2-(4-chlorophenyl)-1,3-dithiane, 12.0 mmol aryl bromide 2r and 20.0 mmol LiN(SiMe3)2 in the presence of 2.5 mol % [PdCl(C3H5)]2 and 5.0 mol % NiXanthphos in THF at room temperature for 3 h. Once the arylation was complete (as judged by TLC), 4.5 equiv. of NBS were added to the arylation reaction mixture and the reaction stirred for 1.5 h at rt before workup and purification of the product on silica gel. This one-pot procedure resulted in the isolation of over 3 g fenofibrate in 86% yield.
Conclusions
Umpolung carbonyl reactivity with metallated dithianes has evolved over 40 years and is now mainstay in synthetic chemistry. In particular, recent applications of metallated dithianes as acyl anion equivalents have achieved new levels of sophistication through “anion relay chemistry (ARC),” enabling the synthesis of architecturally complex molecular frameworks. Despite the remarkable advances in dithiane chemistry, high yielding applications of dithianes as masked acyl anion equivalents in transition metal catalyzed reactions have not been previously achieved.
Herein, we disclose a versatile method for the reversed polarity synthesis of diaryl ketones through palladium-catalyzed direct sp3-C–H arylation of 2-aryl-1,3-dithianes. Keys to the success of this method are the in situ deprotonation of dithianes under catalytic reaction conditions and the identification of a catalyst to couple the resulting lithio- and sodio-based metallated dithianes with aryl bromides. The benefits of this method are that it proceeds at room temperature, is usually complete in 1–3 h, requires simple experimental techniques, and provides various dithianes in good to excellent yields. Moreover, the arylation reaction can be performed in a tandem fashion to release the carbonyl functionality in the presence of readily available oxidants. This tandem process enables the synthesis of diaryl ketones on multigram scales without use of a drybox. Based on these attributes, we expect that this method will be suitable for applications in medicinal chemistry.
Experimental Section
General Procedure A: Preparation of 2-aryl-1,3-dithianes 5, 7, 8 and 15 according to modified literature procedure:[28a]
An oven-dried 10 mL reaction vial equipped with a stirring bar was charged with a benzaldehyde derivative (8.4 mmol, 1.0 equiv.) and Cu(BF4)2.xH2O (0.169 mmol, 2.0 mol %). To the neat stirred aldehyde, 1,3-propanedithiol (10 mmol, 1.0 mL) was added and the vial was closed with a rubber septa. The vial was placed into the preheated (60 °C) oil bath and the reaction mixture stirred at this temperature for 1 h. After cooling to room temperature, the reaction mixture was diluted with in 20 mL CH2Cl2 and washed with water (2 × 10 mL). The organic phase was dried over MgSO4, filtrated and the solvent was removed in a rotatory evaporator. The solid white residue was recrystallized from hexanes to give 2,2-diaryl-1,3-dithianes.
General Procedure B: Arylation of 2-Aryl-1,3-dithianes
An oven-dried 10 mL reaction vial equipped with a stirring bar was charged with 2-aryl-1,3-dithiane (0.2 mmol, 1.0 equiv.) and aryl bromide -if it was solid- (0.4 mmol, 2.0 equiv.) and the vial was brought into a glovebox. The reaction vial was charged with base [LiN(SiMe3)2 or NaN(SiMe3)2] (0.4 mmol, 2.0 equiv.), 1.4 mL THF, 0.4 mL THF solution (0.025 M) of NiXantphos and 0.2 mL THF solution of (0.025 M) [PdCl(allyl)]2, respectively. The vial was sealed with a rubber septa, wrapped with a strip of Parafilm, and taken out of the glovebox. The sealed vial was charged with aryl bromide -if it was liquid- (0.4 mmol, 2.0 equiv.) by a syringe. The resulting solution was stirred for the given time at the stated temperature. The reaction mixture was quenched with 0.1 mL of water and filtered through a small pad of Celite. The pad was then rinsed additional CH2Cl2. The combined organic solution was mixed with 0.5 g of deactivated silica gel and the solvent was removed in a rotatory evaporator. The remaining solid residue was loaded onto a deactivated silica gel column and purified by flash chromatography. The silica gel was deactivated by flushing with 5% triethylamine/hexanes solution (3 times) followed by 20:1 hexanes/ethyl acetate solution (3 times) to remove excess triethylamine.
