

Abstract
Pre-existing maternal diabetes is a high risk factor of diabetic embryopathy, such as neural tube defects (NTDs) and congenital heart defects (CHD). Maternal diabetes significantly increases the production of reactive oxygen species (ROS) resulting in oxidative stress and diabetic embryopathy. Multiple cellular and metabolic factors contribute to these processes. FoxO3a has been demonstrated as a key transcription factor in the signaling transduction pathways responsible for maternal diabetes-induced birth defects. ASK1 activated by oxidative stress stimulates nuclear translocation of FoxO3a, resulting in over-expression of TRADD, which, in turn, leads to caspase 8 activation and apoptosis. Maternal diabetes-activated JNK1/2, downstream effectors of ASK1, can be blocked by SOD1 overexpression, suggesting that oxidative stress is responsible for JNK1/2 signaling activation. Deletion of JNK1/2 significantly suppressed the activity of FoxO3a. These obersvations indicate that maternal diabetes-induced oxidative stress stimulates the activation of ASK1, JNK1/2, FoxO3a, TRADD, caspase 8 cleavage, finally, apoptosis and diabetic embryopathy.
Keywords: maternal diabetes, birth defects, oxidative stress, apoptosis, kinase signaling
Introduction
Pregnancy with pre-existing maternal diabetes significantly increases the risk of excess apoptosis occurs in target tissues of the developing embryos resulting in diabetes-induced birth defects, such as neural tube defects (NTDs) and congenital heart defects (CHDs) 1-8. Each year, 10% of babies of diabetic mothers are born with a major congenital malformation 9. Mechanistic studies demonstrate that maternal diabetes alters multiple cellular and metabolic factors contributing to diabetic embryopathy 1, 4, 10-13. We propose that these cellular and metabolic aberrations occur through a single transcriptional mechanism, a transcription factor and its responsive gene, leading to apoptosis in embryonic cells.
We have determined that the transcription factor, FoxO3a, is activated in diabetic embryopathy 7. FoxO factors are functionally diversified in the induction of apoptosis-related pathogenesis 7, 14. The transcription factor FoxO3a is a key target of PI3K/AKT pathway, in which AKT inactivates FoxO3a by phosphorylation 15. Maternal diabetes activates FoxO3a by using several different manners: inhibited AKT function and activated ASK1-JNK1/2 pathway 3, 7.
The expression of TRADD, an apoptotic gene, is up-regulated in diabetic embryopathy and we propose that TRADD is a FoxO3a responsive gene which initiates caspase dependent apoptosis in diabetic embryopathy 7. Maternal diabetes-induced embryonic cell apoptosis is caspase dependent 1-3, 7, 16.
Previous work by our group and others has suggested that the pro-apoptotic JNK pathway, which is downstream of ASK1 pathway, plays a causative role in the induction of diabetic embryopathy 2, 3. Activated ASK1 stimulates JNK1/2 activation 17, 18, and subsequent mitochondrial dysfunction and cell apoptosis, resulting in diabetic embryopathy. Deletion of both JNK1 and JNK2 gene could inhibit nuclear translocation of FoxO3a 3. Thus, we propose a link between the JNK pathway and FoxO3a activation.
In this review, we will discuss the general function, possible clinical application and crosstalk relationship of molecules downstream of oxidative stress-induced kinase signaling in diabetic embryopathy.
Pathogenesis in the induction of diabetic embryopathy
Hyperglycemia-induced oxidative stress
A variety of antioxidants have been shown to effectively suppress hyperglycemia-induced dysmorphogenesis both in vivo and in vitro 1, 4, 12, 14, 19. Conversely, induction of oxidative stress by depletion of GSH 20, 21, by exposure to xathine/xanthine which directly generate ROS 22, or by treatment with antimycin 23, a mitochondrial complex III inhibitor that stimulates superoxide production, significantly increase embryonic anomalies. Therefore, we hypothesize that oxidative stress is the primary cause of diabetic embryopathy due to enhanced ROS production and weakening of the cellular antioxidant systems (Fig. 1).
Figure 1.

Hyperglycemia produces oxidative stress, resulting in the induction of diabetic embryopathy. Maternal diabetes induces oxidative stress through enhanced ROS production and weakened cellular antioxidant systems. Enhanced ROS production stimulates lipid peroxidation and protein carboxylation leading to overall oxidative stress in developing embryos under maternal diabetic conditions. Maternal diabetes elevates NO production in the embryos, which interacts with ROS to produce peroxynitrite inducing nitrasative stress, finally resulting in diabetic embryopathy. SOD: superoxide dismutase; CAT catalase; GSH: glutathione; GSSG: glutathione disulfide; NO: nitric oxide. ⇩: downregulated; ⇧: upregulated.
In maternal diabetes, increased levels of cellular glucose in embryonic tissues may enhance mitochondrial oxidative glucose metabolism and thus increase mitochondrial ROS production. Enhanced ROS production facilitates lipid peroxidation and protein carboxylation contributing to overall oxidative stress in embryos under maternal diabetic conditions 24, 25. Markers of lipid peroxidation, 8-iso-PGE2 25-28 and malondialdehyde 29, are dramatically elevated in embryos cultured in vitro under hyperglycemic conditions as well as in diabetic patients (Fig. 1).
Cells possess a wide range of antioxidant systems to protect themselves from the toxic effects of excessive levels of ROS. Diabetic conditions profoundly influence cellular antioxidant potential. A significant decrease in the intracellular ROS scavenging enzyme activities of superoxide dismutase (SOD) and catalase (CAT) are seen when rat embryos and their yolk sacs are maintained under diabetic condition 30. In addition, the levels of SOD and CAT mRNA decrease under maternal hyperglycemic conditions correlating inversely to an increase in embryonic anomalies 31, 32. The above evidence supports our assertion that cellular antioxidant defense systems are severely compromised in embryos and the yolk sac in response to maternal hyperglycemia, thereby contributing to cellular oxidative stress during the critical stages of organogenesis (Fig. 1).
The role of nitric oxide
Nitric oxide (NO), a critical signaling molecule involving in many processes 33, is produced from L-arginine by a family of three nitric oxide synthases (NOS). NO plays an important role in early embryonic development by regulating cell survival, apoptosis and differentiation 34-37. Because NO synthesis and function are critical during period of organogenesis, appropriate intracellular NO concentrations is a prerequisite for normal embryonic development and deregulated NO concentrations has been linked to abnormal embryonic outcomes. NO production that is elevated during early organogenesis in embryos from rat models of mild and severe diabetes leading to malformations 38, 39.
Elevated NO may directly interact with ROS generated under hyperglycemic conditions to form potent oxidant peroxynitrite leading to nitrosative stress 40, 41 (Fig. 1). The peroxynitrite anion inhibits mitochondrial electron transport, oxidizes important proteins and initiates lipid peroxidation, thus affecting many signal transduction pathways 42. The mechanism underlying maternal diabetes-increased NO production is not clear and need to be investigated further. Nitrosative stress resulting from elevated NO levels may be one of the mechanisms in the induction of diabetic embryopathy through the JNK pathway because nitrosative stress leads to JNK activation 43. The role of JNK in diabetic embryopathy will be discussed later in this review.
Aberrant signaling pathways
The protein kinase C (PKC) family of serine/threonine protein kinases consists of 12 members, involved in a number of cellular activities, including proliferation, migration, apoptosis, differentiation, and secretion 44, 45 Each member plays its own unique role in cell physiology, although overlapping functions may exist for some isoenzymes. Dregulated PKC activity may be mechanistically involved in diabetic embryopathy.
The diacylglycerol (DAG)-PKC pathway has been implicated in diabetic embryopathy. Maternal hyperglycemia stimulates DAG production in embryonic cells, which, in turn stimulates PKC activity 46. Some PKC isoforms (α, β2, and δ) are up-regulated, while others (ε and ξ) are down-regulated in diabetic embryopathy 47. Inhibiting the activity of some PKC isoforms significantly decreases malformation rate 47 (Fig. 2).
Figure 2.

