

Abstract
The receptor for advanced glycation endproducts (RAGE) was first described as a signal transduction receptor for advanced glycation endproducts (AGEs), the products of nonenzymatic glycation and oxidation of proteins and lipids that accumulate in diabetes and in inflammatory foci. The discovery that RAGE was a receptor for inflammatory S100/calgranulins and high mobility group box 1 (HMGB1) set the stage for linking RAGE to both the consequences and causes of types 1 and 2 diabetes. Recent discoveries regarding the structure of RAGE as well as novel intracellular binding partner interactions advance our understanding of the mechanisms by which RAGE evokes pathological consequences and underscore strategies by which antagonism of RAGE in the clinic may be realized. Finally, recent data tracking RAGE in the clinic suggest that levels of soluble RAGEs and polymorphisms in the gene encoding RAGE may hold promise for the identification of patients who are vulnerable to the complications of diabetes and/or are receptive to therapeutic interventions designed to prevent and reverse the damage inflicted by chronic hyperglycemia, irrespective of its etiology.
Keywords: diabetes, complications, RAGE, inflammation, signal transduction
Introduction
The problem of types 1 and 2 diabetes is a growing one. The incidence of type 1 diabetes, an autoimmune disorder whose pathogenesis is strongly rooted in genetic risk factors, is increasing world-wide at a rate not possibly explained solely by genetic factors.1 Environmental influences such as diet and gut microbiota, pollution, practices of food preparation and preservation, and increased use of antibiotics, as examples, are being investigated as putative underlying mechanisms accounting for this recent acceleration.2 Similarly, the global increase in obesity and greater physical inactivity has been suggested to underlie the alarming rise in the incidence of type 2 diabetes. In addition to the increased overall incidence of type 2 diabetes, it is apparent that an earlier age of onset, particularly in adolescents, is a contributing factor. According to the International Diabetes Federation, the number of adults with impaired glucose tolerance will rise from 344 million in 2010 to a projected 472 million by 2030.3,4 The escalation of type 2 diabetes in the young has led to school-based initiatives to both identify the etiology and find solutions for the increase in type 2 diabetes in young people under 21 years of age to address the crisis.5
In this paper, we will discuss fundamental mechanisms triggered as a consequence of hyperglycemia underlying the pathogenesis of both macro- and microvascular complications in diabetes. In particular, we will focus on recent advances in the biology of the receptor for advanced glycation endproducts (RAGE), an immunoglobulin superfamily molecule whose multiple ligands have been shown to accumulate in diabetic tissues. RAGE was discovered as a receptor for advanced glycation endproducts (AGEs), such as carboxymethyl lysine (CML).6 AGEs, the products of nonenzymatic glycation and oxidation of proteins, form to an accelerated degree in hyperglycemia. AGEs, largely via RAGE, activate signaling mechanisms that cause cell stress, contribute to cellular dysfunction, and damage target organs, leading to complications. The findings that RAGE interacts with non-AGE ligands, such as S100/calgranulins and high mobility group box 1 (HMGB1),7,8 underscore the possibility that RAGE is involved not only in diabetes complications, but in the causes of types 1 and 2 diabetes as well.
RAGE and the cardiovascular complications of diabetes
The chief cause of morbidity and mortality in diabetes is cardiovascular disease, particularly heart attacks and strokes.9 Hence, the mechanisms underlying accelerated atherosclerosis are essential to uncover in order that targeted therapeutic strategies might be developed. In type 1 diabetes, epidemiologic studies of the Diabetes Control and Complications Trials/Epidemiology of Diabetes Interventions and Complications (DCCT/EDIC) showed that intensive glycemic control was protective—even years after levels of glycosylated hemoglobin became indistinguishable from those of the control treated group—against cardiovascular events and death.10 In type 2 diabetes, studies from the United Kingdom demonstrated that incremental rises in glycosylated hemoglobin level were associated with increased risk of cardiovascular disease.11
Prompted by the findings of these large-scale trials, three recent clinical trials addressed whether intensive control of glycemia would afford a beneficial impact in type 2 diabetes by reduction of cardiovascular risk. The outcomes of these three studies were somewhat surprising. The Action to Control Cardiovascular Risk in Diabetes (ACCORD) trial was interrupted prematurely because of more deaths in the intensive versus the standard treatment group. Further, all cause and cardiovascular mortality was not lower in the intensive treatment group versus the standard treatment group.12 A recent follow-up study reported that even after glycosylated hemoglobin levels in the intensive treatment group rose to levels more similar to those in the control treatment groups (6.4–7.2%), five-year mortality was still higher in the previously intensively treated group.13 In two other trials, the Action in Diabetes and Vascular Disease: Preterax and Diamicron Modified Release Evaluation (ADVANCE) and Glucose Control and Vascular Complications in Veterans with Type 2 Diabetes (VADT), although premature deaths were not reported in the intensive treatment group, no benefits of such treatment on cardiovascular disease were observed.14,15 Hence, one conclusion of these studies was that significant hypoglycemia may have been associated with adverse outcomes. Indeed, the investigators of ADVANCE recently published findings on examination of the associations between severe hypoglycemia and diabetic micro- and macrovascular complications. They concluded that severe hypoglycemia was associated with a range of adverse clinical outcomes and could contribute to cardiac ischemia.16
Taken together, these studies suggest that intensive treatment of hyperglycemia and the associated risk of severe hypoglycemia might have been detrimental and did not result in reduction in macrovascular complications, particularly in older subjects. Such findings strongly suggest that strategies directed at blocking the adverse effects of hyperglycemia are likely to be more effective and better tolerated. It is in this context that we propose that RAGE may be one such logical target.
RAGE in cardiovascular disease: studies in animal models
Atherosclerotic plaques retrieved from human subjects reveal that RAGE is expressed in these lesions, and to an enhanced degree in diabetes. RAGE expression colocalizes to smooth muscles (SMCs) and macrophages and to markers of oxidative stress.17 Experiments in animal models of diabetes and accelerated atherosclerosis revealed that blockade of RAGE, using soluble RAGE (the extracellular lig- and binding domain of the receptor) suppressed accelerated atherosclerosis in diabetic apolipoprotein E null mice.18–20 Irrespective of the means by which diabetes was induced—chemical (e.g., streptozotocin) type 1 diabetes model or genetic (e.g., breeding into the db/db background) type 2 diabetes model—soluble RAGE prevented acceleration of diabetic atherosclerosis and suppressed the increased vascular inflammation associated with diabetes. Treatment with soluble RAGE had no effect on levels of glucose or lipids in the diabetic mice,18–20 but it did suppress AGE levels in plasma and tissues,18 suggesting that blocking RAGE attenuated inflammatory and oxidative stresses contributing to acceleration of vascular plaques and to perpetuation of ligand generation.
In other studies, genetically modified animals were used to test the hypothesis that RAGE contributed to acceleration of diabetic atherosclerosis. In apolipoprotein E null mice bred into the RAGE null background, induction of diabetes resulted in less atherosclerosis compared to RAGE-expressing apolipoprotein E null mice with diabetes, despite equal degrees of hyperglycemia and hyperlipidemia. Because we observed that AGEs and inflammatory ligands were present even in nondiabetic atherosclerosis, but to lesser degrees, we tested the effects of RAGE deletion in nondiabetic apolipoprotein E null mice and found that atherosclerosis lesion area and complexity were reduced by deletion of RAGE even in the absence of diabetes.21,22 Consistent with key roles for endothelial RAGE signaling in the absence of diabetes on vascular inflammation, transgenic mice expressing cytoplasmic domain-deleted RAGE (dominant negative or DN RAGE) on the preproendothelin 1 promoter (PPET, largely expressed but not exclusively in endothelial cells), revealed less atherosclerosis in the apolipoprotein E null background versus control apolipoprotein E null mice. We retrieved endothelial cells from RAGE null mice, transgenic PPET DN RAGE mice, or littermate C57BL/6 mice, and found that stimulation with RAGE ligands and with AGE-containing oxidized low density lipoprotein (LDL) stimulated cytokine and adhesion molecule expression in a manner dependent on JNK signaling, and that these effects were significantly reduced in endothelial cells retrieved from RAGE null mice or transgenic PPET DN RAGE mice.21 Of note, when RAGE null mice were bred into a distinct genetic model of hypercholesterolemia and atherosclerosis, the low density lipoprotein receptor (LDL receptor) null background, significantly less atherosclerosis was observed compared to LDL receptor null mice expressing RAGE.23 These studies were performed in the absence of diabetes and solidified that, irrespective of the means by which hypercholesterolemia was induced, RAGE contributed to the acceleration of vascular inflammation and cellular stress leading to atherosclerosis.