General Procedure C: One-pot Umpolung Synthesis of Diaryl Ketones
An oven-dried 10 mL reaction vial equipped with a stirring bar was charged with 2-aryl-1,3-dithiane (0.2 mmol, 1.0 equiv.) and aryl bromide -if it was solid- (0.4 mmol, 2.0 equiv.) and then the vial was brought into a glovebox. The reaction vial was charged with base [LiN(SiMe3)2 or NaN(SiMe3)2] (0.4 mmol, 2.0 equiv.), 1.4 mL THF, 0.4 mL THF solution (0.025 M) of NiXantphos and 0.2 mL THF solution of (0.025 M) [PdCl(allyl)]2, respectively. The vial was sealed with a rubber septum wrapped with a strip of Parafilm and removed from the glovebox. The sealed vial was charged with aryl bromide -if it was liquid- (0.4 mmol, 2.0 equiv.) by syringe. After having stirred the mixture for the given time at the stated temperature, the vial was cooled to rt (if the reaction was carried out 60 °C) and, opened to air and 0.5 mL H2O, I2 (1.2 mmol, 0.3 g), and CaCO3 (1.6 mmol, 0.16 g) were added (Method A) [or 0.5 mL H2O and NBS (1.0 mmol, 0.18 g) (Method B); or 0.5 mL H2O and Selectfluor™ (0.79 mmol, 0.28 g) (Method C)]. The resulting mixture stirred at rt for 1 h. The reaction mixture was taken up in 10 mL CH2Cl2 and washed with water (2 × 5 mL). The aqueous phase was extracted with 10 mL CH2Cl2, and the combined organic phases were dried (MgSO4), filtrated and mixed with 0.5 g of silica gel. The solvent was then removed in a rotatory evaporator. The remaining solid residue was loaded onto a silica gel column and purified by flash chromatography.
General Procedure D: Preparation of 10.0 mmol Scale One-pot Umpolung Synthesis of Fenofibrate
An oven-dried 250 mL two-neck round-bottomed reaction flask equipped with a stirring bar and a glass stopcock adapter connected to a Schlenk line was charged with 2-(4-chlorophenyl)-1,3-dithiane (14, 10 mmol, 2.31 g), 2-(4-bromophenoxy)-2-methylpropanoate (2r, 12 mmol, 3.61 g), NiXantphos (5.0 mol %, 276 mg) and [PdCl(allyl)]2 (2.5 mol %, 91 mg), respectively. The open neck was closed with a rubber septum and sealed with a strip of Parafilm. The flask was evacuated by vacuum and then refilled with nitrogen gas. This process was repeated 3 times and 80 mL dry THF was added through the septum by syringe. The resulting solution was stirred at room temperature for 3 min and a solution of LiN(SiMe3)2 (20 mmol, 20 mL of a 1.0 M of THF solution) was added by syringe under a nitrogen atmosphere. The flask was removed from the Schlenk line and the reaction mixture was stirred under a nitrogen atmosphere at room temperature. After complete consumption (3 hours) of 2-(4-chlorophenyl)-1,3-dithiane (14), 25 mL H2O and NBS (45 mmol, 8.0 g) were added and the flask was closed with a rubber septum. The reaction mixture was stirred for 1.5 h at rt and added to a sepratory funnel with 250 mL CH2Cl2 and washed with water (2 × 50 mL). The aqueous phase was extracted with 250 mL CH2Cl2, and combined organic phases were dried (MgSO4), filtrated and mixed with 5 g of silica gel. The solvent was removed in a rotatory evaporator. The remaining solid residue was loaded onto a silica gel column and purified by flash chromatography on silica gel using 20:1 hexanes/ethyl acetate as eluent to yield the product fenofibrate (3.1 g, 86%) as a white solid. The NMR spectral data match with the previously published data.[30]
Supplementary Material
Acknowledgements
P.J.W. acknowledges the NIH (National Institute of General Medical Sciences NIGMS 104349). B.Y thanks the Scientific and Technical Research Council of Turkey for a TUBITAK-2219 fellowship.
