

Summary
Both human cytomegalovirus (HCMV) and murine cytomegalovirus (MCMV) establish persistent infections that induce the accumulation of virus-specific T cells over time in a process called memory inflation. It has been proposed that T cells expressing T cell receptors (TCR) with high affinity for HCMV-derived peptides are preferentially selected after acute HCMV infection. To test this in the murine model, small numbers of OT-I transgenic T cells, which express a TCR with high affinity for the SIINFEKL peptide, were transferred into congenic mice and recipients were challenged with recombinant MCMV expressing SIINFEKL. OT-Is were selectively enriched during the first three weeks of infection. Similarly, in the absence of OT-Is, the functional avidity of SIINFEKL-specific T cells increased from early to late times post infection. However, even when exceedingly small numbers of OT-Is were transferred, their inflation limited the inflation of host-derived T cells specific for SIINFEKL. Importantly, subtle minor histocompatibility differences led to late rejection of the transferred OT-I T cells in some mice, which allowed host-derived T cells to inflate substantially. Thus, T cells with a high functional avidity are selected shortly after MCMV infection and continuously sustain their clonal dominance in a competitive manner.
Introduction
Cytomegalovirus (CMV) is a ubiquitous β-herpesvirus that infects the majority of people in the world. Infection typically occurs in childhood and is asymptomatic in healthy hosts. However, the virus is never cleared and continuous immune control is essential to keep the host disease free. Thus, CMV is a major infectious complication of immune compromised patients and can be lethal if not controlled (recently reviewed in [1]). Clinical CMV disease in the context of bone marrow transplantation can be controlled by adoptively transferred human CMV (HCMV)-specific CD8+ T cells alone [2, 3]. Moreover, in mice, murine CMV (MCMV)-specific CD8+ T cells directly suppress viral reactivation from latency [4].
The continuous surveillance by CMV-specific CD8+ T cells promotes their accumulation in both humans and mice with impressive results: approximately 5% of all circulating T cells are CMV-specific in the average healthy adult infected with CMV, and this value increases with age [5, 6] in a process called “memory inflation” [7]. In both humans and mice, CMV-specific T cells accumulate without undergoing substantial proliferation during the persistent phase of infection [8-10]. The homeostasis that enables the maintenance of these large populations is not understood.
Several studies have suggested that T cells with high affinity for human (H)CMV are preferentially selected during HCMV infection, especially during periods of viral activity. Specifically, during persistent infection, the HCMV-specific T cells tend to be relatively oligoclonal populations of T cells with high affinity T cell receptors (TCRs), and both viral reactivation and chronic inflammation correlate with a focusing of the CMV-specific T cell repertoire toward individual clones with high affinity for antigen [11-14]. These clones with high affinity for HCMV, tend to display a more differentiated phenotype [12, 13] which could suggest that they more frequently respond to antigen. Interestingly, one study [11] revealed a loss of T cell clones bearing low affinity TCRs early after HCMV infection. The authors of this latter study suggested that the affinity of the TCR may dictate the selection and relative clonal abundance of an individual T cell during HCMV infection. However, it is clear that T cells expressing TCRs with low affinity can persist, and that clones with both high and low affinity TCRs remain comparably functional throughout infection [12, 13].
We sought to use the MCMV model and an adoptive transfer system to directly test whether T cells with a high affinity for MCMV-derived peptides are selected during MCMV infection. The adoptive transfer of transgenic T cells is commonly used to study the T cell response elicited by a wide array of pathogens, including MCMV [15]. Transfers are generally performed into recipient mice that differ from the donor cells at the CD45 or Thy1 loci, enabling donor cells to be identified and monitored in recipients. In published work, donor T cells are generally well tolerated by recipient mice. Nevertheless, there are a few documented cases of immune responses elicited by transferred cells that differed at the CD45 or Thy1 loci [16-20]. While such donor/recipient pairs are often considered to be “congenic”, it is clear that multiple genes are included in the congenic interval. For example, the congenic interval in B6.SJL mice, which contains CD45.1 variant of the CD45 molecule, is approximately 42 mbp in length and encompasses over 300 genes, at least 45 of which harbor polymorphisms that differ from CD45.2+ B6 mice [21]. One study found that immunizing B6.SJL mice (CD45.1+) with a peptide derived from CD45.2 itself, elicited CD45.2-specific T cells [22], raising a long-standing concern that differences in the CD45-molecule may be targeted by immune responses. On top of these differences, genetic drift of inbred strains can be a common occurrence [23] and polymorphisms generated by genetic drift have been shown to induce alloreactivity [24]. Notably, all of these examples of immunity induced by cell transfers are derived from hematopoietic stem cell transplant or tumor transplant studies. There are no published examples of immunity raised by cell transfers after a pathogen infection.
We injected very small numbers of OT-I transgenic T cells into recipient mice that differed at the CD45 locus to assess the selection and maintenance of T cells expressing a TCR with high affinity for an MCMV-derived peptide. OT-I T cells express a TCR with high affinity for the SIINFEKL peptide derived from ovalbumin (ova), presented in the context of H-2Kb MHC (Kd ≈ 5-6 μM [25, 26]). Thus, recipient mice were infected with MCMV expressing the SIINFEKL peptide. Our results show that OT-I T cells were selectively enriched over the first three weeks of infection, and uniformly dominated the total SIINFEKL-specific T cell population, even after extremely small numbers of OT-Is were transferred. However, OT-I T cells were rejected from some recipients as a result of subtle minor histocompatibility differences that likely resulted from genetic drift.
Importantly, this enabled a direct assessment of sub-dominant T cells in mice that retained or lost the dominant clone. We found that robust inflation of the host-derived SIINFEKL-specific T cells only occurred in mice that rejected their OT-I competitors, showing that OT-Is restricted the inflation of host-derived T cells continuously and in a competitive manner. These data support the notion that MCMV, like HCMV, selects T cells expressing TCRs with a high affinity for viral antigen and further suggest that competition for antigen continuously dictates the clonal abundance of T cells in the inflationary pool during MCMV infection.
