

Inflammation is the physiological response to a variety of injuries or insults, including heat, chemical agents or bacterial infection. In the acute phase of inflammation, the response is rapid and of short duration. If the insult or injury is not resolved, the response becomes chronic, which can be considered as nonphysiologic or pathologic. When inflammation becomes chronic, the adaptive immune response is activated with involvement of the cellular and non-cellular mechanisms of acquired immunity. Immune mechanisms play further roles in the resolution of inflammation and in the healing process, including the repair and the regeneration of lost or damaged tissues. Thus, innate (inflammatory) immunity and acquired immunity must be coordinated to return the injured tissue to homeostasis (85).
The etiology of periodontal diseases is bacteria. The human oral cavity harbors a substantial and continuously evolving load of microbial species. The ecological interactions between the host and microbes determine the severity of the disease. Unlike many infectious diseases, periodontal diseases appear to be infections mediated by the overgrowth of commensal organisms, rather than by the acquisition of an exogenous pathogen. As microorganisms evolve more rapidly than their mammalian hosts, immune mechanisms that determine the ecological balance of commensal organisms also need to change to preserve homeostasis (65).
Knowledge of how immune mechanisms and inflammatory responses are regulated is critical for understanding the pathogenesis of complex diseases, such as periodontitis. The pathogenesis of periodontal diseases is mediated by the inflammatory response to bacteria in the dental biofilm (Fig. 1). However, identification of the true ‘pathogens’ in periodontitis has been elusive. There is evidence that specific microbes are associated with the progressive forms of the disease; however, the presence of these microorganisms in individuals with no evidence of disease progression suggests that the disease is the net effect of the immune response and the inflammatory processes, not the mere presence of the bacteria. Regulation of immune–inflammatory mechanisms governs patient susceptibility and is modified by environmental factors (219, 220, 241). This review will address the pathways of inflammation in periodontal diseases by focusing on immunologic mechanisms to elucidate sites of regulation. Clinical features of the periodontal diseases are beyond the scope of this work but are within the context of the pathogenic mechanisms. Possible clinical outcomes will be discussed in relation to the inflammatory–immunologic changes throughout the disease process.
Fig. 1.
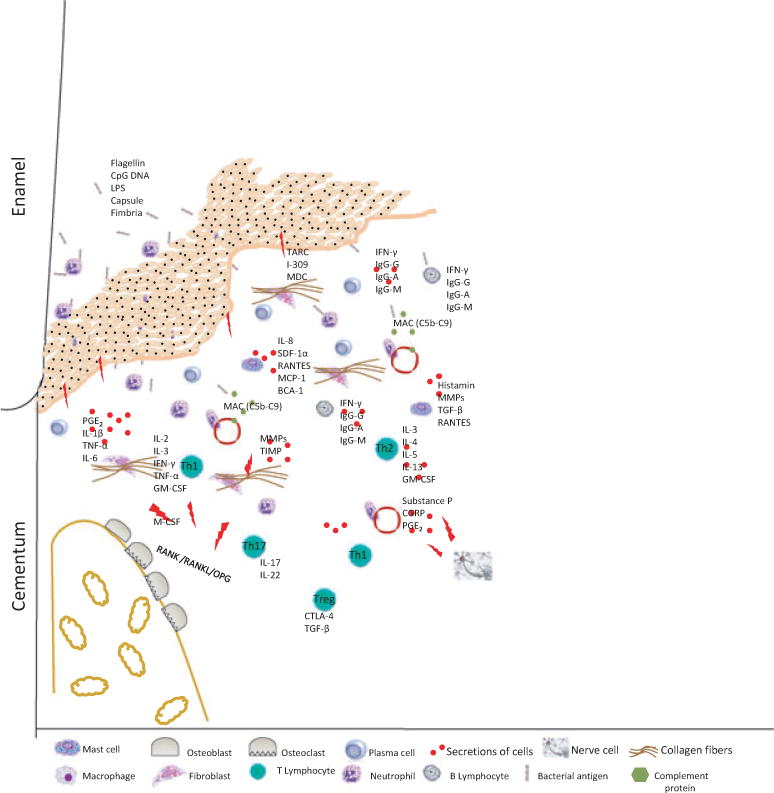
The immune inflammatory response in periodontitis is complex and involves both innate and acquired immunity. This diagram presents an overview of the effector molecules and effector cells in the pathogenesis of periodontitis based on our current understanding of disease pathways. BCA-1, B cell-attracting chemokine 1; CGRP, calcitonin gene-related peptide; CTLA-4, cytotoxic T-lymphocyte-associated antigen 4; GM-CSF, granulocyte– macrophage colony-stimulating factor; IFN-γ, interferon gamma; Ig-A, immunglobulin A; Ig-G, immunglobulin G; Ig-M, immunglobulin M; IL-1β, interleukin-1beta; IL-2, interleukin-2; IL-3, interleukin-3; IL-4, interleukin-4; IL-5, interleukin-5; IL-6, interleukin-6; IL-8, interleukin-8; IL- 13, interleukin-13; IL-17, interleukin-17; IL-22, interleukin-22; LPS, lipopolysaccharide; M-CSF, macrophage colony-stimulating factor; MAC, membrane attack complex; MCP-1, macrophage chemotactic protein-1; MDC, macrophage-derived chemokine; MMPs, matrix metalloproteinases; OPG, osteoprotegerin; PGE2, prostaglandin E2; RANTES, regulated and normal T cell expressed and secreted; SDF-1a, stromal cell-derived factor-1alpha; TARC, thymus and activation-regulated chemokine; TGF-β, transforming growth factor beta; Th1, T-helper 1 cell; Th2, T-helper 2 cell; Th17, T-helper 17 cell; TIMP, tissue inhibitor of matrix metalloproteinases; TNF-α, tumor necrosis factor alpha; Treg T-regulatory cell.
Periodontal diseases: what do we know?
There are two common diseases affecting the periodontium. The first is gingivitis, which is defined as inflammation of the gingiva in which the connective tissue attachment to the tooth remains at its original level. The disease is limited to the soft-tissue compartment of the gingival epithelium and connective tissue (12). The second is periodontitis, which is an inflammation of the supporting tissues of the teeth with progressive attachment loss and bone destruction (55). Both diseases and their symptoms are very common in populations worldwide. In the USA, adolescents have gingivitis and signs of gingival bleeding, whereas 54% of the adult population in the USA exhibits gingival bleeding (99). Thirty-seven per cent of the adult population in the USA suffers from severe periodontitis (160). In both cases, the disease is associated with the accumulation of bacteria at the dento–gingival margin, while the causal relationship of specific organisms is not fully clear. The host responds to microbial challenge by generating an inflammatory cell infiltrate in the tissue subjacent to the periodontal pocket (186).
The initial inflammation in the periodontal tissues should be considered a physiologic defense mechanism against the microbial challenge, rather than pathology. The clinical findings of the disease at this stage include supragingival and subgingival plaque formation, which are usually accompanied by calculus formation and gingival inflammation (154). If plaque is removed, there is resolution with return to homeostasis; if the lesion persists, it becomes pathology. For convenience, we will use the well-known stages of gingivitis and periodontitis, described by Page & Schroeder, in 1976 (186), for descriptive purposes: the initial lesion; the early lesion; the established lesion; and the advanced lesion. The advanced lesion is also called the destructive phase, because it represents the transition from gingivitis to periodontitis. What makes inflammation and immunologic events underlying periodontal diseases confusing is that the immunologic events overlap in different phases of disease. It should be emphasized that division of the immune response into various systems, such as innate immunity and adaptive immunity, is a rather arbitrary distinction imposed by immunologists (12). Although it is easier to describe the inflammation in compartments, the mechanisms involved in inflammation, resolution and healing include all components of the immune system that interact cooperatively to protect the periodontium (266). It is important to bear in mind that as the lesion progresses, the preceding pathways still function.
The initial lesion is the response of resident leukocytes and endothelial cells to the bacterial biofilm. At this stage, there are no signs of clinical inflammation, but the changes in the tissues can be observed histologically. The metabolic products of bacteria trigger junctional epithelium cells to produce cytokines and stimulate neutrons to produce neuropeptides, which cause vasodilatation of local blood vessels. Neutrophils leave the vessel and migrate toward the site of inflammation in response to chemokines. The early lesion follows, with increased numbers of neutrophils in the connective tissue and the appearance of macrophages, lymphocytes, plasma cells and mast cells. Complement proteins are activated. The epithelium proliferates to form rete pegs, observed histologically, and clinical signs of gingival inflammation, such as bleeding, can be seen. Gingival crevice fluid flow is increased.
The following stage is the established lesion. This can be considered as the period of transition from the innate immune response to the acquired immune response. Macrophages, plasma cells, and T and B lymphocytes are dominant, with IgG1 and IgG3 subclasses of B lymphocytes also present. Blood flow is impaired, and the collagenolytic activity is increased. There is also increased collagen production by fibroblasts. Clinically, this stage is a moderate to severe gingivitis with gingival bleeding and color and contour changes. The final stage is the transition to periodontitis: the advanced lesion. Irreversible attachment loss and bone loss are observed histologically and clinically. The inflammatory lesion extends deeper, affecting the alveolar bone (51).
Cells and mediators of periodontal inflammation
The innate immune system includes cells of nonhematopoietic origin, especially epithelial cells; myeloid cells of hematopoietic origin (phagocytes); and the innate humoral defense, the complement cascade (266). Neuropeptides contribute to this initial, immediate response to microbial challenge (236). The initial response, acute inflammation, is the physiologic response to the microbial challenge to recruit adequate cells to sites of infection through the production of cytokines and chemokines (Fig. 1). If the infection fails to clear, the chronic lesion is initiated with transition to the early lesion. Still an innate immune response pathways are stimulated that will activate the adaptive immune response. Innate immunity was formerly thought to be nonspecific, characterized by the phagocytosis and digestion of microorganisms and foreign substances by macrophages and neutrophils (97, 166). Phagocytes such as macrophages and neutrophils have surface receptors that recognize and bind surface molecules of bacteria (266). These pattern recognition receptors, including the toll-like receptors, distinguish between the host and the bacteria (197). After recognition of microorganisms and foreign substances, chemokines are secreted to attract phagocytes. The complement system also generates biologically active proteins, including the anaphylotoxins C3a, C4a and C5a that attract the different host immune cells monocytes, lymphocytes and neutrophils, respectively. Complement proteins can also directly kill certain bacteria. Histamine-induced vasodilatation by mast cells increases blood flow and recruitment of phagocytes.
Complement
The complement cascade can be activated through three pathways: the classical pathway; the lectin pathway; and the alternative pathway.
The classical pathway is activated by immunoglobulin – IgG or IgM – which binds the first complement protein, C1q, to a domain on its Fc tail. Bound C1q binds other C1 proteins to form a complex, C1qrs, which initiates a series of enzymatic reactions, cleaving C4 to C4a and C4b, and C2 to C2a and C2b. C4b and C2a become part of the C1 complex, forming a C3 convertase that cleaves C3 to C3a and C3b. C3b binds to the bacterial surface and, with several accessory proteins, forms a new enzyme to cleave C5 to C5a and C5b. C5b interacts with the terminal complement proteins, C6 to C9, to form the membrane attack complex that inserts C8 and C9 into the bacterial membrane, forming pores to disrupt the membrane.
The lectin pathway employs a mannose-binding lectin to bind carbohydrate on the bacterial cell-surface to form mannose-associated serine protease-2. This molecule has the capacity to interact with the complement proteins C4 and C2 to convert C3 to C3a and C3b, as in the classical pathway.
The alternative pathway is activated by bacterial polysaccharides, such as zymosan, lipopolysaccharide or aggregated IgA, through factor P (properdin) to cleave C3. C3b, and factors B and D convert C5 into C5a and C5b and the cascade continues to completion.
Both gingivitis and chronic periodontitis have been characterized primarily as activators of the alternative pathway. This is of some interest because it suggests that even though pathogen-specific antibodies are formed in chronic periodontitis, most of the complement activation in this disease is still via the alternative pathway (256). It is also known that other than these very well-studied pathways, there are proteins that can interact directly with C3 and C5: plasmin can cleave C3 into C3a and C3b; and thrombin has the ability to cleave C5.
Neuropeptides
During the innate immune response to periodontal pathogens, another element of the human defense system is also activated. Neurons generate electric impulses in response to chemical or mechanical stimuli, conduct the impulse and translate the electrical activity into a chemical signal. Alternatively, peptide neurotransmitters – neuropeptides – can be secreted into the extracellular fluid, where they act locally through receptors on other neurons or immune cells. Most neuropeptides act on nonneuronal targets (90), such as receptors for substance P and calcitonin gene-related peptide, found on immune cells, suggesting that the paracrine action of neuropeptides has important immunomodulatory roles (80, 150, 165) (Fig. 1). Recently, the nervous system has been identified as a critical regulator of inflammation in periodontal diseases (236). Furthermore, under pathological conditions some neuropeptides are synthesized and released from inflammatory cells (168). Therefore, the identification of neuropeptide receptors on immune cells suggests that communication exists between the immune and neurological systems, which may result in modulation of the inflammatory response (80, 165). Neuropeptides signal nonneuronal cells through G protein-coupled receptors located on the cell membrane (150). Vanilloid receptor-1, a neuropeptide receptor, is upregulated in inflammatory bowel disease, suggesting a possible role of this receptor in chronic inflammation (262).
The contribution of the nervous system to inflammation is not limited to vasodilatation and immune-cell recruitment. Cytokines and other products of immune cells can modulate the action, differentiation and survival of nerve cells, while neuropeptides released from neurons play pivotal roles in influencing the immune response. The interaction relies on the receptor-sensitizing characteristics of the cytokines. When a cytokine engages a neuron receptor, it initiates the release of neuropeptides (184). The discovery of protease-activated receptors revealed that protease-activated receptor 2 has an especially important role in chronic inflammation (246). Protease-activated receptor 2 is co-expressed with substance P and calcitonin gene-related peptide on sensory nerves, where it is believed to mediate inflammation (53, 222) (Fig. 1). In addition, cytokines have been shown to regulate the expression of substance P and to facilitate the lipopolysaccharide-induced release of calcitonin gene-related peptide in sympathetic neurons (92, 113). Also, calcitonin gene-related peptide enhances interleukin-1-induced accumulation of neutrophils and induces T-cell cytokine secretion (26, 137). Neural growth factor has been shown to directly regulate the synthesis of calcitonin gene-related peptide by B-cells as it does in sensory neurons (22). In the course of inflammation, kininogens are degraded to form kinins, including bradykinin, which together are important mediators of inflammation. Kinins are known to be proinflammatory, leading to vasodilatation, plasma extravasation and the release of other inflammatory mediators, notably the neuropeptides substance P and calcitonin gene-related peptide (67).
With the identification of neuropeptides in gingival crevice fluid, it is becoming increasingly evident that periodontitis and other orofacial inflammatory disorders may be modulated by imbalances in certain neuropeptides (78, 141, 142, 151, 152). In addition to the presence of neuropeptides in gingival crevice fluid, fibers innervating the periodontal tissues in humans have been shown to be immunoreactive to a number of neuropeptides, including substance P, calcitonin gene-related peptide, vasoactive intestinal peptide and neuropeptide Y (153).
The major function of neuropeptides in inflammation is vasodilatation, vasoconstriction and the recruitment and regulation of immune cells (6, 28, 126). Three major neuropeptides have modulatory effects in periodontal inflammation: substance P, calcitonin gene-related peptide and vasoactive intestinal peptide.
Substance P
Substance P is a member of the tachykinin family of neuropeptides, also known as the neurokinins (150). Substance P evokes a rapid response upon release and causes increased microvascular permeability, edema formation and subsequent plasma protein extravasation. Vasodilatation caused by substance P occurs indirectly by stimulating histamine release from mast cells (29, 150). Accordingly, edema induced by substance P is primarily caused by increased vascular permeability mediated through its action on neurokinin 1 receptors on endothelial cells (136). Several studies have shown the presence of substance P in human gingival tissues and in gingival crevice fluid (11, 151). The levels of substance P in the gingival crevice fluid are reduced after periodontal treatment, supporting the view of a local source of tachykinins in the gingival tissues (151).
The actions of substance P on immune cells are also important. Substance P limits the production of transforming growth factor beta by macrophages (161) and induces the synthesis of interleukin-6 by monocytes (140). Interestingly, substance P is also synthesized by immune cells. Sources of substance P, in addition to neurons, include monocytes, dendritic cells, eosinophils, T-lymphocytes and mast cells (130, 150). Mononuclear phagocytes and dendritic cells produce substance P when activated with lipopolysaccharide in vitro (131).
Calcitonin gene-related peptide
Calcitonin gene-related peptide has potent vasodilator activity and is frequently co-localized with substance P, which has been shown to regulate the vasodilator activity of calcitonin gene-related peptide (23, 38). In addition to its known vasodilator activity, calcitonin gene-related peptide has immunosuppressive actions that down-regulate the inflammatory response (232), such as suppressing interleukin-2 production and the proliferation of murine T-cells (252). It inhibits hydrogen peroxide production by macrophages in response to interferon gamma and presenting antigen (177, 185). Calcitonin gene-related peptide also impacts bone metabolism, thus inhibiting osteoclastic bone resorption and stimulating osteogenesis (123).
Vasoactive intestinal peptide
Vasoactive intestinal peptide is an important immune-modulatory peptide that is capable of regulating the production of both proinflammatory and anti-inflammatory mediators (15, 57, 193). The major nonneuronal sources of vasoactive intestinal peptide are neutrophils and mast cells (36, 178). While vasoactive intestinal peptide inhibits lipopolysaccharide-induced production of tumor necrosis factor alpha, interleukin-6 and interleukin-12 in activated macrophages, it stimulates the production of the potent anti-inflammatory cytokine, interleukin-10, and suppresses T-cell proliferation (57). The levels of vasoactive intestinal peptide are significantly elevated in periodontitis sites compared with clinically healthy sites, and nonsurgical periodontal treatment results in a clinical improvement along with a concomitant reduction in the levels of vasoactive intestinal peptide (142).
Toll-like receptors
In the oral epithelium, complementary defense mechanisms are present. Epithelial cells have tight intercellular junctions that impede the entry of bacteria and their metabolites. Lipopolysaccharide is a cell-wall component of all gram-negative microorganisms (197). Once exposed to lipopolysaccharide, a series of complex mechanisms are triggered, which lead to extracellular matrix degradation and the initiation of osteoclastogenesis. The main function of dendritic cells is presentation of antigen to other cells of the immune system. Recognition of innate immune signals by dendritic cells relies on a limited number of pathogen-related receptors. These include toll-like receptors and related proteins that regulate apoptosis, inflammation and immune responses (3, 95) (Fig. 1).
