

Abstract
Platelets are anucleated cell fragments derived from mature megakaryocytes and function in hemostasis when the endothelium is injured. Hemostasis involving platelets can be divided into four phases: adhesion, activation, secretion, and aggregation. Platelet activation requires a rise in intracellular Ca2+ concentrations and results in both a morphological change and the secretion of platelet granule contents. Na+/H+ exchanger isoform 1 (NHE1) regulates the intracellular pH (pHi) and the volume of platelets. In addition, NHE1 plays a large role in platelet activation. Thrombus generation involves NHE1 activation and an increase in [Ca2+]i, which results from NHE1-mediated Na+ overload and the reversal of the Na+/Ca2+ exchanger. Cariporide (HOE-642), a potent NHE1 inhibitor, has inhibitory effects on the degranulation of human platelets, the formation of platelet–leukocyte-aggregates, and the activation of the GPIIb/IIIa receptor (PAC-1). However, despite the demonstrated protection of myocardial infarction as mediated by cariporide in patients undergoing coronary artery bypass graft surgery, the EXPEDITION clinical trial revealed that cariporide treatment increased mortality due to thromboembolic stroke. These findings suggest that a better understanding of NHE1 and its effect on platelet function and procoagulant factor regulation is warranted in order to develop therapies using NHE inhibitors.
Keywords: Cariporide, EXPEDITION trial, HOE 642, ischemia reperfusion, NHE1, platelet aggregation
1. Introduction
Na+/H+ exchanger isoform 1 (NHE1) is the most abundantly expressed isoform of a family of proteins with nine members, NHE1-NHE9 (Huber et al., 2012). NHE1 plays an important role in regulating H+ homeostasis and cell volume under physiological conditions via H+ extrusion and Na+ influx (Sarigianni et al., 2010). NHE1 has emerged as a therapeutic target molecule for several diseases, which include cardiac ischemia reperfusion injury after heart failure (Mentzer, Jr. et al., 2008), myocardium ischemia (Avkiran, 1999), cerebral ischemic reperfusion injury of ischemic stroke (Leng et al., 2014) and hypoxic ischemic injury of neonatal immature brain injury (Cengiz et al., 2011). The beneficial effects of the blockade of NHE1 function are attributed to the reduction of NHE1-mediated intracellular Na+ overload, an increase in Ca2+ extrusion via Na+/Ca2+ exchange, and a decrease in cell injury after ischemia and reperfusion (Avkiran, 1999; Leng et al., 2014; Mentzer, Jr. et al., 2008).
In light of the roles of NHE1 in myocardial ischemic injury, several clinical trials with various NHE1 inhibitors have been conducted. A trial evaluating zoniporide in patients at risk for coronary disease undergoing non-cardiac surgery showed no benefit in reducing composite cardiovascular end point (Fleisher et al., 2005). ESCAMI (Evaluation of the Safety and Cardioprotective Effects of Eniporide in Acute Myocardial Infarction) evaluated the inhibitor, eniporide, in patients undergoing thrombolytic therapy or angioplasty surgery, which did not limit myocardium infarction (MI) size or improve clinical outcome (Zeymer et al., 2001). Two clinical trials were conducted to assess cariporide (HOE-642): GUARDIAN (Guard During Ischemia Against Necrosis) and EXPEDITION (Na+/H+ Exchange Inhibition to Prevent Coronary Events in Acute Cardiac Condition). In the GUARDIAN study, patients undergoing coronary artery bypass graft surgery (CABG) receiving doses of 120 mg of cariporide had a decreased rate of all-cause mortality and MI (Chaitman, 2003). In the EXPEDITION trial, patients undergoing CABG received cariporide in a 180 mg dose 1 h prior to CABG, 40mg/h for 24 h after CABG, and 20mg/h over the subsequent 24 h. Cariporide significantly decreased rates of MI in the treated group (Mentzer, Jr. et al., 2008). However, despite cariporide’s ability to reduce ischemic reperfusion injury, the clinical trial was terminated because of a high mortality due to ischemic embolic stroke. The increase in ischemic stroke has been hypothesized to result from a reduced procoagulant response and the stimulation of platelet function after administration of NHE1 inhibitor, cariporide, at a high dosage (Mentzer, Jr. et al., 2008). The mechanisms underlying the adverse effects of cariporide are unknown. These findings prompt us to review the current research of platelet biology and regulation, in particular, the roles of NHE1 in the regulation of platelet function. A better understanding of NHE1’s role in platelet function is warranted, which will benefit the development of new strategies to overcome the adverse effects of NHE inhibitors and future applications for protection of ischemic reperfusion injury.
2. Platelet function and regulation
2.1. Platelet biology and function
Platelets are anucleated cell fragments derived from mature megakaryocytes (MK) found in the bone marrow (Schulze and Shivdasani, 2005). The main function of platelets is hemostasis when the endothelium is injured (Ruggeri and Mendolicchio, 2007). Even though the normal role of platelets is to stop bleeding, because of its role in forming blood clots, platelets are involved in various arterial diseases such as heart attack and stroke (Ruggeri, 2002).
In mature MK, microtubules extend the cytoplasm into long processes called “proplatelets” and it is at the tip of these processes that platelets are filled, assembled, and released. In order to increase the number of platelets generated per proplatelet, proplatelet shafts are bifurcated in an event mediated by actin to increase the number of available tips (Hartwig and Italiano, Jr., 2003). The mechanisms by which platelets are released from their proplatelet tips have not been completely elucidated, but an intermediate stage between proplatelets and platelets called the ‘preplatelet’ has been suggested (Thon et al., 2010). Despite our incomplete knowledge regarding this mechanism, it is known that a single MK can lead to the genesis of hundreds or even thousands of individual platelets which culminate in a suicidal event for the original MK (Schulze and Shivdasani, 2005).
Each platelet is about 7 μm3 in volume and 300 nm in diameter and is discoid in shape (Cimmino and Golino, 2013). In its resting state, the platelet’s shape is maintained by a cytoskeleton composed of tubulin and actin polymers (Hartwig and Italiano, Jr., 2003). Despite being only a cell fragment and not a complete cell, platelets have a complex structure. They possess a membrane and cytoskeleton, many types of surface receptors, mitochondria, and dense granules and α-granules, which are capable of secreting various compounds (Cimmino and Golino, 2013).