Oxidative stress induces aberrant signaling pathways. Maternal diabetes-induced oxidative stress activates PKCα, -βII and -δ, stimulates lipid peroxidation, which aggravates oxidative stress; and induces apoptosis and diabetic embryopathy. The ERK1/2 and PI3K/ATK pathway is suppressed by oxidative stress induced by maternal diabetes, which further inhibits cell proliferation and survival signaling. Another pathway affected by maternal diabetes is the ASK-JNK1/2 signaling pathway, which is activated and subsequently induces ER stress and apoptosis events. These aberrant signaling pathways suppresses proliferation, which may contribute to diabetic embryopathy. DAG: diacyl glycerol; PKC: protein kinase C; ERK1/2: extracellular signal-regulated kinase 1/2.
We have also found that activity of extracellular signal-regulated kinase 1/2 (ERK1/2) is down-regulated in diabetic embryopathy 48. The activity of a pro-survival kinase, Akt, is reduced in diabetic embryopathy 49. Akt is the key mediator in the phosphatidylinositol-3 kinase (PI3K) pathway, a central regulator of the mammalian target of rapamycin (mTOR). Downregulation of Akt by maternal diabetes results in the activation of FoxO3a and downstream TRADD and caspase 8 cleavage, contributing to diabetic embryopathy 7 (Fig. 2).
Our work has shown that ASK1-JNK1/2 signaling pathway plays important role in diabetic embryopathy 2, 3, 7, 16. Under hyperglycemic condition, ASK1 is phosphorylated and activated through its dissociation from oxidized Trx 7, 50. On one hand, ASK1 phosphorylation at Thr845 activates JNK1/2 kinase by phosphorylation, which initiates proapoptotic signaling pathways which play key roles in the diabetic embryopahthy 3, 7; on the other hand, ASK1 phosphorylation initiates the UPR (unfolded protein response) and ER stress, which induces diabetic embryopathy by triggering beta-cell dysfunction and apoptosis 16, 51 (Fig. 2).
The crosstalk between the deregulated PKC and ASK1 signaling pathways with the JNK pathway seems to contribute to the induction of diabetic embryopathy.
Altered glucose metabolic pathways
Activity of the hexosamine biosynthetic pathway (HBP) is increased in embryos during diabetic pregnancy, which may contribute to hyperglycemia-induced oxidative stress 52. Increased glycolytic flux can stimulate glucose flux through the HBP pathway, in which fructose-6-phosphate and glutamine are converted to glucosamine-6-phosphate and glutamate (Fig. 3). Experimental activation of the HBP pathway by glucosamine administration mimics the effects of maternal diabetes to inhibit the pentose shunt pathway, a main glucose metabolism pathway in early embryonic development, and to induce oxidative stress by inhibiting information of reduced glutathione 52 (Fig. 3).
Figure 3.

Maternal diabetes-induced oxidative stress alters glucose metabolic pathways. During glycolysis, glucose is converted into fructose-6-phosphate with the help of phosphoglucose isomerase, and then further converted to fructose-1,6-biosphosphate by phosphofructolinase. Under maternal hyperglycemic condition, increased glycolytic flux in the embryo can stimulate flux through the HBP pathway. The fructose-6-phosphate will be transformed to glusamine-6-phosphte or glutamate, during which process, oxidative stress will be further enhanced. HBP: hexosamine biosynthetic pathway.
We propose that all these hyperglycemia-triggered upstream events converge on the Foxo3a central transcription mechanism leading to hyperglycemia-induced apoptosis in the embryonic neural epithelium cells.
Maternal diabetes-induced apoptosis—primary mechanism of diabetic embryopathy
Compelling evidence demonstrates that maternal hyperglycemia increases apoptosis in the embryo 49, 53-55. Apoptosis is specifically seen in neuroepithelial cells which are particularly susceptible to hyperglycemic damage 55. Multiple studies have confirmed that excess cell death, at least in the central nervous system, contributes to the abnormal development of structures in the embryos of diabetic animals 53, 55-58. Hyperglycemia-induced apoptosis involves altered regulation of Bcl-2 family members and caspase activation, critical events in the mitochondrial apoptotic pathway (Fig. 4). In addition, hyperglycemia-associated oxidative stress increases the Bax:Bcl-2 ratio which is associated with cytochrome c release and activation of caspase 3 in embryonic cells 59 (Fig. 4). An increase of Bax expression and release of cytochrome c and activated caspase 3 are characteristics of embryonic cells undergoing apoptosis under maternal hyperglycemic conditions 49, 55 (Fig. 4). These observations strongly suggest that high glucose concentrations cause damage to the neural progenitor cells, leading to excessive apoptosis and abnormal organogenesis. Moreover, our recent studies, in which we used antioxidants to neutralize oxidative stress, suggest a direct connection between hyperglycemia-induced oxidative stress and apoptosis 14.
Figure 4.

Maternal diabetes induces diabetic embryopathy through apoptosis. Oxidative induces the expression of Bax, leading to the increased ratio of Bax/Bcl2. The overexpressed Bax stimulates cytochrome c releasing from mitochondria and activates downstream caspase 3, finally inducing apoptosis and abnormal development. : downregulated; ⇩: upregulated. ⇧
Induction of apoptosis by the ASK1-MKK4-JNK signaling pathway
MAP kinase (MAPK) mediates a range of cellular processes, including apoptotic cell death 60. MAPKs are members of a superfamily of serine/threonine kinases that are activated in response to a variety of extracellular stimuli, including ROS 61, 62. Three distinct MAPK pathways regulate extracellular-signal-regulated-kinases (ERK1 and ERK2), c-Jun NH2-terminal kinases (JNK1, JNK2 and JNK3), and p38 MAPKs (p38α, p38β, p38γ and p38δ). These pathways control a variety of cellular functions, including gene expression, mitosis, and apoptosis through the phosphorylation of specific serine and/or threonine residues of target proteins.
The basic assembly of the MAPK signaling pathway is three-component module: via sequential activation of MAPK kinase kinase (MAP3K), MAPK kinase (MAPKK), and MAPK 63, 64. MAP3K phosphorylates and activates MAPKK, and activated MAPKK hosphorylates and activates MAPK. Because the activation status of MAPKs is largely dependent on MAP3Ks, it is important to understand how MAP3Ks are regulated. Fourteen different MAP3K have been identified. Among them, several MAP3Ks, including ASK1, TAK1 and MLK3, are known to activate the JNK pathway in response to diverse stimuli 63-65. Those MAP3Ks phosphorylate and activate the dual specific kinases, MKK4 (SEK1) and MKK7, which in turn, phosphorylate and activate JNKs 66.
Oxidative stress is one of the most potent activators of ASK1, which is essential for oxidative stress-induced apoptosis through the activation of MKK4/MKK7-JNK cascade 18. Oxidative stress induces phosphorylation of Thr-845 in the activation loop of ASK1, correlating with enhanced ASK1 activity and increased apoptosis 65 (Fig. 5). ASK1-mediated apoptosis is involved in pathogenesis of several oxidative stress-related diseases such as brain ischemia 67, ischemic heart disease 68 and Alzheimer’s disease 69.
Figure 5.

The activity change and interaction of key molecular targets downstream of oxidative stress induced by maternal diabetes. Maternal diabetes-induced oxidative stress activates ASK1 signaling pathway through dissociation with Trx and then phosphorylation. Activated ASK1 further activates JNK1/2 by phosphorylation, resulting in mitochondrial dysfunction and ER stress. Activated ASK1/JNK1/2 stimulates FoxO3a nucleus translocation and subsequent TRADD expression, caspase 8 activation, which induces apoptosis and diabetic embryopathy. AKT pathway may inactivate FoxO3a function by phosphorylation. Oxidative stress inhibits AKT activity by dephosphorylation, which will increase levels of dephosphorylated and activated FoxO3a to translocate to nucleus as transcription factor inducing TRADD over-expression. Thus, the transcription factor FoxO3a is key mechanism of diabetic embryopathy induced by oxidative stress. mitochondrial dysfunction and apoptosis.  : dephosphorylation;
: dephosphorylation; 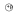 : phosphorylation; ⇩: downregulated; ⇧: upregulated.