The toll-like receptors family of proteins is the best-characterized class of pattern-recognition receptors. Dendritic cells express toll-like receptors, and different dendritic-cell subsets express distinct toll-like receptors that are associated with particular functions in innate responses and the generation of distinct T-cell subsets (98, 103). Toll-like receptors are also expressed on lymphocytes and osteoclast precursors, as well as on macrophages, osteoblasts and stromal and epithelial cells, each of which has different toll-like-receptor expression profiles (83, 96, 107, 228). Toll-like receptors are unique receptors that recognize molecules that are broadly shared by microorganisms, but are distinguishable from host molecules; these are collectively referred to as ‘pathogen-associated molecular patterns’. Toll-like receptors detect multiple pathogen-associated molecular patterns, including lipopolysaccharide, bacterial lipoproteins and lipoteichoic acids, flagellin, CpG DNA of bacteria and viruses, double-stranded RNA and single-stranded viral RNA (96). To date, 11 different toll-like-receptor molecules have been identified in human periodontal tissues, and their expression, distribution and ligand specificities have been characterized (125, 145, 195, 228) (Table 1).
Table 1.
The source cell, location and associated bacteria for toll-like receptors.
| Receptor | Location | Cells | Bacteria |
|---|---|---|---|
| Toll-like receptor 1 | Cell membrane | Myeloid dendritic cells, monocytes | Not specified |
| Toll-like receptor 2 | Cell membrane | Monocytes, natural killer cells, myeloid dendritic cells,mast cells, T-cells, epithelial cells |
Porphyromonas gingivalis Escherichia coli Tannerella forsythia Prevotella intermedia Prevotella nigrescens Treponema denticola |
| Toll-like receptor 3 | Intracellular | Myeloid dendritic cells, natural killer cells, epithelial cells | Not specified |
| Toll-like receptor 4 | Cell membrane | Monocytes, mast cells, neutrophils, T cells, epithelial cells, endothelial cells |
Aggregatibacter actinomycetemcomitans, Veillonella parvula |
| Toll-like receptor 5 | Cell membrane | Monocytes, natural killer cells, myeloid dendritic cells epithelial cells | Not specified |
| Toll-like receptor 6 | Cell membrane | Myeloid cells, mast cells, B-cells, myeloid dendritic cells | Escherichia coli |
| Toll-like receptor 7 | Intracellular | Plasmacytoid dendritic cells, B-cells, eosinophils | Not specified |
| Toll-like receptor 8 | Intracellular | Natural killer cells, T-cells, myeloid cells, myeloid dendritic cells | Not specified |
| Toll-like receptor 9 | Intracellular | Plasmacytoid dendritic cells, B-cells, natural killer cells |
Porphyromonas gingivalis Aggregatibacter actinomycetemcomitans |
| Toll-like receptor 10 | Cell membrane | B-cells, plasmacytoid dendritic cells, myeloid dendritic cells | Not specified |
| Toll-like receptor 11 | Intracellular | Macrophages, dendritic cells, epithelial cells | Not specified |
When toll-like receptors bind pathogen-associated molecular patterns, a series of intracellular events are initiated, leading to the production of cytokines, chemokines and antimicrobial peptides (104). The toll-like-receptor domain can bind four different adapter proteins and has the potential to induce various cytokines through nuclear factor of kappa light polypeptide gene enhancer in B-cells pathways in the nucleus of the cell. Known adapter proteins of toll-like receptors are myeloid differentiation primary response protein (Myd88), toll / interleukin-1 receptor domain-containing adapter protein, toll / interleukin-1 receptor domain containing adapter-inducing interferon beta and toll / interleukin-1 receptor domain containing adapter-inducing interferon beta-related adapter molecule. Different toll-like receptors induce different responses. For example, in dendritic cells, the interaction of toll-like receptor 4 and lipopolysaccharide results in the production of proinflammatory cytokines such as interleukin-12, and the interaction of toll-like receptor 3 with lipopolysaccharide results in the production of type-I interferon. It is known that toll-like receptor 2 and toll-like receptor 4 predominate in periodontal tissues (82). Interestingly, the same toll-like receptor can trigger different responses depending upon the intracellular adapter protein. For example, when lipopolysaccharide binds to toll-like receptor 4 and uses myeloid differentiation primary response protein (MyD88) and toll / interleukin-1 receptor domain-containing adapter protein as adapters, the result is the production of tumor necrosis factor alpha, interleukin-6 and interleukin-12. If the adapters toll / interleukin-1 receptor domain-containing adapter-inducing interferon beta-related adapter molecule and toll / interleukin-1 receptor domain-containing adapter-inducing interferon beta with the same toll-like receptor type are used, release of interferon alpha / beta and activation of interferon regulatory factor 3 follows (1, 35, 107, 125, 253).
Toll-like receptors 1, 2, 4, 5 and 6 recognize mainly products that are unique to bacteria and not made by the host. This gives them the specificity to differentiate microorganisms from the host (96). Recognition by toll-like receptor pathways is a crucial phase in inflammation. For a complete review of toll-like receptors and their pathways, see Uehara & Takada (239).
Antigen presentation and activation of acquired immunity
If the early lesion persists without resolution, bacterial antigens are processed and presented by lymphocytes, macrophages and dendritic cells. Broadly, two different subsets of lymphocytes have evolved to recognize extracellular and intracellular pathogens after being presented with antigens by the innate immune cells: T-lymphocytes and B-lymphocytes. B-lymphocytes bear immunoglobulin molecules on their surface, which function as antigen receptors. Antibody, which is a soluble form of immunoglobulin, is secreted following activation of B-cells to bind pathogens and foreign material in the extracellular spaces (humoral immunity). T-cells are the effectors of cell-mediated immunity (delayed hypersensitivity). The T-cell antigen receptor is a membrane-bound molecule, similar to immunoglobulin, which recognizes peptide fragments of pathogens. Activation of the T-cell receptor requires the major histocompatibility complex, which is also a member of the immunoglobulin superfamily. Two classes of major histocompatibility complex molecules are required for the activation of distinct subsets of T-cells. Various T-cell subsets kill infected target cells and activate macrophages, B-cells and other T-cells. Thus, T-cells are essential for the regulation of both humoral and cell-mediated responses.
Classically, T-lymphocytes have been classified into subsets based on the cell-surface expression of CD4 or CD8 molecules. CD4+ T-cells (T-helper cells) were initially subdivided into two subsets, designated T-helper 1 and T-helper 2, on the basis of their pattern of cytokine production (172). T-helper 1 cells secrete interleukin-2 and interferon gamma, whereas T-helper 2 cells produce interleukins 4, 5, 6, 10 and 13. Both cell types produce interleukin-3, tumor necrosis factor alpha and granulocyte–macrophage colony-stimulating factor (112, 266). The major role of the T-helper 1 cytokines interleukin-2 and interferon gamma is to enhance cell-mediated responses, whereas the T-helper 2 signature cytokine, interleukin-4, suppresses cell-mediated responses (170). T-cell subsets are also important in the behavior of B-cells. For example, T-helper 1 cells direct B-cell secretion of IgG2, whereas T-helper 2 cells up-regulate IgG1 secretion. CD8 T-cells (cytotoxic T-cells) are immune effector cells that also secrete cytokines which are characteristic of either T-helper 1 or T-helper 2 cells (266).
More recent studies have described two new well-defined CD4 T-cell subsets, T-helper 17 and T-regulatory T-cells, which play antagonistic roles as effector and suppressor cells, respectively (4, 207, 254). T-helper 17 cells are named for their unique production of interleukin-17. T-helper 17 cells also produce interleukin-22. T-helper 17 lymphocytes, like T-helper 1 cells, are also noted for their stimulatory role in osteoclastogenesis (258). T-helper 17 cells are observed in chronic periodontitis sites, and T-helper 17-related cytokines are produced in periodontal lesions (180, 226, 247).
T-regulatory cells have a protective role in periodontal tissue damage. Natural T-regulatory cells are CD4- and CD25-expressing T-cells that specifically regulate the activation, proliferation and effector functions of activated conventional T-cells (4, 14, 207). T-regulatory cells are found in periodontal disease sites (30, 175). The cytokines produced by T-regulatory cells are transforming growth factor beta and T-lymphocyte-associated molecule 4 (cytotoxic T-lymphocyte antigen 4), which down-regulate inflammation. Interleukin-10, transforming growth factor beta and cytotoxic T-lymphocyte-associated antigen 4 are reported to decrease periodontal disease progression (30).
New data suggest the existence of an antigen-presenting cell type from the follicular Th-cell lineage that produces interleukin-21 (118). This type of antigen-presenting cell is characterized by the expression of the chemokine receptor, chemokine (C-X-C motif) receptor 5 (24, 50, 211).
The other important antigen-presenting cell is the macrophage. Macrophages, which are phagocytic cells from the myeloid lineage, efficiently ingest particulate antigen and express the major histocompatibility complex class II molecules to induce costimulatory activity on T-cells. Macrophages are widely distributed cells that play an indispensible role in homeostasis and defense. Macrophages can be phenotypically polarized by the microenvironment. The classic inflammatory macrophage (M1) is activated by interferon gamma and lipopolysaccharide. Alternatively activated macrophages (M2) are important cells in the resolution of inflammation; they have reduced capacity to produce proinflammatory cytokines (18).
The transition from the established lesion, dominated by T- and B-cells, to the advanced lesion (progressive periodontitis) is not well understood. We know that dendritic cells also express the major histocompatibility complex class II molecules and have costimulatory activity. The unique ability of B-cells to bind and internalize antigens via their immunoglobulin receptors may be important in activating T-lymphocytes, pointing out that costimulatory molecules are present on B-cells. Costimulation can be thought of as the mechanism by which antigen-presenting cells inform T-cells that the antigen requires a proliferative response preventing T-cell apoptosis or anergy (266). It has become clear that CD4 T-cells and certain innate immune cells, such as dendritic cells, monocytes and neutrophils, are in perfect communication through cytokine networks (7, 66, 108, 220, 231).
It is evident that innate and adaptive systems are coordinately involved in the inflammatory response and tissue destruction, although we lack a complete understanding of the mechanism. In the case of periodontal disease, where all elements of the immune system are involved, inflammatory mechanisms and signals are dysregulated. For instance, T-cells extracted from diseased periodontal tissues exhibit a reduced response to stimuli, which suggests that the cell-mediated response is suppressed in patients with periodontal disease (34); following periodontal therapy, lymphocyte reactivity has been reported to return to normal (225).
Activation of B-cells is an important step in the maturation of the antibody response. This event is mediated mainly by the tumor necrosis factor family of proteins and their receptors. In addition to their role in presenting antigen, B-cells also function as effectors, through cytokine secretion, lysosomal components, reduced oxygen metabolites, nitric oxide and antibodies. This is also important because in severe periodontal lesions, B-cells are the predominant antigen-presenting cells, suggesting that B-cell antigen presentation may allow further activation and clonal expansion of already activated T-cells (63, 266).
The role of antibody and cell-mediated immunity in the pathogenesis of periodontal diseases is beyond the scope of this review. For a recent detailed review of acquired immune mechanisms in periodontitis, see Berglundh & Donati (17). The role of cytokines and other mediators from cells of the acquired immune network is discussed in subsequent sections of this review.
Cytokines and chemokine networks
Cytokines and chemokines (chemotactic cytokines) are the messages between cells. The immune response to infection is regulated by cytokine and chemokine signals. Cytokines are low-molecular-weight proteins involved in the initiation and further stages of inflammation, in which they regulate the amplitude and the duration of the response. The genetic regulation leading to the secretion of proinflammatory cytokines from a variety of cells is generally dependent on the activation of nuclear factor kappa-B transcription (8, 75). The nuclear factor kappa-B regulated pathways are activated by pathogen-associated molecular patterns, such as lipopolysaccharide, through the toll-like receptor pathway (75).
Cytokines are produced by resident cells, such as epithelial cells and fibroblasts, and by phagocytes (neutrophils and macrophages) in the acute and early chronic phases of inflammation, and by immune cells (lymphocytes) in established and advanced lesions (5). After recognition and presentation of microbes to the appropriate cells, cytokines of the innate response, including tumor necrosis factor alpha, interleukin-1beta and interleukin-6, are the first to appear in the periodontal disease pathogenesis pathways (58). Interleukin-1beta and interleukin-6 are signature innate cytokines and have been characteristically associated with inflammatory cell migration and osteoclastogenesis (56, 73). Tumor necrosis factor alpha is a multi-effect cytokine that has many functions, from cell migration to tissue destruction. Tumor necrosis factor alpha impacts cell migration by inducing the up-regulation of adhesion molecules to promote rolling and adhesion of neutrophils to the vessel wall, leading to extravasation. It also stimulates the production of chemokines involved in cell migration to infected and inflamed sites (42, 117, 190, 251). Tumor necrosis factor alpha upregulates the production of interleukin-1beta and interleukin-6 (42, 59, 73, 128, 173, 182, 251). Tumor necrosis factor alpha is also correlated with extracellular matrix degradation and bone resorption through actions promoting the secretion of matrix metalloproteinases and RANKL (62, 72, 73) and coupled bone formation (13). Accordingly, experimental periodontitis in tumor necrosis factor alpha p55 receptor-deficient mice was characterized by a significant decrease in matrix metalloproteinase and RANKL expression and resistance to periodontitis (59).
Chemokines are chemotactic cytokines that play a very important role in the migration of phagocytic cells to the site of infection. Once blood leukocytes exit a vessel, they are attracted, by functional gradients of chemotactic factors, to the site of infection (200, 267). Chemokines are synthesized by a variety of cells including endothelial, epithelial and stromal cells, as well as leukocytes. Functionally, chemokines can be grouped as homeostatic or inflammatory (171). In addition to their cell-trafficking role, chemokines provide messages leading to other biological processes, such as angiogenesis, cell proliferation, apoptosis, tumor metastasis and host defense (48, 171, 200, 201, 267). Bacterial peptides are also chemotactic for inflammatory cells, but the discussion herein will focus upon host-derived chemokines.
Chemokines are small, heparin-binding proteins ranging from 7 to 15 kDa. They are classified into four subfamilies according to the configuration of cysteine residues near the N-terminus. The nomenclature is as follows: chemokines are designated CXC and CX3C if two cysteines are separated and as CC and C if they are not. Their receptors are named by adding ‘R’ to the end of the particular chemokine, for example, ‘CXCR’ or ‘CCR’. Detailed information on the classification codes, the designated names of the chemokines, their receptors and the target cells they are affecting is presented in Table 2 (21, 37, 64, 74, 101, 110, 146, 147, 200, 208, 209, 249, 264, 267). Binding of a chemokine to its respective receptor initiates the cell-migration process, beginning with integrin-dependent adhesion and diapedesis. Chemokines target leukocytes of the innate immune system, as well as lymphocytes of the adaptive immune system (233).
Table 2.
Chemokine classification codes, designated names and affected cell types.
| Receptor (classification code) | Cell type affected | Ligand (classification code) | Ligand (designated name) |
|---|---|---|---|
| CCR1 | Monocytes/macrophages | CCL3 | MIP-1α |
| Neutrophils | CXCL8 CXCL6 CXCL1 CCL23 |
IL-8 GCP-2 GROα CKβ8 |
|
| Osteoclast precursors and mature osteoclasts | CCL5 CCL7 CCL9 |
RANTES MCP-3 MIP-1γ |
|
| CCR2 | Monocytes/macrophages and osteoclast precursors | CCL2 | MCP-1 |
| CCR3 | Th1 lymphocytes | CXCL9 | MIG |
| Th2 lymphocytes and osteoblasts | CXCL10 CXCL11 CCL7 CCL11 CCL13 CCL15 |
IP-10 I-TAC MCP-3 Eotaxin MCP-4 HCC-2 |
|
| CCR4 | Th2 lymphocytes and osteoblasts | CCL22 CCL17 CXCL12 |
MDC TARC SDF-1 |
| CCR5 | Monocytes/macrophages, Th1 lymphocytes and osteoblasts | CCL5 | RANTES |
| B lymphocytes | CXCL13 | BCA-1 | |
| CCR8 | Th2 lymphocytes | CCL1 | I-309 |
| CXCR1 | Osteoclast precursors and osteoblasts | CXCL8 | IL-8 |
| CXCR3 | Osteoclast precursors and Osteoblasts | CXCL9 | MIG |
| CXCR4 | Osteoclast precursors, mature osteoclasts and osteoblasts | CXCL12 | SDF-1 |
| CXCR5 | Osteoblasts | CCL5 CXCL13 |
RANTES BCA-1 |
BCA-1, B cell-attracting chemokine 1; CCL, chemokine (C-C motif) ligand; CCR, chemokine (C-C motif) receptor; CKb8, transcript variant CKb8; CXCL, chemokine (C-X-C motif) ligand; CXCR, chemokine (C-X-C motif) receptor; GCP-2, granulocyte chemotactic protein 2; GROa, melanoma growth stimulatory factor; HCC-2, hemofiltrate CC-chemokine-2; IL-8, interleukin-8; I-TAC, interferon-inducible T-cell chemoattractant; MCP-3, macrophage chemotactic protein-3; MCP-4, macrophage chemotactic protein-4; MDC, macrophage-derived chemokine; MIG, monokine induced by gamma interferon; MIP-1a, macrophage inflammatory protein 1alpha; MIP-1c, macrophage inflammatory protein 1gamma; RANTES, regulated and normal T cell expressed and secreted; SDF-1, stromal cell-derived factor-1; TARC, thymus and activation-regulated chemokine; Th1, T-helper 1; Th2, T-helper 2.
The first cytokine identified to have chemotactic activity was interleukin-8 / chemokine (C-X-C motif) ligand 8. In the periodontium, this cytokine is produced primarily by gingival fibroblasts, gingival epithelial cells and endothelial cells (227, 229, 265). Interleukin-8 is a polymorphonuclear leukocyte chemoattractant. It is detectable in healthy and diseased periodontal tissues and has been associated with subclinical inflammation of the initial lesion, which is comprised of polymorphonuclear neutrophils (163, 188, 263). Interleukin-8 / chemokine (C-X-C motif) ligand also has an important role in bone metabolism. It has direct actions on osteoclast differentiation and activity by signaling through the specific receptor, chemokine (C-X-C motif) receptor 1 (16).