2.2. Platelet activation
Hemostasis involving platelets can be divided into four phases: adhesion, activation, secretion, and aggregation (Cimmino and Golino, 2013). Adhesion is the process in which platelets attach to damaged endothelial cells, and this process is mediated by von Willebrand factor (vWF) and collagen. When the vessel wall becomes damaged, collagen beneath endothelial cells becomes exposed to platelets. Collagen is crucial because it can bind to Glycoprotein VI (GPVI), a collagen receptor on platelets, and it is also necessary for immobilizing vWF. Under high shear conditions, vWF is necessary for platelet adhesion via the Glycoprotein Ib-V-IX complex, but this receptor is incapable of binding plasma vWF; it can only bind vWF immobilized by collagen (Ruggeri and Mendolicchio, 2007). Adhesion occurs 1–3 seconds after injury and results in a monolayer of activated platelets (Kickler et al., 2006a).
Platelet activation results in a morphological change and the secretion of platelet granule contents. With the help of actin and myosin in the cytoplasm, the platelet’s shape changes from a disc to a compact sphere, which causes the granules to become centralized. This centralization is thought to lead to more efficient secretion of granule contents (George, 2000). Platelets contain dense granules and α-granules. Dense granules secrete small non-protein molecules such as ADP, serotonin, and Ca2+, while α-granules secrete larger proteins such as vWF and fibrinogen (George, 2000). While activation is often initiated by subendothelial collagen, other agonists include serotonin, thrombin, thromboxane A2 (TXA2), and ADP (Angiolillo et al., 2010). Since many of the agonists are also secreted by the platelets upon activation, there is a positive feedback loop in clot formation.
Platelet activation also leads to a rise in intracellular Ca2+ concentrations ([Ca2+]i). Ca2+ is an essential second messenger, and the increase in [Ca2+]i contributes to both the change in morphology and the secretion of the granule contents. Upon activation, the platelet can increase [Ca2+]i via Ca2+ entry through the plasma membrane or intracellularly via the release of compartmentalized Ca2+. In platelets, the majority of Ca2+ is stored in the endoplasmic reticulum (Varga-Szabo et al., 2009).
The final step in hemostasis is the aggregation of platelets. Whereas adhesion is the attachment of platelets to the damaged vessel wall, aggregation is the attachment of platelets to other platelets. Platelet activation activates the Glycoprotein IIb/IIIa (GPIIb/IIIa) receptor, the most abundant receptor on the platelet surface, which is necessary for aggregation. The activated GPIIb/IIIa on one platelet can bind a molecule of fibrinogen. The free end of the fibrinogen molecule is then available to bind to GPIIb/IIIa on another platelet. This results in a fibrinogen link connecting two platelets (Kickler et al., 2006b).
Clot formation is essential in hemostasis but would be problematic if it were not controlled. However, the intact endothelium produces various substances that prevent clot formation. Nitric oxide (NO) is one such product, and it can prevent platelet activation in several ways. As mentioned, TXA2 is a platelet agonist involved in platelet activation, and NO can inhibit the TXA2 receptor. It does so by activating cGMP-dependent protein kinase, which serves as a catalyst for the phosphorylation of the TXA2 receptor. Once the receptor is phosphorylated, TXA2 can no longer participate in platelet activation (Wang et al., 1998). NO can also diminish activation by lowering [Ca2+]i which is essential in platelet activation. NO causes an increase in NO-stimulated guanylyl cyclase which inhibits intracellular Ca2+ release, decreases Ca2+ entry, and increases the extrusion rate of Ca2+. Furthermore, NO can also decrease platelet aggregation by reducing the number of GPIIb/IIIa receptors on the surface of platelets (Aszodi et al., 1999).
Another product made by endothelial cells is prostacyclin (PGI2). Similar to the effects of NO, PGI2 can prevent aggregation and can reduce Ca2+. When the PGI2 receptor becomes activated, production of cyclic adenosine monophosphate (cAMP) increases which causes the inhibition of both Ca2+ mobilization and granule release. Additionally, increased cAMP leads to the phosphorylation of vasodilator-stimulated phosphoprotein (VASP). The phosphorylation of VASP influences intracellular actin which helps to regulate platelet structure, and it also inactivates the GPIIb/IIIa receptor. The structural change and the inhibition of Ca2+ and GPIIb/IIIa receptors not only prevents platelet activation but also returns activated platelets to their resting state (Jin et al., 2005).
Endothelial cells also produce plasminogen activators which convert circulating plasminogen into plasmin. Plasmin, a serine protease prevents platelet aggregation via the proteolysis of platelet surface GPIIb/IIIa receptors and the fibrinogen molecules themselves (Jin et al., 2005). Finally, endothelial cells possess CD39, a membrane glycoprotein. By neutralizing ADP secreted by activated platelets, CD39 has an inhibitory effect on ADP-induced platelet aggregation (Jin et al., 2005).
2.3 The Paradoxical Effect of LDL on NHE and Platelet function
High concentrations of low density lipoproteins (LDL) have commonly been linked to increased platelet sensitivity to agonists, which enhances the platelets’ response to stimuli and promotes the positive feedback loop (Nofer et al., 2006). Oversensitivity leads to an increased risk of atherosclerosis and stroke (Hackeng et al., 1999). Native LDLs are thought to work benignly, but elevated levels of native LDLs act as risk factors when defective apoB/E receptors fail to remove them from circulation, which allows them to become oxidized. Once oxidized in sufficient concentrations, LDLs are known to accumulate within the walls of arteries and form foam cells, which induces further aggregation (Korporaal et al., 2007). Studies have shown that oxidized LDLs will inhibit the plasma membrane Ca2+ ATPase, which causes increased Ca2+ levels needed for platelet activation. Native LDLs do not display this effect (Zhao et al., 1996). Thus, oxidation may drastically change the biological properties and function of LDLs.
The common theory proposes that the formation of oxidized LDLs in the subendothelial space of arterial walls is a cause for atherogenesis. The role of oxidized LDLs in inducing platelet aggregation, however, is unclear. Oxidized LDLs have been shown to increase platelet aggregation in some but not all studies, which may be due to different methodology in approaching oxidation, isolation, and concentrations (Chou et al., 2006). In some reports, highly oxidized LDLs failed to activate aggregation, whereas others have shown that mildly oxidized LDLs induced aggregation (Vlasova and Reznikova, 2000). If LDLs do play a role in inducing platelet aggregation, then there appears to exist a paradoxical effect because LDLs inhibit the Na+/H+ exchanger, which should reduce platelet aggregation as explained in the following section (Nofer et al., 2006).