Footnotes
Supporting information for this article is available on the WWW under http://dx.doi.org/10.1002/adsc.201######.((Please delete if not appropriate))
References
- 1.a Bugaut X, Glorius F. Chem. Soc. Rev. 2012;41:3511–3522. doi: 10.1039/c2cs15333e. [DOI] [PubMed] [Google Scholar]; b Opatz T. Synthesis. 2009:1941–1959. [Google Scholar]; c Marion N, Diez-Gonzáles S, Nolan SP. Angew. Chem. Int. Ed. 2007;46:2988–3000. doi: 10.1002/anie.200603380. [DOI] [PubMed] [Google Scholar]; d Johnson JS. Angew. Chem. Int. Ed. 2004;43:1326–1328. doi: 10.1002/anie.200301702. [DOI] [PubMed] [Google Scholar]; e Seebach D. Angew. Chem. Int. Ed. Engl. 1979;18:239–336. [Google Scholar]
- 2.a Eymur S, Göllü M, Tanyeli C. Turk. J. Chem. 2013;37:586–609. [Google Scholar]; b Shen B, Makley DM, Johnston JN. Nature. 2010;465:1027–1032. doi: 10.1038/nature09125. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Enders D, Balensiefer T. Acc. Chem. Res. 2004;37:534–541. doi: 10.1021/ar030050j. [DOI] [PubMed] [Google Scholar]; d Pohl M, Lingen B, Müller M. Chem. Eur. J. 2002;8:5288–5295. doi: 10.1002/1521-3765(20021202)8:23<5288::AID-CHEM5288>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- 3.a Miyata O, Miyoshi T, Ueda M. ARKIVOC. 2013;ii:60–81. [Google Scholar]; b Katrizky AR, Kirichenko K. ARKIVOC. 2006;iv:119–151. [Google Scholar]; c Gröbel B-T, Seebach D. Synthesis. 1977:357–402. [Google Scholar]
- 4.a Lapworth A. J. Chem. Soc., Trans. 1903;83:995–1005. [Google Scholar]; b Lapworth A. J. Chem. Soc., Trans. 1904;85:1206–1214. [Google Scholar]
- 5.a Corey EJ, Seebach D. Angew. Chem. Int. Ed. Engl. 1965;4:1075–1077. [Google Scholar]; b Corey EJ, Seebach D. Angew. Chem. Int. Ed. Engl. 1965;4:1077–1078. [Google Scholar]; c Seebach D. Synthesis. 1969;1:17–36. [Google Scholar]
- 6.a Geum S, Lee H-Y. Org. Lett. 2014;16:2466–2469. doi: 10.1021/ol500849m. [DOI] [PubMed] [Google Scholar]; b Chen ZM, Gutierrez O, Smith AB., III Angew. Chem. Int. Ed. 2014;53:1279–1282. doi: 10.1002/anie.201309270. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Melillo B, Smith AB., III Org. Lett. 2013;15:2282–2285. doi: 10.1021/ol400857k. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Smith AB, III, Han H, Kim W-S. Org. Lett. 2011;13:3328–3331. doi: 10.1021/ol2010598. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Smith AB, III, Tong R. Org. Lett. 2010;12:1260–1263. doi: 10.1021/ol100130x. [DOI] [PMC free article] [PubMed] [Google Scholar]; f Smith AB, III, Folyey MA, Dong S, Orbin A. J. Org. Chem. 2009;74:5987–6001. doi: 10.1021/jo900765p. [DOI] [PMC free article] [PubMed] [Google Scholar]; g Smith AB, III, Cox JM, Furuichi N, Kenesky CS, Zheng J, Wuest WM. Org. Lett. 2008;10:5501–5504. doi: 10.1021/ol8019132. [DOI] [PMC free article] [PubMed] [Google Scholar]; h Smith AB, III, Wuest WM. Chem. Commun. 2008:5883–5895. doi: 10.1039/b810394a. [DOI] [PMC free article] [PubMed] [Google Scholar]; i Smith AB, III, Kim W-S, Wuest WM. Angew. Chem. Int. Ed. 2008;47:7082–7086. doi: 10.1002/anie.200802301. [DOI] [PMC free article] [PubMed] [Google Scholar]; j Smith AB, III, Tomioka T, Risatti CA, Sperry JB, Sfouggatakis C. Org. Lett. 2008;10:4359–4362. doi: 10.1021/ol801792k. [DOI] [PMC free article] [PubMed] [Google Scholar]; k Smith AB, III, Lee D. J. Am. Chem. Soc. 2007;129:10957–10962. doi: 10.1021/ja073329u. [DOI] [PubMed] [Google Scholar]; l Smith AB, III, Adams CM. Acc. Chem. Res. 2004;37:365–377. doi: 10.1021/ar030245r. [DOI] [PubMed] [Google Scholar]; m Smith AB, III, Condon SM, McCauley JA. Acc. Chem. Res. 1998;31:35–46. [Google Scholar]; n Yus M, Nájera C, Foubelo F. Tetrahedron. 2003;59:6147–6212. [Google Scholar]
- 7.Metallated dithianes having cations other than lithium are rarely applied in the literature, see; Seebach D, Leitz HF, Ehrig V. Chem. Ber. 1975;108:1924–1945. Weil R, Collignon N. Bull. Soc. Chim. Fr. 1974:253. Eliel EL, Hartmann AA. J. Org. Chem. 1972;37:505–506.