Another crucial chemokine of innate immunity is macrophage chemotactic protein-1 / chemokine (C-C motif) ligand 2. Macrophage chemotactic protein-1 mediates the recruitment of monocytes / macrophages, the second wave of the innate response to bacteria (76, 183). Macrophage inflammatory protein 1 alpha / chemokine (C-C motif) ligand 3 is the most abundantly expressed chemokine in periodontitis tissues, with its expression localized in the connective tissue subjacent to the pocket epithelium of inflamed gingival tissues. Together with regulated and normal T cell expressed and secreted / chemokine (C-C motif) ligand 5, macrophage inflammatory protein 1 alpha / chemokine (C-C motif) ligand 3 may also be involved in the migration of macrophages to periodontal tissues (64, 102). Chemokine (C-X-C motif) receptor 3 and its ligand, interferon gamma-induced protein 10 / chemokine (C-X-C motif) ligand 10, are also expressed in diseased periodontal tissues, (61, 102) and are associated with higher levels of interferon gamma in inflammation. Chemokine (C-C motif) receptor 4 is expressed at higher levels in chronic periodontitis and it is associated with higher levels of interleukin-4 and interleukin-10 in the periodontium (61, 62).
It has become increasingly clear that chemokines are multipurpose ligands in mediating repair and angiogenesis. The time and the place of secretion are of utmost importance. In a study by DiPietro et al., macrophage chemotactic protein-1 / chemokine (C-C motif) ligand 2-deficient mice demonstrated delayed wound re-epithelialization (43, 149). Chemokines are equally crucial in guiding adaptive immunity and also play a critical role in bone metabolism. For instance, chemokines such as macrophage-derived chemokine / chemokine (C-C motif) ligand 22, thymus and activation-regulated chemokine / chemokine (C-C motif) ligand 17 and I-309 / chemokine (C-C motif) ligand 1 attract T-helper 2 and T-regulatory cells binding to chemokine (C-X-C motif) receptor 4 and chemokine (C-X-C motif) receptor 8, respectively (37, 74, 208). It has been suggested that the expression of T-helper 2 and T-regulatory cell chemoattractants, such as macrophage-derived chemokine / chemokine (C-C motif) ligand 22, thymus and activation-regulated chemokine / chemokine (C-C motif) ligand 17 and I-309 / chemokine (C-C motif) ligand 1, reduce periodontal disease severity (175). B cell-attracting chemokine 1 / chemokine (C-X-C motif) ligand 13, an important B-cell chemoattractant, is expressed in diseased tissues, suggesting a role for the accumulation of these cells in the periodontium. The expression of B cell-attracting chemokine 1 / chemokine (C-X-C motif) ligand 13 may be important in the local humoral response to periodontal pathogens (119).
Chemokines are involved in both the physiology and the pathology of bone metabolism. They are essential signals for the trafficking of osteoblast and osteoclast precursors, and consequently as potential modulators of bone homeostasis (16, 257). The chemokines implicated in regulating bone metabolism are identified through expression of receptors including chemokine (C-C motif) receptors 1 and 2, and chemokine (C-X-C motif) receptors 3 and 4; these receptors are expressed on osteoclast precursors, mature osteoclasts and osteoblasts. The potential chemokine ligands include stromal cell-derived factor-1 / chemokine (C-X-C motif) ligand 12, macrophage inflammatory protein 1 alpha / chemokine (C-C motif) ligand 3, regulated and normal T cell expressed and secreted / chemokine (C-C motif) ligand 5, macrophage inflammatory protein 1 gamma / chemokine (C-C motif) ligand 9, macrophage chemotactic protein-1 beta / chemokine (C-C motif) ligand 2, macrophage chemotactic protein-3 / chemokine (C-C motif) ligand 7, monokine induced by gamma interferon / chemokine (C-X-C motif) ligand 9 and transcript variant CKβ8 / chemokine (C-C motif) ligand 23 (115, 116, 127, 134, 183, 249, 257, 259, 264). Interferon gamma-induced protein 10 / chemokine (C-X-C motif) ligand 10 induces osteoblast proliferation through chemokine (C-C motif) receptor 3 (71, 143), while stromal cell-derived factor-1 alpha / chemokine (C-X-C motif) ligand 12 and B cell-attracting chemokine 1 / chemokine (C-X-C motif) ligand 13 induce both proliferation and collagen type I mRNA expression in osteoblasts through chemokine (C-C motif) receptors 4 and 5 (144). In addition to a role in osteoclastogenesis, chemokines also impact osteoclast functions. Stromal cell-derived factor-1 alpha / chemokine (C-X-C motif) ligand 12 increases the activity of matrix metalloproteinase 9 in human osteoclasts, resulting in increased bone resorption (70).
There is some evidence that regulated and normal T cell expressed and secreted / chemokine (C-C motif) ligand 5 acts on osteoblasts, resulting in chemotaxis and promoting cell survival (260). Interestingly, RANKL also induces the production of macrophage chemotactic protein-1 / chemokine (C-C motif) ligand 2, macrophage inflammatory protein 1 / chemokine (C-C motif) ligand 3, regulated and normal T cell expressed and secreted / chemokine (C-C motif) ligand 5 and monokine induced by gamma interferon / chemokine (C-X-C motif) ligand 9 by osteoclasts, suggesting a coupling contribution to bone resorption (115). Taken together, these studies suggest that chemokines can effectively contribute to bone remodeling by driving osteoblast migration and activation.
Interferon gamma is a signature cytokine of the adaptive immune response. Its main function is to promote antigen-presenting cell binding of antigen by up-regulating major histocompatibility complex class I and class II expression (199, 238). Interferon gamma also plays a major role in B-cell maturation, and, accordingly, in immunoglobulin secretion (179). In periodontal disease, interferon gamma is present at high levels in periodontal lesions, and is associated with progressive lesions or severe forms of periodontitis (46, 61, 88).
Interleukin-4 is another important adaptive-immunity cytokine that induces proliferation of T-cells and regulates B-cell immunoglobulin secretion. It is considered an anti-inflammatory cytokine. Interleukin-4 has known antitumor actions. Interleukin-4 inhibits the activity of proinflammatory cytokines, such as the interleukin-2-induced generation of natural killer cells and the activation of macrophages (179). Interleukin-4 can also block nitric oxide generation by macrophages (170). Studies also suggest that interleukin-4 down-regulates the production of other cytokines, including interleukin-1beta, tumor necrosis factor alpha and interleukin-6, by human peripheral blood monocytes and T-helper 1 cells (44, 49), inhibiting the transcription of these proinflammatory cytokines and interferon gamma. Additionally, interleukin-4 inhibits the production of matrix metalloproteinases and RANKL, and concomitantly induces the up-regulation of tissue inhibitor of metalloproteinases and osteoprotegerin (93, 204), reinforcing its potential protective role in periodontal diseases (68). Interleukin-4 also induces the production of interleukin-10, another anti-inflammatory cytokine (191). Interleukin-10 plays a major role in suppressing immune responses by inhibiting the antigen-presenting capacity of macrophages (40, 52). Interleukin-10 is a potent effector for activated human B-cells (202). It is widely expressed in inflamed periodontal tissues, where it is thought to limit disease severity (60, 62, 133). Interleukin-10 interferes directly with the production of interferon gamma and interleukin-17 by T-helper 17 cells (100, 176). Interleukin-10 plays a direct protective role in tissue destruction by down-regulating both matrix metalloproteinases and RANKL. Interleukin-10 characteristically induces the up-regulation of tissue inhibitor of metalloproteinases, which inhibit the matrix metalloproteinase family of proteins (31, 33, 62).
Interleukin-12 was originally described as a factor that promotes the activity of natural killer cells and CD8 T-cells (132). It has the capacity to enhance T-cell and natural killer cell proliferation after activation by other stimuli (121, 189). Natural killer cells appear to be most effective at preventing early infection, but T- and B-cells and their products are required to resolve the infection (9). Interleukin-4 and interleukin-10 are powerful inhibitors of the production of interleukin-12. It has been suggested that these two cytokines may determine the balance between T-helper 1 and T-helper 2 cells in the periodontal lesion (237).
Interleukin-13 is another potent modulator of human monocyte / macrophage and B-cell function. Monocyte / macrophage cell-surface major histocompatibility complex class II and several integrin molecules are up-regulated by interleukin-13 (39). The monocyte / macrophage-related production of interleukin-1alpha, interleukin-1beta, interleukin-6, interleukin-8 and tumor necrosis factor alpha is inhibited by interleukin-13, and interleukin-1 receptor antagonist secretion is enhanced (39, 269) suggesting an anti-inflammatory role along with interleukin-4 and interleukin-10 (269).
Transforming growth factor beta is a growth factor that regulates cell growth, differentiation and matrix production, and is also a potent immunosuppressive factor that down-regulates the transcription of proinflammatory factors (such as interleukin-1beta and tumor necrosis factor alpha) and matrix metalloproteinases (181, 223). In active periodontal lesions, the levels of transforming growth factor beta are negatively correlated with the levels of RANKL, reinforcing its protective role against tissue destruction (45, 46, 223).
Lipid mediators of inflammation
Prostaglandins are derived from the hydrolysis of membrane phospholipids. Phospholipase A2 cleaves the sn-2 position of membrane phospholipids to generate arachidonic acid, a precursor of a group of small lipids known as eicosanoids (139). Arachidonic acid is metabolized by two major enzyme pathways: (i) lipoxygenases, which catalyze the formation of hydroxyeicosatetraenoic acids, leading to the formation of leukotrienes; and (ii) cyclooxygenases 1 and 2, which catalyze the conversion of arachidonic acid into prostaglandins, prostacyclins and thromboxanes. Prostaglandins have 10 subclasses, of which D, E, F G, H and I are the most important (65). Inflamed gingiva synthesizes significantly larger amounts of prostaglandins when incubated with arachidonic acid than does healthy gingiva (167). Prostaglandin E2 is a potent stimulator of alveolar bone resorption (41, 69). Within gingival lesions, prostaglandin E2 is mainly localized to macrophage-like cells and is secreted when stimulated with bacterial lipopolysaccharide (148). Periodontal ligament cells also produce prostaglandin E2, even when unstimulated. This secretion is enhanced by interleukin-1beta, tumor necrosis factor alpha and parathyroid hormone (196, 205, 206). It is important to note that prostaglandin E2 has biphasic actions on immune function. In high doses, it decreases the levels of IgG, but at low doses it has the potential to increase IgG. When combined with interleukin-4, low doses of prostaglandin E2 induce a synergistic rise in IgG production, suggesting an immune-regulatory role for prostaglandin E2 (79).
Destruction of periodontal tissues
Destruction of bone
It is now generally accepted that disruption of the balance between osteoblast and osteoclast activities by bacterial products and inflammatory cytokines constitutes the main underlying causes of inflammation-induced bone loss (145). Lipopolysaccharide directly stimulates bone resorption when added to osteoclast precursor cultures containing osteoblasts and / or stromal cells (94). A toll-like receptor and inflammation-induced osteoclastogenesis pathway is implicated in the initiation of bone loss (187, 192). Inflammation-induced bone loss in response to periodontal infection has been well studied. Complex inflammatory signals and cytokine networks regulate osteoclastogenesis through RANKL, interleukin-1beta, interleukin-6, tumor necrosis factor alpha and prostaglandin E2 (84) (Fig. 1).
Before the discovery of RANK, its ligand (RANKL) and its antagonist (osteoprotegerin), the development and the formation of osteoclasts were thought to be controlled by factors produced by osteoblasts and bone marrow stromal cells (162, 198). It is now clear that RANKL and osteoprotegerin are the key regulators of bone remodeling and are directly involved in the differentiation, activation and survival of osteoclasts and osteoclast precursors (2, 129, 261). RANKL is expressed by osteoblasts, stromal cells, chondrocytes and other mesenchymal cells. In addition, activated T-cells and B-cells can also express RANKL (111, 158, 235). RANK is expressed by osteoclast progenitors, mature osteoclasts, chondrocytes, monocytes / macrophages and dendritic cells (2, 91). The decoy receptor, osteoprotegerin, is known to be expressed by periodontal tissue cells, including fibroblasts and periodontal ligament cells (145). Blocking RANKL activity with osteoprotegerin significantly inhibits bone loss in rheumatoid arthritis, osteoporosis, cancer-related bone metastasis and diabetes-associated alveolar bone destruction (25, 87, 89, 122, 158, 169), confirming the critical role of the RANKL / RANK / osteoprotegerin triad in osteoclastogenesis. However, it is not that simple. Macrophage colony-stimulating factor is also required, which is produced by osteoblasts / stromal cells (155, 230). The periodontal implication of the macrophage colony-stimulating factor requirement is that the pathogen, stress or pathology that influences the production of macrophage colony-stimulating factor via proinflammatory cytokines will have a significant influence on subsequent osteoclast activity. For example, toll-like receptor 2 activation in human gingival fibroblasts up-regulates the expression of macrophage colony-stimulating factor (27).
It is known that lipopolysaccharide from different pathogens stimulates bone resorption in vitro and in animal models, as in primary mouse calvarial osteoblasts. The activation of toll-like receptor 2 and toll-like receptor 6 by lipopolysaccharide causes enhanced expression of RANKL through a myeloid differentiation primary response protein (MyD88)-dependent mechanism (210). Also, osteoclasts and their precursors have been shown to express toll-like receptors, especially toll-like receptors 2, 4 and 9 (83, 86). Moreover, in mouse calvarial osteoblasts, expression of toll-like receptors 4 and 9 results in the activation of nuclear factor of kappa light polypeptide gene enhancer in B-cells and increased secretion of tumor necrosis factor alpha and macrophage colony-stimulating factor (268). Lipopolysaccharide-induced production of interleukin-1beta through toll-like-receptor pathways can up-regulate RANKL and can also inhibit the expression of osteoprotegerin by osteoblasts, resulting in osteoclast formation in a prostaglandin E2-dependent manner (224). The crucial role of toll-like receptor 2 is that it substantially decreases the responses to lipopolysaccharide (27). These findings point out that lipopolysaccharide, directly via toll-like receptor pathways, induces osteoclast development and activity. Thus, it is believed that toll-like receptors influence the inflammatory response in the bone microenvironment and may play a critical role in modulating inflammation-induced osteoclastogenesis and bone loss. It is also interesting that recent evidence also points to important roles for resident cells in periodontal bone loss because periodontal ligament fibroblasts and osteoclast precursors synergistically increase the expression of genes related to osteoclastogenesis (20).
Destruction of extracellular matrix
There is significant evidence showing that collagenases, along with other matrix metalloproteinases, play an important role in periodontal tissue destruction. Matrix metalloproteinases are a family of structurally related, but genetically distinct, enzymes that degrade extracellular matrix and basement membrane components. This group of 23 human enzymes is classified into collagenases, gelatinases, stromelysins, membrane-type matrix metalloproteinases and other matrix metalloproteinases, mainly based on the substrate specificity and the molecular structure. Matrix metalloproteinases are involved in physiological processes such as tissue development, remodeling and wound healing. Matrix metalloproteinase activity is controlled by changes in the delicate balance between the expression and synthesis of matrix metalloproteinases and their major endogenous inhibitors, tissue inhibitor of matrix metalloproteinases. It is clear that matrix metalloproteinases are up-regulated in periodontal inflammation (241). Matrix metalloproteinase activation involves tissue and plasma proteinases and bacterial proteinases, together with oxidative stress (174, 242).
The expression and pathologic release of matrix metalloproteinases was originally thought to be limited to neutrophils (241), but it is now clear that a broad range of cell types present in normal and diseased human periodontium (including gingival epithelial cells, fibroblasts, endothelial cells, monocytes / macrophages and plasma cells) express distinct matrix metalloproteinases (77, 114, 234, 250).
Transcription of matrix metalloproteinase genes is very low in healthy periodontal tissue. In periodontal disease, secretion of specific matrix metalloproteinases is stimulated or down-regulated by various cytokines. The main stimulatory cytokines for matrix metalloproteinases are tumor necrosis factor alpha, interleukin-1 and interleukin-6. It is also known that active matrix metalloproteinases are capable of activating other matrix metalloproteinases in a mutual activation cascade (248). Certain cytokines are specifically related to particular matrix metalloproteinases. For example, interleukin-1beta and tumor necrosis factor alpha can stimulate the secretion of matrix metalloproteinases 3, 8 and 9 from gingival fibroblasts and the secretion of matrix metalloproteinase-13 from osteoblasts. Transforming growth factor beta suppresses the transcription of matrix metalloproteinase-1, -3 and -8 genes, but induces matrix metalloproteinase-2 and matrix metalloproteinase-13, mainly in keratinocytes (19, 106, 124).
The involvement of matrix metalloproteinases in inflammation is an active area of investigation. A good example of the interaction is interleukin-8 secretion in response to bacterial biofilm. Interleukin-8 recruits neutrophils to the site containing biofilm. The neutrophils will secrete cytokines, as well as matrix metalloproteinases 8 and 9 (221), resulting in the degradation of the extracellular matrix and the signaling other effector cells to produce matrix metalloproteinases. The major collagen-degrading enzyme in periodontitis is matrix metalloproteinase-8, which is mainly produced by neutrophils. This enzyme is found in gingival crevice fluid and saliva in diseased periodontal tissue. The main function of matrix metalloproteinase-8 is the degradation of interstitial collagens (221).
Matrix metalloproteinase-1 (collagenase-1) from mononuclear phagocytes, fibroblasts and epithelial cells has a wide range of substrates. It digests interstitial collagen, extracellular matrix components and soluble nonmatrix mediators (221). Matrix metalloproteinase-9 (gelatinase B) is a gelatinolytic enzyme that degrades several types of extracellular matrix, including basement membrane type IV collagen (135). Matrix metalloproteinase-9 is expressed by neutrophils, but also by cultured epithelial cells. The production of matrix metalloproteinase-9 is stimulated by several cytokines, especially tumor necrosis factor alpha, epidermal growth factor and by some bacterial products such as lipopolysaccharide and phospholipase C (54, 194).
Matrix metalloproteinase-2 (gelatinase A) has been shown to be strongly expressed in inflamed pocket epithelium and to be important in epithelial cell migration (159). Matrix metalloproteinase-13 (collagenase 3) is expressed by the basal cells of the gingival pocket epithelium (240); it degrades type I, type III and type IV collagens, as well as fibronectin, tenascin and some proteoglycans (105, 120). Matrix metalloproteinase-13 plays an important role in the growth of pocket epithelium into periodontal connective tissue. Some oral bacterial species, especially Fusobacterium nucleatum, induce matrix metalloproteinase-13 (241). Matrix metalloproteinase-7 (matrilysin) is another epithelial matrix metalloproteinase with a broad spectrum of substrates. It degrades fibronectin, laminin, type IV collagen, gelatin, elastin, entactin, tenascin and proteoglycans. The enzyme is not commonly secreted by the gingival tissues and has not been reported in human gingival epithelium or in the pocket epithelium of periodontitis patients. It is expressed constitutively in many adult epithelial cells, most notably in the salivary glands, and its secretion is observed in suprabasal epithelial cells (203, 255). Some periodontal pathogens, including F. nucleatum, Fusobacterium necrophorum, Porphyromonas endodontalis and Prevotella denticola, were found to induce the expression of matrix metalloproteinase-7 in porcine gingival epithelial cells (241). Overall, the role of matrix metalloproteinase-7 in periodontal disease is not clear.