3. Na+/H+ exchanger (NHE) in platelet regulation
There are nine discovered Na+/H+ exchanger (NHE) isoforms. NHE1 is ubiquitously expressed, and its main role is the regulation of intracellular pH (pHi) and volume control (Rosskopf, 1999);. Under acidic conditions, NHE1 becomes activated to export 1 H+ ion in exchange with 1 Na+ ion, thereby raising pHi (Huber et al., 2012). NHE1 can also be activated due to a decrease in cell volume. Upon Na+ influx from NHE1 activation, cell volume increases due to the entry of water via osmosis (Rosskopf, 1999).
NHE1 is of significant importance in platelets as it is expressed in high amounts relative to other blood cells (Rosskopf, 1999). Like in other cells, platelet NHE1 responds to acidic conditions. When sodium-propionate was added to platelets in order to induce intracellular acidification, NHE1 became activated to restore pHi (De et al., 2005). Platelet NHE1 also plays a role in volume control in which the addition of Na+ leads to an increase in volume (Rosskopf, 1999). To further support this notion, in the presence of an NHE inhibitor, platelet swelling is reduced (Klinkhardt et al., 2003); (Tomasiak et al., 2005); (Sarigianni et al., 2010); (Xu et al., 2009).
In addition to regulating pH and volume, NHE1 plays a large role in platelet activity. Thrombus generation involves an increase in [Ca2+]i, and NHE1 activation results in such an increase. When Na+ is overloaded in a cell, the Na+/Ca+ exchanger (NCX) becomes reversed to expel Na+ ions in exchange for Ca2+ ions (Huber et al., 2012). It has been demonstrated that NHE inhibitors reduce Ca2+ mobilization and the formation of platelet-leukocyte aggregates (Siffert et al., 1987). Platelet-leukocyte aggregates are a good marker of platelet activation (Michelson et al., 2001). Additionally, thrombin-induced aggregation of platelets is decreased in a Na+-free medium, as compared to a medium containing Na+. Since Na+ influx is mediated by NHE1, this result indicates that NHE1 is involved in platelet aggregation (Bucki et al., 2006). Furthermore, in a study conducted by Roberts and colleague, when collagen was administered to platelets, there was an increase in both [Ca2+]i and aggregation. Aggregation depended on an increase in [Ca2+]i, but the change in [Ca2+]i was independent of NHE activity since the addition of EIPA, an NHE inhibitor, had no significant effect on either [Ca2+]i or aggregation (Roberts et al., 2004). However, a study performed by Samson and colleagues revealed the opposite effect in which EIPA did reduce collagen-induced Na+ influx (Samson et al., 2001). Thus, while Na+ influx and subsequent aggregation is mediated by NHE1 for platelet agonists such as thrombin, there are some contradictory reports as to whether this result applies to collagen. NHE1 is involved in thrombin-induced platelet activity and in ADP-induced platelet activity. Cariporide, an NHE1 inhibitor, decreases the activation of the GPIIb/IIIa receptor when platelets were induced by ADP under acidic conditions (Klinkhardt et al., 2003). NHE1 affects platelet activity via Na+ influx, but an elevation in pHi plays a role as well. In the presence of ionomycin, an ionophore that is capable of causing platelet aggregation by increasing [Ca2+]i, NH4Cl caused an increase in the size of platelet aggregates. In addition to the increase in size, the number of large aggregates increased as well. Furthermore, NH4Cl was found to augment ionomycin-induced Ca2+ entry into platelets. Without the presence of an inducer, the addition of NH4Cl yielded no effect, but the results reveal that alkalinity is a factor in platelet activity (Marumo et al., 2002).
4. The role of platelets in ischemic stroke
Platelets assume a role in the development of focal cerebral ischemia by virtue of their participation in thromboemboli that may initiate stroke symptoms (del Zoppo, 1998), usually due to arterial thromboembolism (Merwick et al., 2010); (Liu et al., 2014); (Fernandez-Lopez et al., 2013); (Ansar et al., 2014).
Platelets are activated by a number of stimuli, such as exposure of the vascular sub-endothelium, fibrin deposition, and abnormal surfaces, e.g., atheromata. A number of observations, including the appearance of platelet thrombi on atheromata in situ, indicate that platelet activation is relevant to stroke. In addition, certain anti-platelet agents (e.g., aspirin) significantly reduce the incidence of ischemic stroke after initial transient ischemic attacks (del Zoppo, 1998); (Marumo et al., 2002). Plasma levels of the platelet α-granule components, PF4 (platelet factor 4) and P-TG (plasma triglyceride), were released into plasma upon platelet activation and can increase ischemic stroke incidents. Iwamoto and colleagues have shown that the ratio of P-TG concentration between the antecubital vein and the internal jugular vein was elevated in the chronic phase of lacunar stroke and in athero-thrombotic stroke patients, which suggests that platelet activation may occur in the ischemic brain (Legrand et al., 1991). In addition, in vitro studies by Abulencia and colleagues demonstrated that platelets obtained from patients with atherosclerotic stroke exhibit increased shear-induced platelet aggregation compared with those from healthy controls (Abulencia et al., 2001).
4.1. Platelet inhibitors and ischemic stroke protection
Platelets play an important role in ischemic stroke and can lead to thrombosis and ischemic infarct. Therefore, inhibition of platelet activation can protect against ischemic stroke. Aspirin and clopidogrel, as platelet inhibitors, have been used clinically for stroke prevention. Aspirin’s efficacy in secondary stroke prevention was demonstrated in clinical trials in the late 1970s (Thomson and Anderson, 2013). The use of long-term, oral anticoagulants to prevent recurrent non-cardioembolic ischemic stroke was halted when warfarin was found to be no more efficacious and more dangerous than aspirin (Fuster et al., 2007). Clopidogrel’s efficacy in the prevention of ischemic events in patients with documented atherothrombotic disease has been well documented, and the drug gained U.S. FDA approval for this application in 1998 (Thomson and Anderson, 2013). Recently, it has been reported that dual antiplatelet therapy (clopidogrel and aspirin) can be more effective than single antiplatelet therapy in preventing early recurrent stroke in patients with acute symptomatic atherothrombosis (predominantly intracranial) of the brain (Hankey, 2013). Furthermore, Wang and colleagues published findings that support dual antiplatelet therapy. Stroke occurred in 8.2% of patients taking both clopidogrel and aspirin, whereas it occurred in 11.7% of patients taking only aspirin (p < 0.001). Rates of hemorrhagic stroke or moderate or severe hemorrhage were similar in the clopidogrel-aspirin group (0.3%) and in the aspirin alone group (0.3%, p = 0.73) (Wang et al., 2013). These findings are consistent with the report by Hankey and colleagues (Hankey, 2013).