Results
SIINFEKL-specific CD8+ T cells dominate memory inflation after infection with MCMV-GFP-SL8
A recombinant strain of MCMV was generated expressing a fusion construct of GFP-SIINFEKL under the control of the endogenous MCMV IE2 promoter (MCMV-GFP-SL8, Figure 1A). This virus consistently grew with marginally delayed kinetics in a multistep growth assay in vitro (Figure 1B). However, after infection of C57BL/6 (B6) mice with MCMV-GFP-SL8, SIINFEKL-specific CD8+ T cell responses steadily inflated (i.e. increased in frequency) over time, becoming the dominant inflationary T cell population in these animals (Figure 1C). As expected, T cells specific for the viral protein M45 contracted after primary clonal expansion (Figure 1C), similar to their behavior after infection with wild-type MCMV (Figure 1C, and [10, 27]). However, unlike the response elicited by wild-type MCMV (Figure 1C, right panel), M38-specific T cells failed to inflate after infection by MCMV-GFP-SL8. These data mirrored our previous observations with a second strain of MCMV expressing ova (K181-tfr-ova) [10], which expresses the full-length ova protein [28].
Figure 1.

Construction and characterization of MCMV-GFP-SL8. A) Schematic of recombinant strain MCMV-GFP-SL8 in which the SIINFEKL peptide was fused to GFP and cloned into the IE2 region of the MCMV genome. B) The growth of MCMV-GFP-SL8 was compared to the parental wild-type BAC-derived MCMV. Error bars represent the standard deviation from 2 to 3 plaque assays. Data is representative of 2 independent assays. C) CD8+ T cell responses for immunodominant peptides were measured in MCMV-GFP-SL8 and MCMV-WT infected mice at the indicated weeks post infection. Peripheral blood CD8+ T cells were analyzed by ex vivo intracellular cytokine staining for IFN-γ at indicated days post infection (n=3 mice per group).
OT-I T cells undergo normal memory inflation in recipients
To test whether T cells expressing a TCR with high affinity for MCMV-derived peptides would be selected during memory inflation, we used OT-I transgenic T cells and an adoptive transfer model. The TCR of OT-I T cells has a high affinity for the SIINFEKL peptide presented by H-2Kb (Kd ≈ 5-6 μM [25, 26]). Thus, these cells provide a good model to evaluate the selection, persistence and inflation of an individual T cell clone with high affinity for its antigen in comparison with host-derived SIINFEKL-specific responses in the same animals. Small numbers of OT-I T cells (600 T cells) on the B6 background (CD45.2+) were transferred into mice that expressed both CD45.1 and CD45.2 (B6 × B6.SJL). One day after adoptive transfer, recipient mice were infected with MCMV-GFP-SL8, and donor OT-I T cells were tracked in the peripheral blood by flow cytometry (Figure 2). Donor OT-I T cells were readily evident in the peripheral blood of recipients after MCMV-GFP-SL8 infection and were able to both persist and inflate (Figure 2B). There was substantial mouse-to-mouse variation in the overall frequency of OT-I T cells over time (Figure 2B), which is consistent with memory inflation in general [29]. Moreover, the proportion of OT-Is expressing an effector phenotype (KLRG-1pos, IL-7Rαlow) was indistinguishable from SIINFEKL-specific T cells in B6 mice that did not receive OT-Is (Figure 2C and not shown). Thus, OT-I T cells are able to become representative members of SIINFEKL-specific inflationary populations. Interestingly however, host-derived SIINFEKL-specific T cells were slightly less likely to express an effector phenotype when compared with OT-Is in the same animal.
Figure 2.

OT-Is form a representative antigen-specific inflationary population. A) Schematic of adoptive transfer. CD45.1/CD45.2 double positive mice received 600 CD45.2+ OT-I CD8+ cells prior to infection with MCMV-GFP-SL8. B) A representative FACS plot of donor OT-I and host CD8+ cells is shown. The frequency of CD45.2+ OT-I T cells was analyzed in the peripheral blood over time for mice in which OT-Is inflated or declined. Each line represents an individual animal. In one mouse, the transferred OT-I population fell below the limit of detection for several weeks and then re-appeared at a chronic time point (grey line). Data are combined from two independent experiments. C) Donor or host-derived SIINFEKL-specific T cells were measured by pentamer staining. Shown is the percentage of pentamer-binding cells expressing an effector phenotype (KLRG-1pos, IL-7Ralow). Each symbol represents an individual mouse. Donor and host phenotype data is derived from recipients shown in “B” (n=9, two independent experiments) or from B6 mice that did not receive an OT-I transfer (n=6, two independent experiments). Significance was assessed using a paired t-test.
Reduced inflation of host-derived SIINFEKL-specific T cells in mice that received OT-Is
Previous work showed that when large numbers of OT-Is are present prior to vaccination, they dominate and suppress B6 host-derived T cells specific for SIINFEKL or other ova-derived peptides [30, 31]. We transferred 600 OT-I T cells and we wondered whether their presence would affect host-derived SIINFEKL-specific immune responses. Indeed, the host-derived SIINFEKL-specific population approximately equaled the frequency of the donor population in only one animal (asterisks in Figure 3A) and in this animal, OT-I T cells declined to undetectable levels prior to re-expanding (grey line in Figure 2B). In addition, the overall frequency of host-derived SIINFEKL-specific T cells was significantly reduced in these mice, compared to mice that never received OT-I T cells (Figure 3B). Thus, the presence of OT-Is correlated with significantly reduced inflation from the host-derived SIINFEKL-specific T cells. In contrast, all animals made comparably poor responses to the MCMV-derived M38 antigen (Figure 3C). Together, these data show that high affinity OT-I T cells can undergo robust memory inflation, and could suggest that such inflation inhibits the host-derived SIINFEKL-specific T cell populations.
Figure 3.
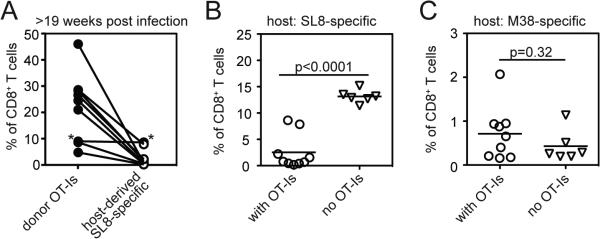
Inflation of OT-I cells correlates with inhibition of the host SIINFEKL-specific responses. A) The frequency of CD8+ T cells that are SIINFEKL-specific (pentamer-binding) and either donor or host derived was determined by flow cytometry. The lines connect the donor and host populations from the same animals (n=9). Asterisks refer to the mouse in which OT-I T cells were temporarily undetectable (grey line in Figure 2C). B and C) The frequency of host-derived SIINFEKL-specific T cells (B) or M38-specific T cells (C) are shown for mice that never received OT-Is or in which OT-Is inflated (as in Figure 2). Significance was assessed using an unpaired t-test.