Matrix metalloproteinase-3 (stromelysin-1) does not digest interstitial collagen. The main substrates of matrix metalloproteinase-3 are basement membrane components such as laminins and type IV collagen. Matrix metalloproteinase-3 is found in gingival crevice fluid and gingival tissue during periodontal inflammation (47, 164, 248).
Regulation of matrix metalloproteinase activity is a function of tissue inhibitor of metalloproteinases. The tissue inhibitor of metalloproteinases class of enzymes function in the regulation of extracellular matrix metabolism. Four members of the family of tissue inhibitor of matrix metalloproteinases (tissue inhibitor of matrix metalloproteinases 1–4) have been identified to date. Although the main function of tissue inhibitor of matrix metalloproteinases is to inhibit matrix metalloproteinases, they also regulate matrix metalloproteinase transportation, stabilization and localization in the extracellular matrix. Tissue inhibitor of matrix metalloproteinases 1, 2 and 4 are secreted extracellular proteins, whereas tissue inhibitor of matrix metalloproteinase 3 is an extracellular matrix-bound molecule (248).
Resolution of inflammation
Periodontal inflammation begins as a protective response to bacterial biofilm. In susceptible individuals, periodontal inflammation fails to resolve and chronic inflammation becomes the periodontal pathology. Periodontal disease results from excess inflammation and may be considered a failure of resolution pathways. An essential goal of interventions in inflammatory disease is the return of tissue to homeostasis, defined as an absence of inflammation. Hence, the rapid and complete elimination of invading leukocytes from a lesion is the ideal outcome following an inflammatory event (243). Accordingly, inadequate resolution and failure to return tissue to homeostasis results in neutrophil-mediated destruction and chronic inflammation (245), with destruction of both extracellular matrix and bone, and scarring and fibrosis (244). Scarring and fibrosis in periodontitis prevent the return to homeostasis (243).
To date, the efforts to control inflammation have been focused on the use of pharmacologic agents that inhibit proinflammatory mediator pathways (e.g. nonsteroidal anti-inflammatory drugs) (214). Nonsteroidal anti-inflammatory drugs target cyclooxygenase 1- and cyclooxygenase 2-dependent pathways, inhibiting the generation of prostanoids. Newer classes of inhibitors target lipoxygenase pathways and leukotriene production, or tumor necrosis factor alpha. The side-effect profiles of these agents prohibit their extended use in periodontal therapy.
More recent discoveries have uncovered the natural proresolving pathways, which are an extension of the same eicosanoid pathways that produce proinflammatory mediators. The physiologic end of the acute inflammatory phase occurs when there is a “class switch” of eicosanoid pathways in neutrophils (138, 243). This class switch is mediated by the up-regulation of 15-lipoxygenase by neutrophils late in inflammation. Neutrophils in the early acute phase produce only 5-lipoxygenase for the production of leukotrienes. 15-lipoxygenase catalyzes a second reaction with hydroxyeicosatetraenoic acid products generated earlier by neutrophils or other cells (213). The series of enzymatic reactions starts with the oxidation of arachidonic acid by a lipoxygenase (5-, 12-or 15-lipoxygenase, depending on the cell of origin). A 5-, 12- or 15-S-hydroxy-(p)-eicosatetraenoic acid intermediate is produced, which is then further acted on by 15-lipoxygenase to induce the synthesis of doubly substituted intermediates (5, 15 hydroxy-(p)-eicosatetraenoic acids, for example) that are further metabolized into lipoxins, such as lipoxins A4 and B4 (109, 245). Lipoxins are receptor agonists that stimulate the resolution of inflammation and promote the restoration of tissue homeostasis through a number of mechanisms. These include limiting the migration of polymorphonuclear neutrophils into sites of inflammation and modulating the phenotype of macrophages to stimulate the uptake of apoptotic polymorphonuclear neutrophils without secreting proinflammatory cytokines (156, 157, 217).
Unlike other nonsteroidal anti-inflammatory drugs, aspirin has unique characteristics. Aspirin acetylates cyclooxygenase 2 to inhibit further production of prostanoids from arachidonic acid metabolism, but the acetylated cyclooxygenase 2 has new enzyme activity as a 15-epi-lipoxygenase. This alternative pathway leads to the synthesis of 15-R-hydroxy-(p)-eicosatetraenoic acid. This molecule is transformed into 5(6)-epoxytetraene with the help of 5-lipoxygenase, and the product is 15-epi-LXs or aspirin-triggered lipoxins (245). Aspirin-triggered lipoxin, the epimer of native lipoxin, possesses more powerful proresolving properties (32, 218, 245).
Lipoxins are the natural proresolving molecules derived from endogenous fatty acids (arachidonic acid). Dietary fatty acids of the omega-3 class are also metabolized by similar pathways, and the products (resolvins and their aspirin-triggered derivatives) have similar biologic activity to lipoxins (215, 243). Resolvins stimulate the resolution of inflammation through multiple mechanisms, including preventing neutrophil penetration, the phagocytosis of apoptotic neutrophils to clear the lesion and enhancing the clearance of inflammation within the lesion to promote tissue regeneration (10, 81, 212). Interestingly, the classic inflammatory eicosanoids (i.e. prostaglandins and leukotrienes), in addition to activating and amplifying the cardinal signs of inflammation, are responsible for inducing the production of mediators that have both anti-inflammatory and proresolution activities, reinforcing the active nature of the resolution process (216). In an animal model of periodontitis, treatment with resolvin-E1 completely eliminated the signs of inflammation, enabling the regeneration of lost tissues (81).
Conclusion
Periodontal diseases are inflammatory diseases in which microbial etiologic factors induce a series of host responses that mediate inflammatory events (Fig. 1). In susceptible individuals, dysregulation of inflammatory and immune pathways leads to chronic inflammation, tissue destruction and disease. Physiologic inflammation is a well-orchestrated network of cells, mediators and tissues. It is very important to consider the inflammatory / immune response as a whole, rather than many different modules working separately. As disease appears to be the result of loss of regulation and a failure to return to homeostasis, it is important to achieve a more complete understanding of the molecular and cellular events in this complex system.
The paradigm shift in our understanding of inflammatory disease, such as periodontitis, is that resolution of inflammation is an active, rather than a passive, process that activates specific biochemical programs of resolution. Precursor fatty-acid substrates from cells (arachidonic acid) and dietary sources (omega-3 fatty acids) yield lipid mediators (lipoxins and resolvins, respectively) that counter-regulate proinflammatory signals. It is increasingly evident that future care of periodontal infections and periodontal surgical patients will rely on clinicians having a detailed map and molecular appreciation of the resolution programs for inflammation and tissue injury. Systematic temporal study of infection and resolution in human tissues is of paramount importance in the treatment of bacterially initiated disease. Studies of models of disease suggest that the shift to chronicity of the infection and the persistence of the pathogen results from increased inflammation and a failure of innate mucosal antibacterial systems. Susceptibility to chronic inflammatory disorders may therefore result from uncontrolled resolution of the inflammatory process. Considering the limited and semi-successful treatment options for periodontitis, research on the orchestration of this complex system can bring us one step closer for better treatment opportunities. As many current and widely used drugs have been developed without knowledge of their impact in resolution circuits, some agents, such as selective cyclooxygenase 2 inhibitors and certain lipoxygenase inhibitors, have proven to be toxic to the resolution programs (216). It will be important to learn whether resolution pharmacology leads to new treatments for human disease.
References
- 1.Alexopoulou L, Holt AC, Medzhitov R, Flavell RA. Recognition of double-stranded RNA and activation of NF-kappaB by toll-like receptor 3. Nature. 2001;413:732–738. doi: 10.1038/35099560. [DOI] [PubMed] [Google Scholar]
- 2.Anderson DM, Maraskovsky E, Billingsley WL, Dougall WC, Tometsko ME, Roux ER, Teepe MC, DuBose RF, Cosman D, Galibert L. A homologue of the TNF receptor and its ligand enhance T-cell growth and dendritic-cell function. Nature. 1997;390:175–179. doi: 10.1038/36593. [DOI] [PubMed] [Google Scholar]
- 3.Anderson KV. Toll signaling pathways in the innate immune response. Curr Opin Immunol. 2000;12:13–19. doi: 10.1016/s0952-7915(99)00045-x. [DOI] [PubMed] [Google Scholar]
- 4.Appay V, van Lier RA, Sallusto F, Roederer M. Phenotype and function of human T lymphocyte subsets: consensus and issues. Cytometry A. 2008;73:975–983. doi: 10.1002/cyto.a.20643. [DOI] [PubMed] [Google Scholar]
- 5.Ara T, Kurata K, Hirai K, Uchihashi T, Uematsu T, Imamura Y, Furusawa K, Kurihara S, Wang PL. Human gingival fibroblasts are critical in sustaining inflammation in periodontal disease. J Periodontal Res. 2009;44:21–27. doi: 10.1111/j.1600-0765.2007.01041.x. [DOI] [PubMed] [Google Scholar]
- 6.Awawdeh L, Lundy FT, Shaw C, Lamey PJ, Linden GJ, Kennedy JG. Quantitative analysis of substance P, neurokinin A and calcitonin gene-related peptide in pulp tissue from painful and healthy human teeth. Int Endod J. 2002;35:30–36. doi: 10.1046/j.1365-2591.2002.00451.x. [DOI] [PubMed] [Google Scholar]
- 7.Baker PJ, Dixon M, Roopenian DC. Genetic control of susceptibility to Porphyromonas gingivalis-induced alveolar bone loss in mice. Infect Immun. 2000;68:5864–5868. doi: 10.1128/iai.68.10.5864-5868.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Baldwin AS., Jr The NF-kappa B and I kappa B proteins: new discoveries and insights. Annu Rev Immunol. 1996;14:649–683. doi: 10.1146/annurev.immunol.14.1.649. [DOI] [PubMed] [Google Scholar]
- 9.Bancroft GJ. The role of natural killer cells in innate resistance to infection. Curr Opin Immunol. 1993;5:503–510. doi: 10.1016/0952-7915(93)90030-v. [DOI] [PubMed] [Google Scholar]
- 10.Bannenberg GL, Chiang N, Ariel A, Arita M, Tjonahen E, Gotlinger KH, Hong S, Serhan CN. Molecular circuits of resolution: formation and actions of resolvins and protectins. J Immunol. 2005;174:4345–4355. doi: 10.4049/jimmunol.174.7.4345. [DOI] [PubMed] [Google Scholar]
- 11.Bartold PM, Kylstra A, Lawson R. Substance P: an immunohistochemical and biochemical study in human gingival tissues. A role for neurogenic inflammation? J Periodontol. 1994;65:1113–1121. doi: 10.1902/jop.1994.65.12.1113. [DOI] [PubMed] [Google Scholar]
- 12.Beck J, Arbes SJ., Jr . Epidemiology of gingival and periodontal diseases. In: Newman MG, Takei HH, Klokkevold PR, editors. Carranza’s clinical periodontology. St Louis: Saunders/Elsevier; 2006. pp. 110–132. [Google Scholar]
- 13.Behl Y, Siqueira M, Ortiz J, Li J, Desta T, Faibish D, Graves DT. Activation of the acquired immune response reduces coupled bone formation in response to a periodontal pathogen. J Immunol. 2008;181:8711–8718. doi: 10.4049/jimmunol.181.12.8711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Belkaid Y, Tarbell K. Regulatory t cells in the control of host-microorganism interactions (*) Annu Rev Immunol. 2009;27:551–589. doi: 10.1146/annurev.immunol.021908.132723. [DOI] [PubMed] [Google Scholar]
- 15.Bellinger DL, Lorton D, Brouxhon S, Felten S, Felten DL. The significance of vasoactive intestinal polypeptide (VIP) in immunomodulation. Adv Neuroimmunol. 1996;6:5–27. doi: 10.1016/s0960-5428(96)00008-3. [DOI] [PubMed] [Google Scholar]
- 16.Bendre MS, Montague DC, Peery T, Akel NS, Gaddy D, Suva LJ. Interleukin-8 stimulation of osteoclastogenesis and bone resorption is a mechanism for the increased osteolysis of metastatic bone disease. Bone. 2003;33:28–37. doi: 10.1016/s8756-3282(03)00086-3. [DOI] [PubMed] [Google Scholar]
- 17.Berglundh T, Donati M. Aspects of adaptive host response in periodontitis. J Clin Periodontol. 2005;32(Suppl 6):87–107. doi: 10.1111/j.1600-051X.2005.00820.x. [DOI] [PubMed] [Google Scholar]
- 18.Bhatavadekar NB, Williams RC. Modulation of the host inflammatory response in periodontal disease management: exciting new directions. Int Dent J. 2009;59:305–308. [PubMed] [Google Scholar]
- 19.Birkedal-Hansen H. Role of matrix metalloproteinases in human periodontal diseases. J Periodontol. 1993;64:474–484. doi: 10.1902/jop.1993.64.5s.474. [DOI] [PubMed] [Google Scholar]
- 20.Bloemen V, Schoenmaker T, de Vries TJ, Everts V. Direct cell-cell contact between periodontal ligament fibroblasts and osteoclast precursors synergistically increases the expression of genes related to osteoclastogenesis. J Cell Physiol. 2009;222:565–573. doi: 10.1002/jcp.21971. [DOI] [PubMed] [Google Scholar]
- 21.Bonecchi R, Bianchi G, Bordignon PP, D’Ambrosio D, Lang R, Borsatti A, Sozzani S, Allavena P, Gray PA, Mantovani A, Sinigaglia F. Differential expression of chemokine receptors and chemotactic responsiveness of type 1 T helper cells (Th1s) and Th2s. J Exp Med. 1998;187:129–134. doi: 10.1084/jem.187.1.129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bracci-Laudiero L, Aloe L, Buanne P, Finn A, Stenfors C, Vigneti E, Theodorsson E, Lundeberg T. NGF modulates CGRP synthesis in human B-lymphocytes: a possible anti-inflammatory action of NGF? J Neuroimmunol. 2002;123:58–65. doi: 10.1016/s0165-5728(01)00475-1. [DOI] [PubMed] [Google Scholar]
- 23.Brain SD, Williams TJ, Tippins JR, Morris HR, MacIntyre I. Calcitonin gene-related peptide is a potent vasodilator. Nature. 1985;313:54–56. doi: 10.1038/313054a0. [DOI] [PubMed] [Google Scholar]
- 24.Breitfeld D, Ohl L, Kremmer E, Ellwart J, Sallusto F, Lipp M, Forster R. Follicular B helper T cells express CXC chemokine receptor 5, localize to B cell follicles, and support immunoglobulin production. J Exp Med. 2000;192:1545–1552. doi: 10.1084/jem.192.11.1545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Brown JM, Zhang J, Keller ET. OPG, RANKL, and RANK in cancer metastasis: expression and regulation. Cancer Treat Res. 2004;118:149–172. doi: 10.1007/978-1-4419-9129-4_7. [DOI] [PubMed] [Google Scholar]
- 26.Buckley TL, Brain SD, Collins PD, Williams TJ. Inflammatory edema induced by interactions between IL-1 and the neuropeptide calcitonin gene-related peptide. J Immunol. 1991;146:3424–3430. [PubMed] [Google Scholar]
- 27.Burns E, Bachrach G, Shapira L, Nussbaum G. Cutting edge: TLR2 is required for the innate response to Porphyromonas gingivalis: activation leads to bacterial persistence and TLR2 deficiency attenuates induced alveolar bone resorption. J Immunol. 2006;177:8296–8300. doi: 10.4049/jimmunol.177.12.8296. [DOI] [PubMed] [Google Scholar]
- 28.Byers MR, Taylor PE. Effect of sensory denervation on the response of rat molar pulp to exposure injury. J Dent Res. 1993;72:613–618. doi: 10.1177/00220345930720031001. [DOI] [PubMed] [Google Scholar]
- 29.Cao YQ, Mantyh PW, Carlson EJ, Gillespie AM, Epstein CJ, Basbaum AI. Primary afferent tachykinins are required to experience moderate to intense pain. Nature. 1998;392:390–394. doi: 10.1038/32897. [DOI] [PubMed] [Google Scholar]