: phosphorylation; ⇩: downregulated; ⇧: upregulated.
Most recently, it has been shown that high glucose-activated ASK1 mediates hyperglycemia-induced endothelial cell senescence 70. These findings are consistent with our hypothesis that ASK1 functions as a mediator of diabetes-related embryo malformation. Indeed, we found that deletion of the Ask1 gene significantly reduces maternal diabetes-induced capase cleavage, neuroepithelial cell apoptosis and NTD formation 7. This suggests that ASK1 plays an essential role in the induction of apoptosis leading to NTD formation (Fig. 5).
The JNK pathway specifically responds to stress-induced signals that drive apoptosis 71, 72. JNK has three isoforms (JNK1, JNK2, and JNK3) encoded by three different genes. The Jnk1 and Jnk2 genes are ubiquitously expressed, whereas Jnk3 is found to be neural tissue specific 66. The specific molecular targets of JNK include transcription factor AP-1 (mainly c-Jun, JunB, and ATF-2), Foxo factors 73, 74, as well as many other non-transcription factors, such as Bcl-2 proteins, which are closely related to apoptotic cell death factors 75. Both exogenous and endogenous ROS increase JNK1/2 activity 76-80. Substantial genetic and pharmacological evidence suggests that JNK serves as a key pro-apoptotic mediator during oxidative stress 18, 81-85. Mice having null mutations in any single JNK gene develop normally 86, as do JNK1/JNK3 or JNK2/JNK3 double mutants.85. Although JNK1/JNK2 null mutants die in utero due to abnormal apoptosis in the brain 87. These mice are useful models for delineating apoptotic pathways involving JNKs.
The ASK1-JNK pathway appears to play mutual causation role with ER stress signaling. Maternal diabetes induces ER stress and ASK1-JNK signaling pathway (Fig. 5). ASK1-JNK is a key component of the unfolded protein response signalosome, which leads to ER stress 3, 17, 88. We also found deletion of Ask1, Jnk1 or Jnk2 gene could abolish maternal diabetes-induced ER stress and subsequent apoptosis in the neuroepithelial cell 3, 16.
JNK1/2 activation-- a critical role in diabetic embryopathy
MAPK activity is altered in diabetic patients and in cells cultured in high glucose, suggesting that MAPKs may be involved in hyperglycemia-induced complications 48, 89, 90. Less, however, is known about the activity of MAPKs in embryos under maternal diabetic conditions. An increase in JNK1/2 activity is associated with increased apoptosis in the yolk sacs of malformed embryos from diabetic mothers 19, 48. We have reported that supplementation with antioxidants reduces JNK1/2 activity and the embryonic malformation rate in embryos 14, suggesting that hyperglycemia-induced oxidative stress is responsible for JNK1/2 activation. These observations indicate that increased JNK1/2 activity may play an important role in diabetic embryopathy (Fig. 5). In addition, an increase in phosphorylated MKK4 coincides with JNK1/2 activation in diabetic embryopathy 8.
Treatment with a JNK1/2 inhibitor, SP600125, prevents hyperglycemia-induced embryopathy 8. Additionally, maternal diabetes-induced embryonic anomalies are significantly reduced in the JNK2 null background. It is also revealing that sorbitol, a potent JNK1/2 activator 91, mimics the teratogenic effect of hyperglycemia. This evidence strongly suggests that JNK1/2 activation is crucial for diabetic embryopathy. The neural tube and yolk sac of the conceptus are extremely vulnerable to the negative effects of maternal hyperglycemia. We have demonstrated that hyperglycemia induces yolk sac vasculopathy and embryo malformation and that blockade of JNK1/2 activation reverses these effects. Thus, pharmacologic and genetic evidences strongly suggest that JNK1/2 activation mediates the deleterious effect of hyperglycemia on embryonic development and yolk sac vasculature.
FoxO factors induces apoptosis by transcriptionally up-regulating apoptotic genes
The FoxO subfamily of Forkhead transcription factors is composed of three functionally related members, FoxO1a, FoxO3a and FoxO4 92. FoxO proteins are evolutionarily conserved transcriptional activators of genes involved in apoptosis 93, cell cycle inhibition and DNA repair 94. FoxO factors interact with a core consensus DNA sequence (5′-AAAA(C/T)AAA-3′) to modulate target gene expression 95-97. FoxOs drive apoptotic responses in stress-exposed cells 98, 99 by regulating expression of apoptosis-relevant target genes including Bim 100, TRAIL 101, Fas ligand (FasL) 102 and TRADD 95.
The transcriptional activity of FoxO factors is controlled by subcellular localization and phosphorylation 103. Phosphorylation of FoxO factors at Thr-24 and Thr-32 by kinases prevents nuclear translocation thereby inhibiting FoxO-dependent transcription 104. FoxO factors mediate JNK1/2 activity-induced apoptosis 73 (Fig. 5). Phosphorylation of FoxO proteins by JNK1/2 results in FoxoO nuclear accumulation and enhanced transcription 74 (Fig. 5). The 14-3-3 protein, an adaptor protein that interacts with proteins having modifications of phosphoserine or phosphothreonine, binds to FoxO to sequester it in the cytoplasm. JNK1/2 phosphorylates the 14-3-3 chaperone protein causing the release of FoxO and its translocation to the nucleus 105 (Fig. 5).
Emerging evidence suggest that FoxO factors play a tumor suppressor role in a variety of cancers 106. FoxO3a suppresses prostate cancer progression in mice 107. In vivo overexpression of FoxO3a increases the expression level of p27kip1 and inhibits cell proliferation in vascular smooth muscle cells 108. FoxO3a promotes tumor cell death. FoxO3a activator STI571 treatment in leukemia cells and breast cancer cells inhibits tumor growth. FoxO3a inhibits T cell proliferation and induces T cell apoptosis, resulting in autoimmunity prevention 109. Genetic variation within the FoxO3a gene was strongly associated with human longevity 110. Based on its clinical function, FoxO3a could be as a molecular target for treatment of diabetic embryopathy. The pro-apoptotic effect of FoxO3a may be implicated in diabetic embryopathy.
Indeed, FOXO3a gene deletion reduces maternal diabetes-induced apoptosis and NTD formation 7. In previous study, the NTD rate of FoxO3a−/− embryos from diabetic mice was 3.1%, significantly lower than that of wild-type embryos (25.6%) from diabetic dams. Moreover, FoxO3a deletion was associated with a reduction in the maternal diabetes-increased mRNA and protein abundance for TRADD, cleavage of caspase 3 and 8 and neuroepithelial cell apoptosis 7 (Fig. 5).
TRADD--a critical mediator of apoptosis in the TNFα pathway
TRADD is a key adaptor protein in the tumor necrosis factor (TNF) signaling cascade that mediates TNF-induced apoptosis 111, 112. TRADD contains a death domain which is a homotypic protein interaction module that signals cell death 111. TRADD interacts with FADD via corresponding death domains, and this interaction triggers caspase 8 activation 113. FADD is an adapter protein that was originally isolated as a transducer of apoptotic signals for death domain-containing receptors 114. In addition to a death domain, FADD contains a death effector domain. TRADD overexpression leads to apoptosis in a FADD dependent manner 111, 112. Dominant-negative FADD (DN-FADD), which lacks amino acids 1-79, can inhibit TRADD-induced apoptosis in both in vivo and in vitro systems 112. In vitro study demonstrates that TRADD can be used for Glioblastoma Multiforme tumors by using its dual function of directly inducing rapid apoptosis and sensitizing Glioblastoma Multiforme cells to standard anti-neoplastic therapy 115. Based on its clinical function, the apoptosis inducer TRADD could be as aother molecular target for treatment of diabetic embryopathy.
Maternal diabetes increases the expression level of TRADD, and deletion of foxo3a gene ameliorates maternal diabetes-increased TRADD abundance, demonstrating that the TRADD gene is responsive to the FoxO3a transcription factor, which inhibits maternal diabetes-induced NTD formation 7 (Fig. 5). Furthermore, deletion of Ask1 gene significantly abrogates maternal diabetes-induced TRADD expression. These results suggest that maternal diabetes-induced ASK1 activation is responsible for TRADD stimulation and subsequent apoptosis induction 7.