4.2. NHE inhibition reduces cerebral ischemia-reperfusion injury
Ionic dysregulation mediated by multiple ion channels and exchanger proteins plays an important role in cerebral ischemic reperfusion injury (Khanna et al., 2014; Pignataro et al., 2014; Shah and Aizenman, 2014; Song and Yu, 2014). Among the nine NHE isoforms, NHE1 is the most intensively studied due to its over-activation in a series of pathological processes such as essential hypertension, myocardial ischemic-reperfusion injury, post-ischemic dysfunction and cancer cell progression. Genetic ablation of NHE1 attenuates intracellular Na+ and Ca2+ accumulation in neuronal cultures after oxygen and glucose deprivation and results in less cell death (Luo and Sun, 2007). Cariporide is neuroprotective in the mouse model of transient focal ischemia (Wang et al., 2008). The efficacy of NHE1 inhibition by cariporide in neuroprotection is also documented with a magnetic resonance imaging (MRI) study to assess reduced brain lesion during reperfusion after cariporide treatment (Ferrazzano et al., 2011). Moreover, NHE1 is involved in microglial migration. Chemoattractant, bradykinin (BK), stimulated microglial migration by increasing lamellipodial area and protrusion rate, but reduced lamellipodial persistence time. Blocking NHE1 activity with cariporide not only acidified microglia and abolished the BK-triggered dynamic changes of lamellipodia, but it also reduced microglial motility and microchemotaxis in response to BK (Shi et al., 2013). Beneficial effects of blocking NHE1 are also well documented in perinatal brain injury (Uria-Avellanal and Robertson, 2014).
4.3 Cariporide reduces ischemic heart injury but increases embolic stroke
There has been strong support for NHE1 blockade in the protection against ischemic cardiac injury. An initial study with amiloride, a prototypical nonspecific NHE inhibitor, in rats demonstrated enhanced ventricular recovery and diminished enzyme efflux after myocardium ischemic reperfusion (Karmazyn, 1998). Since this study, several highly specific and selective inhibitors for the NHE1 isoform have been developed in order to support the investigation of cardioprotective strategies. These agents have been shown to possess cardioprotective properties in a variety of experimental models and animal species (Wang et al., 2007). Beneficial results of blocking NHE1 include the delay of ischemia-induced rise of cytosolic Ca2+ using amiloride (Murphy et al., 1991), the reduction of Na+ overload using cariporide (Hartmann and Decking, 1999), and the reduction of infarct size in pigs (Rohmann et al., 1995) and in rabbits with cariporide (Linz et al., 1998).
The cardioprotective effects of NHE1 inhibition in animal models of myocardial infarction has led to multiple clinical assessments of NHE1 inhibitors in the treatment of cardiovascular disease. Cariporide is an NHE1 inhibitor which has been clinically investigated for the treatment of ischemia reperfusion injury and acute myocardial infarction (Huber et al., 2012). The outcomes of these trials have shown some positive results in regards to the GUARDIAN trial, which demonstrated decreased mortality and myocardium infarct (MI) for 36 hours postoperatively and with beneficial effects lasting 6 months (Chaitman, 2003). The EXPEDITION study, which investigated the effect of cariporide on a cohort of patients undergoing coronary artery bypass surgery (n=5761), demonstrated positive results in the decreased rate of MI, but there were adverse effects of cerebrovascular blockage leading to ischemic stroke (Table 1). For this trial, 180 mg of cariporide was administered intravenously for 1 h prior to surgery, 40 mg/h over 24 h after surgery, and 20 mg/h over the subsequent 24 h. The rate of myocardial infarction decreased (18.9% in the placebo group vs. 14.4% in the cariporide group, p < 0.001) and yet mortality rates increased (1.5% vs. 2.2%, p=0.02) (Mentzer, Jr. et al., 2008). Despite the robust cardioprotective effects from NHE1 inhibition as demonstrated in experimental studies, clinical trials involving patients at risk for coronary heart disease have provided mixed results (Fleisher et al., 2005); (Zeymer et al., 2001); (Chaitman, 2003); (Mentzer, Jr. et al., 2008). The cardioprotective effect of cariporide was negated by increased cerebrovascular complications leading to ischemic stroke.
Table 1.
Clinical trials evaluating NHE1 inhibitors
| Clinical Trial | NHE1 Inhibitor | Patient Population | N | Dosage | Main Findings |
|---|---|---|---|---|---|
| PMIIa | Zoniporide | Patients with known or multiple risk factors for coronary artery disease undergoing noncardiac vascular surgery | 769 | 3, 6, 12 mg/kg or placebo delivered as 1-hr loading dose immediately before surgery and followed by a continuous intravenous infusion for up to 7 days | Failed to demonstrate efficacy in reducing the proportion of patients who experience a composite cardiovascular endpoint |
| ESCAMIb | Eniporide | Patients undergoing thrombolytic therapy or primary angioplasty for acute ST-elevation MI | 1,389 | 50, 100, 150, 200 mg or placebo delivered as a 10-min infusion before reperfusion therapy | Failed to demonstrate a reduction in enzymatic infarct size and an improvement in clinical outcome |
| GUARDIANc | Cariporide | Patients with acute coronary syndrome or undergoing high-risk PCI or CABG | 11,590 | 20, 80, 120 mg or placebo delivered as a 1-hr infusion every 8 hr for 2 to 7 days | 120 mg reduced the rate of death and MI in patients undergoing CABG |
| EXPEDITIONd | Cariporide | Patients undergoing high-risk CABG | 5,761 | 180 mg or placebo delivered as a 1-hr preoperative loading dose, then 40 mg/hr over 24 hr and 20 mg/hr for the subsequent 24 hr | Reduced incidence of MI but increased mortality associated with an increase in CVE |
. (Chaitman, 2003)
CABG = coronary artery bypass grafting
CVE = cerebrovascular events
MI = myocardial infarction
PCI = percutaneous coronary intervention
PMII = perioperative myocardial ischemic injury
These results appear to be puzzling considering the aforementioned NHE1 function in platelet activation. Increased platelet activation is associated with ischemic stroke, while platelet inhibitors have been demonstrated to reduce the occurrence of stroke. Therefore, cariporide would be expected to inhibit platelet activation. The paradoxical nature of the EXPEDITION results may be in part explained by the different protocols in administering cariporide (Sarigianni et al., 2010). The dose used in the EXPEDITION trial was administered continuously with nearly double the highest dose used in the GUARDIAN study (Leng et al., 2014).