- 30.Cardoso CR, Garlet GP, Moreira AP, Junior WM, Rossi MA, Silva JS. Characterization of CD4+ CD25+ natural regulatory T cells in the inflammatory infiltrate of human chronic periodontitis. J Leukoc Biol. 2008;84:311–318. doi: 10.1189/jlb.0108014. [DOI] [PubMed] [Google Scholar]
- 31.Chou WY, Lu CN, Lee TH, Wu CL, Hung KS, Concejero AM, Jawan B, Wang CH. Electroporative interleukin-10 gene transfer ameliorates carbon tetrachloride-induced murine liver fibrosis by MMP and TIMP modulation. Acta Pharmacol Sin. 2006;27:469–476. doi: 10.1111/j.1745-7254.2006.00304.x. [DOI] [PubMed] [Google Scholar]
- 32.Claria J, Lee MH, Serhan CN. Aspirin-triggered lipoxins (15-epi-LX) are generated by the human lung adenocarcinoma cell line (A549)-neutrophil interactions and are potent inhibitors of cell proliferation. Mol Med. 1996;2:583–596. [PMC free article] [PubMed] [Google Scholar]
- 33.Claudino M, Trombone AP, Cardoso CR, Ferreira SB, Jr, Martins W, Jr, Assis GF, Santos CF, Trevilatto PC, Campanelli AP, Silva JS, Garlet GP. The broad effects of the functional IL-10 promoter-592 polymorphism: modulation of IL-10, TIMP-3, and OPG expression and their association with periodontal disease outcome. J Leukoc Biol. 2008;84:1565–1573. doi: 10.1189/jlb.0308184. [DOI] [PubMed] [Google Scholar]
- 34.Cole KL, Seymour GJ, Powell RN. Phenotypic and functional analysis of T cells extracted from chronically inflamed human periodontal tissues. J Periodontol. 1987;58:569–573. doi: 10.1902/jop.1987.58.8.569. [DOI] [PubMed] [Google Scholar]
- 35.Cook DN, Pisetsky DS, Schwartz DA. Toll-like receptors in the pathogenesis of human disease. Nat Immunol. 2004;5:975–979. doi: 10.1038/ni1116. [DOI] [PubMed] [Google Scholar]
- 36.Cutz E, Chan W, Track NS, Goth A, Said SI. Release of vasoactive intestinal polypeptide in mast cells by histamine liberators. Nature. 1978;275:661–662. doi: 10.1038/275661a0. [DOI] [PubMed] [Google Scholar]
- 37.D’Ambrosio D, Iellem A, Bonecchi R, Mazzeo D, Sozzani S, Mantovani A, Sinigaglia F. Selective up-regulation of chemokine receptors CCR4 and CCR8 upon activation of polarized human type 2 Th cells. J Immunol. 1998;161:5111–5115. [PubMed] [Google Scholar]
- 38.de Hoon JN, Pickkers P, Smits P, Struijker-Boudier HA, Van Bortel LM. Calcitonin gene-related peptide: exploring its vasodilating mechanism of action in humans. Clin Pharmacol Ther. 2003;73:312–321. doi: 10.1016/s0009-9236(03)00007-9. [DOI] [PubMed] [Google Scholar]
- 39.de Waal Malefyt R, Figdor CG, Huijbens R, Mohan-Peterson S, Bennett B, Culpepper J, Dang W, Zurawski G, de Vries JE. Effects of IL-13 on phenotype, cytokine production, and cytotoxic function of human monocytes. Comparison with IL-4 and modulation by IFN-gamma or IL-10. J Immunol. 1993;151:6370–6381. [PubMed] [Google Scholar]
- 40.de Waal Malefyt R, Yssel H, de Vries JE. Direct effects of IL-10 on subsets of human CD4+ T cell clones and resting T cells. Specific inhibition of IL-2 production and proliferation. J Immunol. 1993;150:4754–4765. [PubMed] [Google Scholar]
- 41.Dietrich JW, Goodson JM, Raisz LG. Stimulation of bone resorption by various prostaglandins in organ culture. Prostaglandins. 1975;10:231–240. doi: 10.1016/0090-6980(75)90042-8. [DOI] [PubMed] [Google Scholar]
- 42.Dinarello CA. Proinflammatory cytokines. Chest. 2000;118:503–508. doi: 10.1378/chest.118.2.503. [DOI] [PubMed] [Google Scholar]
- 43.Dipietro LA, Reintjes MG, Low QE, Levi B, Gamelli RL. Modulation of macrophage recruitment into wounds by monocyte chemoattractant protein-1. Wound Repair Regen. 2001;9:28–33. doi: 10.1046/j.1524-475x.2001.00028.x. [DOI] [PubMed] [Google Scholar]
- 44.Donnelly RP, Fenton MJ, Finbloom DS, Gerrard TL. Differential regulation of IL-1 production in human monocytes by IFN-gamma and IL-4. J Immunol. 1990;145:569–575. [PubMed] [Google Scholar]
- 45.Dutzan N, Gamonal J, Silva A, Sanz M, Vernal R. Overexpression of forkhead box p3 and its association with receptor activator of nuclear factor-kappa B ligand, interleukin (IL)-17, IL-10 and transforming growth factor-beta during the progression of chronic periodontitis. J Clin Periodontol. 2009;36:396–403. doi: 10.1111/j.1600-051X.2009.01390.x. [DOI] [PubMed] [Google Scholar]
- 46.Dutzan N, Vernal R, Hernandez M, Dezerega A, Rivera O, Silva N, Aguillon JC, Puente J, Pozo P, Gamonal J. Levels of interferon-gamma and transcription factor T-bet in progressive periodontal lesions in patients with chronic periodontitis. J Periodontol. 2009;80:290–296. doi: 10.1902/jop.2009.080287. [DOI] [PubMed] [Google Scholar]
- 47.Egeblad M, Werb Z. New functions for the matrix metalloproteinases in cancer progression. Nat Rev Cancer. 2002;2:161–174. doi: 10.1038/nrc745. [DOI] [PubMed] [Google Scholar]
- 48.Esche C, Stellato C, Beck LA. Chemokines: key players in innate and adaptive immunity. J Invest Dermatol. 2005;125:615–628. doi: 10.1111/j.0022-202X.2005.23841.x. [DOI] [PubMed] [Google Scholar]
- 49.Essner R, Rhoades K, McBride WH, Morton DL, Economou JS. IL-4 down-regulates IL-1 and TNF gene expression in human monocytes. J Immunol. 1989;142:3857–3861. [PubMed] [Google Scholar]
- 50.Fazilleau N, Mark L, McHeyzer-Williams LJ, McHeyzer-Williams MG. Follicular helper T cells: lineage and location. Immunity. 2009;30:324–335. doi: 10.1016/j.immuni.2009.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Fiorellini JP, Ishikawa SO, Kim DM. Gingival inflammation. In: Carranza FA, editor. Clinical periodontology. Philadelphia: Elsevier Saunders; 2006. pp. 355–361. [Google Scholar]
- 52.Fiorentino DF, Zlotnik A, Vieira P, Mosmann TR, Howard M, Moore KW, O’Garra A. IL-10 acts on the antigen-presenting cell to inhibit cytokine production by Th1 cells. J Immunol. 1991;146:3444–3451. [PubMed] [Google Scholar]
- 53.Fiorucci S, Distrutti E. Role of PAR2 in pain and inflammation. Trends Pharmacol Sci. 2002;23:153–155. doi: 10.1016/s0165-6147(00)01932-5. [DOI] [PubMed] [Google Scholar]
- 54.Firth JD, Putnins EE, Larjava H, Uitto VJ. Bacterial phospholipase C upregulates matrix metalloproteinase expression by cultured epithelial cells. Infect Immun. 1997;65:4931–4936. doi: 10.1128/iai.65.12.4931-4936.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Flemmig TF. Periodontitis. Ann Periodontol. 1999;4:32–38. doi: 10.1902/annals.1999.4.1.32. [DOI] [PubMed] [Google Scholar]
- 56.Fonseca JE, Santos MJ, Canhao H, Choy E. Interleukin-6 as a key player in systemic inflammation and joint destruction. Autoimmun Rev. 2009;8:538–542. doi: 10.1016/j.autrev.2009.01.012. [DOI] [PubMed] [Google Scholar]
- 57.Ganea D, Delgado M. Vasoactive intestinal peptide (VIP) and pituitary adenylate cyclase-activating polypeptide (PACAP) as modulators of both innate and adaptive immunity. Crit Rev Oral Biol Med. 2002;13:229–237. doi: 10.1177/154411130201300303. [DOI] [PubMed] [Google Scholar]
- 58.Garlet GP. Destructive and protective roles of cytokines in periodontitis: a re-appraisal from host defense and tissue destruction viewpoints. J Dent Res. 2010;89:1349–1363. doi: 10.1177/0022034510376402. [DOI] [PubMed] [Google Scholar]
- 59.Garlet GP, Cardoso CR, Campanelli AP, Ferreira BR, Avila-Campos MJ, Cunha FQ, Silva JS. The dual role of p55 tumour necrosis factor-alpha receptor in Actinobacillus actinomycetemcomitans-induced experimental periodontitis: host protection and tissue destruction. Clin Exp Immunol. 2007;147:128–138. doi: 10.1111/j.1365-2249.2006.03260.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Garlet GP, Cardoso CR, Campanelli AP, Martins W, Jr, Silva JS. Expression of suppressors of cytokine signaling in diseased periodontal tissues: a stop signal for disease progression? J Periodontal Res. 2006;41:580–584. doi: 10.1111/j.1600-0765.2006.00908.x. [DOI] [PubMed] [Google Scholar]
- 61.Garlet GP, Martins W, Jr, Ferreira BR, Milanezi CM, Silva JS. Patterns of chemokines and chemokine receptors expression in different forms of human periodontal disease. J Periodontal Res. 2003;38:210–217. doi: 10.1034/j.1600-0765.2003.02012.x. [DOI] [PubMed] [Google Scholar]
- 62.Garlet GP, Martins W, Jr, Fonseca BA, Ferreira BR, Silva JS. Matrix metalloproteinases, their physiological inhibitors and osteoclast factors are differentially regulated by the cytokine profile in human periodontal disease. J Clin Periodontol. 2004;31:671–679. doi: 10.1111/j.1600-051X.2004.00545.x. [DOI] [PubMed] [Google Scholar]
- 63.Gemmell E, Carter CL, Hart DN, Drysdale KE, Seymour GJ. Antigen-presenting cells in human periodontal disease tissues. Oral Microbiol Immunol. 2002;17:388–393. doi: 10.1034/j.1399-302x.2002.170609.x. [DOI] [PubMed] [Google Scholar]
- 64.Gemmell E, Carter CL, Seymour GJ. Chemokines in human periodontal disease tissues. Clin Exp Immunol. 2001;125:134–141. doi: 10.1046/j.1365-2249.2001.01511.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Gemmell E, Marshall RI, Seymour GJ. Cytokines and prostaglandins in immune homeostasis and tissue destruction in periodontal disease. Periodontol 2000. 1997;14:112–143. doi: 10.1111/j.1600-0757.1997.tb00194.x. [DOI] [PubMed] [Google Scholar]
- 66.Gemmell E, Yamazaki K, Seymour GJ. Destructive periodontitis lesions are determined by the nature of the lymphocytic response. Crit Rev Oral Biol Med. 2002;13:17–34. doi: 10.1177/154411130201300104. [DOI] [PubMed] [Google Scholar]
- 67.Geppetti P. Sensory neuropeptide release by bradykinin: mechanisms and pathophysiological implications. Regul Pept. 1993;47:1–23. doi: 10.1016/0167-0115(93)90268-d. [DOI] [PubMed] [Google Scholar]
- 68.Giannopoulou C, Kamma JJ, Mombelli A. Effect of inflammation, smoking and stress on gingival crevicular fluid cytokine level. J Clin Periodontol. 2003;30:145–153. doi: 10.1034/j.1600-051x.2003.300201.x. [DOI] [PubMed] [Google Scholar]
- 69.Goodson JM, Dewhirst FE, Brunetti A. Prostaglandin E2 levels and human periodontal disease. Prostaglandins. 1974;6:81–85. doi: 10.1016/s0090-6980(74)80043-2. [DOI] [PubMed] [Google Scholar]
- 70.Grassi F, Cristino S, Toneguzzi S, Piacentini A, Facchini A, Lisignoli G. CXCL12 chemokine up-regulates bone resorption and MMP-9 release by human osteoclasts: CXCL12 levels are increased in synovial and bone tissue of rheumatoid arthritis patients. J Cell Physiol. 2004;199:244–251. doi: 10.1002/jcp.10445. [DOI] [PubMed] [Google Scholar]
- 71.Grassi F, Piacentini A, Cristino S, Toneguzzi S, Cavallo C, Facchini A, Lisignoli G. Human osteoclasts express different CXC chemokines depending on cell culture substrate: molecular and immunocytochemical evidence of high levels of CXCL10 and CXCL12. Histochem Cell Biol. 2003;120:391–400. doi: 10.1007/s00418-003-0587-3. [DOI] [PubMed] [Google Scholar]
- 72.Graves DT, Cochran D. The contribution of interleukin-1 and tumor necrosis factor to periodontal tissue destruction. J Periodontol. 2003;74:391–401. doi: 10.1902/jop.2003.74.3.391. [DOI] [PubMed] [Google Scholar]
- 73.Graves DT, Fine D, Teng YT, Van Dyke TE, Hajishengallis G. The use of rodent models to investigate host-bacteria interactions related to periodontal diseases. J Clin Periodontol. 2008;35:89–105. doi: 10.1111/j.1600-051X.2007.01172.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Gu L, Tseng S, Horner RM, Tam C, Loda M, Rollins BJ. Control of Th2 polarization by the chemokine monocyte chemoattractant protein-1. Nature. 2000;404:407–411. doi: 10.1038/35006097. [DOI] [PubMed] [Google Scholar]
- 75.Hanada T, Yoshimura A. Regulation of cytokine signaling and inflammation. Cytokine Growth Factor Rev. 2002;13:413–421. doi: 10.1016/s1359-6101(02)00026-6. [DOI] [PubMed] [Google Scholar]
- 76.Hanazawa S, Kawata Y, Takeshita A, Kumada H, Okithu M, Tanaka S, Yamamoto Y, Masuda T, Umemoto T, Kitano S. Expression of monocyte chemoattractant protein 1 (MCP-1) in adult periodontal disease: increased monocyte chemotactic activity in crevicular fluids and induction of MCP-1 expression in gingival tissues. Infect Immun. 1993;61:5219–5224. doi: 10.1128/iai.61.12.5219-5224.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Hanemaaijer R, Sorsa T, Konttinen YT, Ding Y, Sutinen M, Visser H, van Hinsbergh VW, Helaakoski T, Kainulainen T, Ronka H, Tschesche H, Salo T. Matrix metalloproteinase-8 is expressed in rheumatoid synovial fibroblasts and endothelial cells. Regulation by tumor necrosis factor-alpha and doxycycline. J Biol Chem. 1997;272:31504–31509. doi: 10.1074/jbc.272.50.31504. [DOI] [PubMed] [Google Scholar]
- 78.Hanioka T, Takaya K, Matsumori Y, Matsuse R, Shizukuishi S. Relationship of the substance P to indicators of host response in human gingival crevicular fluid. J Clin Periodontol. 2000;27:262–266. doi: 10.1034/j.1600-051x.2000.027004262.x. [DOI] [PubMed] [Google Scholar]
- 79.Harrell JC, Stein SH. Prostaglandin E2 regulates gingival mononuclear cell immunoglobulin production. J Periodontol. 1995;66:222–227. doi: 10.1902/jop.1995.66.3.222. [DOI] [PubMed] [Google Scholar]
- 80.Hartung HP, Wolters K, Toyka KV. Substance P: binding properties and studies on cellular responses in guinea pig macrophages. J Immunol. 1986;136:3856–3863. [PubMed] [Google Scholar]
- 81.Hasturk H, Kantarci A, Goguet-Surmenian E, Blackwood A, Andry C, Serhan CN, Van Dyke TE. Resolvin E1 regulates inflammation at the cellular and tissue level and restores tissue homeostasis in vivo. J Immunol. 2007;179:7021–7029. doi: 10.4049/jimmunol.179.10.7021. [DOI] [PubMed] [Google Scholar]
- 82.Hatakeyama J, Tamai R, Sugiyama A, Akashi S, Sugawara S, Takada H. Contrasting responses of human gingival and periodontal ligament fibroblasts to bacterial cell-surface components through the CD14 / toll-like receptor system. Oral Microbiol Immunol. 2003;18:14–23. doi: 10.1034/j.1399-302x.2003.180103.x. [DOI] [PubMed] [Google Scholar]
- 83.Hayashi S, Yamada T, Tsuneto M, Yamane T, Takahashi M, Shultz LD, Yamazaki H. Distinct osteoclast precursors in the bone marrow and extramedullary organs characterized by responsiveness to toll-like receptor ligands and TNF-alpha. J Immunol. 2003;171:5130–5139. doi: 10.4049/jimmunol.171.10.5130. [DOI] [PubMed] [Google Scholar]
- 84.Henderson B, Nair SP, Ward JM, Wilson M. Molecular pathogenicity of the oral opportunistic pathogen Actinobacillus actinomycetemcomitans. Annu Rev Microbiol. 2003;57:29–55. doi: 10.1146/annurev.micro.57.030502.090908. [DOI] [PubMed] [Google Scholar]
- 85.Trowbridge HO, Emling RC. Inflammation: a review of the process. Illinois: Quintessence Publishing Co, Inc.; 1997. Introduction; pp. IX–X. [Google Scholar]
- 86.Hirschfeld M, Weis JJ, Toshchakov V, Salkowski CA, Cody MJ, Ward DC, Qureshi N, Michalek SM, Vogel SN. Signaling by toll-like receptor 2 and 4 agonists results in differential gene expression in murine macrophages. Infect Immun. 2001;69:1477–1482. doi: 10.1128/IAI.69.3.1477-1482.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Hofbauer LC, Schoppet M. Clinical implications of the osteoprotegerin / RANKL / RANK system for bone and vascular diseases. JAMA. 2004;292:490–495. doi: 10.1001/jama.292.4.490. [DOI] [PubMed] [Google Scholar]
- 88.Honda T, Domon H, Okui T, Kajita K, Amanuma R, Yamazaki K. Balance of inflammatory response in stable gingivitis and progressive periodontitis lesions. Clin Exp Immunol. 2006;144:35–40. doi: 10.1111/j.1365-2249.2006.03028.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Honore P, Luger NM, Sabino MA, Schwei MJ, Rogers SD, Mach DB, O’Keefe PF, Ramnaraine ML, Clohisy DR, Mantyh PW. Osteoprotegerin blocks bone cancer-induced skeletal destruction, skeletal pain and pain-related neurochemical reorganization of the spinal cord. Nat Med. 2000;6:521–528. doi: 10.1038/74999. [DOI] [PubMed] [Google Scholar]