Clinical significance and future prospective
About 1.85 million American women of reproductive age (18-44 years) have diabetes, and the number is continuing to increase due to the obesity epidemic 116. Unfortunately euglycemic control by insulin administration is difficult to achieve as even transient exposure to maternal hyperglycemia causes embryonic malformation. Thus, maternal diabetes-induced birth defects remain a huge health problem. Development of accessible, convenient and effective prevention strategies is an urgent task. To achieve this goal, understanding the mechanism of maternal diabetes-induced malformations is an essential and key step. Towards this goal, we propose studies that will define the mechanism of diabetic embryopathy at both the cellular and transcriptional levels.
We have used animal models to delineate the mechanism of maternal diabetes-induced malformations 1-5, 7, 8, 16 and specifically focused on the developing neural tube. We have previously demonstrated that apoptosis is implicated in diabetic embryopathy 2, 3, 5, 7, 16. Our goal is to identify molecular intermediates of the apoptotic cascade responsible for diabetic embryopathy, with ultimately to provide a mechanistic basis for development of therapeutic agents.
Observation of excessive apoptosis in neural stem cells of malformed neural tubes provides the basis for the hypothesis that maternal hyperglycemia-induced apoptosis in neural tube cells results in NTDs 2, 3, 5, 7, 16. However, the mechanism of maternal hyperglycemia-induced apoptosis in target tissues remains elusive. Thus, we have been seeking to systemically dissect the mechanism responsible for maternal diabetes-induced NTDs from the signal transduction levels to transcriptional mechanism. Furthermore, we have revealed which apoptotic control proteins mediate maternal diabetes-induced apoptosis based on animal models. By unraveling the apoptotic mechanisms leading to diabetic embryopathy, we will provide a mechanistic basis for the use of cutting-edge, mechanism-based therapeutic strategies designed to prevent diabetes-associated birth defects.
Our animal studies have demonstrated that SOD1 overexpression in transgenic mice suppresses maternal diabetes-induced pro-apoptotic kinase signaling, endoplasmic reticulum stress and nitrosative stress in the developing embryo1, 4, 117. Besides structural birth defects, oxidative stress is also involved in the etiology of other pregnancy complications118. Studies using human maternal blood samples have revealed several important oxidative stress markers for early diagnosis of adverse pregnancy outcomes119-121. The use of antioxidants in preventing human birth defects produces conflict results. Folic acid supplementation reduces the incidences of NTDs122; however, periconceptional use of vitamins or supplements that contain folic acid does not reduce the incidence of congenital heart defects in pregestational diabetic pregnancies123, 124. Our animal studies have revealed many candidates that may be effective in prevention of human structural birth defects.
Acknowledgments
This study is supported by NIH R01DK083243, R01DK101972 (P. Y.) and R01DK103024 (to P. Y. and E. A. R), and an American Diabetes Association Basic Science Award (1-13-BS-220). The authors are grateful to Dr. Julie Wu at the University of Maryland School of Medicine for critical reading and editing.
Source of financial support: This research is supported by NIH R01DK083243, R01DK101972 and R56 DK095380 (P.Y.), and R01DK103024 (to P.Y and E.A.R).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Disclosure: None of the authors have a conflict of interest.
REFERENCES
- 1.Li X, Weng H, Reece EA, Yang P. SOD1 overexpression in vivo blocks hyperglycemia-induced specific PKC isoforms: substrate activation and consequent lipid peroxidation in diabetic embryopathy. American journal of obstetrics and gynecology. 2011;205:84 e1–6. doi: 10.1016/j.ajog.2011.02.071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Li X, Weng H, Xu C, Reece EA, Yang P. Oxidative stress-induced JNK1/2 activation triggers proapoptotic signaling and apoptosis that leads to diabetic embryopathy. Diabetes. 2012;61:2084–92. doi: 10.2337/db11-1624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Li X, Xu C, Yang P. c-Jun NH2-terminal kinase 1/2 and endoplasmic reticulum stress as interdependent and reciprocal causation in diabetic embryopathy. Diabetes. 2013;62:599–608. doi: 10.2337/db12-0026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wang F, Reece EA, Yang P. Superoxide dismutase 1 overexpression in mice abolishes maternal diabetes-induced endoplasmic reticulum stress in diabetic embryopathy. American journal of obstetrics and gynecology. 2013;209:345 e1–7. doi: 10.1016/j.ajog.2013.06.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Xu C, Li X, Wang F, Weng H, Yang P. Trehalose prevents neural tube defects by correcting maternal diabetes-suppressed autophagy and neurogenesis. American journal of physiology Endocrinology and metabolism. 2013;305:E667–78. doi: 10.1152/ajpendo.00185.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yang P, Cao Y, Li H. Hyperglycemia induces inducible nitric oxide synthase gene expression and consequent nitrosative stress via c-Jun N-terminal kinase activation. American journal of obstetrics and gynecology. 2010;203:185 e5–11. doi: 10.1016/j.ajog.2010.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yang P, Li X, Xu C, et al. Maternal hyperglycemia activates an ASK1-FoxO3acaspase 8 pathway that leads to embryonic neural tube defects. Science signaling. 2013;6:ra74. doi: 10.1126/scisignal.2004020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yang P, Zhao Z, Reece EA. Involvement of c-Jun N-terminal kinases activation in diabetic embryopathy. Biochemical and biophysical research communications. 2007;357:749–54. doi: 10.1016/j.bbrc.2007.04.023. [DOI] [PubMed] [Google Scholar]
- 9.Feig DS, Palda VA. Type 2 diabetes in pregnancy: a growing concern. Lancet. 2002;359:1690–2. doi: 10.1016/S0140-6736(02)08599-9. [DOI] [PubMed] [Google Scholar]
- 10.Hagay ZJ, Weiss Y, Zusman I, et al. Prevention of diabetes-associated embryopathy by overexpression of the free radical scavenger copper zinc superoxide dismutase in transgenic mouse embryos. American journal of obstetrics and gynecology. 1995;173:1036–41. doi: 10.1016/0002-9378(95)91323-8. [DOI] [PubMed] [Google Scholar]
- 11.Reece EA, Wu YK. Prevention of diabetic embryopathy in offspring of diabetic rats with use of a cocktail of deficient substrates and an antioxidant. American journal of obstetrics and gynecology. 1997;176:790–7. doi: 10.1016/s0002-9378(97)70602-1. discussion 97-8. [DOI] [PubMed] [Google Scholar]