Selection and dominance of OT-Is even after comparable primary clonal expansion
When 600 OT-I T cells were transferred, the donor cells outnumbered the host-derived SIINFEKL-specific T cells as early as 7 days post infection (Figure 4B and not shown), which is consistent with previous studies [32]. However, dominance of OT-I T cells within this first week might mask any subsequent selection of these T cells during the persistent phase of infection. B6 mice are reported to contain ~100 to 200 SIINFEKL-specific T cells [33]. Thus, to more rigorously examine selection of the OT-I T cells, we transferred 60 or fewer congenically marked OT-I T cells prior to infection with MCMV-GFP-SL8. In these experiments donor OT-Is expressed both CD45.1 and CD45.2 (double-positive OT-Is, DP-OT-Is) and recipients were B6 (CD45.2+) mice. By reducing the number of transferred T cells, donor OT-Is and host-derived SIINFEKL-specific T cells were present in approximately equal proportions 7 days after infection (Figure 4B), thereby eliminating any acute-phase advantage that OT-Is might have. Nevertheless, memory inflation led to the uniform dominance of OT-Is within the SIINFEKL-specific population, and this was sustained throughout the experimental time course (Figure 4C). Thus, high affinity OT-I T cells still dominated memory inflation even when very low numbers of precursors were seeded into the naïve pool prior to infection. When the proportion of all SIINFEKL-specific T cells that were donor- or host-derived was assessed over time, it was evident that OT-Is were preferentially enriched during the first several weeks after infection (Figure 4D). However, after the 3 week time point, the ratio between the donor and host populations remained largely stable. Thus, while OT-I T cells were preferentially selected during the first 3 weeks of infection, equilibrium between donor and host-populations was eventually achieved and maintained throughout the experimental period.
Figure 4.

Selection of OT-Is after comparable primary expansion. A) Schematic of adoptive transfer. B6 (CD45.2+) mice received 600 (n=5), 60, 20 or 6 (n=19) OT-I CD8+ cells expressing both CD45.1 and CD45.2 (DP-OT-Is) prior to infection with MCMV-GFP-SL8. A representative FACS plot of donor/host discrimination is shown. B) Host and donor SIINFEKL-specific T cells in the peripheral blood were measured 7 days post infection by intracellular cytokine staining for IFN-γ production. Data are combined from three independent experiments. C) Inflation of donor OT-Is and host-derived SIINFEKL-specific T cells in the peripheral blood is shown for mice that received ≤ 60 OT-Is and OT-Is inflated. D) The total SIINFEKL-specific response was measured by IFN-γ production from peripheral blood T cells and the percentage of all responding cells that were donor-derived OT-Is was plotted over time for the mice shown in “C” (n=7). Data are combined from the three experiments. The shaded area visually denotes the period of time during which OT-Is increased relative to host-derived SIINFEKL-specific T cells.
Selection of T cells with increased functional avidity after infection
The selection of OT-Is during the first several weeks of MCMV-GFP-SL8 infection might reflect the selection of high affinity T cells in general. If so, we should be able to detect a general increase in the overall avidity of the SIINFEKL-specific T cell population. To test this, we measured the amount of SIINFEKL peptide required to stimulate 50% of the IFN-γ producing cells per animal (EC50). This corresponds to the “functional avidity” of the population and is an established method to assess the overall sensitivity of a T cell population [34]. In mice that did not receive OT-I T cells the functional avidity of the SIINFEKL-specific cells was quite broad 7 days after infection, with a range of avidities that differed by more than 20-fold between mice (Figure 5A). Such variation between animals is consistent with differences in the precursor pool of cells specific for SIINFEKL prior to infection. By late times post infection however (> 3 months), the average functional avidity of the population increased by 2.5-fold and narrowed greatly (Figure 5A). The populations with a low functional avidity were no longer evident and the overall functional avidity was comparable to that of OT-I T cells themselves (Figure 5A and 5B). These data suggest that shortly after infection with MCMV-GFP-SL8, the SIINFEKL-specific population contained cells with low functional avidity that required more peptide to respond. Over time, however, infection induced the selection of T cells with a high functional avidity for the SIINFEKL peptide.
Figure 5.
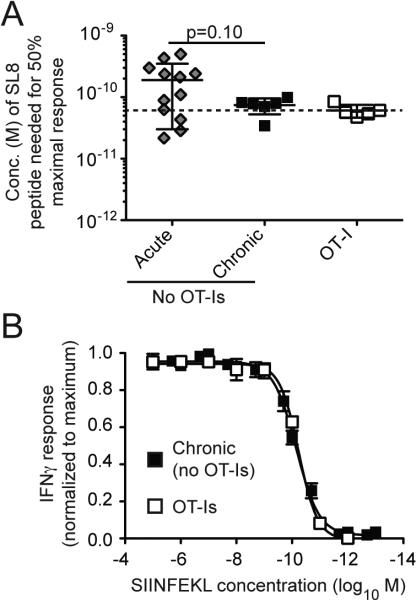
Comparable functional avidity of OT-Is and host-derived SIINFEKL-specific T cells. A) Functional avidity of splenocytes was measured after 7 days (acute, n=12), or more than 3 months (chronic, n=6). The avidity of OT-Is was measured after they had undergone inflation for more than 3 months (n=5). The concentration of SIINFEKL peptide necessary to achieve 50% of the maximal IFNγ response (EC50) was assessed for each animal. Note that populations with lower functional avidity required more SIINFEKL peptide to achieve the EC50, and thus appear higher on the y-axis of the graph. The data is combined from 1 to 4 independent experiments. Significance was assessed using an unpaired t-test. B) Shown are the normalized response curves used to calculate the EC50 values shown in A. Responses to the SIINFEKL peptide were normalized to the percent of maximal IFN-γ and averaged for OT-Is after undergoing inflation (n=5) or SIINFEKL-specific T cells after inflation in mice that never received OT-Is (n=6).