- 90.Hoyle CHV. Neuropeptides, essential data. Chichester, UK: John Wiley & and Sons Ltd; 1996. [Google Scholar]
- 91.Hsu H, Lacey DL, Dunstan CR, Solovyev I, Colombero A, Timms E, Tan HL, Elliott G, Kelley MJ, Sarosi I, Wang L, Xia XZ, Elliott R, Chiu L, Black T, Scully S, Capparelli C, Morony S, Shimamoto G, Bass MB, Boyle WJ. Tumor necrosis factor receptor family member RANK mediates osteoclast differentiation and activation induced by osteoprotegerin ligand. Proc Natl Acad Sci USA. 1999;96:3540–3545. doi: 10.1073/pnas.96.7.3540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Hua XY, Chen P, Fox A, Myers RR. Involvement of cytokines in lipopolysaccharide-induced facilitation of CGRP release from capsaicin-sensitive nerves in the trachea: studies with interleukin-1beta and tumor necrosis factor-alpha. J Neurosci. 1996;16:4742–4748. doi: 10.1523/JNEUROSCI.16-15-04742.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Ihn H, Yamane K, Asano Y, Kubo M, Tamaki K. IL-4 up-regulates the expression of tissue inhibitor of metalloproteinase-2 in dermal fibroblasts via the p38 mitogen-activated protein kinase dependent pathway. J Immunol. 2002;168:1895–1902. doi: 10.4049/jimmunol.168.4.1895. [DOI] [PubMed] [Google Scholar]
- 94.Iino Y, Hopps RM. The bone-resorbing activities in tissue culture of lipopolysaccharides from the bacteria Actinobacillus actinomycetemcomitans, Bacteroides gingivalis and Capnocytophaga ochracea isolated from human mouths. Arch Oral Biol. 1984;29:59–63. doi: 10.1016/0003-9969(84)90043-8. [DOI] [PubMed] [Google Scholar]
- 95.Inohara N, Nunez G. NODS: intracellular proteins involved in inflammation and apoptosis. Nat Rev Immunol. 2003;3:371–382. doi: 10.1038/nri1086. [DOI] [PubMed] [Google Scholar]
- 96.Iwasaki A, Medzhitov R. Toll-like receptor control of the adaptive immune responses. Nat Immunol. 2004;5:987–995. doi: 10.1038/ni1112. [DOI] [PubMed] [Google Scholar]
- 97.Janeway CA, Jr, Medzhitov R. Innate immune recognition. Annu Rev Immunol. 2002;20:197–216. doi: 10.1146/annurev.immunol.20.083001.084359. [DOI] [PubMed] [Google Scholar]
- 98.Jarrossay D, Napolitani G, Colonna M, Sallusto F, Lanzavecchia A. Specialization and complementarity in microbial molecule recognition by human myeloid and plasmacytoid dendritic cells. Eur J Immunol. 2001;31:3388–3393. doi: 10.1002/1521-4141(200111)31:11<3388::aid-immu3388>3.0.co;2-q. [DOI] [PubMed] [Google Scholar]
- 99.Fiorellini JP, Ishikawa SO, Kim DM. Clinical features of gingivitis. In: Carranza FA, editor. Clinical periodontology. Philadelphia: Saunders Elsevier; 2006. pp. 362–372. [Google Scholar]
- 100.Jovanovic DV, Di Battista JA, Martel-Pelletier J, Jolicoeur FC, He Y, Zhang M, Mineau F, Pelletier JP. IL-17 stimulates the production and expression of proinflammatory cytokines, IL-beta and TNF-alpha, by human macrophages. J Immunol. 1998;160:3513–3521. [PubMed] [Google Scholar]
- 101.Kabashima H, Yoneda M, Nagata K, Hirofuji T, Ishihara Y, Yamashita M, Maeda K. The presence of chemokine receptor (CCR5, CXCR3, CCR3)-positive cells and chemokine (MCP1, MIP-1alpha, MIP-1beta, IP-10)-positive cells in human periapical granulomas. Cytokine. 2001;16:62–66. doi: 10.1006/cyto.2001.0947. [DOI] [PubMed] [Google Scholar]
- 102.Kabashima H, Yoneda M, Nagata K, Hirofuji T, Maeda K. The presence of chemokine (MCP-1, MIP-1alpha, MIP-1beta, IP-10, RANTES)-positive cells and chemokine receptor (CCR5, CXCR3)-positive cells in inflamed human gingival tissues. Cytokine. 2002;20:70–77. doi: 10.1006/cyto.2002.1985. [DOI] [PubMed] [Google Scholar]
- 103.Kadowaki N, Ho S, Antonenko S, Malefyt RW, Kastelein RA, Bazan F, Liu YJ. Subsets of human dendritic cell precursors express different toll-like receptors and respond to different microbial antigens. J Exp Med. 2001;194:863–869. doi: 10.1084/jem.194.6.863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Kagnoff MF, Eckmann L. Epithelial cells as sensors for microbial infection. J Clin Invest. 1997;100:6–10. doi: 10.1172/JCI119522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Kahari VM, Saarialho-Kere U. Matrix metalloproteinases in skin. Exp Dermatol. 1997;6:199–213. doi: 10.1111/j.1600-0625.1997.tb00164.x. [DOI] [PubMed] [Google Scholar]
- 106.Kahari VM, Saarialho-Kere U. Matrix metalloproteinases and their inhibitors in tumour growth and invasion. Ann Med. 1999;31:34–45. doi: 10.3109/07853899909019260. [DOI] [PubMed] [Google Scholar]
- 107.Kaisho T, Akira S. Toll-like receptors as adjuvant receptors. Biochim Biophys Acta. 2002;1589:1–13. doi: 10.1016/s0167-4889(01)00182-3. [DOI] [PubMed] [Google Scholar]
- 108.Kantarci A, Oyaizu K, Van Dyke TE. Neutrophil-mediated tissue injury in periodontal disease pathogenesis: findings from localized aggressive periodontitis. J Periodontol. 2003;74:66–75. doi: 10.1902/jop.2003.74.1.66. [DOI] [PubMed] [Google Scholar]
- 109.Kantarci A, Van Dyke TE. Lipoxins in chronic inflammation. Crit Rev Oral Biol Med. 2003;14:4–12. doi: 10.1177/154411130301400102. [DOI] [PubMed] [Google Scholar]
- 110.Kaplan G, Luster AD, Hancock G, Cohn ZA. The expression of a gamma interferon-induced protein (IP-10) in delayed immune responses in human skin. J Exp Med. 1987;166:1098–1108. doi: 10.1084/jem.166.4.1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Kawai T, Matsuyama T, Hosokawa Y, Makihira S, Seki M, Karimbux NY, Goncalves RB, Valverde P, Dibart S, Li YP, Miranda LA, Ernst CW, Izumi Y, Taubman MA. B and T lymphocytes are the primary sources of RANKL in the bone resorptive lesion of periodontal disease. Am J Pathol. 2006;169:987–998. doi: 10.2353/ajpath.2006.060180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Kelso A. Th1 and Th2 subsets: paradigms lost? Immunol Today. 1995;16:374–379. doi: 10.1016/0167-5699(95)80004-2. [DOI] [PubMed] [Google Scholar]
- 113.Kessler JA, Freidin MM, Kalberg C, Chandross KJ. Cytokines regulate substance P expression in sympathetic neurons. Regul Pept. 1993;46:70–75. doi: 10.1016/0167-0115(93)90014-y. [DOI] [PubMed] [Google Scholar]
- 114.Kiili M, Cox SW, Chen HY, Wahlgren J, Maisi P, Eley BM, Salo T, Sorsa T. Collagenase-2 (MMP-8) and collagenase-3 (MMP-13) in adult periodontitis: molecular forms and levels in gingival crevicular fluid and immunolocalisation in gingival tissue. J Clin Periodontol. 2002;29:224–232. doi: 10.1034/j.1600-051x.2002.290308.x. [DOI] [PubMed] [Google Scholar]
- 115.Kim MS, Day CJ, Selinger CI, Magno CL, Stephens SR, Morrison NA. MCP-1-induced human osteoclast-like cells are tartrate-resistant acid phosphatase, NFATC1, and calcitonin receptor-positive but require receptor activator of NFkappaB ligand for bone resorption. J Biol Chem. 2006;281:1274–1285. doi: 10.1074/jbc.M510156200. [DOI] [PubMed] [Google Scholar]
- 116.Kim MS, Magno CL, Day CJ, Morrison NA. Induction of chemokines and chemokine receptors CCR2b and CCR4 in authentic human osteoclasts differentiated with rankl and osteoclast like cells differentiated by MCP-1 and RANTES. J Cell Biochem. 2006;97:512–518. doi: 10.1002/jcb.20649. [DOI] [PubMed] [Google Scholar]
- 117.Kindle L, Rothe L, Kriss M, Osdoby P, Collin-Osdoby P. Human microvascular endothelial cell activation by IL-1 and TNF-alpha stimulates the adhesion and transendothelial migration of circulating human CD14+ monocytes that develop with RANKL into functional osteoclasts. J Bone Miner Res. 2006;21:193–206. doi: 10.1359/JBMR.051027. [DOI] [PubMed] [Google Scholar]
- 118.King C, Tangye SG, Mackay CR. T follicular helper (TFH) cells in normal and dysregulated immune responses. Annu Rev Immunol. 2008;26:741–766. doi: 10.1146/annurev.immunol.26.021607.090344. [DOI] [PubMed] [Google Scholar]
- 119.Klausen B, Hougen HP, Fiehn NE. Increased periodontal bone loss in temporarily B lymphocyte-deficient rats. J Periodontal Res. 1989;24:384–390. doi: 10.1111/j.1600-0765.1989.tb00887.x. [DOI] [PubMed] [Google Scholar]
- 120.Knauper V, Cowell S, Smith B, Lopez-Otin C, O’Shea M, Morris H, Zardi L, Murphy G. The role of the C-terminal domain of human collagenase-3 (MMP-13) in the activation of procollagenase-3, substrate specificity, and tissue inhibitor of metalloproteinase interaction. J Biol Chem. 1997;272:7608–7616. doi: 10.1074/jbc.272.12.7608. [DOI] [PubMed] [Google Scholar]
- 121.Kobayashi M, Fitz L, Ryan M, Hewick RM, Clark SC, Chan S, Loudon R, Sherman F, Perussia B, Trinchieri G. Identification and purification of natural killer cell stimulatory factor (NKSF), a cytokine with multiple biologic effects on human lymphocytes. J Exp Med. 1989;170:827–845. doi: 10.1084/jem.170.3.827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Kong YY, Feige U, Sarosi I, Bolon B, Tafuri A, Morony S, Capparelli C, Li J, Elliott R, McCabe S, Wong T, Campagnuolo G, Moran E, Bogoch ER, Van G, Nguyen LT, Ohashi PS, Lacey DL, Fish E, Boyle WJ, Penninger JM. Activated T cells regulate bone loss and joint destruction in adjuvant arthritis through osteoprotegerin ligand. Nature. 1999;402:304–309. doi: 10.1038/46303. [DOI] [PubMed] [Google Scholar]
- 123.Konttinen Y, Imai S, Suda A. Neuropeptides and the puzzle of bone remodeling. State of the art. Acta Orthop Scand. 1996;67:632–639. doi: 10.3109/17453679608997772. [DOI] [PubMed] [Google Scholar]
- 124.Konttinen YT, Salo T, Hanemaaijer R, Valleala H, Sorsa T, Sutinen M, Ceponis A, Xu JW, Santavirta S, Teronen O, Lopez-Otin C. Collagenase-3 (MMP-13) and its activators in rheumatoid arthritis: localization in the pannus-hard tissue junction and inhibition by alendronate. Matrix Biol. 1999;18:401–412. doi: 10.1016/s0945-053x(99)00030-x. [DOI] [PubMed] [Google Scholar]
- 125.Krutzik SR, Modlin RL. The role of toll-like receptors in combating mycobacteria. Semin Immunol. 2004;16:35–41. doi: 10.1016/j.smim.2003.10.005. [DOI] [PubMed] [Google Scholar]
- 126.Kvinnsland I, Heyeraas KJ. Effect of traumatic occlusion on CGRP and SP immunoreactive nerve fibre morphology in rat molar pulp and periodontium. Histochemistry. 1992;97:111–120. doi: 10.1007/BF00267300. [DOI] [PubMed] [Google Scholar]
- 127.Kwak HB, Lee SW, Jin HM, Ha H, Lee SH, Takeshita S, Tanaka S, Kim HM, Kim HH, Lee ZH. Monokine induced by interferon-gamma is induced by receptor activator of nuclear factor kappa B ligand and is involved in osteoclast adhesion and migration. Blood. 2005;105:2963–2969. doi: 10.1182/blood-2004-07-2534. [DOI] [PubMed] [Google Scholar]
- 128.Kwan Tat S, Padrines M, Theoleyre S, Heymann D, Fortun Y. IL-6, RANKL, TNF-alpha / IL-1: interrelations in bone resorption pathophysiology. Cytokine Growth Factor Rev. 2004;15:49–60. doi: 10.1016/j.cytogfr.2003.10.005. [DOI] [PubMed] [Google Scholar]
- 129.Lacey DL, Timms E, Tan HL, Kelley MJ, Dunstan CR, Burgess T, Elliott R, Colombero A, Elliott G, Scully S, Hsu H, Sullivan J, Hawkins N, Davy E, Capparelli C, Eli A, Qian YX, Kaufman S, Sarosi I, Shalhoub V, Senaldi G, Guo J, Delaney J, Boyle WJ. Osteoprotegerin ligand is a cytokine that regulates osteoclast differentiation and activation. Cell. 1998;93:165–176. doi: 10.1016/s0092-8674(00)81569-x. [DOI] [PubMed] [Google Scholar]
- 130.Lai JP, Douglas SD, Ho WZ. Human lymphocytes express substance P and its receptor. J Neuroimmunol. 1998;86:80–86. doi: 10.1016/s0165-5728(98)00025-3. [DOI] [PubMed] [Google Scholar]
- 131.Lambrecht BN, Germonpre PR, Everaert EG, Carro-Muino I, De Veerman M, de Felipe C, Hunt SP, Thielemans K, Joos GF, Pauwels RA. Endogenously produced substance P contributes to lymphocyte proliferation induced by dendritic cells and direct TCR ligation. Eur J Immunol. 1999;29:3815–3825. doi: 10.1002/(SICI)1521-4141(199912)29:12<3815::AID-IMMU3815>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- 132.Lamont AG, Adorini L. IL-12: a key cytokine in immune regulation. Immunol Today. 1996;17:214–217. doi: 10.1016/0167-5699(96)30011-x. [DOI] [PubMed] [Google Scholar]
- 133.Lappin DF, MacLeod CP, Kerr A, Mitchell T, Kinane DF. Anti-inflammatory cytokine IL-10 and T cell cytokine profile in periodontitis granulation tissue. Clin Exp Immunol. 2001;123:294–300. doi: 10.1046/j.1365-2249.2001.01448.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Lean JM, Murphy C, Fuller K, Chambers TJ. CCL9 / MIP-1gamma and its receptor CCR1 are the major chemokine ligand / receptor species expressed by osteoclasts. J Cell Biochem. 2002;87:386–393. doi: 10.1002/jcb.10319. [DOI] [PubMed] [Google Scholar]
- 135.Lee W, Aitken S, Sodek J, McCulloch CA. Evidence of a direct relationship between neutrophil collagenase activity and periodontal tissue destruction in vivo: role of active enzyme in human periodontitis. J Periodontal Res. 1995;30:23–33. doi: 10.1111/j.1600-0765.1995.tb01249.x. [DOI] [PubMed] [Google Scholar]
- 136.Lembeck F, Donnerer J, Tsuchiya M, Nagahisa A. The non-peptide tachykinin antagonist, CP-96,345, is a potent inhibitor of neurogenic inflammation. Br J Pharmacol. 1992;105:527–530. doi: 10.1111/j.1476-5381.1992.tb09013.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Levite M. Neuropeptides, by direct interaction with T cells, induce cytokine secretion and break the commitment to a distinct T helper phenotype. Proc Natl Acad Sci USA. 1998;95:12544–12549. doi: 10.1073/pnas.95.21.12544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Levy BD, Clish CB, Schmidt B, Gronert K, Serhan CN. Lipid mediator class switching during acute inflammation: signals in resolution. Nat Immunol. 2001;2:612–619. doi: 10.1038/89759. [DOI] [PubMed] [Google Scholar]
- 139.Lewis RA. Interactions of eicosanoids and cytokines in immune regulation. Adv Prostaglandin Thromboxane Leukot Res. 1990;20:170–178. [PubMed] [Google Scholar]
- 140.Lieb K, Fiebich BL, Busse-Grawitz M, Hull M, Berger M, Bauer J. Effects of substance P and selected other neuropeptides on the synthesis of interleukin-1 beta and interleukin-6 in human monocytes: a re-examination. J Neuroimmunol. 1996;67:77–81. doi: 10.1016/0165-5728(96)00034-3. [DOI] [PubMed] [Google Scholar]
- 141.Linden GJ, McKinnell J, Shaw C, Lundy FT. Substance P and neurokinin A in gingival crevicular fluid in periodontal health and disease. J Clin Periodontol. 1997;24:799–803. doi: 10.1111/j.1600-051x.1997.tb01192.x. [DOI] [PubMed] [Google Scholar]
- 142.Linden GJ, Mullally BH, Burden DJ, Lamey PJ, Shaw C, Ardill J, Lundy FT. Changes in vasoactive intestinal peptide in gingival crevicular fluid in response to periodontal treatment. J Clin Periodontol. 2002;29:484–489. doi: 10.1034/j.1600-051x.2002.290602.x. [DOI] [PubMed] [Google Scholar]
- 143.Lisignoli G, Cristino S, Toneguzzi S, Grassi F, Piacentini A, Cavallo C, Facchini A, Mariani E. IL1beta and TNFalpha differently modulate CXCL13 chemokine in stromal cells and osteoblasts isolated from osteoarthritis patients: evidence of changes associated to cell maturation. Exp Gerontol. 2004;39:659–665. doi: 10.1016/j.exger.2003.09.030. [DOI] [PubMed] [Google Scholar]
- 144.Lisignoli G, Toneguzzi S, Piacentini A, Cristino S, Grassi F, Cavallo C, Facchini A. CXCL12 (SDF-1) and CXCL13 (BCA-1) chemokines significantly induce proliferation and collagen type I expression in osteoblasts from osteoarthritis patients. J Cell Physiol. 2006;206:78–85. doi: 10.1002/jcp.20435. [DOI] [PubMed] [Google Scholar]
- 145.Liu YC, Lerner UH, Teng YT. Cytokine responses against periodontal infection: protective and destructive roles. Periodontol 2000. 2010;52:163–206. doi: 10.1111/j.1600-0757.2009.00321.x. [DOI] [PubMed] [Google Scholar]
- 146.Loetscher P, Pellegrino A, Gong JH, Mattioli I, Loetscher M, Bardi G, Baggiolini M, Clark-Lewis I. The ligands of cxc chemokine receptor 3, i-tac, mig, and ip10, are natural antagonists for CCR3. J Biol Chem. 2001;276:2986–2991. doi: 10.1074/jbc.M005652200. [DOI] [PubMed] [Google Scholar]