- 12.Yang P, Li H. Epigallocatechin-3-gallate ameliorates hyperglycemia-induced embryonic vasculopathy and malformation by inhibition of Foxo3a activation. American journal of obstetrics and gynecology. 2010;203:75 e1–6. doi: 10.1016/j.ajog.2010.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yang P, Reece EA. Role of HIF-1alpha in maternal hyperglycemia-induced embryonic vasculopathy. American journal of obstetrics and gynecology. 2011;204:332 e1–7. doi: 10.1016/j.ajog.2011.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Reece EA, Wu YK, Zhao Z, Dhanasekaran D. Dietary vitamin and lipid therapy rescues aberrant signaling and apoptosis and prevents hyperglycemia-induced diabetic embryopathy in rats. American journal of obstetrics and gynecology. 2006;194:580–5. doi: 10.1016/j.ajog.2005.08.052. [DOI] [PubMed] [Google Scholar]
- 15.Santo EE, Stroeken P, Sluis PV, Koster J, Versteeg R, Westerhout EM. FOXO3a is a major target of inactivation by PI3K/AKT signaling in aggressive neuroblastoma. Cancer research. 2013;73:2189–98. doi: 10.1158/0008-5472.CAN-12-3767. [DOI] [PubMed] [Google Scholar]
- 16.Wang F, Wu Y, Gu H, et al. Ask1 gene deletion blocks maternal diabetes-induced endoplasmic reticulum stress in the developing embryo by disrupting the unfolded protein response signalosome. Diabetes. 2014 doi: 10.2337/db14-0409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nishitoh H, Saitoh M, Mochida Y, et al. ASK1 is essential for JNK/SAPK activation by TRAF2. Molecular cell. 1998;2:389–95. doi: 10.1016/s1097-2765(00)80283-x. [DOI] [PubMed] [Google Scholar]
- 18.Tobiume K, Matsuzawa A, Takahashi T, et al. ASK1 is required for sustained activations of JNK/p38 MAP kinases and apoptosis. EMBO reports. 2001;2:222–8. doi: 10.1093/embo-reports/kve046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Reece EA, Wu YK, Wiznitzer A, et al. Dietary polyunsaturated fatty acid prevents malformations in offspring of diabetic rats. American journal of obstetrics and gynecology. 1996;175:818–23. doi: 10.1016/s0002-9378(96)80005-6. [DOI] [PubMed] [Google Scholar]
- 20.Sakamaki H, Akazawa S, Ishibashi M, et al. Significance of glutathione-dependent antioxidant system in diabetes-induced embryonic malformations. Diabetes. 1999;48:1138–44. doi: 10.2337/diabetes.48.5.1138. [DOI] [PubMed] [Google Scholar]
- 21.Trocino RA, Akazawa S, Ishibashi M, et al. Significance of glutathione depletion and oxidative stress in early embryogenesis in glucose-induced rat embryo culture. Diabetes. 1995;44:992–8. doi: 10.2337/diab.44.8.992. [DOI] [PubMed] [Google Scholar]
- 22.Jenkinson PC, Anderson D, Gangolli SD. Malformations induced in cultured rat embryos by enzymically generated active oxygen species. Teratogenesis, carcinogenesis, and mutagenesis. 1986;6:547–54. doi: 10.1002/tcm.1770060608. [DOI] [PubMed] [Google Scholar]
- 23.Chang TI, Horal M, Jain SK, Wang F, Patel R, Loeken MR. Oxidant regulation of gene expression and neural tube development: Insights gained from diabetic pregnancy on molecular causes of neural tube defects. Diabetologia. 2003;46:538–45. doi: 10.1007/s00125-003-1063-2. [DOI] [PubMed] [Google Scholar]
- 24.Eriksson UJ, Wentzel P, Minhas HS, Thornalley PJ. Teratogenicity of 3-deoxyglucosone and diabetic embryopathy. Diabetes. 1998;47:1960–6. doi: 10.2337/diabetes.47.12.1960. [DOI] [PubMed] [Google Scholar]
- 25.Wentzel P, Welsh N, Eriksson UJ. Developmental damage, increased lipid peroxidation, diminished cyclooxygenase-2 gene expression, and lowered prostaglandin E2 levels in rat embryos exposed to a diabetic environment. Diabetes. 1999;48:813–20. doi: 10.2337/diabetes.48.4.813. [DOI] [PubMed] [Google Scholar]
- 26.Morrow JD, Hill KE, Burk RF, Nammour TM, Badr KF, Roberts LJ., 2nd A series of prostaglandin F2-like compounds are produced in vivo in humans by a non-cyclooxygenase, free radical-catalyzed mechanism. Proceedings of the National Academy of Sciences of the United States of America. 1990;87:9383–7. doi: 10.1073/pnas.87.23.9383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gopaul NK, Anggard EE, Mallet AI, Betteridge DJ, Wolff SP, Nourooz-Zadeh J. Plasma 8-epi-PGF2 alpha levels are elevated in individuals with non-insulin dependent diabetes mellitus. FEBS letters. 1995;368:225–9. doi: 10.1016/0014-5793(95)00649-t. [DOI] [PubMed] [Google Scholar]
- 28.Decsi T, Minda H, Hermann R, et al. Polyunsaturated fatty acids in plasma and erythrocyte membrane lipids of diabetic children. Prostaglandins, leukotrienes, and essential fatty acids. 2002;67:203–10. doi: 10.1054/plef.2002.0420. [DOI] [PubMed] [Google Scholar]
- 29.Li R, Chase M, Jung SK, Smith PJ, Loeken MR. Hypoxic stress in diabetic pregnancy contributes to impaired embryo gene expression and defective development by inducing oxidative stress. American journal of physiology Endocrinology and metabolism. 2005;289:E591–9. doi: 10.1152/ajpendo.00441.2004. [DOI] [PubMed] [Google Scholar]
- 30.Ornoy A, Zaken V, Kohen R. Role of reactive oxygen species (ROS) in the diabetes-induced anomalies in rat embryos in vitro: reduction in antioxidant enzymes and low-molecular-weight antioxidants (LMWA) may be the causative factor for increased anomalies. Teratology. 1999;60:376–86. doi: 10.1002/(SICI)1096-9926(199912)60:6<376::AID-TERA10>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- 31.Zaken V, Kohen R, Ornoy A. Vitamins C and E improve rat embryonic antioxidant defense mechanism in diabetic culture medium. Teratology. 2001;64:33–44. doi: 10.1002/tera.1045. [DOI] [PubMed] [Google Scholar]
- 32.Cederberg J, Eriksson UJ. Decreased catalase activity in malformation-prone embryos of diabetic rats. Teratology. 1997;56:350–7. doi: 10.1002/(SICI)1096-9926(199712)56:6<350::AID-TERA2>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- 33.Moncada S, Palmer RM, Higgs EA. Nitric oxide: physiology, pathophysiology, and pharmacology. Pharmacological reviews. 1991;43:109–42. [PubMed] [Google Scholar]
- 34.Lee QP, Juchau MR. Dysmorphogenic effects of nitric oxide (NO) and NO-synthase inhibition: studies with intra-amniotic injections of sodium nitroprusside and NG-monomethyl-L-arginine. Teratology. 1994;49:452–64. doi: 10.1002/tera.1420490605. [DOI] [PubMed] [Google Scholar]
- 35.Plachta N, Traister A, Weil M. Nitric oxide is involved in establishing the balance between cell cycle progression and cell death in the developing neural tube. Experimental cell research. 2003;288:354–62. doi: 10.1016/s0014-4827(03)00215-5. [DOI] [PubMed] [Google Scholar]
- 36.Feng Q, Song W, Lu X, et al. Development of heart failure and congenital septal defects in mice lacking endothelial nitric oxide synthase. Circulation. 2002;106:873–9. doi: 10.1161/01.cir.0000024114.82981.ea. [DOI] [PubMed] [Google Scholar]
- 37.Topel I, Stanarius A, Wolf G. Distribution of the endothelial constitutive nitric oxide synthase in the developing rat brain: an immunohistochemical study. Brain research. 1998;788:43–8. doi: 10.1016/s0006-8993(97)01506-0. [DOI] [PubMed] [Google Scholar]
- 38.Jawerbaum A, Gonzalez ET, Novaro V, Faletti A, Sinner D, Gimeno MA. Increased prostaglandin E generation and enhanced nitric oxide synthase activity in the non-insulin-dependent diabetic embryo during organogenesis. Reproduction, fertility, and development. 1998;10:191–6. doi: 10.1071/r97077. [DOI] [PubMed] [Google Scholar]