To determine the mechanisms underlying the beneficial effects of RAGE deletion in apolipoprotein E null mice, we performed Affymetrix genomic arrays on aortas retrieved from diabetic and nondiabetic apolipoprotein E null mice expressing or devoid of RAGE. Experiments were performed at age nine weeks, after two weeks of established hyperglycemia, in order to determine the initiating factors linked to atherosclerosis and its RAGE- and diabetes-dependence. We found that a major pathway impacted both by diabetes and RAGE was the ROCK1 branch of the transforming growth factor-β (TGF-β) pathway and that smooth muscle cells principally expressed three molecules associated with this family: thrombospondin-1, ROCK1, and TGF-β.24 Consistent with key roles for this pathway in mediating smooth muscle cell migration and proliferation, pretreatment of wild-type smooth muscle cells retrieved from mouse aorta with two different inhibitors of ROCK (fasudil and Y27632) resulted in decreased migration and proliferation stimulated by the RAGE ligand S100B.24
In other studies, generation of chimeric apolipoprotein E null mice (nondiabetic) after lethal irradiation and reconstitution with RAGE expressing or RAGE null bone marrow revealed that mice reconstituted with RAGE null bone marrow had decreased atherosclerosis and necrotic cores particularly at later stages, suggesting important roles for RAGE in atherosclerosis progression. Expression of key inflammatory molecules and the ligand HMGB1 in the lesions was also reduced by deletion of RAGE.25
In summary, these studies to date have suggested that RAGE expression in endothelial cells, macrophages, and smooth muscle cells contributes to the pathogenesis of atherosclerosis, and particularly its acceleration in diabetes. Certainly, pathogenic roles for RAGE expression in distinct cell types, such as lymphocytes, dendritic cells, stem, or progenitor cells, cannot be excluded at this time. Studies are in progress to rigorously test the signaling mechanisms impacted by RAGE in distinct cell types linked to atherosclerosis.
In addition to roles in macrovascular disease and atherosclerosis, RAGE also contributes to the pathogenesis of myocardial dysfunction, especially in diabetes. Work by Ramasamy et al. showed that hearts of diabetic mice subjected to ischemia/reperfusion in the isolated perfused mode displayed increased damage as assessed by release of higher levels of lactic dehydrogenase (LDH), reduced ATP levels in the heart, and higher left ventricular developed pressure (LVDP), the latter a marker of cardiac dysfunction, compared to nondiabetic mice, and that in the presence of soluble RAGE or by RAGE deletion, these parameters were greatly improved.26 Studies in vitro in isolated primary adult ventricular cardiomyocytes revealed that cardiomyocytes devoid of RAGE were more resistant to apoptosis and oxidative stress compared to wild-type cardiomyocytes exposed to in vitro applied hypoxia.27 Other studies have confirmed the importance of RAGE in cardiac ischemia.28–31
Studies in the isolated perfused heart or in isolated cardiomyocytes were complemented by experiments in which mice were subjected to transient ligation of the left anterior descending coronary artery, a model of myocardial infarction. Compared to wild-type mice, mice devoid of RAGE displayed highly significant reductions in infarct volume and increased cardiac function, as assessed by echocardiography, compared to wild-type control animals.32,33
Taken together, these findings support the potential benefits of RAGE antagonism in heart disease, especially in diabetes, and suggest that roles for RAGE blockade in cardioprotection are not limited to its potential impact on macrovascular disease and atherosclerosis, but to innate cardiac dysfunction as well.
RAGE and cardiovascular disease: studies in human subjects
In addition to the studies described above on expression of RAGE in human atherosclerotic plaques, the study of soluble levels of RAGE in cardiovascular disease has become a highly studied area of research. Two different forms of soluble RAGE may be detected in human plasma: “total” soluble RAGE (cell surface cleaved form) and endogenous secretory or esRAGE (derived from a splice variant of RAGE). Although mixed results have been reported suggesting either low34 or high35 levels of soluble RAGE as putative biomarkers of the presence or extent of cardiovascular disease, a very recent study examined total sRAGE and esRAGE levels at the prerandomization phase of a study testing atorvastatin in type 2 diabetic subjects. In this nested case–control study, the findings revealed that sRAGE and esRAGE were higher in those with low body mass index, higher adiponectin levels, lower estimated glomerular filtration rate, and white ethnicity. Independent of the treatment arm, sRAGE and esRAGE were associated with incident cardiovascular disease, but there was no association with stroke. Of note, treatment with atorvastatin had no effect on sRAGE levels.35 It is important to note that two other studies in diabetic and nondiabetic subjects showed that treatment with statins (atorvastatin) raised sRAGE levels.36,37 Certainly, as recently reviewed by Wilson,38 further analysis is needed to discern the predictive nature of sRAGE levels in cardiovascular disease. Indeed, in this context, it is critical to consider the status of renal function, as significantly impaired renal function has been linked to higher sRAGE levels.39 Hence, interpretation of sRAGE levels must be made in the context of renal status, particularly in subjects with diabetes.
Lastly, in addition to effects of medications commonly used in cardiovascular or metabolic disease on plasma levels of soluble RAGE, these medications also may exert their benefit, at least in part, via suppression of cellular/tissue levels of RAGE. For example, treatment with simvastatin reduced atherosclerotic plaque expression of RAGE in human subjects,40 treatment of streptozotocindiabetic mice with the peroxisome proliferator activator receptor delta agonist L-165041 reduced RAGE expression in the kidneys,41 treatment of mesangial cells with glucagon-like peptide 1 (GLP-1) inhibited RAGE expression,42 and treatment of experimental animals with atorvastatin protects against middle cerebral artery occlusion, at least in part, via reduced expression of RAGE in the brain consequent to stroke.43
Taken together, evidence is mounting linking RAGE to mechanisms of atherosclerosis, especially in diabetes. Importantly, measurement of soluble RAGEs, under active investigation in cardiovascular disease, holds promise as a means of biomarking the state of diabetic macrovessels. In addition to macrovascular disease of diabetes, RAGE is also implicated in microvascular complications. In this review, we will discuss recent advances on the role of RAGE in one such complication, diabetic nephropathy.
RAGE and diabetic nephropathy
In the Western world, diabetes is the leading cause of end-stage renal disease, surpassing other etiologies, such as hypertension.44,45 Likely support for the contribution of AGEs to the pathogenesis of diabetic kidney disease was obtained by studies in which pimagedine (aminoguanidine) was administered to types 1 and 2 diabetic subjects with nephropathy. Despite toxicity associated with the agent (glomerulonephritis), the use of pimagedine was associated with decreased 24-hour proteinuria in the treated subjects.46 Notably, however, the outcome of estimated glomerular filtration rate was marginally but not statistically significantly improved in the treated group (P = 0.05).46 Although pimagedine did not advance in clinical development for these reasons, its use nevertheless supported roles for AGEs in the pathogenesis of diabetic kidney disease.
It was in this context that we postulated roles for the chief AGE receptor RAGE in diabetic nephropathy. In human subjects, D’Agati et al. showed that AGEs and RAGE were both present and expressed in human diabetic kidneys to increased degrees compared to nondiabetic control subjects, particularly in the glomerulus and especially in glomerular epithelial cells (podocytes) and endothelial cells.47 Based on these findings suggesting roles for AGE–RAGE in diabetic kidney disease, multiple pharmacological strategies were tested in vivo in animal models; examples of which include administration of soluble RAGE and antibodies to RAGE. In each case, targeting RAGE resulted in attenuation of functional and pathological endpoints of nephropathy in mouse models of types 1 and 2 diabetes.48–50
Studies in homozygous RAGE null mice confirmed important roles for RAGE in the pathogenesis of diabetic nephropathy.48 Although earlier studies employed mice vulnerable to the earlest changes in the diabetic kidney but without frank loss of glomerular filtration rate (GFR), recent experiments in OVE26 mice, a mouse model of type 1 diabetes in the FVB genetic background, reveal significant loss of GFR as measured by inulin clearance. In OVE26/RAGE-null (OVE26 RKO) mice, significant protection from glomerular sclerosis, thickening of the glomerular basement membrane, podocyte effacement, and loss of GFR were noted compared to OVE26 littermates expressing RAGE.51 Experiments performed on these animals revealed two key insights into the mechanisms by which RAGE deletion exerted beneficial effects in the diabetic kidney.