- 8.For some applications of dithianes in catalytic reactions where they do not behave as acyl anion equivalents, see: Du W, Tian L, Lai J, Huo X, Xie X, She X, Tang S. Org. Lett. 2014;16:2470–2473. doi: 10.1021/ol500850d. Zhao X, Zhong Z, Peng L, Zhang W, Wang J. Chem. Commun. 2009:2535–2537. doi: 10.1039/b903028j. Morita E, Iwasaki M, Yoshida S, Yorimitsu H, Oshima K. Chem. Lett. 2009;38:624–625. Luh T-Y, Lee C-F. Eur. J. Org. Chem. 2005:3875–3885. Breit B. Angew. Chem. Int. Ed. 1998;37:453–456. doi: 10.1002/(SICI)1521-3773(19980302)37:4<453::AID-ANIE453>3.0.CO;2-M. Horikawa Y, Watanabe M, Fujiwara T, Takeda T. J. Am. Chem. Soc. 1997;119:1127–1128. Luh T-Y. Acc. Chem. Res. 1991;24:257–263. For non-transition metal catalyzed reactions see: Denmark SE, Cullen LR. Org. Lett. 2014;16:70–73. doi: 10.1021/ol403041k. Michida M, Mukaiyama T. Chem. Asian J. 2008;3:1592–1600. doi: 10.1002/asia.200800091.
- 9.Malanga C, Aronica LA, Lardicci L. Tetrahedron Lett. 1995;36:9185–9188. [Google Scholar]
- 10.a Luque-Ortega JR, Reuther P, Rivas L, Dardonville C. J. Med. Chem. 2010;53:1788–1798. doi: 10.1021/jm901677h. [DOI] [PubMed] [Google Scholar]; b Vooturi SK, Cheung CM, Rybak MJ, Firestine SM. J. Med. Chem. 2009;52:5020–5031. doi: 10.1021/jm900519b. [DOI] [PubMed] [Google Scholar]; c Deng Y, Young-Won C, Chai H, Keller WJ, Kinghorn AD. J. Nat. Prod. 2007;70:2049–2052. doi: 10.1021/np070501z. [DOI] [PubMed] [Google Scholar]; d Pecchio M, Solís PN, López-Pérez JL, Vásquez Y, Rodríguez N, Olmedo D, Correa M, Feliciano AS, Gupta MP. J. Nat. Prod. 2006;69:400–413. doi: 10.1021/np050338c. [DOI] [PubMed] [Google Scholar]; e Lampe JW, Biggers CK, Defauw JM, Foglesong RJ, Hall SE, Heerding JM, Hollinshead SP, Hu H, Hughes PF, Jagdmann GE, Jr., Johnson MG, Lai Y-S, Lowden CT, Lynch MP, Mendoza JS, Murphy MM, Wilson JW, Ballas LM, Carter K, Darges JW, Davis JE, Hubbard FR, Stamper ML. J. Med. Chem. 2002;45:2624–2643. doi: 10.1021/jm020018f. [DOI] [PubMed] [Google Scholar]
- 11.a Khan AA, Kamena F, Timmer MSM, Stocker BL. Org. Biomol. Chem. 2013;11:881–885. doi: 10.1039/c2ob27257a. [DOI] [PubMed] [Google Scholar]; b Poupaert JH, Depreux P, McCurdy CR. Monatshefte für Chemie. 2003;134:823–830. [Google Scholar]; c Ushijima S, Dohi S, Moriyama K, Togo H. Tetrahedron. 2012;68:1436–1442. [Google Scholar]; d Dohi S, Moriyama K, Togo H. Tetrahedron. 2012;68:6557–6564. [Google Scholar]
- 12.a Hajipour AR, Pourkaveh R. Synlett. 2014;25:1101–1105. [Google Scholar]; b Li Y, Lu W, Xue D, Wang C, Liu Z-T. J. Xia, Synlett. 2014;25:1097–1100. [Google Scholar]; c Jiang T-S, Wang G-W. Adv. Synt. Catal. 2014;356:369–373. [Google Scholar]; d Maji A, Rana S, Akansha, Maiti D. Angew. Chem. Int. Ed. 2014;53:2428–2432. doi: 10.1002/anie.201308785. [DOI] [PubMed] [Google Scholar]; e Kunchithapatham K, Eichman CC, Stambuli JP. Chem. Commun. 