NHE1 remains a likely effective therapeutic target for several diseases despite the results from the EXPEDITION study. Therefore, developing novel and selective NHE inhibitors with a longer half-life and high potency are desirable. Dose-dependent pharmacokinetics of NHE1 inhibitor, KR-33028, were evaluated in rats after intravenous and oral administration. KR-33028 was efficiently absorbed across the gastrointestinal membrane and demonstrated increased bioavailability at high oral doses due to saturation of the first-pass metabolism (Kim et al., 2007). More recent efforts to optimize the NHE1 inhibitors for future clinical studies resulted in the identification of compound 60, based on modification of the NHE1 inhibitor sabiporide (Huber et al., 2012). Compound 60 displays a pharmokinetic profile suitable for chronic administration in humans, and demonstrates remarkable efficacy in reducing ischemia reperfusion injury in rat MI models (Huber et al., 2012). Further evaluation of efficacy of these new inhibitors in reducing ischemic reperfusion injury in stroke is warranted.
5. Conclusion
The main function of platelets is to initiate thrombus formation that permits wound healing in response to vascular injury. Platelets assume a role in the development of ischemic disease by virtue of their participation in thromboembolism that may initiate stroke or myocardial infarction. NHE1 is important in the regulation of H+ and Na+ homeostasis in platelets (Figure 1). Coordinated function of NHE1 and NCX leads to increased Na+ and Ca2+ levels and platelet-leukocyte aggregation. Cariporide, an NHE1 inhibitor, has inhibitory effects on the degranulation of human platelets and the formation of platelet-leukocyte aggregates. Despite the cardio- or neuroprotective effects of cariporide in the preclinical studies, clinical trials provided mixed results. Clinical evaluation of cariporide in the EXPEDITION trial demonstrated protection of myocardial infarction, but revealed increased incidence of cerebrovascular events of thromboembolic origin. The significant adverse effects observed in the EXPEDITION trial have been suggested to be attributed to cariporide–mediated effects on platelet and/or other procoagulant factors. Additional research is warranted to better understand the role of NHE1 in platelet regulation. NHE1 remains as an important therapeutic target for cardiovascular diseases. Therefore, development of NHE1 inhibitors with improved potency and selectivity has great potentials for clinical usage.
Figure 1. Schematic illustration of NHE1 in platelet activation.
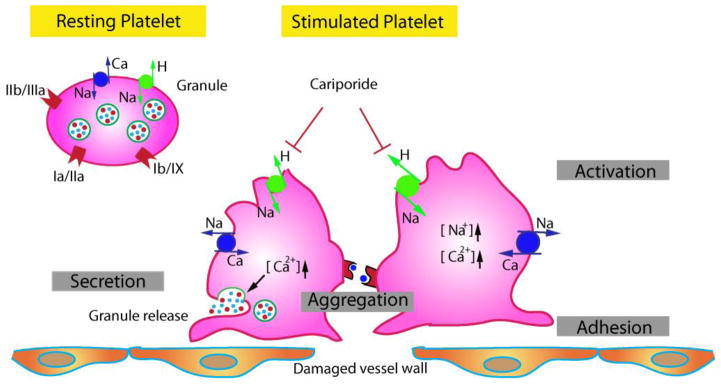
Hemostasis involving platelets can be divided into four phases: adhesion, activation, secretion, and aggregation. NHE1 is important in the regulation of H+ and Na+ homeostasis in platelets. In addition to receptor-mediated signaling cascade (glycoprotein IIa, IIb, etc), coordinated function of NHE1 and Na+/Ca2+ exchange leads to increased Na+ and Ca2+ levels and platelet-leukocyte aggregation. Cariporide, an NHE1 inhibitor, has inhibitory effects on the degranulation of human platelets and the formation of platelet-leukocyte aggregates.
Highlights.
Overview of current research of platelet biology and regulation.
Summary of the roles of Na+/H+ exchanger 1 in the regulation of platelet function.
Insight for NHE1 as a therapeutic target for ischemic reperfusion injury.
Paradoxical effects of cariporide on platelet activation and ischemic stroke.
A need to better understand NHE1 in platelet function and procoagulant regulation.
Acknowledgments
This work was supported in part by NIH grant R01 NS048216 (DS) and Technology Cooperation of Province and Chinese Academy of Sciences Program 2014 YS14C11 (SH).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Reference List
- Abulencia JP, Tien N, McCarty OJ, Plymire D, Mousa SA, Konstantopoulos K. Comparative antiplatelet efficacy of a novel, nonpeptide GPIIb/IIIa antagonist (XV454) and abciximab (c7E3) in flow models of thrombosis. Arterioscler Thromb Vasc Biol. 2001;21:149–156. doi: 10.1161/01.atv.21.1.149. [DOI] [PubMed] [Google Scholar]
- Angiolillo DJ, Ueno M, Goto S. Basic principles of platelet biology and clinical implications. Circ J. 2010;74:597–607. doi: 10.1253/circj.cj-09-0982. [DOI] [PubMed] [Google Scholar]
- Ansar S, Chatzikonstantinou E, Thiagarajah R, Tritschler L, Fatar M, Hennerici MG, Meairs S. Pro-inflammatory mediators and apoptosis correlate to rt-PA response in a novel mouse model of thromboembolic stroke. PLoS One. 2014;9:e85849. doi: 10.1371/journal.pone.0085849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aszodi A, Pfeifer A, Ahmad M, Glauner M, Zhou XH, Ny L, Andersson KE, Kehrel B, Offermanns S, Fassler R. The vasodilator-stimulated phosphoprotein (VASP) is involved in cGMP- and cAMP-mediated inhibition of agonist-induced platelet aggregation, but is dispensable for smooth muscle function. EMBO J. 1999;18:37–48. doi: 10.1093/emboj/18.1.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avkiran M. Rational basis for use of sodium-hydrogen exchange inhibitors in myocardial ischemia. Am J Cardiol. 1999;83:10G–17G. doi: 10.1016/s0002-9149(99)00215-5. [DOI] [PubMed] [Google Scholar]