First, our data revealed the intriguing finding that despite equivalent degrees of hyperglycemia in OVE26 and OVE26 RKO mice, the levels of the AGE precursor methylglyoxal (MG) were lower in the kidneys of mice devoid of RAGE, and, indeed, indistinguishable from those in the kidneys of nondiabetic FVB mice. We reported that compared to OVE26 mice, kidneys from OVE26 RKO mice displayed higher levels of glyoxalase1 mRNA, protein and activity.51 Hence, RAGE-dependent regulation of key pathways linked to AGE detoxification may explain these findings and add further support for the role of AGEs in the pathogenesis of nephropathy.
Second, in addition to RAGE-dependent regulation of glyoxalase1, additional studies using Affymetrix gene arrays were performed on isolated glomeruli from OVE26 and OVE26 RKO mice at two months of age. We reasoned that at such a time point, although microalbuminuria was evident, highly advanced glomerular lesions were not yet present. We identified significant changes in expression of Serpine1 (the gene for plasminogen activator inhibitor 1 or PAI 1) in the glomeruli. Serpine 1 levels were increased by 1.46-fold in OVE26 glomeruli versus wild-type FVB; this was verified by real-time PCR experiments. At seven months of age, kidney cortex from OVE26 mice revealed a 4.3-fold increase in levels of Serpine1 mRNA versus wild-type FVB mice. These levels were significantly lower in OVE26 RKO cortex. In parallel, levels of TGF-β, TGF-β–induced (active) and α I (IV) collagen were significantly higher in the cortex of OVE26 versus OVE26 RKO mice.51 Lastly, ROCK1 activity, linked to TGF-β, was significantly higher in the kidney cortex of OVE26 versus OVE26 RKO mice (Fig. 1).51 Interestingly, in renal tubular cells, others have shown that AGE-mediated induction of connective tissue growth factor (CTGF) occurs in a TGF-β–dependent manner via Smad3 signaling.52
Figure 1.

PAI-1 (Serpine1), Tgf-β1, Tgf-β–induced, and α1-(IV) collagen mRNA transcripts and ROCK1 activity are lower in OVE26 RKO kidney cortex than in OVE26 kidney cortex at age seven months. Real-time PCR for PAI-1 (A), Tgf-β1 (B), Tgf-β1–induced (C), and α 1-(IV) collagen (D) gene products was performed, normalized to 18s transcript levels, and expressed as fold-change compared with the FVB or OVE26 group, as indicated in the figure (**P < 0.01, ***P < 0.005, ##P < 0.0005, ###P < 0.0001). n = at least 6 per group. (E) ROCK1 activity was measured as the amount of phosphorylated MYPT1 compared with total ROCK1 (*** P = 0.005). n = at least 3 per group. Reprinted with permission from Ref. 51.
Recent findings implicate RAGE in the angiotensin II (angII) axis. In cultured podocytes, angiotensin II induces RAGE expression in a manner dependent on the AT2 receptor. In mice devoid of AT2 receptor, angII treatment did not upregulate RAGE to the same degree as that observed in wild-type mice.53 Such findings add further support to the concept that AT1 receptor blockade alone may not be sufficient for the treatment of diabetic nephropathy, and suggest that complementary strategies to block RAGE may impart benefit to the diabetic kidney.
Work from our laboratory and others has suggested that RAGE-dependent inflammation contributes to the pathogenesis of diabetic complications—both in the macro- and microvasculature. The discovery that RAGE bound S100/calgranulins and HMGB1 ligands suggested direct roles for RAGE in inflammation. In the section to follow, we discuss recent insights into the role of RAGE in propagation of inflammation.
RAGE and the inflammatory response: new twists
The earliest studies examining the effects of ligands on the RAGE gene promoter shed first light on the concept that RAGE upregulation and action is sustained in a ligand-enriched environment; contrary to other settings in which ligands downregulate expression of their receptors, RAGE ligands upregulate expression of RAGE.54 In other studies, it has been shown that RAGE stimulation upregulates two key transcription factors implicated in inflammatory responses, NF-κB and early growth response-1 (Egr-1).55,56 In diabetic tissues and in chronic inflammation, RAGE is implicated in the sustained activation of NF-KB that likely contributes to the chronicity and unrelenting nature of diabetic target cell stress and dysfunction.57,58
Given roles for RAGE in inflammatory mechanisms, experiments have been performed illustrating the effects of RAGE deletion or antagonism in a range of infectious settings, such as cecal ligation and puncture (sepsis), influenza A viral pneumonia, pneumococcal pneumonia, Escherichia coli pneumonia and sepsis, and Listeria monocytogenes, as examples.59–63 In these settings, blocking RAGE action was beneficial and resulted in either improved survival and/or markedly reduced tissue damage.
In the particular context of periodontal disease, it is known that subjects with diabetes experience increased severity of periodontal disease; Lamster et al. showed that RAGE and AGEs were expressed in human diabetic gingival tissue retrieved at the time of surgery.64 In murine models of accelerated alveolar bone loss and gingival inflammation in diabetes, administration of soluble RAGE suppressed exaggerated gingival inflammation, matrix metalloproteinase activity, and alveolar bone loss in mice treated by oral and anal gavage with the periodontal pathogen Porphryomonas gingivalis (Pg) strain 381.65 Recently, Lalla et al. addressed the direct question, does RAGE play a role in the pathogenicity of Pg? Their intriguing findings linked RAGE directly to endothelial cell stress. Endothelial cells were retrieved from the aortas of wild-type or RAGE null mice and infected with Pg strain 381 or a fimbriae-deficient mutant strain known as DPG3. A number of findings emerged; first, in wild-type endothelial cells, Pg 381 resulted in increased expression of RAGE; second, levels of AGEs and monocyte chemoattractant peptide-1 (MCP-1) were increased by Pg 381 in wild-type but not in RAGE null endothelial cells, and not by DPG3 in wild-type endothelial cells; and third, treatment of human aortic endothelial cells with Pg 381 upregulated RAGE expression in a manner blocked by antioxidants or AGE blockade.66
Hence, these experiments suggested direct links of RAGE to endothelial stress induced by the pathogen Pg 381; the finding that DPG3 had no effect on AGE, RAGE, or MCP-1 production strongly suggested that invasion was required for the effects of RAGE in endothelial cells, and perhaps that RAGE influences this invasion process. Further experimentation is required to address this point. At this stage, however, such data provide further potential benefits to strategies aimed at pharmacological blockade of RAGE.
Taken together, these data reinforce roles for RAGE at multiple levels of the inflammatory response and support the concept of a RAGE-dependent gene regulatory network in inflammation. In mouse models, skin inflammation was shown to be mediated via key transcription factors regulated by RAGE, including Sp1, Tcfap2, E2f, myc, and Egr-1.67 In human subjects, recent studies have shown direct correlations between RAGE polymorphism G82S and serum levels of C-reactive protein in the Chinese Han population.68 From mouse to human, the role of RAGE as a key contributor to the inflammatory response is being confirmed.
Therefore, although our work began in the context of complications of diabetes, it is not surprising that RAGE is linked to the pathogenesis of diabetes as well.