- 147.Loetscher P, Uguccioni M, Bordoli L, Baggiolini M, Moser B, Chizzolini C, Dayer JM. Ccr5 is characteristic of th1 lymphocytes. Nature. 1998;391:344–345. doi: 10.1038/34814. [DOI] [PubMed] [Google Scholar]
- 148.Loning T, Albers HK, Lisboa BP, Burkhardt A, Caselitz J. Prostaglandin e and the local immune response in chronic periodontal disease. Immunohistochemical and radioimmunological observations. J Periodontal Res. 1980;15:525–535. doi: 10.1111/j.1600-0765.1980.tb00310.x. [DOI] [PubMed] [Google Scholar]
- 149.Low QE, Drugea IA, Duffner LA, Quinn DG, Cook DN, Rollins BJ, Kovacs EJ, DiPietro LA. Wound healing in MIP-1alpha −/−) and MCP-1 −/−) mice. Am J Pathol. 2001;159:457–463. doi: 10.1016/s0002-9440(10)61717-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Lundy FT, Linden GJ. Neuropeptides and neurogenic mechanisms in oral and periodontal inflammation. Crit Rev Oral Biol Med. 2004;15:82–98. doi: 10.1177/154411130401500203. [DOI] [PubMed] [Google Scholar]
- 151.Lundy FT, Mullally BH, Burden DJ, Lamey PJ, Shaw C, Linden GJ. Changes in substance P and neurokinin A in gingival crevicular fluid in response to periodontal treatment. J Clin Periodontol. 2000;27:526–530. doi: 10.1034/j.1600-051x.2000.027007526.x. [DOI] [PubMed] [Google Scholar]
- 152.Lundy FT, Shaw C, McKinnell J, Lamey PJ, Linden GJ. Calcitonin gene-related peptide in gingival crevicular fluid in periodontal health and disease. J Clin Periodontol. 1999;26:212–216. doi: 10.1034/j.1600-051x.1999.260403.x. [DOI] [PubMed] [Google Scholar]
- 153.Luthman J, Johansson O, Ahlstrom U, Kvint S. Immunohistochemical studies of the neurochemical markers, CGRP, enkephalin, galanin, gamma-MSH, NPY, PHI, proctolin, PTH, somatostatin, SP, VIP, tyrosine hydroxylase and neurofilament in nerves and cells of the human attached gingiva. Arch Oral Biol. 1988;33:149–158. doi: 10.1016/0003-9969(88)90039-8. [DOI] [PubMed] [Google Scholar]
- 154.Novak JM, Novak KF. Chronic periodontitis. In: Carranza FA, editor. Clinical periodontology. Philadelphia: Saunders Elsevier; 2006. pp. 494–499. [Google Scholar]
- 155.MacDonald KP, Rowe V, Bofinger HM, Thomas R, Sasmono T, Hume DA, Hill GR. The colony-stimulating factor 1 receptor is expressed on dendritic cells during differentiation and regulates their expansion. J Immunol. 2005;175:1399–1405. doi: 10.4049/jimmunol.175.3.1399. [DOI] [PubMed] [Google Scholar]
- 156.Maddox JF, Hachicha M, Takano T, Petasis NA, Fokin VV, Serhan CN. Lipoxin A4 stable analogs are potent mimetics that stimulate human monocytes and THP-1 cells via a G-protein-linked lipoxin A4 receptor. J Biol Chem. 1997;272:6972–6978. doi: 10.1074/jbc.272.11.6972. [DOI] [PubMed] [Google Scholar]
- 157.Maddox JF, Serhan CN. Lipoxin A4 and B4 are potent stimuli for human monocyte migration and adhesion: selective inactivation by dehydrogenation and reduction. J Exp Med. 1996;183:137–146. doi: 10.1084/jem.183.1.137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Mahamed DA, Marleau A, Alnaeeli M, Singh B, Zhang X, Penninger JM, Teng YT. G(−) anaerobes-reactive CD4+ T-cells trigger RANKL-mediated enhanced alveolar bone loss in diabetic NOD mice. Diabetes. 2005;54:1477–1486. doi: 10.2337/diabetes.54.5.1477. [DOI] [PubMed] [Google Scholar]
- 159.Makela M, Larjava H, Pirila E, Maisi P, Salo T, Sorsa T, Uitto VJ. Matrix metalloproteinase 2 (gelatinase A) is related to migration of keratinocytes. Exp Cell Res. 1999;251:67–78. doi: 10.1006/excr.1999.4564. [DOI] [PubMed] [Google Scholar]
- 160.Ryan ME, Preshaw PM. Host modulation. In: Carranza FA, editor. Clinical periodontology. Philadelphia: Saunders Elsevier; 2006. pp. 275–283. [Google Scholar]
- 161.Marriott I, Bost KL. Substance P diminishes lipopolysaccharide and interferon-gamma-induced TGF-beta 1 production by cultured murine macrophages. Cell Immunol. 1998;183:113–120. doi: 10.1006/cimm.1998.1248. [DOI] [PubMed] [Google Scholar]
- 162.Martin TJ, Sims NA. Osteoclast-derived activity in the coupling of bone formation to resorption. Trends Mol Med. 2005;11:76–81. doi: 10.1016/j.molmed.2004.12.004. [DOI] [PubMed] [Google Scholar]
- 163.Mathur A, Michalowicz B, Castillo M, Aeppli D. Interleukin-1 alpha, interleukin-8 and interferon-alpha levels in gingival crevicular fluid. J Periodontal Res. 1996;31:489–495. doi: 10.1111/j.1600-0765.1996.tb01414.x. [DOI] [PubMed] [Google Scholar]
- 164.McCawley LJ, Matrisian LM. Matrix metalloproteinases: they’re not just for matrix anymore! Curr Opin Cell Biol. 2001;13:534–540. doi: 10.1016/s0955-0674(00)00248-9. [DOI] [PubMed] [Google Scholar]
- 165.McGillis JP, Humphreys S, Reid S. Characterization of functional calcitonin gene-related peptide receptors on rat lymphocytes. J Immunol. 1991;147:3482–3489. [PubMed] [Google Scholar]
- 166.Medzhitov R, Janeway CA., Jr Innate immunity: the virtues of a nonclonal system of recognition. Cell. 1997;91:295–298. doi: 10.1016/s0092-8674(00)80412-2. [DOI] [PubMed] [Google Scholar]
- 167.Mendieta CF, Reeve CM, Romero JC. Biosynthesis of prostaglandins in gingiva of patients with chronic periodontitis. J Periodontol. 1985;56:44–47. doi: 10.1902/jop.1985.56.1.44. [DOI] [PubMed] [Google Scholar]
- 168.Metwali A, Blum AM, Ferraris L, Klein JS, Fiocchi C, Weinstock JV. Eosinophils within the healthy or inflamed human intestine produce substance P and vasoactive intestinal peptide. J Neuroimmunol. 1994;52:69–78. doi: 10.1016/0165-5728(94)90164-3. [DOI] [PubMed] [Google Scholar]
- 169.Mizuno A, Amizuka N, Irie K, Murakami A, Fujise N, Kanno T, Sato Y, Nakagawa N, Yasuda H, Mochizuki S, Gomibuchi T, Yano K, Shima N, Washida N, Tsuda E, Morinaga T, Higashio K, Ozawa H. Severe osteoporosis in mice lacking osteoclastogenesis inhibitory factor / osteoprotegerin. Biochem Biophys Res Commun. 1998;247:610–615. doi: 10.1006/bbrc.1998.8697. [DOI] [PubMed] [Google Scholar]
- 170.Modlin RL, Nutman TB. Type 2 cytokines and negative immune regulation in human infections. Curr Opin Immunol. 1993;5:511–517. doi: 10.1016/0952-7915(93)90031-m. [DOI] [PubMed] [Google Scholar]
- 171.Moser B, Wolf M, Walz A, Loetscher P. Chemokines: multiple levels of leukocyte migration control. Trends Immunol. 2004;25:75–84. doi: 10.1016/j.it.2003.12.005. [DOI] [PubMed] [Google Scholar]
- 172.Murphy KM, Reiner SL. The lineage decisions of helper T cells. Nat Rev Immunol. 2002;2:933–944. doi: 10.1038/nri954. [DOI] [PubMed] [Google Scholar]
- 173.Musacchio E, Valvason C, Botsios C, Ostuni F, Furlan A, Ramonda R, Modesti V, Sartori L, Punzi L. The tumor necrosis factor-{alpha}-blocking agent infliximab inhibits interleukin 1beta (IL-1beta) and IL-6 gene expression in human osteoblastic cells. J Rheumatol. 2009;36:1575–1579. doi: 10.3899/jrheum.081321. [DOI] [PubMed] [Google Scholar]
- 174.Nagase H. Activation mechanisms of matrix metalloproteinases. Biol Chem. 1997;378:151–160. [PubMed] [Google Scholar]
- 175.Nakajima T, Ueki-Maruyama K, Oda T, Ohsawa Y, Ito H, Seymour GJ, Yamazaki K. Regulatory T-cells infiltrate periodontal disease tissues. J Dent Res. 2005;84:639–643. doi: 10.1177/154405910508400711. [DOI] [PubMed] [Google Scholar]
- 176.Naundorf S, Schroder M, Hoflich C, Suman N, Volk HD, Grutz G. IL-10 interferes directly with TCR-induced IFN-gamma but not IL-17 production in memory T cells. Eur J Immunol. 2009;39:1066–1077. doi: 10.1002/eji.200838773. [DOI] [PubMed] [Google Scholar]
- 177.Nong YH, Titus RG, Ribeiro JM, Remold HG. Peptides encoded by the calcitonin gene inhibit macrophage function. J Immunol. 1989;143:45–49. [PubMed] [Google Scholar]
- 178.O’Dorisio MS, O’Dorisio TM, Cataland S, Balcerzak SP. Vasoactive intestinal polypeptide as a biochemical marker for polymorphonuclear leukocytes. J Lab Clin Med. 1980;96:666–672. [PubMed] [Google Scholar]
- 179.O’Garra A. Interleukins and the immune system 2. Lancet. 1989;1:1003–1005. doi: 10.1016/s0140-6736(89)92640-8. [DOI] [PubMed] [Google Scholar]
- 180.Ohyama H, Kato-Kogoe N, Kuhara A, Nishimura F, Nakasho K, Yamanegi K, Yamada N, Hata M, Yamane J, Terada N. The involvement of IL-23 and the Th17 pathway in periodontitis. J Dent Res. 2009;88:633–638. doi: 10.1177/0022034509339889. [DOI] [PubMed] [Google Scholar]
- 181.Okada H, Murakami S. Cytokine expression in periodontal health and disease. Crit Rev Oral Biol Med. 1998;9:248–266. doi: 10.1177/10454411980090030101. [DOI] [PubMed] [Google Scholar]
- 182.Okada N, Kobayashi M, Mugikura K, Okamatsu Y, Hanazawa S, Kitano S, Hasegawa K. Interleukin-6 production in human fibroblasts derived from periodontal tissues is differentially regulated by cytokines and a glucocorticoid. J Periodontal Res. 1997;32:559–569. doi: 10.1111/j.1600-0765.1997.tb00932.x. [DOI] [PubMed] [Google Scholar]
- 183.Okamatsu Y, Kim D, Battaglino R, Sasaki H, Spate U, Stashenko P. MIP-1 gamma promotes receptor-activator- of-NF-kappa-B-ligand-induced osteoclast formation and survival. J Immunol. 2004;173:2084–2090. doi: 10.4049/jimmunol.173.3.2084. [DOI] [PubMed] [Google Scholar]
- 184.Opree A, Kress M. Involvement of the proinflammatory cytokines tumor necrosis factor-alpha, IL-1 beta, and IL-6 but not IL-8 in the development of heat hyperalgesia: effects on heat-evoked calcitonin gene-related peptide release from rat skin. J Neurosci. 2000;20:6289–6293. doi: 10.1523/JNEUROSCI.20-16-06289.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Owan I, Ibaraki K. The role of calcitonin gene-related peptide (CGRP) in macrophages: the presence of functional receptors and effects on proliferation and differentiation into osteoclast-like cells. Bone Miner. 1994;24:151–164. doi: 10.1016/s0169-6009(08)80152-3. [DOI] [PubMed] [Google Scholar]
- 186.Page RC, Schroeder HE. Pathogenesis of inflammatory periodontal disease. A summary of current work. Lab Invest. 1976;34:235–249. [PubMed] [Google Scholar]
- 187.The American Academy of Periodontology. The pathogenesis of periodontal diseases. J Periodontol. 1999;70:457–470. doi: 10.1902/jop.1999.70.4.457. [DOI] [PubMed] [Google Scholar]
- 188.Payne JB, Reinhardt RA, Masada MP, DuBois LM, Allison AC. Gingival crevicular fluid IL-8: correlation with local IL-1 beta levels and patient estrogen status. J Periodontal Res. 1993;28:451–453. [PubMed] [Google Scholar]
- 189.Perussia B, Chan SH, D’Andrea A, Tsuji K, Santoli D, Pospisil M, Young D, Wolf SF, Trinchieri G. Natural killer (NK) cell stimulatory factor or IL-12 has differential effects on the proliferation of TCR-alpha beta+, TCR-gamma delta+ T lymphocytes, and NK cells. J Immunol. 1992;149:3495–3502. [PubMed] [Google Scholar]
- 190.Peschon JJ, Torrance DS, Stocking KL, Glaccum MB, Otten C, Willis CR, Charrier K, Morrissey PJ, Ware CB, Mohler KM. TNF receptor-deficient mice reveal divergent roles for p55 and p75 in several models of inflammation. J Immunol. 1998;160:943–952. [PubMed] [Google Scholar]
- 191.Pestka S, Krause CD, Sarkar D, Walter MR, Shi Y, Fisher PB. Interleukin-10 and related cytokines and receptors. Annu Rev Immunol. 2004;22:929–979. doi: 10.1146/annurev.immunol.22.012703.104622. [DOI] [PubMed] [Google Scholar]
- 192.Pihlstrom BL, Michalowicz BS, Johnson NW. Periodontal diseases. Lancet. 2005;366:1809–1820. doi: 10.1016/S0140-6736(05)67728-8. [DOI] [PubMed] [Google Scholar]
- 193.Pozo D, Delgado M, Martinez M, Guerrero JM, Leceta J, Gomariz RP, Calvo JR. Immunobiology of vasoactive intestinal peptide (VIP) Immunol Today. 2000;21:7–11. doi: 10.1016/s0167-5699(99)01525-x. [DOI] [PubMed] [Google Scholar]
- 194.Putnins EE, Firth JD, Uitto VJ. Stimulation of collagenase (matrix metalloproteinase-1) synthesis in histiotypic epithelial cell culture by heparin is enhanced by keratinocyte growth factor. Matrix Biol. 1996;15:21–29. doi: 10.1016/s0945-053x(96)90123-7. [DOI] [PubMed] [Google Scholar]
- 195.Quesniaux V, Fremond C, Jacobs M, Parida S, Nicolle D, Yeremeev V, Bihl F, Erard F, Botha T, Drennan M, Soler MN, Le Bert M, Schnyder B, Ryffel B. Toll-like receptor pathways in the immune responses to mycobacteria. Microbes Infect. 2004;6:946–959. doi: 10.1016/j.micinf.2004.04.016. [DOI] [PubMed] [Google Scholar]
- 196.Richards D, Rutherford RB. The effects of interleukin 1 on collagenolytic activity and prostaglandin-E secretion by human periodontal-ligament and gingival fibroblast. Arch Oral Biol. 1988;33:237–243. doi: 10.1016/0003-9969(88)90184-7. [DOI] [PubMed] [Google Scholar]
- 197.Rietschel ET, Brade H. Bacterial endotoxins. Sci Am. 1992;267:54–61. doi: 10.1038/scientificamerican0892-54. [DOI] [PubMed] [Google Scholar]
- 198.Rodan GA, Martin TJ. Role of osteoblasts in hormonal control of bone resorption–a hypothesis. Calcif Tissue Int. 1981;33:349–351. doi: 10.1007/BF02409454. [DOI] [PubMed] [Google Scholar]
- 199.Rosa FM, Mellows M. Effect of gamma interferon on MHC antigens. Immunol Today. 1984;9:261–262. doi: 10.1016/0167-5699(84)90135-X. [DOI] [PubMed] [Google Scholar]
- 200.Rossi D, Zlotnik A. The biology of chemokines and their receptors. Annu Rev Immunol. 2000;18:217–242. doi: 10.1146/annurev.immunol.18.1.217. [DOI] [PubMed] [Google Scholar]
- 201.Rot A, von Andrian UH. Chemokines in innate and adaptive host defense: basic chemokinese grammar for immune cells. Annu Rev Immunol. 2004;22:891–928. doi: 10.1146/annurev.immunol.22.012703.104543. [DOI] [PubMed] [Google Scholar]
- 202.Rousset F, Garcia E, Defrance T, Peronne C, Vezzio N, Hsu DH, Kastelein R, Moore KW, Banchereau J. Interleukin 10 is a potent growth and differentiation factor for activated human B lymphocytes. Proc Natl Acad Sci USA. 1992;89:1890–1893. doi: 10.1073/pnas.89.5.1890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Saarialho-Kere UK. Patterns of matrix metalloproteinase and TIMP expression in chronic ulcers. Arch Dermatol Res. 1998;290(Suppl):S47–S54. doi: 10.1007/pl00007453. [DOI] [PubMed] [Google Scholar]
- 204.Saidenberg-Kermanac’h N, Bessis N, Lemeiter D, de Vernejoul MC, Boissier MC, Cohen-Solal M. Interleukin-4 cellular gene therapy and osteoprotegerin decrease inflammation-associated bone resorption in collagen-induced arthritis. J Clin Immunol. 2004;24:370–378. doi: 10.1023/B:JOCI.0000029116.12371.bf. [DOI] [PubMed] [Google Scholar]
- 205.Saito S, Rosol TJ, Saito M, Ngan PW, Shanfeld J, Davidovitch Z. Bone-resorbing activity and prostaglandin E produced by human periodontal ligament cells in vitro. J Bone Miner Res. 1990;5:1013–1018. doi: 10.1002/jbmr.5650051004. [DOI] [PubMed] [Google Scholar]
- 206.Saito S, Saito M, Ngan P, Lanese R, Shanfeld J, Davidovitch Z. Effects of parathyroid hormone and cytokines on prostaglandin E synthesis and bone resorption by human periodontal ligament fibroblasts. Arch Oral Biol. 1990;35:845–855. doi: 10.1016/0003-9969(90)90010-8. [DOI] [PubMed] [Google Scholar]
- 207.Sallusto F, Lanzavecchia A. Heterogeneity of CD4+ memory T cells: functional modules for tailored immunity. Eur J Immunol. 2009;39:2076–2082. doi: 10.1002/eji.200939722. [DOI] [PubMed] [Google Scholar]
- 208.Sallusto F, Lanzavecchia A, Mackay CR. Chemokines and chemokine receptors in T-cell priming and Th1 / Th2-mediated responses. Immunol Today. 1998;19:568–574. doi: 10.1016/s0167-5699(98)01346-2. [DOI] [PubMed] [Google Scholar]