Rejection of OT-I T cells from some recipients
Through the course of these experiments, it became apparent that OT-I T cells were lost from a subset of our recipient mice. This was most evident when OT-I T cells on the B6 (CD45.2+) background were transferred into B6.SJL mice expressing CD45.1 (Figure 6A). In this scenario, donor OT-I T cells expanded and were readily detectible in the peripheral blood 7 days after MCMV-GFP-SL8 infection (Figure 6A), but disappeared from nearly all recipients and were virtually undetectable by day 21 post infection (Figure 6B and Table I). Host-derived SIINFEKL-specific T cells eventually inflated in these animals (Figure 6C), demonstrating the persistence of the infection and the antigen. Loss of OT-Is occurred after infection with either MCMV-GFP-SL8 (Figure 6B) or the MCMV strain K181-tfr-ova (Table I). To demonstrate that donor OT-Is were being rejected from these mice, we established an in vivo killing assay. Splenocytes from CD45.2+ OT-I mice (targets) and CD45.1+ mice (controls) were labeled with CFSE, mixed, and co-injected into CD45.1+ mice that had previously lost their transferred OT-I population. Mice that retained their donor OT-Is and naïve mice that never received OT-Is were used as controls. Two days after transfer, the ratio of target to control cells within the CFSE labeled population was compared. These data revealed that loss of OT-Is from the initial transfer correlated with specific killing of splenocytes derived from OT-Is (Figures 6D), indicating immune rejection. Importantly, within the target splenocytes from OT-I mice, both T cells and non-T cells were killed (not shown), indicating that the rejection was not the result of an immune response targeting the OT-I TCR idiotype.
Figure 6.

CD45.2+ OT-I T cells are frequently rejected from CD45.1 mice following robust primary expansion. A) Schematic of adoptive transfer and representative FACS plot of donor/host discrimination. B) The frequency of donor OT-I T cells in the peripheral blood was determined 7 and 21 days after infection with MCMV-GFP-SL8. Shown are CD45.1+ mice that received 600 (n=13) or 2,600 (n=3) CD45.2+ OT-I T cells. C) Host-derived SIINFEKL-specific CD8+ T cell responses were measured in CD45.1+ mice that received 600 CD45.2+ OT-I T cells (n=7). Shown is the host-derived IFN-γ production after stimulation of peripheral blood T cells with the SIINFEKL peptide in vitro. Asterisks indicate the two mice that retained their OT-I population at day 21 (Fig. 6B). Data from B and C are representative of three independent experiments. D) Representative FACS plots from an in vivo killing assay performed with CD45.2+ OT-I targets and CD45.1+ control splenocytes. Data are representative of 3 independent experiments. E) In vivo killing assays were conducted as in “D” except that some mice simultaneously received control cells (CD45.1+) as well as OT-I targets (expressing both CD45.1 and CD45.2) and B6 targets (CD45.2+). All 3 populations were enumerated as in D. Specific killing was calculated as described in the materials and methods. Data are combined from 2 independent killing assays.
Table I.
Summary of OT-I transfer experimentsa
| recipient mice | source | # of OT-I transferred | genotype of OT-I | Virus | pfu | # of mice | # of rejectorsb | % rejected |
|---|---|---|---|---|---|---|---|---|
| B6.SJL (CD45.1+) | Taconic | 600+ | CD45.2 | GFP-SL8 | 2×105 | 33 | 28 | 84.85% |
| B6.SJL (CD45.1+) | Taconic | 600 | CD45.2 | K181-ova | 2×105 | 7 | 6 | 85.71% |
| B6.SJL (CD45.1+) | Jackson | 600+ | CD45.2 & CD45.1/.2 | GFP-SL8 | 2×105 | 25 | 4 | 16% |
| CD45.1/.2 | in house | 600 | CD45.2 | GFP-SL8 | 2×105 | 19 | 7 | 36.84% |
| B6 (CD45.2+) | Jackson | 6 - 3,600 | CD45.1/.2 | GFP-SL8 | 2×105 | 32 | 11 | 34.38% |
| B6 (CD45.2+) | Jackson | 600 | CD45.1/.2 | GFP-SL8 | 2×104 | 4 | 3 | 75% |
| B6 (CD45.2+) | Jackson | 600 | CD45.1/.2 | GFP-SL8 | 50 | 7 | 2 | 28.57% |
| μMT (CD45.2+) | Jackson | 600 | CD45.1/.2 | GFP-SL8 | 2×105 | 6 | 4 | 66.67% |
Shown is a summary of OT-I transfer experiments including the genotype of the donor and recipient animals, the origin of recipient mice, the number and genotype of OT-Is transferred, the virus and titer used to infect recipients and the number and frequency of mice that retained or lost their OT-Is.
Experiments were not always carried for the same amount of time. It is possible that more mice would have lost OT-Is given more time.
Several studies have suggested that allelic differences between B6 (CD45.2+) and B6.SJL (CD45.1+) mice can elicit immune responses that may cause rejection of transferred hematopoietic cells [16, 18-22]. To test this, we included splenocytes from non-transgenic B6 (CD45.2+) mice in our killing assay. As shown in Figure 6E, B6 splenocytes were poorly killed in mice that lost OT-Is, showing that the CD45.2 molecule itself is not the primary target for rejection. In support of this, we also observed loss of CD45.2+ OT-I T cells from a subset of CD45.1+/CD45.2+ double positive recipients (Table I). In these experiments, the recipients expressed the CD45.2 molecule, as well as any other polymorphisms included in the CD45.1 congenic interval, demonstrating that differences between B6 and B6.SJL could not be responsible for loss of the OT-Is. Overall, these results imply that more subtle minor histocompatibility differences between the donor and recipient strains were responsible for rejection of the OT-Is. Such polymorphisms may develop from genetic drift [23, 24]. If true, we might expect that using independently maintained lines of donor or recipient mice would result in different rates of rejection. The experiments described thus far were conducted with donor and recipient mice purchased from Taconic Farms. Notably, while B6.SJL mice from Taconic Farms are maintained on the C57BL/6J background, OT-Is from Taconic Farms have been bred into the C57BL/6N background. However, OT-I's purchased from Jackson Laboratories, which are maintained on the C57BL/6J background, were also rapidly rejected from B6.SJL mice purchased from Taconic Farms (not shown). Thus, differences between C57BL/6N and C57BL/6J strains [35] could not account for rejection in our experiments. Remarkably however, regardless of whether OT-Is were maintained on the C57BL/6J or C57BL/6N background, their rejection was rare in B6.SJL recipients purchased from Jackson Laboratories (Table I and not shown). To summarize several experiments, we considered mice to have lost the transferred cells when their OT-I frequencies fell below 0.3% of CD8+ T cells in the peripheral blood within 91 days of infection. With these criteria, 34 of 40 B6.SJL recipients purchased from Taconic Farms (85%), but only 4 of 25 recipients purchased from Jackson Laboratories (16%), lost the transferred OT-I T cells (Table I and not shown). Thus, two independently maintained lines of B6.SJL mice rejected the transferred OT-Is at markedly different rates. Together, these data suggest that genetic drift within the B6.SJL strains has resulted in the generation of subtle minor histocompatibility polymorphisms that cause rejection of the transferred OT-I T cells.