RAGE and the pathogenesis of diabetes
Previous studies demonstrated that administration of soluble RAGE to NOD/scid mice subjected to adoptive transfer of diabetogenic splenocytes from NOD mice delayed the time to diabetes compared to treatment with vehicle, murine serum albumin.69 In parallel, levels of RAGE, S100/calgranulin, and T cells were significantly decreased in the islets of soluble RAGE treated mice.69 Furthermore, levels of key cytokines implicated in inflammatory damage to the islets, TNF-α and IL-1β, were significantly reduced by treatment with sRAGE (Fig. 2).69 On account of these findings, further studies were performed in a murine model of allogeneic, orthotopic heart transplantation in a mouse model, and revealed that administration of soluble RAGE significantly prolonged the time to allograft rejection.70 These findings led to the ultimate testing of RAGE in T cell responses and revealed that RAGE is inducibly upregulated during T cell activation. Clynes et al. showed that transfer of RAGE null OT II T cells (express T cell receptors recognizing ovalbumin) into OVA-immunized hosts resulted in reduced proliferative responses that were further diminished when RAGE null OT II T cells were transferred into RAGE null recipients. Although RAGE null dendritic cells displayed no overt abnormalities in antigen recognition responses, RAGE null T cells showed significantly impaired proliferative responses in vitro to nominal and alloantigens, in parallel with decreased production of interferon-γ and interleukin 2. Those findings indicated for the first time that RAGE expressed on T cells is required for efficient priming of T cells, and suggest that RAGE may play direct roles in the pathogenesis of autoimmune disorders such as type 1 diabetes.71
Figure 2.
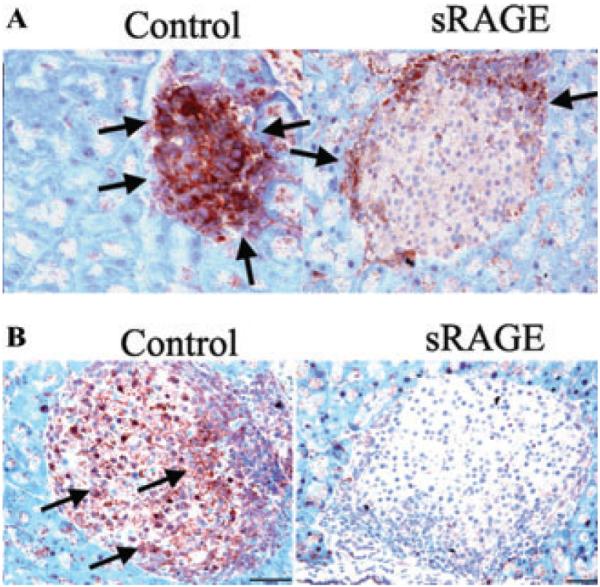
Treatment with sRAGE reduces the expression of IL-1βand TNF-α in islets in NOD/scid mice subjected to adoptive transfer of diabetogenic splenocytes from NOD mice. The islets of NOD/scid mice that received splenocytes from diabetic NOD mice were studied for expression of IL-1β (A) and TNF-α (B) by immunohistochemistry. The area of cells staining with the anticytokine antibodies is indicated with arrows and was determined for each treatment group (control versus soluble RAGE). Representative sections from three separate recipients in each category are shown. The expression levels of both IL-1β and TNF-α were reduced by treatment with sRAGE. Reprinted with permission from Ref. 69.
In the direct context of pancreatic islet β cell toxicity, two recent papers have suggested that RAGE may be implicated directly in this process, thereby further linking RAGE to the pathogenesis of diabetes. Lee et al. showed that RAGE was expressed on INS-1 cells (rat pancreatic β cell line) and on human islets. They showed that RAGE ligands S100B and HMGB1 induced apoptotic death of INS-1 cells and islets in a manner suppressed by an NADPH oxidase inhibitor.72 In other studies, Zhu et al. showed that RAGE ligand-glycated serum induced upregulation of RAGE in INS-1 cells and induced apoptosis of these cells in parallel with Bcl-2 expression in a time- and dose-dependent manner. Both antibody to RAGE and RAGE knockdown blocked these adverse effects of glycated serum, thereby directly implicating RAGE in β cell death.73 Those authors noted, however, that although glycated serum blocked glucose stimulated insulin secretion in rat islets, blockade of RAGE had no protective effects.73 However, in further work, Shu et al. showed that AGEs decrease insulin secretion via repression of Pdx-1 protein expression and that antibodies to RAGE restored Pdx-1 expression and expression of insulin mRNA in INS-1 cells.74 Taken together, these data suggest that RAGE ligand–RAGE interaction may play multiple roles in the steps causing and perpetuating islet dysfunction in types 1 and 2 diabetes. Given the varied effects of RAGE signaling in a range of disease-like settings, a question that arose was, how do the diverse ligands of RAGE all recognize this receptor?
RAGE structure and ligand identification: insights from structural biology
Prototypic of members of the immunoglobulin superfamily, RAGE is composed of immunoglobulin-like domains. Its extracellular region contains one V-type immunoglobulin domain followed by two distinct C-type immunoglobulin domains; this region of the molecule is followed by a single transmembrane domain, and then in the intracellular space, a short and highly charged cytoplasmic domain.75
We generated individual V, C1, and C2 domains and first showed that CML-AGEs bound selectively to the V-domain;6 subsequent studies indicated that the S100/calgranulin, HMGB1, and amyloid-β peptide ligands all cross-competed with each other, suggesting that the V-domain was the key site of binding action.76 Yet, despite these findings strongly suggesting a key characteristic of the V-domain, which yielded the multiligand nature of the receptor, the precise means by which these ligands might bind RAGE remained elusive.
In 2010, two papers appeared reporting the crystal structure of RAGE; remarkably, the findings were highly similar and laid the foundation for a much crisper understanding of RAGE and ligand binding.77,78 In the first paper, Koch et al. studied the ligand S100B and RAGE. The X-ray crystal structure of the V-C1 domain of human RAGE was resolved at 1.85Å, and the V-C1 ligand-binding surface was mapped onto the structure from titrations with S100B monitored by heteronuclear NMR spectroscopy. A highly basic surface in the V-C1 domain was identified that accounted for the ligand-binding characteristics.77 In the second work, Park et al. resolved the structure of RAGE to 1.5 Å; their work revealed that RAGE was a highly elongated molecule with a large basic patch and a large hydrophobic patch. They found that binding of RAGE to S100B was dependent on calcium and on residues in the C’D loop (residues 54–67) of the first domain. Interestingly, they showed that AGE binding was dependent on the recognition of negative charges on the AGE proteins. These authors also reported that RAGE binds to dsDNA and dsRNA.78
In recent work, Xue et al. reported on the mechanisms by which carboxyethyl lysine (CEL) ligands of RAGE interact with the receptor.79 Their work showed that the CEL moiety fits inside a positively charged cavity of the V domain and that peptide backbone atoms make specific contacts with the V domain.79 Importantly, they showed that the geometry of the bound CEL peptide was compatible with many CML- and CEL-modified sites within plasma proteins, thereby explaining how patterned ligands such as these specific AGEs may bind to RAGE.
Sarkany et al. recently reported on the structure of the soluble form of RAGE. These authors demonstrated that soluble RAGE displays concentration-dependent oligomerization behavior mediated by the presence of Ca2+ ions. They employed synchrotron small-angle X-ray scattering and determined the solution structure of human soluble RAGE in both the monomeric and dimeric forms. The model for the monomer is reported to display a J-like shape, and the dimer to be formed through the association of the two N-terminal domains with an elongated structure.80
Certainly, now that a crystal structure of RAGE is available, it is likely that such knowledge will accelerate the development of a range of chemical antagonists. In this context, exciting recent discoveries point to new targets for antagonizing RAGE in the intracellular space.