- 209.Sallusto F, Lenig D, Mackay CR, Lanzavecchia A. Flexible programs of chemokine receptor expression on human polarized T helper 1 and 2 lymphocytes. J Exp Med. 1998;187:875–883. doi: 10.1084/jem.187.6.875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Sato N, Takahashi N, Suda K, Nakamura M, Yamaki M, Ninomiya T, Kobayashi Y, Takada H, Shibata K, Yamamoto M, Takeda K, Akira S, Noguchi T, Udagawa N. MyD88 but not TRIF is essential for osteoclastogenesis induced by lipopolysaccharide, diacyl lipopeptide, and IL-1alpha. J Exp Med. 2004;200:601–611. doi: 10.1084/jem.20040689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Schaerli P, Willimann K, Lang AB, Lipp M, Loetscher P, Moser B. CXC chemokine receptor 5 expression defines follicular homing T cells with B cell helper function. J Exp Med. 2000;192:1553–1562. doi: 10.1084/jem.192.11.1553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Schwab JM, Chiang N, Arita M, Serhan CN. Resolvin E1 and protectin D1 activate inflammation-resolution programmes. Nature. 2007;447:869–874. doi: 10.1038/nature05877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Serhan CN. A search for endogenous mechanisms of anti-inflammation uncovers novel chemical mediators: missing links to resolution. Histochem Cell Biol. 2004;122:305–321. doi: 10.1007/s00418-004-0695-8. [DOI] [PubMed] [Google Scholar]
- 214.Serhan CN, Brain SD, Buckley CD, Gilroy DW, Haslett C, O’Neill LA, Perretti M, Rossi AG, Wallace JL. Resolution of inflammation: state of the art, definitions and terms. FASEB J. 2007;21:325–332. doi: 10.1096/fj.06-7227rev. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Serhan CN, Chiang N. Endogenous pro-resolving and anti-inflammatory lipid mediators: a new pharmacologic genus. Br J Pharmacol. 2008;153(Suppl 1):S200–S215. doi: 10.1038/sj.bjp.0707489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Serhan CN, Chiang N, Van Dyke TE. Resolving inflammation: dual anti-inflammatory and pro-resolution lipid mediators. Nat Rev Immunol. 2008;8:349–361. doi: 10.1038/nri2294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Serhan CN, Fiore S, Brezinski DA, Lynch S. Lipoxin A4 metabolism by differentiated HL-60 cells and human monocytes: conversion to novel 15-oxo and dihydro products. Biochemistry. 1993;32:6313–6319. doi: 10.1021/bi00076a002. [DOI] [PubMed] [Google Scholar]
- 218.Serhan CN, Maddox JF, Petasis NA, Akritopoulou-Zanze I, Papayianni A, Brady HR, Colgan SP, Madara JL. Design of lipoxin A4 stable analogs that block transmigration and adhesion of human neutrophils. Biochemistry. 1995;34:14609–14615. doi: 10.1021/bi00044a041. [DOI] [PubMed] [Google Scholar]
- 219.Seymour GJ. Importance of the host response in the periodontium. J Clin Periodontol. 1991;18:421–426. doi: 10.1111/j.1600-051x.1991.tb02310.x. [DOI] [PubMed] [Google Scholar]
- 220.Seymour GJ, Gemmell E. Cytokines in periodontal disease: where to from here? Acta Odontol Scand. 2001;59:167–173. doi: 10.1080/000163501750266765. [DOI] [PubMed] [Google Scholar]
- 221.Sorsa T, Tjaderhane L, Konttinen YT, Lauhio A, Salo T, Lee HM, Golub LM, Brown DL, Mantyla P. Matrix metalloproteinases: contribution to pathogenesis, diagnosis and treatment of periodontal inflammation. Ann Med. 2006;38:306–321. doi: 10.1080/07853890600800103. [DOI] [PubMed] [Google Scholar]
- 222.Steinhoff M, Vergnolle N, Young SH, Tognetto M, Amadesi S, Ennes HS, Trevisani M, Hollenberg MD, Wallace JL, Caughey GH, Mitchell SE, Williams LM, Geppetti P, Mayer EA, Bunnett NW. Agonists of proteinase-activated receptor 2 induce inflammation by a neurogenic mechanism. Nat Med. 2000;6:151–158. doi: 10.1038/72247. [DOI] [PubMed] [Google Scholar]
- 223.Steinsvoll S, Halstensen TS, Schenck K. Extensive expression of TGF-beta1 in chronically-inflamed periodontal tissue. J Clin Periodontol. 1999;26:366–373. doi: 10.1034/j.1600-051x.1999.260606.x. [DOI] [PubMed] [Google Scholar]
- 224.Suda K, Udagawa N, Sato N, Takami M, Itoh K, Woo JT, Takahashi N, Nagai K. Suppression of osteoprotegerin expression by prostaglandin E2 is crucially involved in lipopolysaccharide-induced osteoclast formation. J Immunol. 2004;172:2504–2510. doi: 10.4049/jimmunol.172.4.2504. [DOI] [PubMed] [Google Scholar]
- 225.Suzuki JB, Risom L, Falkler WA, Jr, Collison C, Bowers G. Effect of periodontal therapy on spontaneous lymphocyte response and neutrophil chemotaxis in localized and generalized juvenile periodontitis patients. J Clin Periodontol. 1985;12:124–134. doi: 10.1111/j.1600-051x.1985.tb01371.x. [DOI] [PubMed] [Google Scholar]
- 226.Takahashi K, Azuma T, Motohira H, Kinane DF, Kitetsu S. The potential role of interleukin-17 in the immunopathology of periodontal disease. J Clin Periodontol. 2005;32:369–374. doi: 10.1111/j.1600-051X.2005.00676.x. [DOI] [PubMed] [Google Scholar]
- 227.Takashiba S, Takigawa M, Takahashi K, Myokai F, Nishimura F, Chihara T, Kurihara H, Nomura Y, Murayama Y. Interleukin-8 is a major neutrophil chemotactic factor derived from cultured human gingival fibroblasts stimulated with interleukin-1 beta or tumor necrosis factor alpha. Infect Immun. 1992;60:5253–5258. doi: 10.1128/iai.60.12.5253-5258.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Takeda K, Kaisho T, Akira S. Toll-like receptors. Annu Rev Immunol. 2003;21:335–376. doi: 10.1146/annurev.immunol.21.120601.141126. [DOI] [PubMed] [Google Scholar]
- 229.Takigawa M, Takashiba S, Myokai F, Takahashi K, Arai H, Kurihara H, Murayama Y. Cytokine-dependent synergistic regulation of interleukin-8 production from human gingival fibroblasts. J Periodontol. 1994;65:1002–1007. doi: 10.1902/jop.1994.65.11.1002. [DOI] [PubMed] [Google Scholar]
- 230.Tanaka S, Takahashi N, Udagawa N, Tamura T, Akatsu T, Stanley ER, Kurokawa T, Suda T. Macrophage colony-stimulating factor is indispensable for both proliferation and differentiation of osteoclast progenitors. J Clin Invest. 1993;91:257–263. doi: 10.1172/JCI116179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Taubman MA, Kawai T. Involvement of T-lymphocytes in periodontal disease and in direct and indirect induction of bone resorption. Crit Rev Oral Biol Med. 2001;12:125–135. doi: 10.1177/10454411010120020301. [DOI] [PubMed] [Google Scholar]
- 232.Taylor AW, Yee DG, Streilein JW. Suppression of nitric oxide generated by inflammatory macrophages by calcitonin gene-related peptide in aqueous humor. Invest Ophthalmol Vis Sci. 1998;39:1372–1378. [PubMed] [Google Scholar]
- 233.Terricabras E, Benjamim C, Godessart N. Drug discovery and chemokine receptor antagonists: Eppur si muove! Autoimmun Rev. 2004;3:550–556. doi: 10.1016/j.autrev.2004.07.037. [DOI] [PubMed] [Google Scholar]
- 234.Tervahartiala T, Pirila E, Ceponis A, Maisi P, Salo T, Tuter G, Kallio P, Tornwall J, Srinivas R, Konttinen YT, Sorsa T. The in vivo expression of the collagenolytic matrix metalloproteinases (MMP-2, -8, -13, and -14) and matrilysin (MMP-7) in adult and localized juvenile periodontitis. J Dent Res. 2000;79:1969–1977. doi: 10.1177/00220345000790120801. [DOI] [PubMed] [Google Scholar]
- 235.Theill LE, Boyle WJ, Penninger JM. RANK-l and RANK: T cells, bone loss, and mammalian evolution. Annu Rev Immunol. 2002;20:795–823. doi: 10.1146/annurev.immunol.20.100301.064753. [DOI] [PubMed] [Google Scholar]
- 236.Tracey KJ. The inflammatory reflex. Nature. 2002;420:853–859. doi: 10.1038/nature01321. [DOI] [PubMed] [Google Scholar]
- 237.Trinchieri G. Interleukin-12 and its role in the generation of Th1 cells. Immunol Today. 1993;14:335–338. doi: 10.1016/0167-5699(93)90230-I. [DOI] [PubMed] [Google Scholar]
- 238.Trinchieri G, Perussia B. Immune interferon: a pleiotropic lymphokine with multiple effects. Immunol Today. 1985;6:131–136. doi: 10.1016/0167-5699(85)90080-5. [DOI] [PubMed] [Google Scholar]
- 239.Uehara A, Takada H. Functional TLRs and NODs in human gingival fibroblasts. J Dent Res. 2007;86:249–254. doi: 10.1177/154405910708600310. [DOI] [PubMed] [Google Scholar]
- 240.Uitto VJ, Airola K, Vaalamo M, Johansson N, Putnins EE, Firth JD, Salonen J, Lopez-Otin C, Saarialho-Kere U, Kahari VM. Collagenase-3 (matrix metalloproteinase-13) expression is induced in oral mucosal epithelium during chronic inflammation. Am J Pathol. 1998;152:1489–1499. [PMC free article] [PubMed] [Google Scholar]
- 241.Uitto VJ, Overall CM, McCulloch C. Proteolytic host cell enzymes in gingival crevice fluid. Periodontol 2000. 2003;31:77–104. doi: 10.1034/j.1600-0757.2003.03106.x. [DOI] [PubMed] [Google Scholar]
- 242.van den Steen PE, Dubois B, Nelissen I, Rudd PM, Dwek RA, Opdenakker G. Biochemistry and molecular biology of gelatinase B or matrix metalloproteinase-9 (MMP-9) Crit Rev Biochem Mol Biol. 2002;37:375–536. doi: 10.1080/10409230290771546. [DOI] [PubMed] [Google Scholar]
- 243.Van Dyke TE. Control of inflammation and periodontitis. Periodontol 2000. 2007;45:158–166. doi: 10.1111/j.1600-0757.2007.00229.x. [DOI] [PubMed] [Google Scholar]
- 244.Van Dyke TE. The management of inflammation in periodontal disease. J Periodontol. 2008;79:1601–1608. doi: 10.1902/jop.2008.080173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Van Dyke TE, Serhan CN. Resolution of inflammation: a new paradigm for the pathogenesis of periodontal diseases. J Dent Res. 2003;82:82–90. doi: 10.1177/154405910308200202. [DOI] [PubMed] [Google Scholar]
- 246.Vergnolle N. Proteinase-activated receptor-2-activating peptides induce leukocyte rolling, adhesion, and extravasation in vivo. J Immunol. 1999;163:5064–5069. [PubMed] [Google Scholar]
- 247.Vernal R, Dutzan N, Chaparro A, Puente J, Antonieta Valenzuela M, Gamonal J. Levels of interleukin-17 in gingival crevicular fluid and in supernatants of cellular cultures of gingival tissue from patients with chronic periodontitis. J Clin Periodontol. 2005;32:383–389. doi: 10.1111/j.1600-051X.2005.00684.x. [DOI] [PubMed] [Google Scholar]
- 248.Visse R, Nagase H. Matrix metalloproteinases and tissue inhibitors of metalloproteinases: structure, function, and biochemistry. Circ Res. 2003;92:827–839. doi: 10.1161/01.RES.0000070112.80711.3D. [DOI] [PubMed] [Google Scholar]
- 249.Votta BJ, White JR, Dodds RA, James IE, Connor JR, Lee-Rykaczewski E, Eichman CF, Kumar S, Lark MW, Gowen M. CKbeta-8 [CCL23], a novel CC chemokine, is chemotactic for human osteoclast precursors and is expressed in bone tissues. J Cell Physiol. 2000;183:196–207. doi: 10.1002/(SICI)1097-4652(200005)183:2<196::AID-JCP6>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- 250.Wahlgren J, Salo T, Teronen O, Luoto H, Sorsa T, Tjaderhane L. Matrix metalloproteinase-8 (MMP-8) in pulpal and periapical inflammation and periapical root-canal exudates. Int Endod J. 2002;35:897–904. doi: 10.1046/j.1365-2591.2002.00587.x. [DOI] [PubMed] [Google Scholar]
- 251.Wajant H, Pfizenmaier K, Scheurich P. Tumor necrosis factor signaling. Cell Death Differ. 2003;10:45–65. doi: 10.1038/sj.cdd.4401189. [DOI] [PubMed] [Google Scholar]
- 252.Wang F, Millet I, Bottomly K, Vignery A. Calcitonin gene-related peptide inhibits interleukin 2 production by murine T lymphocytes. J Biol Chem. 1992;267:21052–21057. [PubMed] [Google Scholar]
- 253.Watters TM, Kenny EF, O’Neill LA. Structure, function and regulation of the toll / IL-1 receptor adaptor proteins. Immunol Cell Biol. 2007;85:411–419. doi: 10.1038/sj.icb.7100095. [DOI] [PubMed] [Google Scholar]
- 254.Weaver CT, Hatton RD. Interplay between the Th17 and Treg cell lineages: a (co-)evolutionary perspective. Nat Rev Immunol. 2009;9:883–889. doi: 10.1038/nri2660. [DOI] [PubMed] [Google Scholar]
- 255.Wilson CL, Matrisian LM. Matrilysin: an epithelial matrix metalloproteinase with potentially novel functions. Int J Biochem Cell Biol. 1996;28:123–136. doi: 10.1016/1357-2725(95)00121-2. [DOI] [PubMed] [Google Scholar]
- 256.Wingrove JA, DiScipio RG, Chen Z, Potempa J, Travis J, Hugli TE. Activation of complement components C3 and C5 by a cysteine proteinase (gingipain-1) from Porphyromonas (Bacteroides) gingivalis. J Biol Chem. 1992;267:18902–18907. [PubMed] [Google Scholar]
- 257.Wright LM, Maloney W, Yu X, Kindle L, Collin-Osdoby P, Osdoby P. Stromal cell-derived factor-1 binding to its chemokine receptor CXCR4 on precursor cells promotes the chemotactic recruitment, development and survival of human osteoclasts. Bone. 2005;36:840–853. doi: 10.1016/j.bone.2005.01.021. [DOI] [PubMed] [Google Scholar]
- 258.Yago T, Nanke Y, Ichikawa N, Kobashigawa T, Mogi M, Kamatani N, Kotake S. IL-17 induces osteoclastogenesis from human monocytes alone in the absence of osteoblasts, which is potently inhibited by anti-TNF-alpha antibody: a novel mechanism of osteoclastogenesis by IL-17. J Cell Biochem. 2009;108:947–955. doi: 10.1002/jcb.22326. [DOI] [PubMed] [Google Scholar]
- 259.Yang M, Mailhot G, MacKay CA, Mason-Savas A, Aubin J, Odgren PR. Chemokine and chemokine receptor expression during colony stimulating factor-1-induced osteoclast differentiation in the toothless osteopetrotic rat: a key role for CCL9 (MIP-1gamma) in osteoclastogenesis in vivo and in vitro. Blood. 2006;107:2262–2270. doi: 10.1182/blood-2005-08-3365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260.Yano S, Mentaverri R, Kanuparthi D, Bandyopadhyay S, Rivera A, Brown EM, Chattopadhyay N. Functional expression of beta-chemokine receptors in osteoblasts: role of regulated upon activation, normal T cell expressed and secreted (RANTES) in osteoblasts and regulation of its secretion by osteoblasts and osteoclasts. Endocrinology. 2005;146:2324–2335. doi: 10.1210/en.2005-0065. [DOI] [PubMed] [Google Scholar]
- 261.Yasuda H, Shima N, Nakagawa N, Mochizuki SI, Yano K, Fujise N, Sato Y, Goto M, Yamaguchi K, Kuriyama M, Kanno T, Murakami A, Tsuda E, Morinaga T, Higashio K. Identity of osteoclastogenesis inhibitory factor (OCIF) and osteoprotegerin (OPG): a mechanism by which OPG / OCIF inhibits osteoclastogenesis in vitro. Endocrinology. 1998;139:1329–1337. doi: 10.1210/endo.139.3.5837. [DOI] [PubMed] [Google Scholar]
- 262.Yiangou Y, Facer P, Dyer NH, Chan CL, Knowles C, Williams NS, Anand P. Vanilloid receptor 1 immunore-activity in inflamed human bowel. Lancet. 2001;357:1338–1339. doi: 10.1016/s0140-6736(00)04503-7. [DOI] [PubMed] [Google Scholar]
- 263.Yoshimura T, Matsushima K, Tanaka S, Robinson EA, Appella E, Oppenheim JJ, Leonard EJ. Purification of a human monocyte-derived neutrophil chemotactic factor that has peptide sequence similarity to other host defense cytokines. Proc Natl Acad Sci USA. 1987;84:9233–9237. doi: 10.1073/pnas.84.24.9233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264.Yu X, Huang Y, Collin-Osdoby P, Osdoby P. CCR1 chemokines promote the chemotactic recruitment, RANKL development, and motility of osteoclasts and are induced by inflammatory cytokines in osteoblasts. J Bone Miner Res. 2004;19:2065–2077. doi: 10.1359/JBMR.040910. [DOI] [PubMed] [Google Scholar]
- 265.Yumoto H, Nakae H, Fujinaka K, Ebisu S, Matsuo T. Interleukin-6 (IL-6) and IL-8 are induced in human oral epithelial cells in response to exposure to periodontopathic Eikenella corrodens. Infect Immun. 1999;67:384–394. doi: 10.1128/iai.67.1.384-394.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266.Zadeh HH, Nichols FC, Miyasaki KT. The role of the cell-mediated immune response to Actinobacillus actinomycetemcomitans and Porphyromonas gingivalis in periodontitis. Periodontol 2000. 1999;20:239–288. doi: 10.1111/j.1600-0757.1999.tb00163.x. [DOI] [PubMed] [Google Scholar]
- 267.Zlotnik A, Yoshie O. Chemokines: a new classification system and their role in immunity. Immunity. 2000;12:121–127. doi: 10.1016/s1074-7613(00)80165-x. [DOI] [PubMed] [Google Scholar]
- 268.Zou W, Amcheslavsky A, Bar-Shavit Z. CpG oligodeoxynucleotides modulate the osteoclastogenic activity of osteoblasts via toll-like receptor 9. J Biol Chem. 2003;278:16732–16740. doi: 10.1074/jbc.M212473200. [DOI] [PubMed] [Google Scholar]
- 269.Zurawski G, de Vries JE. Interleukin 13, an interleukin 4-like cytokine that acts on monocytes and B cells, but not on T cells. Immunol Today. 1994;15:19–26. doi: 10.1016/0167-5699(94)90021-3. [DOI] [PubMed] [Google Scholar]