- 39.Jawerbaum A, Sinner D, White V, et al. Modulation of PGE2 generation in the diabetic embryo: effect of nitric oxide and superoxide dismutase. Prostaglandins, leukotrienes, and essential fatty acids. 2001;64:127–33. doi: 10.1054/plef.2001.0251. [DOI] [PubMed] [Google Scholar]
- 40.Jawerbaum A, Higa R, White V, et al. Peroxynitrites and impaired modulation of nitric oxide concentrations in embryos from diabetic rats during early organogenesis. Reproduction (Cambridge, England) 2005;130:695–703. doi: 10.1530/rep.1.00699. [DOI] [PubMed] [Google Scholar]
- 41.Jawerbaum A, Gonzalez E. The role of alterations in arachidonic acid metabolism and nitric oxide homeostasis in rat models of diabetes during early pregnancy. Current pharmaceutical design. 2005;11:1327–42. doi: 10.2174/1381612053507503. [DOI] [PubMed] [Google Scholar]
- 42.Szabo C. Multiple pathways of peroxynitrite cytotoxicity. Toxicology letters. 2003;140-141:105–12. doi: 10.1016/s0378-4274(02)00507-6. [DOI] [PubMed] [Google Scholar]
- 43.Shen HM, Liu ZG. JNK signaling pathway is a key modulator in cell death mediated by reactive oxygen and nitrogen species. Free radical biology & medicine. 2006;40:928–39. doi: 10.1016/j.freeradbiomed.2005.10.056. [DOI] [PubMed] [Google Scholar]
- 44.Dempsey EC, Newton AC, Mochly-Rosen D, et al. Protein kinase C isozymes and the regulation of diverse cell responses. American journal of physiology. 2000;279:L429–38. doi: 10.1152/ajplung.2000.279.3.L429. [DOI] [PubMed] [Google Scholar]
- 45.Wright MM, McMaster CR. Phospholipid synthesis, diacylglycerol compartmentation, and apoptosis. Biological research. 2002;35:223–9. doi: 10.4067/s0716-97602002000200014. [DOI] [PubMed] [Google Scholar]
- 46.Hiramatsu Y, Sekiguchi N, Hayashi M, et al. Diacylglycerol production and protein kinase C activity are increased in a mouse model of diabetic embryopathy. Diabetes. 2002;51:2804–10. doi: 10.2337/diabetes.51.9.2804. [DOI] [PubMed] [Google Scholar]
- 47.Zhiyong Z, Wu YK, Reece EA. Demonstration of the essential role of protein kinase C isoforms in hyperglycemia-induced embryonic malformations. Reproductive sciences (Thousand Oaks, Calif. 2008;15:349–56. doi: 10.1177/1933719108316986. [DOI] [PubMed] [Google Scholar]
- 48.Reece EA, Ma XD, Wu YK, Dhanasekaran D. Aberrant patterns of cellular communication in diabetes-induced embryopathy. I. Membrane signalling. J Matern Fetal Neonatal Med. 2002;11:249–53. doi: 10.1080/jmf.11.4.249.253. [DOI] [PubMed] [Google Scholar]
- 49.Reece EA, Ma XD, Zhao Z, Wu YK, Dhanasekaran D. Aberrant patterns of cellular communication in diabetes-induced embryopathy in rats: II, apoptotic pathways. American journal of obstetrics and gynecology. 2005;192:967–72. doi: 10.1016/j.ajog.2004.10.592. [DOI] [PubMed] [Google Scholar]
- 50.Saitoh M, Nishitoh H, Fujii M, et al. Mammalian thioredoxin is a direct inhibitor of apoptosis signal-regulating kinase (ASK) 1. The EMBO journal. 1998;17:2596–606. doi: 10.1093/emboj/17.9.2596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ozcan U, Cao Q, Yilmaz E, et al. Endoplasmic reticulum stress links obesity, insulin action, and type 2 diabetes. Science. 2004;306:457–61. doi: 10.1126/science.1103160. [DOI] [PubMed] [Google Scholar]
- 52.Horal M, Zhang Z, Stanton R, Virkamaki A, Loeken MR. Activation of the hexosamine pathway causes oxidative stress and abnormal embryo gene expression: involvement in diabetic teratogenesis. Birth defects research. 2004;70:519–27. doi: 10.1002/bdra.20056. [DOI] [PubMed] [Google Scholar]
- 53.Forsberg H, Eriksson UJ, Welsh N. Apoptosis in embryos of diabetic rats. Pharmacology & toxicology. 1998;83:104–11. doi: 10.1111/j.1600-0773.1998.tb01452.x. [DOI] [PubMed] [Google Scholar]
- 54.Phelan SA, Ito M, Loeken MR. Neural tube defects in embryos of diabetic mice: role of the Pax-3 gene and apoptosis. Diabetes. 1997;46:1189–97. doi: 10.2337/diab.46.7.1189. [DOI] [PubMed] [Google Scholar]
- 55.Sun F, Kawasaki E, Akazawa S, et al. Apoptosis and its pathway in early postimplantation embryos of diabetic rats. Diabetes research and clinical practice. 2005;67:110–8. doi: 10.1016/j.diabres.2004.06.008. [DOI] [PubMed] [Google Scholar]
- 56.Moley KH. Hyperglycemia and apoptosis: mechanisms for congenital malformations and pregnancy loss in diabetic women. Trends in endocrinology and metabolism: TEM. 2001;12:78–82. doi: 10.1016/s1043-2760(00)00341-6. [DOI] [PubMed] [Google Scholar]
- 57.Eriksson UJ, Borg LA, Cederberg J, et al. Pathogenesis of diabetes-induced congenital malformations. Upsala journal of medical sciences. 2000;105:53–84. doi: 10.1517/03009734000000055. [DOI] [PubMed] [Google Scholar]
- 58.Fine EL, Horal M, Chang TI, Fortin G, Loeken MR. Evidence that elevated glucose causes altered gene expression, apoptosis, and neural tube defects in a mouse model of diabetic pregnancy. Diabetes. 1999;48:2454–62. doi: 10.2337/diabetes.48.12.2454. [DOI] [PubMed] [Google Scholar]
- 59.Kang BP, Frencher S, Reddy V, Kessler A, Malhotra A, Meggs LG. High glucose promotes mesangial cell apoptosis by oxidant-dependent mechanism. Am J Physiol Renal Physiol. 2003;284:F455–66. doi: 10.1152/ajprenal.00137.2002. [DOI] [PubMed] [Google Scholar]
- 60.Ueda S, Masutani H, Nakamura H, Tanaka T, Ueno M, Yodoi J. Redox control of cell death. Antioxidants & redox signaling. 2002;4:405–14. doi: 10.1089/15230860260196209. [DOI] [PubMed] [Google Scholar]
- 61.Kyosseva SV. Mitogen-activated protein kinase signaling. International review of neurobiology. 2004;59:201–20. doi: 10.1016/S0074-7742(04)59008-6. [DOI] [PubMed] [Google Scholar]
- 62.Torres M, Forman HJ. Redox signaling and the MAP kinase pathways. BioFactors (Oxford, England) 2003;17:287–96. doi: 10.1002/biof.5520170128. [DOI] [PubMed] [Google Scholar]
- 63.Widmann C, Gibson S, Jarpe MB, Johnson GL. Mitogen-activated protein kinase: conservation of a three-kinase module from yeast to human. Physiological reviews. 1999;79:143–80. doi: 10.1152/physrev.1999.79.1.143. [DOI] [PubMed] [Google Scholar]
- 64.Kyriakis JM, Avruch J. Mammalian mitogen-activated protein kinase signal transduction pathways activated by stress and inflammation. Physiological reviews. 2001;81:807–69. doi: 10.1152/physrev.2001.81.2.807. [DOI] [PubMed] [Google Scholar]
- 65.Tobiume K, Saitoh M, Ichijo H. Activation of apoptosis signal-regulating kinase 1 by the stress-induced activating phosphorylation of pre-formed oligomer. Journal of cellular physiology. 2002;191:95–104. doi: 10.1002/jcp.10080. [DOI] [PubMed] [Google Scholar]
- 66.Davis RJ. Signal transduction by the JNK group of MAP kinases. Cell. 2000;103:239–52. doi: 10.1016/s0092-8674(00)00116-1. [DOI] [PubMed] [Google Scholar]
- 67.Zhang Q, Zhang G, Meng F, Tian H. Biphasic activation of apoptosis signal-regulating kinase 1-stress-activated protein kinase 1-c-Jun N-terminal protein kinase pathway is selectively mediated by Ca2+-permeable alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionate receptors involving oxidative stress following brain ischemia in rat hippocampus. Neuroscience letters. 2003;337:51–5. doi: 10.1016/s0304-3940(02)01295-8. [DOI] [PubMed] [Google Scholar]