RAGE, signal transduction, and diaphanous-1
Ligand binding to RAGE in the extracellular space stimulates signal transduction mechanisms that ultimately are responsible for the diverse effects of RAGE on gene expression changes. It is well established that RAGE is expressed on multiple cell types and that ligand binding stimulates signal transduction cascades such as mitogen-activated protein kinases, phosphatidylinositol 3-kinase, Jak/STAT (signal transducers and activators of transcription), and the Rho GTPases Rac-1 and Cdc42.6–8,81–84 To discern the precise mechanisms by which RAGE signals through the cytoplasmic domain, a yeast two hybrid assay in which the human RAGE cytoplasmic domain was used as bait to identify potential binding partners in a lung library; the lung was chosen since RAGE was initially identified from bovine lung extract.85 From the yeast two hybrid assay, multiple clones of the FH1 domain of diaphanous-1 (mDia1) were identified as potential binding partners of the RAGE cytoplasmic domain.86 The FH1 domain of mDia1 is proline-rich and has been reported to interact with mediators of the actin cytoskeleton (profilin) and various signal transduction pathway molecules, such as c-Src.87–88 To confirm the interaction, coimmunoprecipitation studies using his-tagged RAGE cytoplasmic domain and Myc-tagged mDia1 (as well as deletion mutants testing the roles of individual domains of mDia1) were performed and confirmed that the RAGE cytoplasmic domain bound the FH1 domain of mDia1.86 Indeed, a recent publication confirmed this binding in cultured cells.89 In other studies, C6 glioma cells expressing full-length or cytoplasmic domain-deleted or dominant negative (DN) RAGE showed that mDia1 was coimmunoprecipitated with RAGE but mDia1 did not coimmunoprecipitate with RAGE cytoplasmic domain-deleted DN RAGE–expressing C6 glioma cells.86
The key test of the mDia1 finding was whether mDia1 was implicated in RAGE-mediated cellular signaling and functional outcomes, such as cellular migration. In RAGE-expressing cells, but not DN RAGE-expressing cells, both Rac1 and Cdc42 were rapidly activated upon ligand stimulation. To test its role in RAGE ligand-stimulated cellular migration, use of siRNA knockdown mDia1 knock-down suppressed RAGE ligand-stimulated cellular migration but scramble siRNA controls had no in hibitory effects.86 Interestingly, siRNA knockdown of mDia1 expression did not affect migration of cells in response to a non-RAGE ligand such as fetal bovine serum (10%). In parallel, siRNA knockdown of mDia1 blocked RAGE ligand-stimulated activation of rac1 and cd42, whereas scramble siRNAs had no effect.86
In addition to its actions as an effector of Rho GTPase signaling, mDia1 has also been shown to modulate serum response factors (SRFs),90,91 key factors that bind to serum response elements (SRE) in the promoters of a number of genes, thereby regulating critical cellular functions, in a manner dependent on mDia1 effects on the actin cytoskeleton.92 As Egr-1 is an SRF–SRE–dependent gene, we tested the role of mDia1. It is important to note that previous work had shown that in endothelial cells in hypoxia, RAGE upregulates Egr-1 in a manner dependent on rapid activation of protein kinase C-βII and JNK signaling.56 Interestingly, when endothelial cells were placed in hypoxia, rapid (within 10 min) release of RAGE ligand AGE reactive epitopes into the cellular supernatants was observed. In vivo, pretreatment of mice with aminoguanidine, or in vitro, pretreatment of wild-type endothelial cells with anti-AGE IgG, blocked hypoxia-mediated upregulation of Egr-1.56
Hence, tests of human THP-1 cells showed that hypoxia increased expression of mDia1 versus control normoxia treatment of these cells. siRNA knockdown of mDia1 during hypoxia blocked hypoxia-stimulated upregulation of Egr-1 in THP-1 cells, whereas scrambled siRNA had no effect. Furthermore, thioglycollate-elicited murine macrophages retrieved from RAGE null or mDia1 null mice failed to upregulate Egr-1 in hypoxia compared to that observed in wild-type murine macrophages.93(Fig. 3) Taken together, these data demonstrated for the first time key roles for mDia1 in mediating RAGE ligand-stimulated activation of Rho GTPases, cellular migration, and hypoxia-stimulated upregulation of Egr-1.
Figure 3.

Hypoxia induces mDia-1 expression and mDia1 plays key roles in hypoxia-mediated upregulation of Egr-1. THP-1 cells and macrophages from mDia1 null mice were exposed to hypoxia (H; 0.5% of oxygen) or normoxia (N) for the indicated times. (A) Total protein was prepared from THP-1 cells, and immunoblotting with anti-mDia1 IgG was performed on 30 µg/lane of protein from THP-1 cells. Results of multiple experiments were quantified. (B and C) Total RNA was prepared from the indicated cells, and real-time PCR analysis of Egr-1 expression was performed. Data are represented as the relative expression of mRNA for Egr-1 normalized to 18 SirRNA. (B) RNA was prepared from THP-1 cells transfected with siRNA-mDia1 or scramble siRNA and then subjected to hypoxia for 15 min. (C) RNA was prepared from macrophages from wild-type or mDia1 (Drf1) null mice after exposure to hypoxia or normoxia for the indicated times. *P < 0.0001 and **P < 0.001 indicate statistical significance; #indicates no statistical significance. Note that Drf1 indicates mDia1. Adapted from Ref. 93.
Conclusions: what we have learned and where do we proceed from here?
As with any biological target, the chief goal is the translation of the findings to human subjects. In the case of RAGE, there appears to be multiple possible opportunities. From the standpoint of therapeutics, studies using pharmacological antagonists and RAGE null mice support that especially in the case of diabetic macro- and microvascular disease, blocking RAGE is beneficial. As discussed above, potent roles for RAGE in diabetic atherosclerosis, cardiac dysfunction, and nephropathy have been shown. In addition, but not covered in this review, from studies in human subjects suggesting links of RAGE gene polymorphisms and soluble RAGE levels to animal models of these disorders, evidence is mounting linking RAGE to diabetic retinopathy,94–100 peripheral neuropathy,101–104 and impaired wound healing.105–108
Despite this promise, key questions remain on the long-term safety and advisability of blocking RAGE. We and others have shown that even long-term (> 6 months) treatment with soluble RAGE is well tolerated by diabetic mice,100 and that homozygous RAGE null mice display no overt abnormalities in viability or fertility. Rather, they appear to be protected against the chronic complications of diabetes, as discussed above.
These considerations prompt the question, what is the natural function of RAGE? We predict that the ligand families of RAGE, particularly certain members of the S100/calgranulin family and HMGB1, lead dual lives in cellular systems. We propose that in homeostatic systems, free of chronic inflammation, aging, hyperglycemia, or sustained oxidative stress, for example, release of these RAGE ligand families serves, together with the host of other such mediators, as key first lines of defense against infection, inflammation, or injury. However, in settings in which these molecules may accumulate in the microenvironments, both by failure of clearance or perpetuation of signals eliciting their release, the failure to quell these ligand responses tips the biological balance to favor the development of chronic disease.
Hence, it is in this context we speculate that our recent discovery that the RAGE cytoplasmic domain binds to the formin mDia1, and that mDia1 is required for RAGE ligand-stimulated activation of Rac1 and Cdc42 and cellular migration in transformed cells, and for hypoxia-stimulated upregulation of Egr-1 in macrophages, points to the RAGE cytoplasmic domain-mDia1 interaction as a focused target of RAGE signaling. Indeed, others have reported that the RAGE cytoplasmic domain may bind to ERK and to TIRAP (the latter an adaptor protein for toll like receptors 2/4). 89,109 In the latter two studies, however, the functional implications of this apparent binding relationship to in vivo pathobiology have yet to be shown. Is it possible that targeting RAGE cytoplasmic domain/mDia1 will antagonize the pathological impact of RAGE signaling, and that interactions of the RAGE cytoplasmic domain with distinct molecules affect adaptive functions of the molecule? The answers to these fundamental questions are under active investigation and are eagerly awaited.
Footnotes
Conflicts of interest
The authors declare no conflicts of interest.
References
- 1.Forlenza GP, Paradise Black NM, McNamara EG, Sullivan SE. Ankyloglossia, exclusive breastfeeding, and failure to thrive. Pediatrics. 2010;125:e1500–e1504. doi: 10.1542/peds.2009-2101. [DOI] [PubMed] [Google Scholar]
- 2.Peng H, Hagopian W. Environmental factors in the development of Type 1 diabetes. Rev. Endocr. Metab. Disord. 2006;7:149–162. doi: 10.1007/s11154-006-9024-y. [DOI] [PubMed] [Google Scholar]
- 3.Hu FB. Globalization of diabetes: the role of diet, lifestyle, and genes. Diabetes Care. 2011;34:1249–1257. doi: 10.2337/dc11-0442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.International Diabetes Federation IDF Diabetes Atlas. Epidemiology and Morbidity. International Diabetes Federation. Available from: http://www.idf.org.