2011:12679–12681. doi: 10.1039/c1cc16114h. [DOI] [PubMed] [Google Scholar]; f Wu X-F, Neumann H, Beller M. Chem. Soc. Rev. 2011;40:4986–5009. doi: 10.1039/c1cs15109f. [DOI] [PubMed] [Google Scholar]; g Kobayashi K, Nishimura Y, Gao F, Gotoh K, Nishihara Y, Takagi K. J. Org. Chem. 2011;76:1949–1952. doi: 10.1021/jo1025173. [DOI] [PubMed] [Google Scholar]; h Li H, Yang M, Qi Y, Xue J. Eur. J. Org. Chem. 2011:2662–2667. [Google Scholar]; i Qin C, Chen J, Wu H, Cheng J, Zhang Q, Zuo B, Su W, Ding J. Tetrahedron Lett. 2008;49:1884–1888. [Google Scholar]; j Neumann H, Brennführer A, Beller M. Adv. Synth. Catal. 2008;350:2437–2442. [Google Scholar]; k Pucheault M, Darses S, Genet J-P. J. Am. Chem. Soc. 2004;126:15356–15357. doi: 10.1021/ja044749b. [DOI] [PubMed] [Google Scholar]; l Duplais C, Bures F, Sapountzis I, Korn TJ, Cahiez G, Knochel P. Angew. Chem. Int. Ed. 2004;43:2968–2970. doi: 10.1002/anie.200453696. [DOI] [PubMed] [Google Scholar]; m Huang Y-C, Majumdar KK, Cheng C-H. J. Org. Chem. 2002;67:1682–1684. doi: 10.1021/jo010289i. [DOI] [PubMed] [Google Scholar]; n Savarin C, Srogl J, Liebeskind LS. Org. Lett. 2000;2:3229–3231. doi: 10.1021/ol000231a. [DOI] [PubMed] [Google Scholar]; o Liebeskind LS, Srogl J. J. Am. Chem. Soc. 2000;122:11260–11261. [Google Scholar]
- 13.a Hanzawa Y, Narita K, Yabe M, Taguchi T. Tetrahedron. 2002;58:10429–10435. [Google Scholar]; b Hanzawa Y, Tabuchi N, Taguchi T. Tetrahedron Lett. 1998;39:6249–6252. [Google Scholar]
- 14.Obora Y, Nakanishi M, Tokunaga M, Tsuji Y. J. Org. Chem. 2002;67:5835–5837. doi: 10.1021/jo0202482. [DOI] [PubMed] [Google Scholar]
- 15.Lee D, Ryu T, Park Y, Lee HP. Org. Lett. 2014;16:1144–1147. doi: 10.1021/ol500003g. [DOI] [PubMed] [Google Scholar]
- 16.a Capperucci A, Degl'Innocenti A, Faggi C, Reginato G, Ricci A, Dembech P, Seconi G. J. Org. Chem. 1989;54:2966–2968. [Google Scholar]; b Yamamoto K, Hayashi A, Suzuki S, Tsuji J. Organometallics. 1987;6:974–979. [Google Scholar]
- 17.Takemiya A, Hartwig JF. J. Am. Chem. Soc. 2006;128:14800–14801. doi: 10.1021/ja064782t. [DOI] [PubMed] [Google Scholar]
- 18.Schmink JR, Krska SW. J. Am. Chem. Soc. 2011;133:19574–19577. doi: 10.1021/ja2064318. [DOI] [PubMed] [Google Scholar]
- 19.a Zhang J, Bellomo A, Trongsiriwat N, Jia T, Carroll PJ, Dreher SD, Tudge MT, Yin H, Robinson JR, Schelter EJ, Walsh PJ. J. Am. Chem. Soc. 2014;136:6276–6287. doi: 10.1021/ja411855d. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Bellomo A, Zhang J, Trongsiriwat N, Walsh PJ. Chem. Sci. 2013;4:849–857. [Google Scholar]; c Zhang J, Bellomo A, Creamer AD, Dreher SD, Walsh PJ. J. Am. Chem. Soc. 2012;134:13765–13772. doi: 10.1021/ja3047816. [DOI] [PubMed] [Google Scholar]
- 20.Li M, Yücel B, Adrio J, Bellomo A, Walsh PJ. Chem. Sci. 2014;5:2383–2391. doi: 10.1039/C3SC53526F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Frensch G, Hussain N, Marques FA, Walsh PJ. Adv. Synth. Catal. 2014 doi: 10.1002/adsc.201400679. accepted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Montel S, Jia T, Walsh PJ. Org. Lett. 2014;16:130–133. doi: 10.1021/ol403124g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.a Jia T, Bellomo A, EL Baina K, Dreher SD, Walsh PJ. J. Am. Chem. Soc. 2013;135:3740–3743. doi: 10.1021/ja4009776. [DOI] [PubMed] [Google Scholar]; b Zheng B, Jia T, Walsh PJ. Org. Lett. 2013;15:1690–1693. doi: 10.1021/ol400472v. [DOI] [PubMed] [Google Scholar]