- 68.Watanabe T, Otsu K, Takeda T, et al. Apoptosis signal-regulating kinase 1 is involved not only in apoptosis but also in non-apoptotic cardiomyocyte death. Biochemical and biophysical research communications. 2005;333:562–7. doi: 10.1016/j.bbrc.2005.05.151. [DOI] [PubMed] [Google Scholar]
- 69.Kadowaki H, Nishitoh H, Urano F, et al. Amyloid beta induces neuronal cell death through ROS-mediated ASK1 activation. Cell death and differentiation. 2005;12:19–24. doi: 10.1038/sj.cdd.4401528. [DOI] [PubMed] [Google Scholar]
- 70.Yokoi T, Fukuo K, Yasuda O, et al. Apoptosis Signal-Regulating Kinase 1 Mediates Cellular Senescence Induced by High Glucose in Endothelial Cells. Diabetes. 2006;55:1660–65. doi: 10.2337/db05-1607. [DOI] [PubMed] [Google Scholar]
- 71.Wada T, Penninger JM. Mitogen-activated protein kinases in apoptosis regulation. Oncogene. 2004;23:2838–49. doi: 10.1038/sj.onc.1207556. [DOI] [PubMed] [Google Scholar]
- 72.Lin A, Dibling B. The true face of JNK activation in apoptosis. Aging cell. 2002;1:112–6. doi: 10.1046/j.1474-9728.2002.00014.x. [DOI] [PubMed] [Google Scholar]
- 73.Wang MC, Bohmann D, Jasper H. JNK extends life span and limits growth by antagonizing cellular and organism-wide responses to insulin signaling. Cell. 2005;121:115–25. doi: 10.1016/j.cell.2005.02.030. [DOI] [PubMed] [Google Scholar]
- 74.Essers MA, Weijzen S, de Vries-Smits AM, et al. FOXO transcription factor activation by oxidative stress mediated by the small GTPase Ral and JNK. The EMBO journal. 2004;23:4802–12. doi: 10.1038/sj.emboj.7600476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Lin A. Activation of the JNK signaling pathway: breaking the brake on apoptosis. Bioessays. 2003;25:17–24. doi: 10.1002/bies.10204. [DOI] [PubMed] [Google Scholar]
- 76.Torres M. Mitogen-activated protein kinase pathways in redox signaling. Front Biosci. 2003;8:d369–91. doi: 10.2741/999. [DOI] [PubMed] [Google Scholar]
- 77.Sata N, Klonowski-Stumpe H, Han B, Haussinger D, Niederau C. Menadione induces both necrosis and apoptosis in rat pancreatic acinar AR4-2J cells. Free radical biology & medicine. 1997;23:844–50. doi: 10.1016/s0891-5849(97)00064-6. [DOI] [PubMed] [Google Scholar]
- 78.Ding M, Li J, Leonard SS, et al. Differential role of hydrogen peroxide in UV-induced signal transduction. Molecular and cellular biochemistry. 2002;234-235:81–90. [PubMed] [Google Scholar]
- 79.Chan WH, Wu CC, Yu JS. Curcumin inhibits UV irradiation-induced oxidative stress and apoptotic biochemical changes in human epidermoid carcinoma A431 cells. Journal of cellular biochemistry. 2003;90:327–38. doi: 10.1002/jcb.10638. [DOI] [PubMed] [Google Scholar]
- 80.Lee SA, Dritschilo A, Jung M. Role of ATM in oxidative stress-mediated c-Jun phosphorylation in response to ionizing radiation and CdCl2. The Journal of biological chemistry. 2001;276:11783–90. doi: 10.1074/jbc.M004517200. [DOI] [PubMed] [Google Scholar]
- 81.Li Y, Arita Y, Koo HC, Davis JM, Kazzaz JA. Inhibition of c-Jun N-terminal kinase pathway improves cell viability in response to oxidant injury. American journal of respiratory cell and molecular biology. 2003;29:779–83. doi: 10.1165/rcmb.2003-0087RC. [DOI] [PubMed] [Google Scholar]
- 82.Kim YS, Jhon DY, Lee KY. Involvement of ROS and JNK1 in selenite-induced apoptosis in Chang liver cells. Experimental & molecular medicine. 2004;36:157–64. doi: 10.1038/emm.2004.22. [DOI] [PubMed] [Google Scholar]
- 83.Yoon SO, Park SJ, Yoon SY, Yun CH, Chung AS. Sustained production of H(2)O(2) activates pro-matrix metalloproteinase-2 through receptor tyrosine kinases/phosphatidylinositol 3-kinase/NF-kappa B pathway. The Journal of biological chemistry. 2002;277:30271–82. doi: 10.1074/jbc.M202647200. [DOI] [PubMed] [Google Scholar]
- 84.Yoshizumi M, Kogame T, Suzaki Y, et al. Ebselen attenuates oxidative stress-induced apoptosis via the inhibition of the c-Jun N-terminal kinase and activator protein-1 signalling pathway in PC12 cells. British journal of pharmacology. 2002;136:1023–32. doi: 10.1038/sj.bjp.0704808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Dong C, Yang DD, Tournier C, et al. JNK is required for effector T-cell function but not for T-cell activation. Nature. 2000;405:91–4. doi: 10.1038/35011091. [DOI] [PubMed] [Google Scholar]
- 86.She QB, Chen N, Bode AM, Flavell RA, Dong Z. Deficiency of c-Jun-NH(2)-terminal kinase-1 in mice enhances skin tumor development by 12-Otetradecanoylphorbol-13-acetate. Cancer research. 2002;62:1343–8. [PubMed] [Google Scholar]
- 87.Kuan CY, Yang DD, Samanta Roy DR, Davis RJ, Rakic P, Flavell RA. The Jnk1 and Jnk2 protein kinases are required for regional specific apoptosis during early brain development. Neuron. 1999;22:667–76. doi: 10.1016/s0896-6273(00)80727-8. [DOI] [PubMed] [Google Scholar]
- 88.Urano F, Wang X, Bertolotti A, et al. Coupling of stress in the ER to activation of JNK protein kinases by transmembrane protein kinase IRE1. Science. 2000;287:664–6. doi: 10.1126/science.287.5453.664. [DOI] [PubMed] [Google Scholar]
- 89.Srivastava AK. High glucose-induced activation of protein kinase signaling pathways in vascular smooth muscle cells: a potential role in the pathogenesis of vascular dysfunction in diabetes (review) International journal of molecular medicine. 2002;9:85–9. [PubMed] [Google Scholar]
- 90.Kikkawa R, Koya D, Haneda M. Progression of diabetic nephropathy. Am J Kidney Dis. 2003;41:S19–21. doi: 10.1053/ajkd.2003.50077. [DOI] [PubMed] [Google Scholar]
- 91.Bagowski CP, Xiong W, Ferrell JE., Jr. c-Jun N-terminal kinase activation in Xenopus laevis eggs and embryos. A possible non-genomic role for the JNK signaling pathway. The Journal of biological chemistry. 2001;276:1459–65. doi: 10.1074/jbc.M008050200. [DOI] [PubMed] [Google Scholar]
- 92.Anderson MJ, Viars CS, Czekay S, Cavenee WK, Arden KC. Cloning and characterization of three human forkhead genes that comprise an FKHR-like gene subfamily. Genomics. 1998;47:187–99. doi: 10.1006/geno.1997.5122. [DOI] [PubMed] [Google Scholar]
- 93.Kajihara T, Jones M, Fusi L, et al. Differential expression of FOXO1 and FOXO3a confers resistance to oxidative cell death upon endometrial decidualization. Molecular endocrinology (Baltimore, Md. 2006;20:2444–55. doi: 10.1210/me.2006-0118. [DOI] [PubMed] [Google Scholar]
- 94.Tran H, Brunet A, Grenier JM, et al. DNA repair pathway stimulated by the forkhead transcription factor FOXO3a through the Gadd45 protein. Science. 2002;296:530–4. doi: 10.1126/science.1068712. [DOI] [PubMed] [Google Scholar]
- 95.Rokudai S, Fujita N, Kitahara O, Nakamura Y, Tsuruo T. Involvement of FKHR-dependent TRADD expression in chemotherapeutic drug-induced apoptosis. Molecular and cellular biology. 2002;22:8695–708. doi: 10.1128/MCB.22.24.8695-8708.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Chong ZZ, Li F, Maiese K. Oxidative stress in the brain: novel cellular targets that govern survival during neurodegenerative disease. Progress in neurobiology. 2005;75:207–46. doi: 10.1016/j.pneurobio.2005.02.004. [DOI] [PubMed] [Google Scholar]