- 5.Sweat V, Bruzzese JM, Albert S, et al. The Banishing Obesity and Diabetes in Youth (BODY) Project: description and feasibility of a program to halt obesity-associated disease among urban high school students. J. Community Health. 2011 doi: 10.1007/s10900-011-9453-8. In press. [DOI] [PubMed] [Google Scholar]
- 6.Kislinger T, Fu C, Huber B, et al. N (epsilon)-(carboxymethyl)lysine adducts of proteins are ligands for receptor for advanced glycation end products that activate cell signaling pathways and modulate gene expression. J. Biol. Chem. 1999;274:31740–31749. doi: 10.1074/jbc.274.44.31740. [DOI] [PubMed] [Google Scholar]
- 7.Hofmann MA, Drury S, Fu C, et al. RAGE mediates a novel proinflammatory axis: a central cell surface receptor for S100/calgranulin polypeptides. Cell. 1999;97:889–901. doi: 10.1016/s0092-8674(00)80801-6. [DOI] [PubMed] [Google Scholar]
- 8.Taguchi A, Blood DC, del Toro G, et al. Blockade of RAGE-amphoterin signalling suppresses tumour growth and metastases. Nature. 2000;405:354–360. doi: 10.1038/35012626. [DOI] [PubMed] [Google Scholar]
- 9.Moss SE, Klein R, Klein BE. Cause-specific mortality in a population-based study of diabetes. Am. J. Public. Health. 1991;81:1158–1162. doi: 10.2105/ajph.81.9.1158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nathan DM, Cleary PA, Backlund JY, et al. G. Diabetes Control and Complications Trial/Epidemiology of Diabetes Interventions and Complications Study Research Intensive diabetes treatment and cardiovascular disease in patients with type 1 diabetes. N. Engl. J. Med. 2005;353:2643–2653. doi: 10.1056/NEJMoa052187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Khaw KT, Wareham N, Bingham S, et al. Association of hemoglobin A1c with cardiovascular disease and mortality in adults: the European prospective investigation into cancer in Norfolk. Ann. Intern. Med. 2004;141:413–420. doi: 10.7326/0003-4819-141-6-200409210-00006. [DOI] [PubMed] [Google Scholar]
- 12.Action to Control Cardiovascular Risk in Diabetes Study Group. Gerstein HC, Miller ME, Byington RP, et al. Effects of intensive glucose lowering in type 2 diabetes. N. Engl. J. Med. 2008;358:2545–2559. doi: 10.1056/NEJMoa0802743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.ACCORD Study Group. Gerstein HC, Miller ME, Genuth S, et al. Long-term effects of intensive glucose lowering on cardiovascular outcomes. N. Engl. J. Med. 2011;364:818–828. doi: 10.1056/NEJMoa1006524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.ADVANCE Collaborative Group. Patel A, MacMahon S, Chalmers J, et al. Intensive blood glucose control and vascular outcomes in patients with type 2 diabetes. N. Engl. J. Med. 2008;358:2560–2572. doi: 10.1056/NEJMoa0802987. [DOI] [PubMed] [Google Scholar]
- 15.Duckworth W, Abraira C, Moritz T, et al. Glucose control and vascular complications in veterans with type 2 diabetes. N. Engl. J. Med. 2009;360:129–139. doi: 10.1056/NEJMoa0808431. [DOI] [PubMed] [Google Scholar]
- 16.Zoungas S, Patel A, Chalmers J, et al. Severe hypoglycemia and risks of vascular events and death. N. Engl. J. Med. 2010;363:1410–1418. doi: 10.1056/NEJMoa1003795. [DOI] [PubMed] [Google Scholar]
- 17.Cipollone F, Iezzi A, Fazia M, et al. The receptor RAGE as a progression factor amplifying arachidonate-dependent inflammatory and proteolytic response in human atherosclerotic plaques: role of glycemic control. Circulation. 2003;108:1070–1077. doi: 10.1161/01.CIR.0000086014.80477.0D. [DOI] [PubMed] [Google Scholar]
- 18.Park L, Raman KG, Lee KJ, et al. Suppression of accelerated diabetic atherosclerosis by the soluble receptor for advanced glycation endproducts. Nat. Med. 1998;4:1025–1031. doi: 10.1038/2012. [DOI] [PubMed] [Google Scholar]
- 19.Bucciarelli LG, Wendt T, Qu W, et al. RAGE blockade stabilizes established atherosclerosis in diabetic apolipoprotein E-null mice. Circulation. 2002;106:2827–2835. doi: 10.1161/01.cir.0000039325.03698.36. [DOI] [PubMed] [Google Scholar]
- 20.Wendt T, Harja E, Bucciarelli L, et al. RAGE modulates vascular inflammation and atherosclerosis in a murine model of type 2 diabetes. Atherosclerosis. 2006;185:70–77. doi: 10.1016/j.atherosclerosis.2005.06.013. [DOI] [PubMed] [Google Scholar]
- 21.Harja E, Bu DX, Hudson BI, et al. Vascular and inflammatory stresses mediate atherosclerosis via RAGE and its ligands in apoE−/−mice. J. Clin. Invest. 2008;118:183–194. doi: 10.1172/JCI32703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Soro-Paavonen A, Watson AM, Li J, et al. Receptor for advanced glycation end products (RAGE) deficiency attenuates the development of atherosclerosis in diabetes. Diabetes. 2008;57:2461–2469. doi: 10.2337/db07-1808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sun L, Ishida T, Yasuda T, et al. RAGE mediates oxidized LDL-induced pro-inflammatory effects and atherosclerosis in nondiabetic LDL receptor-deficient mice. Cardiovasc. Res. 2009;82:371–381. doi: 10.1093/cvr/cvp036. [DOI] [PubMed] [Google Scholar]
- 24.Bu DX, Rai V, Shen X, et al. Activation of the ROCK1 branch of the transforming growth factor-beta pathway contributes to RAGE-dependent acceleration of atherosclerosis in diabetic ApoE-null mice. Circ. Res. 2010;106:1040–1051. doi: 10.1161/CIRCRESAHA.109.201103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Morris-Rosenfeld S, Blessing E, Preusch MR, et al. Deletion of bone marrow-derived receptor for advanced glycation end products inhibits atherosclerotic plaque progression. Eur. J. Clin. Invest. 2011;41:1164–1171. doi: 10.1111/j.1365-2362.2011.02514.x. [DOI] [PubMed] [Google Scholar]
- 26.Bucciarelli LG, Ananthakrishnan R, Hwang YC, et al. RAGE and modulation of ischemic injury in the diabetic myocardium. Diabetes. 2008;57:1941–1951. doi: 10.2337/db07-0326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shang L, Ananthakrishnan R, Li Q, et al. RAGE modulates hypoxia/reoxygenation injury in adult murine cardiomyocytes via JNK and GSK-3beta signaling pathways. PLoS One. 2010;5:e10092. doi: 10.1371/journal.pone.0010092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang LJ, Lu L, Zhang FR, et al. Increased serum high-mobility group box-1 and cleaved receptor for advanced glycation endproducts levels and decreased endogenous secretory receptor for advanced glycation endproducts levels in diabetic and nondiabetic patients with heart failure. Eur. J. Heart Fail. 2011;13:440–449. doi: 10.1093/eurjhf/hfq231. [DOI] [PubMed] [Google Scholar]
- 29.Ma H, Li SY, Xu P, et al. Advanced glycation end-product (AGE) accumulation and AGE receptor (RAGE) up-regulation contribute to the onset of diabetic cardiomyopathy. J. Cell. Mol. Med. 2009;13:1751–1764. doi: 10.1111/j.1582-4934.2008.00547.x. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 30.Nielsen JM, Kristiansen SB, Nørregaard R, et al. Blockage of receptor for advanced glycation end products prevents development of cardiac dysfunction in db/db type 2 diabetic mice. Eur. J. Heart Fail. 2009;11:638–647. doi: 10.1093/eurjhf/hfp070. [DOI] [PubMed] [Google Scholar]