- Bucki R, Pastore JJ, Giraud F, Janmey PA, Sulpice JC. Involvement of the Na+/H+ exchanger in membrane phosphatidylserine exposure during human platelet activation. Biochim Biophys Acta. 2006;1761:195–204. doi: 10.1016/j.bbalip.2005.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cengiz P, Kleman N, Uluc K, Kendigelen P, Hagemann T, Akture E, Messing A, Ferrazzano P, Sun D. Inhibition of Na+/H+ exchanger isoform 1 is neuroprotective in neonatal hypoxic ischemic brain injury. Antioxid Redox Signal. 2011;14:1803–1813. doi: 10.1089/ars.2010.3468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaitman BR. A review of the GUARDIAN trial results: clinical implications and the significance of elevated perioperative CK-MB on 6-month survival. J Card Surg. 2003;18(Suppl 1):13–20. doi: 10.1046/j.1540-8191.18.s1.3.x. [DOI] [PubMed] [Google Scholar]
- Chou DS, Chan CH, Hsiao G, Shen MY, Tsai YJ, Chen TF, Sheu JR. Inhibitory mechanisms of low concentrations of oxidized low-density lipoprotein on platelet aggregation. J Biomed Sci. 2006;13:333–343. doi: 10.1007/s11373-005-9042-x. [DOI] [PubMed] [Google Scholar]
- Cimmino G, Golino P. Platelet biology and receptor pathways. J Cardiovasc Transl Res. 2013;6:299–309. doi: 10.1007/s12265-012-9445-9. [DOI] [PubMed] [Google Scholar]
- De CE, Lanza GA, Romagnoli E, Ciabattoni G, Sestito A, Pasqualetti P, Crea F, Maseri A, Landolfi R. Abnormal pH-sensing of platelet Na+/H+ exchanger in patients with cardiac syndrome X. Int J Cardiol. 2005;100:371–376. doi: 10.1016/j.ijcard.2004.03.078. [DOI] [PubMed] [Google Scholar]
- del Zoppo GJ. The role of platelets in ischemic stroke. Neurology. 1998;51:S9–14. doi: 10.1212/wnl.51.3_suppl_3.s9. [DOI] [PubMed] [Google Scholar]
- Fernandez-Lopez D, Faustino J, Derugin N, Vexler ZS. Acute and chronic vascular responses to experimental focal arterial stroke in the neonate rat. Transl Stroke Res. 2013;4:179–188. doi: 10.1007/s12975-012-0214-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrazzano P, Shi Y, Manhas N, Wang Y, Hutchinson B, Chen X, Chanana V, Gerdts J, Meyerand ME, Sun D. Inhibiting the Na+/H+ exchanger reduces reperfusion injury: a small animal MRI study. Front Biosci(Elite Ed) 2011;3:81–88. doi: 10.2741/e222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleisher LA, Newman MF, St Aubin LB, Cropp AB, Billing CB, Bonney S, Mackey WC, Poldermans D, Corbalan R, Pereira AH, Coriat P. Efficacy of zoniporide, an Na/H exchange ion inhibitor, for reducing perioperative cardiovascular events in vascular surgery patients. J Cardiothorac Vasc Anesth. 2005;19:570–576. doi: 10.1053/j.jvca.2005.06.012. [DOI] [PubMed] [Google Scholar]
- Fuster V, Ryden LE, Cannom DS, Crijns HJ, Curtis AB, Ellenbogen KA, Halperin JL, Le Heuzey JY, Kay GN, Lowe JE, Olsson SB, Prystowsky EN, Tamargo JL, Wann S, Priori SG, Blanc JJ, Budaj A, Camm AJ, Dean V, Deckers JW, Despres C, Dickstein K, Lekakis J, McGregor K, Metra M, Morais J, Osterspey A, Zamorano JL, Smith SC, Jr, Jacobs AK, Adams CD, Anderson JL, Antman EM, Hunt SA, Nishimura R, Ornato JP, Page RL, Riegel B. ACC/AHA/ESC 2006 guidelines for the management of patients with atrial fibrillation--excutive summary. Rev Port Cardiol. 2007;26:383–446. [PubMed] [Google Scholar]
- George JN. Platelets. Lancet. 2000;355:1531–1539. doi: 10.1016/S0140-6736(00)02175-9. [DOI] [PubMed] [Google Scholar]
- Hackeng CM, Huigsloot M, Pladet MW, Nieuwenhuis HK, van Rijn HJ, Akkerman JW. Low-density lipoprotein enhances platelet secretion via integrin-alphaIIbbeta3-mediated signaling. Arterioscler Thromb Vasc Biol. 1999;19:239–247. doi: 10.1161/01.atv.19.2.239. [DOI] [PubMed] [Google Scholar]
- Hankey GJ. Dual antiplatelet therapy in acute transient ischemic attack and minor stroke. N Engl J Med. 2013;369:82–83. doi: 10.1056/NEJMe1305127. [DOI] [PubMed] [Google Scholar]
- Hartmann M, Decking UK. Blocking Na(+)-H+ exchange by cariporide reduces Na(+)-overload in ischemia and is cardioprotective. J Mol Cell Cardiol. 1999;31:1985–1995. doi: 10.1006/jmcc.1999.1029. [DOI] [PubMed] [Google Scholar]
- Hartwig J, Italiano J., Jr The birth of the platelet. J Thromb Haemost. 2003;1:1580–1586. doi: 10.1046/j.1538-7836.2003.00331.x. [DOI] [PubMed] [Google Scholar]
- Huber JD, Bentzien J, Boyer SJ, Burke J, De LS, Eickmeier C, Guo X, Haist JV, Hickey ER, Kaplita P, Karmazyn M, Kemper R, Kennedy CA, Kirrane T, Madwed JB, Mainolfi E, Nagaraja N, Soleymanzadeh F, Swinamer A, Eldrup AB. Identification of a potent sodium hydrogen exchanger isoform 1 (NHE1) inhibitor with a suitable profile for chronic dosing and demonstrated cardioprotective effects in a preclinical model of myocardial infarction in the rat. J Med Chem. 2012;55:7114–7140. doi: 10.1021/jm300601d. [DOI] [PubMed] [Google Scholar]
- Jin RC, Voetsch B, Loscalzo J. Endogenous mechanisms of inhibition of platelet function. Microcirculation. 2005;12:247–258. doi: 10.1080/10739680590925493. [DOI] [PubMed] [Google Scholar]
- Karmazyn M. The myocardial sodium-hydrogen exchanger (NHE) and its role in mediating ischemic and reperfusion injury. Keio J Med. 1998;47:65–72. doi: 10.2302/kjm.47.65. [DOI] [PubMed] [Google Scholar]
- Khanna A, Kahle KT, Walcott BP, Gerzanich V, Simard JM. Disruption of ion homeostasis in the neurogliovascular unit underlies the pathogenesis of ischemic cerebral edema. Transl Stroke Res. 2014;5:3–16. doi: 10.1007/s12975-013-0307-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kickler TS, Oguni S, Borowitz MJ. A clinical evaluation of high fluorescent platelet fraction percentage in thrombocytopenia. Am J Clin Pathol. 2006a;125:282–287. doi: 10.1309/50H8-JYHN-9JWC-KAM7. [DOI] [PubMed] [Google Scholar]