OT-I T cells restrict inflation of host-derived T cells in a competitive manner
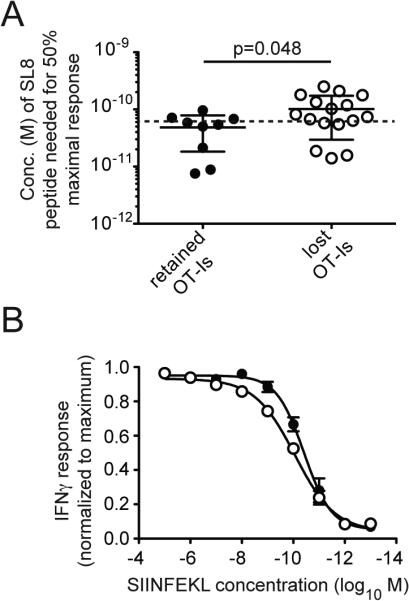
The fact that OT-Is were lost from some, but not all mice after participating in the primary T cell response was an unexpected benefit: it allowed us to explore the fate of suppressed, host-derived T cells after the dominant OT-I clone was removed from the system. We reasoned that if OT-Is were restricting the inflation of host-derived SIINFEKL-specific T cells continuously in a competitive manner, loss of OT-Is should facilitate inflation of the host-derived T cells. For these experiments, we utilized mice that had received 60 or fewer OT-I T cells in the adoptive transfer (as in Figure 4). Overall, transferring OT-Is expressing both CD45.1 and CD45.2 (DP-OT-Is) into B6 (CD45.2+) recipients resulted in loss of donors from 37% of mice (Table I), albeit with somewhat delayed kinetics (Figure 7A). Importantly, loss of OT-I T cells allowed robust inflation from the host-derived SIINFEKL-specific population, even when recipients were stratified into mice that lost OT-Is early or late in infection (compare the host-responses in Figure 7B to that shown in Figure 4C). This was true despite the fact that the transferred OT-I T cells were preferentially enriched prior to their disappearance (Figure 7C, shaded area).
Figure 7.

Rejection of transferred OT-Is allows host-derived SIINFEKL-specific T cells to inflate. A) 60, 20 or 6 DP OT-I T cells were transferred into B6 (CD45.2+) recipients before infection with MCMV-GFP-SL8. Shown is the frequency of DP OT-Is in the peripheral blood in mice in which OT-Is were lost over time. Arrows indicate two mice in which OT-Is declined but still comprised 1.6% and 0.57% of all CD8+ T cells in the peripheral blood at the final time point. B) Inflation of donor OT-Is and host-derived SIINFEKL-specific T cells in the peripheral blood is shown for the mice shown in “A”. Animals were grouped into those in which OT-Is were lost by week 9 (left graph) or declined but comprised more than 0.3% of all CD8 T cells at least 9 weeks post infection (right graph). C) The total SIINFEKL-specific response was measured by IFN-γ production from peripheral blood T cells and the percentage of all responding cells that were donor-derived OT-Is was plotted over time. The shaded area visually denotes the early selection of OT-Is relative to host-derived SIINFEKL-specific T cells. Data for A-C are combined from the three independent experiments. D) In vivo killing assays were conducted as in Figure 6 except that mice received a mixture of CFSE-labeled B6 splenocytes (CD45.2+ controls) and DP OT-I splenocytes. Specific killing was calculated for each animal (n=7). Data are combined from two independent experiments. E) As in “D” except that mice received a CFSE-labeled mixture of B6 splenocytes (CD45.2+ controls), B6.SJL splenocytes (CD45.1+ nontransgenic targets), or DP OT-I splenocytes. Shown are representative FACS plots and specific killing of target populations from one experiment (n=5).
To conclude that OT-Is were suppressing host-derived SIINFEKL-specific T cells and that loss of OT-Is allowed host-derived T cells to inflate, it was critical to show that OT-Is were rejected from recipients. Otherwise, it was possible that host-derived clones were preferentially selected to inflate instead of the OT-Is. Thus, to test whether the loss of DP-OT-I T cells represented immunological rejection, we performed in vivo killing assays using splenocytes from DP-OT-I mice as targets and splenocytes from B6 mice as controls. In all cases, loss of OT-Is from the initial transfer correlated with specific killing of OT-I-derived splenocytes (Figure 7D and E). Notably, non-transgenic cells expressing CD45.1 (B6.SJL) were also killed in mice that had rejected OT-Is, although much less efficiently than transgenic DP-OT-I splenocytes (Figure 7E). These data demonstrate that loss of DP-OT-I T cells in B6 mice was mediated by immune rejection and that their loss was unconnected with their ability to inflate. Thus, despite the early selection of OT-I T cells (Figure 7C), their belated removal from the system allowed host-derived T cells to accumulate. These data suggest that inflation of host-derived SIINFEKL-specific T cells was actively restricted by the presence of OT-I T cells, implying that competition between T cells can account for relative clonal abundance during memory inflation. Overall, these data show that, like HCMV infection, MCMV infection induces the selection of T cells with a high functional avidity for MCMV-derived antigens shortly after infection. Further, these data suggest that relative clonal abundance is actively maintained in a competitive manner.
Discussion
Previous work has suggested that HCMV-specific T cells with high-affinity TCRs were selected shortly after acute HCMV infection [11-14]. Using an adoptive transfer model, our data show that T cells expressing a high-affinity TCR are also selected during the first 3 weeks after acute MCMV infection, even when transferred in exceedingly low numbers. Moreover, selection was not just limited to the transferred transgenic T cells. Indeed, the overall avidity of SIINFEKL-specific T cells was similarly increased after infection in the absence of OT-Is, resulting in populations that had a comparable functional avidity to OT-I T cells themselves (Figure 5).
Despite the early selection of OT-I T cells, we found that OT-Is and host-derived SIINFEKL-specific T cells achieved equilibrium after approximately 3 weeks, and that there was little additional selection of OT-I T cells over host-derived T cells during the remainder of memory inflation (Figure 4D). However, the sporadic, and often belated rejection of OT-I transgenic T cells allowed us to compare host-derived SIINFEKL-specific T cells in mice that retained or lost their high affinity OT-I competitors. We observed that marked inflation of host-derived T cells only occurred when OT-Is were lost, which shows that dominant populations with high functional avidity have the capacity to restrict the inflation of sub-dominant T cells specific for the same antigen. The fact that the late loss of the OT-I T cells enabled the inflation of host-derived SIINFEKL-specific T cells (Figure 7B) suggests that competition between T cells specific for the same peptide is continuous. These data suggest that such competition maintains the diversity and relative clonal abundance of T cells during persistent CMV infections.