- 24.a Zheng B, Jia T, Walsh PJ. Adv. Synth. Catal. 2014;356:165–178. doi: 10.1002/adsc.201300851. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Zheng B, Jia T, Walsh PJ. Org. Lett. 2013;15:4190–4193. doi: 10.1021/ol4019002. [DOI] [PubMed] [Google Scholar]
- 25.Montel S, Raffier L, He Y, Walsh PJ. Org. Lett. 2014;16:1446–1449. doi: 10.1021/ol5002413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hussain N, Frensch G, Zhang J, Walsh PJ. Angew. Chem. Int. Ed. 2014;53:3693–3697. doi: 10.1002/anie.201309084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.a Birkholz M-N, Freixa Z, van Leeuwen PWNM. Chem. Soc. Rev. 2009;38:1099–1118. doi: 10.1039/b806211k. [DOI] [PubMed] [Google Scholar]; b Leeuwen PWNM, Kamer PCJ, Reek JNH, Dierkes P. Chem. Rev. 2000;100:2741–2769. doi: 10.1021/cr9902704. [DOI] [PubMed] [Google Scholar]; c van der Veen LA, Keeven PH, Schoemaker GC, Reek JNH, Kamer PCJ, van Leeuwen PWNM, Lutz M, Speek AL. Organometallics. 2000;19:872–883. [Google Scholar]
- 28.a Besra RC, Rudrawar S, Chakraborti AK. Tetrahedron Lett. 2005;46:6213–6217. [Google Scholar]; b Weng S-S, Chang S-C, Chang T-H, Chyn J-P, Lee S-W, Lin C-H, Chen F-K. Synthesis. 2010:1493–1499. [Google Scholar]; c Rudrawar S, Besra RC, Chakraborti AK. Synthesis. 2006:2767–2771. [Google Scholar]; d Ali MH, Gomes MG. Synthesis. 2005:1326–1332. [Google Scholar]; e Dong D, Ouyang Y, Yu H, Liu Q, Liu J, Wang M, Zhu J. J. Org. Chem. 2005;70:4535–4537. doi: 10.1021/jo050271y. [DOI] [PubMed] [Google Scholar]; f De SK. Synthesis. 2004:2837–2840. [Google Scholar]; g De SK. Tetrahedron Lett. 2004;45:2339–2341. [Google Scholar]; h Firouzabadi H, Iranpoor N, Hazarkhani H. J. Org. Chem. 2001;66:7527–7529. doi: 10.1021/jo015798z. [DOI] [PubMed] [Google Scholar]; i Samajdar S, Basu MK, Becker FF, Banik BK. Tetrahedron Lett. 2001;42:4425–4427. [Google Scholar]
- 29.a Cho H-H, Kim S-H. Bull. Korean Chem. Soc. 2012;33:3083–3086. doi: 10.5012/bkcs.2012.33.12.4041. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Lo Fiego MJ, Silbestri GF, Chopa AB, Lockhart MT. J. Org. Chem. 2011;76:1707–1714. doi: 10.1021/jo102366q. [DOI] [PubMed] [Google Scholar]; c O'Keefe BM, Simmons N, Martin SF. Tetrahedron. 2011;67:4344–4351. doi: 10.1016/j.tet.2011.03.074. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Chuzel O, Roesch A, Genet J-G, Darses S. J. Org. Chem. 2008;73:7800–7802. doi: 10.1021/jo801460w. [DOI] [PubMed] [Google Scholar]; e O'Keefe BM, Simmons N, Martin SF. Org. Lett. 2008;10:5301–5304. doi: 10.1021/ol802202j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.For very recent synthesis of fenofibrate see: Bjerglund KM, Skrydstrup T, Molander GA. Org. Lett. 2014;16:1888–1891. doi: 10.1021/ol5003362.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.