- 97.Finkel T. Redox-dependent signal transduction. FEBS letters. 2000;476:52–4. doi: 10.1016/s0014-5793(00)01669-0. [DOI] [PubMed] [Google Scholar]
- 98.Brunet A, Sweeney LB, Sturgill JF, et al. Stress-dependent regulation of FOXO transcription factors by the SIRT1 deacetylase. Science. 2004;303:2011–5. doi: 10.1126/science.1094637. [DOI] [PubMed] [Google Scholar]
- 99.Kops GJ, Dansen TB, Polderman PE, et al. Forkhead transcription factor FOXO3a protects quiescent cells from oxidative stress. Nature. 2002;419:316–21. doi: 10.1038/nature01036. [DOI] [PubMed] [Google Scholar]
- 100.Gilley J, Coffer PJ, Ham J. FOXO transcription factors directly activate bim gene expression and promote apoptosis in sympathetic neurons. The Journal of cell biology. 2003;162:613–22. doi: 10.1083/jcb.200303026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Modur V, Nagarajan R, Evers BM, Milbrandt J. FOXO proteins regulate tumor necrosis factor-related apoptosis inducing ligand expression. Implications for PTEN mutation in prostate cancer. The Journal of biological chemistry. 2002;277:47928–37. doi: 10.1074/jbc.M207509200. [DOI] [PubMed] [Google Scholar]
- 102.Suhara T, Kim HS, Kirshenbaum LA, Walsh K. Suppression of Akt signaling induces Fas ligand expression: involvement of caspase and Jun kinase activation in Akt-mediated Fas ligand regulation. Molecular and cellular biology. 2002;22:680–91. doi: 10.1128/MCB.22.2.680-691.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Lam EW, Francis RE, Petkovic M. FOXO transcription factors: key regulators of cell fate. Biochemical Society transactions. 2006;34:722–6. doi: 10.1042/BST0340722. [DOI] [PubMed] [Google Scholar]
- 104.Brunet A, Bonni A, Zigmond MJ, et al. Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell. 1999;96:857–68. doi: 10.1016/s0092-8674(00)80595-4. [DOI] [PubMed] [Google Scholar]
- 105.Sunayama J, Tsuruta F, Masuyama N, Gotoh Y. JNK antagonizes Akt-mediated survival signals by phosphorylating 14-3-3. The Journal of cell biology. 2005;170:295–304. doi: 10.1083/jcb.200409117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Greer EL, Brunet A. FOXO transcription factors at the interface between longevity and tumor suppression. Oncogene. 2005;24:7410–25. doi: 10.1038/sj.onc.1209086. [DOI] [PubMed] [Google Scholar]
- 107.Shukla S, Bhaskaran N, Maclennan GT, Gupta S. Deregulation of FoxO3a accelerates prostate cancer progression in TRAMP mice. The Prostate. 2013;73:1507–17. doi: 10.1002/pros.22698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Abid MR, Yano K, Guo S, et al. Forkhead transcription factors inhibit vascular smooth muscle cell proliferation and neointimal hyperplasia. The Journal of biological chemistry. 2005;280:29864–73. doi: 10.1074/jbc.M502149200. [DOI] [PubMed] [Google Scholar]
- 109.Wang Y, Zhou Y, Graves DT. FOXO transcription factors: their clinical significance and regulation. BioMed research international. 2014;2014:925350. doi: 10.1155/2014/925350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Willcox BJ, Donlon TA, He Q, et al. FOXO3A genotype is strongly associated with human longevity. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:13987–92. doi: 10.1073/pnas.0801030105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Hsu H, Xiong J, Goeddel DV. The TNF receptor 1-associated protein TRADD signals cell death and NF-kappa B activation. Cell. 1995;81:495–504. doi: 10.1016/0092-8674(95)90070-5. [DOI] [PubMed] [Google Scholar]
- 112.Hsu H, Shu HB, Pan MG, Goeddel DV. TRADD-TRAF2 and TRADD-FADD interactions define two distinct TNF receptor 1 signal transduction pathways. Cell. 1996;84:299–308. doi: 10.1016/s0092-8674(00)80984-8. [DOI] [PubMed] [Google Scholar]
- 113.Chinnaiyan AM, Tepper CG, Seldin MF, et al. FADD/MORT1 is a common mediator of CD95 (Fas/APO-1) and tumor necrosis factor receptor-induced apoptosis. The Journal of biological chemistry. 1996;271:4961–5. doi: 10.1074/jbc.271.9.4961. [DOI] [PubMed] [Google Scholar]
- 114.Boldin MP, Varfolomeev EE, Pancer Z, Mett IL, Camonis JH, Wallach D. A novel protein that interacts with the death domain of Fas/APO1 contains a sequence motif related to the death domain. The Journal of biological chemistry. 1995;270:7795–8. doi: 10.1074/jbc.270.14.7795. [DOI] [PubMed] [Google Scholar]
- 115.Yount GL, Afshar G, Ries S, et al. Transcriptional activation of TRADD mediates p53-independent radiation-induced apoptosis of glioma cells. Oncogene. 2001;20:2826–35. doi: 10.1038/sj.onc.1204393. [DOI] [PubMed] [Google Scholar]
- 116.Prevention CfDCa CDC to indtroduce book on diabetes in women. 2001 [Google Scholar]
- 117.Weng H, Li X, Reece EA, Yang P. SOD1 suppresses maternal hyperglycemia-increased iNOS expression and consequent nitrosative stress in diabetic embryopathy. American journal of obstetrics and gynecology. 2012;206:448 e1–7. doi: 10.1016/j.ajog.2012.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Zhou X, Zhang GY, Wang J, Lu SL, Cao J, Sun LZ. A novel bridge between oxidative stress and immunity: the interaction between hydrogen peroxide and human leukocyte antigen G in placental trophoblasts during preeclampsia. American journal of obstetrics and gynecology. 2012;206:447 e7–16. doi: 10.1016/j.ajog.2012.03.013. [DOI] [PubMed] [Google Scholar]
- 119.Bahado-Singh RO, Akolekar R, Chelliah A, et al. Metabolomic analysis for first-trimester trisomy 18 detection. American journal of obstetrics and gynecology. 2013;209:65 e1–9. doi: 10.1016/j.ajog.2013.03.028. [DOI] [PubMed] [Google Scholar]
- 120.Bahado-Singh RO, Akolekar R, Mandal R, et al. Metabolomic analysis for first-trimester Down syndrome prediction. American journal of obstetrics and gynecology. 2013;208:371 e1–8. doi: 10.1016/j.ajog.2012.12.035. [DOI] [PubMed] [Google Scholar]
- 121.Bahado-Singh RO, Ertl R, Mandal R, et al. Metabolomic prediction of fetal congenital heart defect in the first trimester. American journal of obstetrics and gynecology. 2014;211:240 e1–40 e14. doi: 10.1016/j.ajog.2014.03.056. [DOI] [PubMed] [Google Scholar]
- 122.Parker SE, Yazdy MM, Tinker SC, Mitchell AA, Werler MM. The impact of folic acid intake on the association among diabetes mellitus, obesity, and spina bifida. American journal of obstetrics and gynecology. 2013;209:239 e1–8. doi: 10.1016/j.ajog.2013.05.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Correa A, Gilboa SM, Botto LD, et al. Lack of periconceptional vitamins or supplements that contain folic acid and diabetes mellitus-associated birth defects. American journal of obstetrics and gynecology. 2012;206:218 e1–13. doi: 10.1016/j.ajog.2011.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Oakley GP., Jr. Failing to prevent birth defects caused by maternal diabetes mellitus. American journal of obstetrics and gynecology. 2012;206:179–80. doi: 10.1016/j.ajog.2011.12.019. [DOI] [PubMed] [Google Scholar]