While previous work has shown that OT-I T cells can out-expand and even suppress endogenous SIINFEKL-specific T cells from B6 mice [30, 31, 36], it is important to note that we transferred between 50-times and 3,300-times fewer OT-I T cells in the experiments described here, which resulted in comparably sized populations 7 days after infection. Thus, we can conclude that suppression of the host-derived T cells specific for SIINFEKL was not due the precursor frequency of the OT-Is. Surprisingly however, when OT-Is were lost after primary expansion, the functional avidities of the remaining SIINFEKL-specific populations were broadly distributed and reduced on average, similarly to early time points (not shown). Thus, host-derived T cells with a low functional avidity persisted within the population even though they were not enriched, and were revealed only after the OT-Is were rejected. Note that functional avidity of the T cells is a measure of responsiveness and not simply affinity of the TCR for MHC-peptide complexes. Together, these data suggest that MCMV infection normally selects T cells with high functional avidity for MCMV-derived antigens during the first several weeks of infection and that these cells achieve equilibrium through their competition for antigen.
Our data confirm that recruitment of naïve T cells at late times post infection is not a pre-requisite of memory inflation, as indicated by several studies [10, 15, 37]. Our data extend these previous observations by demonstrating that even very small numbers of precursor T cells (between 6 and 60 transferred OT-I T cells in Figure 4B) were able to inflate to more than 10% of the total circulating CD8+ T cell pool. Extrapolating these results to natural CMV infection or CMV-based vaccinations suggests that any T cell clone present in the pre-infection repertoire with a high affinity for a CMV-encoded peptide will be selected early during memory inflation, will have a competitive advantage over other clones, and may even suppress the inflation of less avid T cells. Indeed, the exclusive inflation of SIINFEKL-specific T cells after infection with MCMV-GFP-SL8 (Figure 1 and [10]) might be explained by competitive suppression of MCMV-specific T cells. This hypothesis is consistent with previous work showing that removal of two immunodominant epitopes from MCMV enabled the inflation of T cells specific for a new, unrelated antigen [38]. Such dominance of high affinity responses may prove problematic for generating memory inflation against tumor antigens encoded by a CMV-based vector, as few T cells are expected to recognize these quasi-self antigens with high affinity.
The rejection of OT-I T cells in our experiments was unexpectedly frequent. Notably, we also observed rejection of CD45.2+ OT-Is from CD45.1+ recipients (both Taconic derived) challenged with Vaccinia virus expressing the SIINFEKL peptide (not shown). These data demonstrate that rejection of OT-Is is not dependent on MCMV infection exclusively. While it is likely that different T cell stimuli will have different impacts on rejection, it also seems likely that multiple viral infections, or perhaps methods of immunization, will induce a similar rejection of OT-I T cells. Although we suspect that other labs have come across this phenomenon with other pathogens (or even with MCMV), we found no discussion of this in the literature. In contrast, the immunogenicity of polymorphisms in the CD45 locus have been discussed for several years in the context of hematopoietic cell transplants [16, 18-22]. However, our killing assay data directly show that differences between B6 and B6.SJL mice were not the primary determinants of rejection. Instead, the fact that two independently maintained stocks of B6.SJL mice rejected OT-Is at markedly different rates implies that genetic drift of the donor and recipient strains is the major determinant of rejection. However, the mechanism by which OT-I T cells were rejected remains unclear. Rejection was not exclusively antibody mediated as it still occurred in μMT mice, which lack B cells (not shown). Notably however, μMT mice were originally produced on the 129 background [39] and therefore might carry additional minor histocompatibility differences. Regardless, these results clearly show that rejection of transferred cells may be a common confounding issue, at least for studies of MCMV infection, unless donor and recipient pairs are carefully matched.
Overall, our data show that T cell clones specific for the MCMV-encoded peptides compete for antigen during the acute and persistent phases of MCMV infection. Our evidence suggests that the precursor pool of T cells with high functional avidity for viral antigen and long-term competition between T cell clones has the ability to fundamentally shape the diversity of the inflationary T cell pool. Whether this has long-term consequences for the host remains to be seen and a more detailed study of the breadth of T cell clones over time may elucidate this issue.
Materials and Methods
Mice
C57BL/6J mice were purchased from Jackson Laboratory. RAG2 −/− OT-I (B6.129S6-Rag2tm1Fwa Tg(TcraTcrb)1100Mjb) mice were purchased from Taconic Farms and bred in house for use in all experiments. OT-I mice from Taconic are maintained on the B6/N background. OT-I transgenic mice on the B6/J background (C57BL/6-Tg(TcraTcrb)1100Mjb/J) were purchased from the Jackson Laboratories. B6.SJL-CD45.1 congenic mice were purchased from Taconic Farms (B6.SJL-Ptprca/BoyAiTac) or Jackson Laboratories (B6.SJL-Ptprca Pepcb/BoyJ) as indicated in the text. Heterozygous mice were produced by breeding. Donor and recipient mice were gender matched for all adoptive transfers. All protocols were approved by the Thomas Jefferson University Institutional Animal Care and Use Committee.
Virus Strains and Infections
To produce the recombinant strain MCMV-GFP-SL8, the sequence encoding the SIINFEKL peptide (ova250-264) plus 7 amino acids upstream of the peptide was attached to the 3’ end of the sequence encoding GFP using PCR. The fusion construct was recombined with MCMV encoded as a bacterial artificial chromosome (BAC, strain MW97.01 [40]) and targeted to replace the m128 exon (IE2 gene) using established techniques [40]. The multistep growth curve shown in Figure 1 was performed by infecting M2-10B4 cells with a multiplicity of infection (MOI) of 0.1, harvesting lysates at the indicated times and measuring viral growth by plaque assay without centrifugal enhancement on M2-10B4 cells. Mice were infected i.p. with 2×105 plaque forming units (pfu) of the virus.
Adoptive Transfer of OT-I T Cells
Naïve OT-I spleens were harvested, passed through a 70 μm cell strainer and washed twice with RPMI (Cellgro, 1640 with L-glutamine + 10% FBS + 1% penicillin/streptomycin + 5 × 10-5 M β-mercaptoethanol) to form single cell suspensions. Total splenic cells were counted on a Z2 Coulter Particle Count and Size Analyzer (Beckman Coulter) and a sample was assessed for the frequency of CD8+ T cells by flow cytometry. Splenocytes containing the indicated numbers of CD8+ cells were injected retroorbitally into recipients and mice were infected with MCMV-GFP-SL8. In almost all experiments, mice were infected 1 day post OT-I transfer. For some of the mice shown in Figures 4 and 7 (n=8 total mice), recipients were infected 9 days post transfer. For all mice shown that received OT-I T cells, the OT-I populations comprised greater than 1% of all CD8's at some point after infection.