- 31.Petrova R, Yamamoto Y, Muraki K, et al. Advanced glycation endproduct-induced calcium handling impairment in mouse cardiac myocytes. J. Mol. Cell. Cardiol. 2002;34:1425–1431. doi: 10.1006/jmcc.2002.2084. [DOI] [PubMed] [Google Scholar]
- 32.Aleshin A, Ananthakrishnan R, Li Q, et al. RAGE modulates myocardial injury consequent to LAD infarction via impact on JNK and STAT signaling in a murine model. Am. J. Physiol. Heart Circ. Physiol. 2008;294:H1823–H1832. doi: 10.1152/ajpheart.01210.2007. [DOI] [PubMed] [Google Scholar]
- 33.Andrassy M, Volz HC, Igwe JC, et al. High-mobility group box-1 in ischemia-reperfusion injury of the heart. Circulation. 2008;117:3216–3226. doi: 10.1161/CIRCULATIONAHA.108.769331. [DOI] [PubMed] [Google Scholar]
- 34.Falcone C, Emanuele E, D’Angelo A, et al. Plasma levels of soluble receptor for advanced glycation end products and coronary artery disease in nondiabetic men. Arterioscler. Thromb. Vasc. Biol. 2005;25:1032–1037. doi: 10.1161/01.ATV.0000160342.20342.00. [DOI] [PubMed] [Google Scholar]
- 35.Colhoun HM, Betteridge DJ, Durrington P, et al. Total soluble and endogenous secretory receptor for advanced glycation endproducts as predictive biomarkers of coronary heart disease risk in patients with type 2 diabetes: an analysis from the CARDS trial. Diabetes. 2011;60:2379–2385. doi: 10.2337/db11-0291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tam HL, Shiu SW, Wong Y, et al. Effects of atorvastatin on serum soluble receptors for advanced glycation end-products in type 2 diabetes. Atherosclerosis. 2010;209:173–177. doi: 10.1016/j.atherosclerosis.2009.08.031. [DOI] [PubMed] [Google Scholar]
- 37.Santilli F, Bucciarelli L, Noto D, et al. Decreased plasma soluble RAGE in patients with hypercholesterolemia: effects of statins. Free Radic. Biol. Med. 2007;43:1255–1262. doi: 10.1016/j.freeradbiomed.2007.06.017. [DOI] [PubMed] [Google Scholar]
- 38.Wilson C. Cardiovascular endocrinology: RAGE-a biomarker for CHD in T2DM? Nat. Rev. Endocrinol. 2011;7:561. doi: 10.1038/nrendo.2011.136. [DOI] [PubMed] [Google Scholar]
- 39.Kalousovaá M, Hodkovaá M, Kazderovaá M, et al. Soluble receptor for advanced glycation end products in patients with decreased renal function. Am. J. Kidney Dis. 2006;47:406–411. doi: 10.1053/j.ajkd.2005.12.028. [DOI] [PubMed] [Google Scholar]
- 40.Cuccurullo C, Iezzi A, Fazia ML, et al. Suppression of RAGE as a basis of simvastatin-dependent plaque stabilization in type 2 diabetes. Arterioscler. Thromb. Vasc. Biol. 2006;26:2716–2723. doi: 10.1161/01.ATV.0000249630.02085.12. [DOI] [PubMed] [Google Scholar]
- 41.Liang YJ, Chen SA, Jian JH. Peroxisome proliferator-activated receptor delta downregulates the expression of the receptor for advanced glycation end products and pro-inflammatory cytokines in the kidney of streptozotocin-induced diabetic mice. Eur. J. Pharm. Sci. 2011;43:65–70. doi: 10.1016/j.ejps.2011.03.011. [DOI] [PubMed] [Google Scholar]
- 42.Ishibashi Y, Nishino Y, Matsui T, et al. Glucagon-like peptide-1 suppresses advanced glycation end product-induced monocyte chemoattractant protein-1 expression in mesangial cells by reducing advanced glycation end product receptor level. Metabolism. 2011;60:1271–1277. doi: 10.1016/j.metabol.2011.01.010. [DOI] [PubMed] [Google Scholar]
- 43.Wang L, Zhang X, Liu L, et al. Atorvastatin protects rat brains against permanent focal ischemia and down-regulates HMGB1, HMGB1 receptors (RAGE and TLR4), NF-kappaB expression. Neurosci. Lett. 2010;471:152–156. doi: 10.1016/j.neulet.2010.01.030. [DOI] [PubMed] [Google Scholar]
- 44.Ritz E, Orth SR. Nephropathy in patients with type 2 diabetes mellitus. N. Engl. J. Med. 1999;341:1127–1133. doi: 10.1056/NEJM199910073411506. [DOI] [PubMed] [Google Scholar]
- 45.U.S. Renal Data System . USRDS 2004 Annual Data Report: Atlas of End-Stage Renal Disease in the United States. National Institutes of Health, National Institute of Diabetes an Digestive and Kidney Diseases; Bethesda, MD, USA: 2004. [Google Scholar]
- 46.Bolton WK, Cattran DC, Williams ME, et al. Randomized trial of an inhibitor of formation of advanced glycation end products in diabetic nephropathy. Am. J. Nephrol. 2004;24:32–40. doi: 10.1159/000075627. [DOI] [PubMed] [Google Scholar]
- 47.Tanji N, Markowitz GS, Fu C, et al. Expression of advanced glycation end products and their cellular receptor RAGE in diabetic nephropathy and nondiabetic renal disease. J. Am. Soc. Nephrol. 2000;11:1656–1666. doi: 10.1681/ASN.V1191656. [DOI] [PubMed] [Google Scholar]
- 48.Wendt TM, Tanji N, Guo J, et al. RAGE drives the development of glomerulosclerosis and implicates podocyte activation in the pathogenesis of diabetic nephropathy. Am. J. Pathol. 2003;162:1123–1137. doi: 10.1016/S0002-9440(10)63909-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Jensen LJ, Denner L, Schrijvers BF, et al. Renal effects of a neutralising RAGE-antibody in long-term streptozotocin-diabetic mice. J. Endocrinol. 2006;188:493–501. doi: 10.1677/joe.1.06524. [DOI] [PubMed] [Google Scholar]
- 50.Flyvbjerg A, Denner L, Schrijvers BF, et al. Long-term renal effects of a neutralizing RAGE antibody in obese type 2 diabetic mice. Diabetes. 2004;53:166–172. doi: 10.2337/diabetes.53.1.166. [DOI] [PubMed] [Google Scholar]
- 51.Reiniger N, Lau K, McCalla D, et al. Deletion of the receptor for advanced glycation end products reduces glomerulosclerosis and preserves renal function in the diabetic OVE26 mouse. Diabetes. 2010;59:2043–2054. doi: 10.2337/db09-1766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chung AC, Zhang H, Kong YZ, et al. Advanced glycation end-products induce tubular CTGF via TGF-beta-independent Smad3 signaling. J. Am. Soc. Nephrol. 2010;21:249–260. doi: 10.1681/ASN.2009010018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ruüster C, Franke S, Wenzel U, et al. Podocytes of AT2 receptor knockout mice are protected from angiotensin II-mediated RAGE induction. Am. J. Nephrol. 2011;34:309–317. doi: 10.1159/000329321. [DOI] [PubMed] [Google Scholar]
- 54.Li J, Schmidt AM. Characterization and functional analysis of the promoter of RAGE, the receptor for advanced glycation end products. J. Biol. Chem. 1997;272:16498–16506. doi: 10.1074/jbc.272.26.16498. [DOI] [PubMed] [Google Scholar]
- 55.Schmidt AM, Hori O, Chen JX, et al. Advanced glycation endproducts interacting with their endothelial receptor induce expression of vascular cell adhesion molecule-1 (VCAM-1) in cultured human endothelial cells and in mice. A potential mechanism for the accelerated vasculopathy of diabetes. J. Clin. Invest. 1995;96:1395–1403. doi: 10.1172/JCI118175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Chang JS, Wendt T, Qu W, et al. Oxygen deprivation triggers upregulation of early growth response-1 by the receptor for advanced glycation end products. Circ. Res. 2008;102:905–913. doi: 10.1161/CIRCRESAHA.107.165308. [DOI] [PubMed] [Google Scholar]
- 57.Bierhaus A, Schiekofer S, Schwaninger M, et al. Diabetes-associated sustained activation of the transcription factor nuclear factor-kappaB. Diabetes. 2001;50:2792–2808. doi: 10.2337/diabetes.50.12.2792. [DOI] [PubMed] [Google Scholar]