- Kickler TS, Oguni S, Borowitz MJ. A clinical evaluation of high fluorescent platelet fraction percentage in thrombocytopenia. Am J Clin Pathol. 2006b;125:282–287. doi: 10.1309/50H8-JYHN-9JWC-KAM7. [DOI] [PubMed] [Google Scholar]
- Kim YH, Yoo SD, Kim YS, Lee KH, Lee HS. Dose-dependent pharmacokinetics of a new Na+/H+ exchanger inhibitor KR-33028 in rats. Biopharm Drug Dispos. 2007;28:423–429. doi: 10.1002/bdd.571. [DOI] [PubMed] [Google Scholar]
- Klinkhardt U, Kuczka K, Harder S. Effects of the NHE-1 inhibitor cariporide alone or together with the P2Y12 antagonist AR-C 69331 MX on CD62p expression and formation of platelet-leukocyte aggregates. Thromb Res. 2003;111:251–257. doi: 10.1016/j.thromres.2003.09.015. [DOI] [PubMed] [Google Scholar]
- Korporaal SJ, Van EM, Adelmeijer J, Ijsseldijk M, Out R, Lisman T, Lenting PJ, Van Berkel TJ, Akkerman JW. Platelet activation by oxidized low density lipoprotein is mediated by CD36 and scavenger receptor-A. Arterioscler Thromb Vasc Biol. 2007;27:2476–2483. doi: 10.1161/ATVBAHA.107.150698. [DOI] [PubMed] [Google Scholar]
- Legrand C, Woimant F, Haguenau M, Caen J. Platelet surface glycoprotein changes in patients with cerebral ischemia. Nouv Rev Fr Hematol. 1991;33:497–499. [PubMed] [Google Scholar]
- Leng T, Shi Y, Xiong ZG, Sun D. Proton-sensitive cation channels and ion exchangers in ischemic brain injury: new therapeutic targets for stroke? Prog. Neurobiol. 2014;115:189–209. doi: 10.1016/j.pneurobio.2013.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linz W, Albus U, Crause P, Jung W, Weichert A, Scholkens BA, Scholz W. Dose-dependent reduction of myocardial infarct mass in rabbits by the NHE-1 inhibitor cariporide (HOE 642) Clin Exp Hypertens. 1998;20:733–749. doi: 10.3109/10641969809052116. [DOI] [PubMed] [Google Scholar]
- Liu J, Wang Y, Akamatsu Y, Lee CC, Stetler RA, Lawton MT, Yang GY. Vascular remodeling after ischemic stroke: mechanisms and therapeutic potentials. Prog Neurobiol. 2014;115:138–156. doi: 10.1016/j.pneurobio.2013.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo J, Sun D. Physiology and pathophysiology of Na(+)/H(+) exchange isoform 1 in the central nervous system. Curr Neurovasc Res. 2007;4:205–215. doi: 10.2174/156720207781387178. [DOI] [PubMed] [Google Scholar]
- Marumo M, Suehiro A, Kakishita E, Wakabayashi I. Intracellular alkalinization augments platelet aggregation due to increase in cytosolic free-Ca2+ Platelets. 2002;13:159–165. doi: 10.1080/0953371027993. [DOI] [PubMed] [Google Scholar]
- Mentzer RM, Jr, Bartels C, Bolli R, Boyce S, Buckberg GD, Chaitman B, Haverich A, Knight J, Menasche P, Myers ML, Nicolau J, Simoons M, Thulin L, Weisel RD. Sodium-hydrogen exchange inhibition by cariporide to reduce the risk of ischemic cardiac events in patients undergoing coronary artery bypass grafting: results of the EXPEDITION study. Ann Thorac Surg. 2008;85:1261–1270. doi: 10.1016/j.athoracsur.2007.10.054. [DOI] [PubMed] [Google Scholar]
- Merwick A, Albers GW, Amarenco P, Arsava EM, Ay H, Calvet D, Coutts SB, Cucchiara BL, Demchuk AM, Furie KL, Giles MF, Labreuche J, Lavallee PC, Mas JL, Olivot JM, Purroy F, Rothwell PM, Saver JL, Sheehan OC, Stack JP, Walsh C, Kelly PJ. Addition of brain and carotid imaging to the ABCD(2) score to identify patients at early risk of stroke after transient ischaemic attack: a multicentre observational study. Lancet Neurol. 2010;9:1060–1069. doi: 10.1016/S1474-4422(10)70240-4. [DOI] [PubMed] [Google Scholar]
- Michelson AD, Barnard MR, Krueger LA, Valeri CR, Furman MI. Circulating monocyte-platelet aggregates are a more sensitive marker of in vivo platelet activation than platelet surface P-selectin: studies in baboons, human coronary intervention, and human acute myocardial infarction. Circulation. 2001;104:1533–1537. doi: 10.1161/hc3801.095588. [DOI] [PubMed] [Google Scholar]
- Murphy CT, Elmore M, Kellie S, Westwick J. Comparison of the role of protein kinase C in platelet functional responses induced by three different mechanisms, PAF, ionomycin and arachidonic acid. Biochim Biophys Acta. 1991;1133:46–54. doi: 10.1016/0167-4889(91)90240-x. [DOI] [PubMed] [Google Scholar]
- Nofer JR, Noll C, Feuerborn R, Assmann G, Tepel M. Low density lipoproteins inhibit the Na+/H+ antiport in human platelets via activation of p38MAP kinase. Biochem Biophys Res Commun. 2006;340:751–757. doi: 10.1016/j.bbrc.2005.12.070. [DOI] [PubMed] [Google Scholar]
- Pignataro G, Sirabella R, Anzilotti S, Di RG, Annunziato L. Does Na(+)/Ca(2)(+) exchanger, NCX, represent a new druggable target in stroke intervention? Transl. Stroke Res. 2014;5:145–155. doi: 10.1007/s12975-013-0308-8. [DOI] [PubMed] [Google Scholar]
- Roberts DE, McNicol A, Bose R. Mechanism of collagen activation in human platelets. J Biol Chem. 2004;279:19421–19430. doi: 10.1074/jbc.M308864200. [DOI] [PubMed] [Google Scholar]
- Rohmann S, Weygandt H, Minck KO. Preischaemic as well as postischaemic application of a Na+/H+ exchange inhibitor reduces infarct size in pigs. Cardiovasc Res. 1995;30:945–951. [PubMed] [Google Scholar]
- Rosskopf D. Sodium-hydrogen exchange and platelet function. J Thromb Thrombolysis. 1999;8:15–24. doi: 10.1023/a:1008986329267. [DOI] [PubMed] [Google Scholar]