Antibodies, pentamer staining and FACS analysis
At the indicated times post infection, donor and recipient cells were identified by flow cytometry using the CD45.1 (clone A20), CD45.2 (clone 104), antibodies, and cells were co-stained with antibodies specific for CD8α (clone 53-6.7), CD127 (clone A7R34), KLRG-1 (clone 2F1), Vα2 (clone B20.1), CD27 (clone LG.3A10), CD44 (clone IM7), and IFN-γ (clone XMG1.2). In all cases, antibodies were purchased from BD Biosciences, eBioscience, or BioLegend. H-2Kb pentamers loaded with the SIINFEKL peptide (ProImmune) or H-2Kb tetramers (NIH tetramer core facility: http://tetramer.yerkes.emory.edu/) loaded with M38316-323 were used to identify antigen-specific T cells. Pentamer staining was performed following the manufacturer's recommended protocol. Tetramer staining was performed as previously described [10]. Cells were analyzed on an LSR II flow cytometer (BD Biosciences) and using FlowJo software (TreeStar, Ashland, OR).
Intracellular cytokine stimulation assay
In some cases, total SIINFEKL-specific responses were measured by ex vivo intracellular cytokine stimulation, performed as described [10] with a few modifications. In brief, we found that maximal IFN-γ was produced within 3 hours of stimulation and that limited cell death occurred during this time period (not shown). Thus, cells were incubated with 1μg/ml peptide (synthesized by Genemed Synthesis - http://www.genemedsyn.com) in the presence of 1μg/ml brefeldin A (GolgiPlug, BD Biosciences) for 3 hours at 37° C in 96-well U-bottomed plates prior to staining for intracellular IFN-γ. Cells were analyzed by flow cytometry as above. To assess the functional avidity, cells were stimulated with a titration of the SIINFEKL peptide and the percent of maximal response for each animal was calculated and graphed. Non-linear regression curves were fit to the data and EC50 values were calculated using GraphPad Prism. For measuring the functional avidity of acutely infected mice without OT-Is in Figure 5, four of the twelve mice were the transgene-negative offspring of B6.Cg-Tg(Tyr-cre/ERT2)13Bos/J mice (expressing the Cre-recombinase) on the B6/J background.
In vivo killing assay
An equal number of target and contro splenocytes were labeled in PBS with 1μM CFSE for 15 minutes, washed, and adoptively transferred into mice that had either retained or lost OT-Is, or naïve controls. Target cells were OT-I-derived splenocytes or non-transgenic splenocytes with the same CD45 background as the original OT-I donors. Control cells were syngeneic with the host. Two days later, CFSE-labeled cells were enumerated in the spleen by FACS analysis and the various target and control populations were discriminated by CD45 staining as indicated in the figures. Percent specific killing was calculated by comparing the ratio of the target population to the total control syngeneic population per individual experimental or control mouse using the following formula: 100 - [(target cells /control cells in experimental animals)/(average target/average control cells in control animals)]× 100.
Statistical Analyses
Unless otherwise noted, error bars represent standard error of the mean. In all cases, statistical significance was measured by a two-tailed student's t-test and p-values are shown in the figure or legend. All statistical analyses were performed using GraphPad Prism (GraphPad Software).
Supplementary Material
Figure 8.
Acknowledgements
The authors thank Elizabeth Aikens for collecting some of the flow cytometry data depicted in Figure 1. This work was supported by the NIH grant K22AI081866 awarded to C.M.S..
Abbreviations used
- CMV
cytomegalovirus
- MCMV
murine cytomegalovirus
- HCMV
human cytomegalovirus
- TCR
T cell receptor
- ova
ovalbumin
- pfu
plaque forming unit
Footnotes
The authors have no conflicts of interest to declare.
References
- 1.Crough T, Khanna R. Clin Microbiol Rev. 2009;22:76–98. doi: 10.1128/CMR.00034-08. Table of Contents. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Riddell SR, Watanabe KS, Goodrich JM, Li CR, Agha ME, Greenberg PD. Science. 1992;257:238–241. doi: 10.1126/science.1352912. [DOI] [PubMed] [Google Scholar]
- 3.Walter EA, Greenberg PD, Gilbert MJ, Finch RJ, Watanabe KS, Thomas ED, Riddell SR. New England Journal of Medicine. 1995;333:1038–1044. doi: 10.1056/NEJM199510193331603. [DOI] [PubMed] [Google Scholar]
- 4.Simon CO, Holtappels R, Tervo HM, Bohm V, Daubner T, Oehrlein-Karpi SA, Kuhnapfel B, Renzaho A, Strand D, Podlech J, Reddehase MJ, Grzimek NK. J Virol. 2006;80:10436–10456. doi: 10.1128/JVI.01248-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Komatsu H, Sierro S, V Cuero A, Klenerman P. Clin Exp Immunol. 2003;134:9–12. doi: 10.1046/j.1365-2249.2003.02266.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sylwester AW, Mitchell BL, Edgar JB, Taormina C, Pelte C, Ruchti F, Sleath PR, Grabstein KH, Hosken NA, Kern F, Nelson JA, Picker LJ. J Exp Med. 2005;202:673–685. doi: 10.1084/jem.20050882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Karrer U, Sierro S, Wagner M, Oxenius A, Hengel H, Koszinowski UH, Phillips RE, Klenerman P. J Immunol. 2003;170:2022–2029. doi: 10.4049/jimmunol.170.4.2022. [DOI] [PubMed] [Google Scholar]
- 8.Gamadia LE, Remmerswaal EB, Weel JF, Bemelman F, van Lier RA, Ten Berge IJ. Blood. 2003;101:2686–2692. doi: 10.1182/blood-2002-08-2502. [DOI] [PubMed] [Google Scholar]
- 9.Sierro S, Rothkopf R, Klenerman P. European Journal of Immunology. 2005;35:1113–1123. doi: 10.1002/eji.200425534. [DOI] [PubMed] [Google Scholar]
- 10.Snyder CM, Cho KS, Bonnett EL, van Dommelen S, Shellam GR, Hill AB. Immunity. 2008;29:650–659. doi: 10.1016/j.immuni.2008.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Day EK, Carmichael AJ, ten Berge IJ, Waller EC, Sissons JG, Wills MR. J Immunol. 2007;179:3203–3213. doi: 10.4049/jimmunol.179.5.3203. [DOI] [PubMed] [Google Scholar]
- 12.Iancu EM, Corthesy P, Baumgaertner P, Devevre E, Voelter V, Romero P, Speiser DE, Rufer N. J Immunol. 2009;183:319–331. doi: 10.4049/jimmunol.0803647. [DOI] [PubMed] [Google Scholar]
- 13.Price DA, Brenchley JM, Ruff LE, Betts MR, Hill BJ, Roederer M, Koup RA, Migueles SA, Gostick E, Wooldridge L, Sewell AK, Connors M, Douek DC. J Exp Med. 2005;202:1349–1361. doi: 10.1084/jem.20051357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Trautmann L, Rimbert M, Echasserieau K, Saulquin X, Neveu B, Dechanet J, Cerundolo V, Bonneville M. J Immunol. 2005;175:6123–6132. doi: 10.4049/jimmunol.175.9.6123. [DOI] [PubMed] [Google Scholar]
- 15.Torti N, Walton SM, Brocker T, Rulicke T, Oxenius A. PLoS Pathog. 2011;7:e1002313. doi: 10.1371/journal.ppat.1002313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bhattacharya D, Rossi DJ, Bryder D, Weissman IL. J Exp Med. 2006;203:73–85. doi: 10.1084/jem.20051714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.McKenna KC, Vicetti Miguel RD, Beatty KM, Bilonick RA. J Leukoc Biol. 2011;89:291–300. doi: 10.1189/jlb.0610333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Muller AM, Linderman JA, Florek M, Miklos D, Shizuru JA. Proc Natl Acad Sci U S A. 2010;107:14721–14726. doi: 10.1073/pnas.1009220107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.van Os R, Sheridan TM, Robinson S, Drukteinis D, Ferrara JL, Mauch PM. Stem Cells. 2001;19:80–87. doi: 10.1634/stemcells.19-1-80. [DOI] [PubMed] [Google Scholar]
- 20.Xu H, Exner BG, Chilton PM, Schanie C, Ildstad ST. Stem Cells. 2004;22:1039–1048. doi: 10.1634/stemcells.22-6-1039. [DOI] [PubMed] [Google Scholar]
- 21.Waterstrat A, Liang Y, Swiderski CF, Shelton BJ, Van Zant G. Blood. 2010;115:408–417. doi: 10.1182/blood-2008-03-143370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen W, Chatta GS, Rubin WD, Clark JG, Hackman RC, Madtes DK, Ligitt DH, Kusunoki Y, Martin PJ, Cheever MA. J Immunol. 1998;161:909–918. [PubMed] [Google Scholar]
- 23.Taft RA, Davisson M, Wiles MV. Trends Genet. 2006;22:649–653. doi: 10.1016/j.tig.2006.09.010. [DOI] [PubMed] [Google Scholar]
- 24.Fanning SL, Appel MY, Berger SA, Korngold R, Friedman TM. J Immunol. 2009;183:4261–4272. doi: 10.4049/jimmunol.0900971. [DOI] [PubMed] [Google Scholar]
- 25.Alam SM, Travers PJ, Wung JL, Nasholds W, Redpath S, Jameson SC, Gascoigne NR. Nature. 1996;381:616–620. doi: 10.1038/381616a0. [DOI] [PubMed] [Google Scholar]
- 26.Rosette C, Werlen G, Daniels MA, Holman PO, Alam SM, Travers PJ, Gascoigne NR, Palmer E, Jameson SC. Immunity. 2001;15:59–70. doi: 10.1016/s1074-7613(01)00173-x. [DOI] [PubMed] [Google Scholar]
- 27.Munks MW, Cho KS, Pinto AK, Sierro S, Klenerman P, Hill AB. J Immunol. 2006;177:450–458. doi: 10.4049/jimmunol.177.1.450. [DOI] [PubMed] [Google Scholar]
- 28.Cunningham PT, Lloyd ML, Harvey NL, Williams E, Hardy CM, Redwood AJ, Lawson MA, Shellam GR. Vaccine. 2010 [Google Scholar]
- 29.Snyder CM, Loewendorf A, Bonnett EL, Croft M, Benedict CA, Hill AB. J Immunol. 2009;183:3932–3941. doi: 10.4049/jimmunol.0900227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kedl RM, Schaefer BC, Kappler JW, Marrack P. Nat Immunol. 2002;3:27–32. doi: 10.1038/ni742. [DOI] [PubMed] [Google Scholar]
- 31.Kedl RM, Rees WA, Hildeman DA, Schaefer B, Mitchell T, Kappler J, Marrack P. J Exp Med. 2000;192:1105–1113. doi: 10.1084/jem.192.8.1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Badovinac VP, Haring JS, Harty JT. Immunity. 2007;26:827–841. doi: 10.1016/j.immuni.2007.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Obar JJ, Khanna KM, Lefrancois L. Immunity. 2008 doi: 10.1016/j.immuni.2008.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Slifka MK, Whitton JL. Nat Immunol. 2001;2:711–717. doi: 10.1038/90650. [DOI] [PubMed] [Google Scholar]
- 35.Zurita E, Chagoyen M, Cantero M, Alonso R, Gonzalez-Neira A, Lopez-Jimenez A, Lopez-Moreno JA, Landel CP, Benitez J, Pazos F, Montoliu L. Transgenic Res. 2011;20:481–489. doi: 10.1007/s11248-010-9403-8. [DOI] [PubMed] [Google Scholar]
- 36.Zehn D, Lee SY, Bevan MJ. Nature. 2009;458:211–214. doi: 10.1038/nature07657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Loewendorf AI, Arens R, Purton JF, Surh CD, Benedict CA. Viral Immunol. 2011 doi: 10.1089/vim.2010.0140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Holtappels R, Simon CO, Munks MW, Thomas D, Deegen P, Kuhnapfel B, Daubner T, Emde SF, Podlech J, Grzimek NK, Oehrlein-Karpi SA, Hill AB, Reddehase MJ. J Virol. 2008;82:5781–5796. doi: 10.1128/JVI.00155-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kitamura D, Roes J, Kuhn R, Rajewsky K. Nature. 1991;350:423–426. doi: 10.1038/350423a0. [DOI] [PubMed] [Google Scholar]
- 40.Wagner M, Jonjic S, Koszinowski UH, Messerle M. Journal of Virology. 1999;73:7056–7060. doi: 10.1128/jvi.73.8.7056-7060.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cho KS, Hill AB. J Immunol Methods. 2008;330:137–145. doi: 10.1016/j.jim.2007.10.019. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.