- Ruggeri ZM. Platelets in atherothrombosis. Nat Med. 2002;8:1227–1234. doi: 10.1038/nm1102-1227. [DOI] [PubMed] [Google Scholar]
- Ruggeri ZM, Mendolicchio GL. Adhesion mechanisms in platelet function. Circ Res. 2007;100:1673–1685. doi: 10.1161/01.RES.0000267878.97021.ab. [DOI] [PubMed] [Google Scholar]
- Samson J, Stelmach H, Tomasiak M. The importance of Na+/H+ exchanger for the generation of procoagulant activity by porcine blood platelets. Platelets. 2001;12:436–442. doi: 10.1080/09537100120078395. [DOI] [PubMed] [Google Scholar]
- Sarigianni M, Tsapas A, Mikhailidis DP, Kaloyianni M, Koliakos G, Fliegel L, Paletas K. Na+ H+ exchanger-1: a link with atherogenesis? Expert. Opin Investig Drugs. 2010;19:1545–1556. doi: 10.1517/13543784.2010.532123. [DOI] [PubMed] [Google Scholar]
- Schulze H, Shivdasani RA. Mechanisms of thrombopoiesis. J Thromb Haemost. 2005;3:1717–1724. doi: 10.1111/j.1538-7836.2005.01426.x. [DOI] [PubMed] [Google Scholar]
- Shah NH, Aizenman E. Voltage-gated potassium channels at the crossroads of neuronal function, ischemic tolerance, and neurodegeneration. Transl Stroke Res. 2014;5:38–58. doi: 10.1007/s12975-013-0297-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y, Yuan H, Kim D, Chanana V, Baba A, Matsuda T, Cengiz P, Ferrazzano P, Sun D. Stimulation of Na(+)/H(+) exchanger isoform 1 promotes microglial migration. PLoS One. 2013;8:e74201. doi: 10.1371/journal.pone.0074201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siffert W, Siffert G, Scheid P, Riemens T, Gorter G, Akkerman JW. Inhibition of Na+/H+ exchange reduces Ca2+ mobilization without affecting the initial cleavage of phosphatidylinositol 4,5-bisphosphate in thrombin-stimulated platelets. FEBS Lett. 1987;212:123–126. doi: 10.1016/0014-5793(87)81569-7. [DOI] [PubMed] [Google Scholar]
- Song M, Yu SP. Ionic regulation of cell volume changes and cell death after ischemic stroke. Transl Stroke Res. 2014;5:17–27. doi: 10.1007/s12975-013-0314-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomson RM, Anderson DC. Aspirin and clopidogrel for prevention of ischemic stroke. Curr Neurol Neurosci Rep. 2013;13:327. doi: 10.1007/s11910-012-0327-y. [DOI] [PubMed] [Google Scholar]
- Thon JN, Montalvo A, Patel-Hett S, Devine MT, Richardson JL, Ehrlicher A, Larson MK, Hoffmeister K, Hartwig JH, Italiano JE., Jr Cytoskeletal mechanics of proplatelet maturation and platelet release. J Cell Biol. 2010;191:861–874. doi: 10.1083/jcb.201006102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomasiak M, Ciborowski M, Stelmach H. The role of Na+/H+ exchanger in serotonin secretion from porcine blood platelets. Acta Biochim Pol. 2005;52:811–822. [PubMed] [Google Scholar]
- Uria-Avellanal C, Robertson NJ. Na(+)/H(+) exchangers and intracellular pH in perinatal brain injury. Transl Stroke Res. 2014;5:79–98. doi: 10.1007/s12975-013-0322-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varga-Szabo D, Braun A, Nieswandt B. Calcium signaling in platelets. J Thromb Haemost. 2009;7:1057–1066. doi: 10.1111/j.1538-7836.2009.03455.x. [DOI] [PubMed] [Google Scholar]
- Vlasova LF, Reznikova EO. Dependence of responses of the mouth mucosa on physico-chemical characteristics of the surface of the plate denture made of acrylic resins. Biull Eksp Biol Med. 2000;129:109–112. [PubMed] [Google Scholar]
- Wang GR, Zhu Y, Halushka PV, Lincoln TM, Mendelsohn ME. Mechanism of platelet inhibition by nitric oxide: in vivo phosphorylation of thromboxane receptor by cyclic GMP-dependent protein kinase. Proc Natl Acad Sci USA. 1998;95:4888–4893. doi: 10.1073/pnas.95.9.4888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang P, Zaragoza C, Holman W. Sodium-hydrogen exchange inhibition and beta-blockade additively decrease infarct size. Ann Thorac Surg. 2007;83:1121–1127. doi: 10.1016/j.athoracsur.2006.10.039. [DOI] [PubMed] [Google Scholar]
- Wang Y, Johnston SC, Wang Y. Clopidogrel with aspirin in minor stroke or transient ischemic attack. N Engl J Med. 2013;369:1376–1377. doi: 10.1056/NEJMc1309713. [DOI] [PubMed] [Google Scholar]
- Wang Y, Luo J, Chen X, Chen H, Cramer SW, Sun D. Gene inactivation of Na+/H+ exchanger isoform 1 attenuates apoptosis and mitochondrial damage following transient focal cerebral ischemia. Eur J Neurosci. 2008;28:51–61. doi: 10.1111/j.1460-9568.2008.06304.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu WT, Jin N, Xu J, Xu YG, Wang QJ, You QD. Design, synthesis and biological evaluation of novel substituted benzoylguanidine derivatives as potent Na+/H+ exchanger inhibitors. Bioorg Med Chem Lett. 2009;19:3283–3287. doi: 10.1016/j.bmcl.2009.04.079. [DOI] [PubMed] [Google Scholar]
- Zeymer U, Suryapranata H, Monassier JP, Opolski G, Davies J, Rasmanis G, Linssen G, Tebbe U, Schroder R, Tiemann R, Machnig T, Neuhaus KL. The Na(+)/H(+) exchange inhibitor eniporide as an adjunct to early reperfusion therapy for acute myocardial infarction. Results of the evaluation of the safety and cardioprotective effects of eniporide in acute myocardial infarction (ESCAMI) trial. J Am Coll Cardiol. 2001;38:1644–1650. doi: 10.1016/s0735-1097(01)01608-4. [DOI] [PubMed] [Google Scholar]
- Zhao B, Dierichs R, Miller FN, Dean WL. Oxidized low density lipoprotein inhibits platelet plasma membrane Ca(2+)-ATPase. Cell Calcium. 1996;19:453–458. doi: 10.1016/s0143-4160(96)90118-9. [DOI] [PubMed] [Google Scholar]