- 58.Kislinger T, Tanji N, Wendt T, et al. Receptor for advanced glycation end products mediates inflammation and enhanced expression of tissue factor in vasculature of diabetic apolipoprotein E-null mice. Arterioscler. Thromb. Vasc. Biol. 2001;21:905–910. doi: 10.1161/01.atv.21.6.905. [DOI] [PubMed] [Google Scholar]
- 59.Christaki E, Opal SM, Keith JC, Jr., et al. A monoclonal antibody against RAGE alters gene expression and is protective in experimental models of sepsis and pneumococcal pneumonia. Shock. 2011;35:492–498. doi: 10.1097/SHK.0b013e31820b2e1c. [DOI] [PubMed] [Google Scholar]
- 60.van Zoelen MA, van der Sluijs KF, Achouiti A, et al. Receptor for advanced glycation end products is detrimental during influenza A virus pneumonia. Virology. 2009;391:265–273. doi: 10.1016/j.virol.2009.05.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.van Zoelen MA, Schouten M, de Vos AF, et al. The receptor for advanced glycation end products impairs host defense in pneumococcal pneumonia. J. Immunol. 2009;182:4349–4356. doi: 10.4049/jimmunol.0801199. [DOI] [PubMed] [Google Scholar]
- 62.Ramsgaard L, Englert JM, Manni ML, et al. Lack of the receptor for advanced glycation end-products attenuates E. coli pneumonia in mice. PLoS One. 2011;6:e20132. doi: 10.1371/journal.pone.0020132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lutterloh EC, Opal SM, Pittman DD, et al. Inhibition of the RAGE products increases survival in experimental models of severe sepsis and systemic infection. Crit. Care. 2007;11:R122. doi: 10.1186/cc6184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Schmidt AM, Weidman E, Lalla E, et al. Advanced glycation endproducts (AGEs) induce oxidant stress in the gingiva: a potential mechanism underlying accelerated periodontal disease associated with diabetes. J. Periodont. Res. 1996;31:508–515. doi: 10.1111/j.1600-0765.1996.tb01417.x. [DOI] [PubMed] [Google Scholar]
- 65.Lalla E, Lamster IB, Feit M, et al. Blockade of RAGE suppresses periodontitis-associated bone loss in diabetic mice. J. Clin. Invest. 2000;105:1117–1124. doi: 10.1172/JCI8942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Pollreisz A, Hudson BI, Chang JS, et al. Receptor for advanced glycation endproducts mediates proatherogenic responses to periodontal infection in vascular endothelial cells. Atherosclerosis. 2010;212:451–456. doi: 10.1016/j.atherosclerosis.2010.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Riehl A, Bauer T, Brors B, et al. Identification of the Rage-dependent gene regulatory network in a mouse model of skin inflammation. BMC Genom. 2010;11:537. doi: 10.1186/1471-2164-11-537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Gao J, Shao Y, Lai W, et al. Association of polymorphisms in the RAGE gene with serum CRP levels and coronary artery disease in the Chinese Han population. J. Hum. Genet. 2010;55:668–675. doi: 10.1038/jhg.2010.85. [DOI] [PubMed] [Google Scholar]
- 69.Chen Y, Yan SS, Colgan J, et al. Blockade of late stages of autoimmune diabetes by inhibition of the receptor for advanced glycation end products. J. Immunol. 2004;173:1399–1405. doi: 10.4049/jimmunol.173.2.1399. [DOI] [PubMed] [Google Scholar]
- 70.Moser B, Szabolcs MJ, Ankersmit HJ, et al. Blockade of RAGE suppresses alloimmune reactions in vitro and delays allograft rejection in murine heart transplantation. Am. J. Transplant. 2007;7:293–302. doi: 10.1111/j.1600-6143.2006.01617.x. Erratum in Am. J. Transplant. 2007 May; 2007(2005): 1318. [DOI] [PubMed] [Google Scholar]
- 71.Moser B, Desai DD, Downie MP, et al. Receptor for advanced glycation end products expression on T cells contributes to antigen-specific cellular expansion in vivo. J. Immunol. 2007;179:8051–8058. doi: 10.4049/jimmunol.179.12.8051. [DOI] [PubMed] [Google Scholar]
- 72.Lee BW, Chae HY, Kwon SJ, et al. RAGE ligands induce apoptotic cell death of pancreatic beta-cells via oxidative stress. Int. J. Mol. Med. 2010;26:813–818. [PubMed] [Google Scholar]
- 73.Zhu Y, Shu T, Lin Y, et al. Inhibition of the receptor for advanced glycation endproducts (RAGE) protects pancreatic beta-cells. Biochem. Biophys. Res. Commun. 2011;404:159–165. doi: 10.1016/j.bbrc.2010.11.085. [DOI] [PubMed] [Google Scholar]
- 74.Shu T, Zhu Y, Wang H, et al. AGEs decrease insulin synthesis in pancreatic beta-cell by repressing Pdx-1 protein expression at the post-translational level. PLoS One. 2011;6:e18782. doi: 10.1371/journal.pone.0018782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Neeper M, Schmidt AM, Brett J, et al. Cloning and expression of a cell surface receptor for advanced glycosylation end products of proteins. J. Biol. Chem. 1992;267:14998–15004. [PubMed] [Google Scholar]
- 76.Leclerc E, Fritz G, Vetter SW, Heizmann CW. Binding of S100 proteins to RAGE: an update. Biochim. Biophys. Acta. 2009;1793:993–1007. doi: 10.1016/j.bbamcr.2008.11.016. [DOI] [PubMed] [Google Scholar]
- 77.Koch M, Chitayat S, Dattilo BM, et al. Structural basis for ligand recognition and activation of RAGE. Structure. 2010;18:1342–1352. doi: 10.1016/j.str.2010.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Park H, Adsit FG, Boyington JC. The 1.5 A~ crystal structure of human receptor for advanced glycation endproducts (RAGE) ectodomains reveals unique features determining ligand binding. J. Biol. Chem. 2010;285:40762–40770. doi: 10.1074/jbc.M110.169276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Xue J, Rai V, Singer D, et al. Advanced glycation end product recognition by the receptor for AGEs. Structure. 2011;19:722–732. doi: 10.1016/j.str.2011.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Sarkany Z, Ikonen T, Ferreira-da-Silva F, et al. Solution structure of the soluble receptor for advanced glycation end-products (sRAGE) J. Biol. Chem. 2011;286:37,525–37,534. doi: 10.1074/jbc.M111.223438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Huttunen HJ, Fages C, Rauvala H. Receptor for advanced glycation end products (RAGE)-mediated neurite outgrowth and activation of NF-kappaB require the cytoplasmic domain of the receptor but different downstream signaling pathways. J. Biol. Chem. 1999;274:19919–19924. doi: 10.1074/jbc.274.28.19919. [DOI] [PubMed] [Google Scholar]
- 82.Lander HM, Tauras JM, Ogiste JS, et al. Activation of the receptor for advanced glycation end products triggers a p21(ras)-dependent mitogen-activated protein kinase pathway regulated by oxidant stress. J. Biol. Chem. 1997;272:17810–17814. doi: 10.1074/jbc.272.28.17810. [DOI] [PubMed] [Google Scholar]
- 83.Huang JS, Guh JY, Chen HC, et al. Role of receptor for advanced glycation end-product (RAGE) and the JAK/STAT-signaling pathway in AGE-induced collagen production in NRK-49F cells. J. Cell. Biochem. 2001;81:102–113. doi: 10.1002/1097-4644(20010401)81:1<102::aid-jcb1027>3.0.co;2-y. [DOI] [PubMed] [Google Scholar]
- 84.Yeh CH, Sturgis L, Haidacher J, et al. Requirement for p38 and p44/p42 mitogen-activated protein kinases in RAGE-mediated nuclear factor-kappaB transcriptional activation and cytokine secretion. Diabetes. 2001;50:1495–1504. doi: 10.2337/diabetes.50.6.1495. [DOI] [PubMed] [Google Scholar]
- 85.Schmidt AM, Vianna M, Gerlach M, et al. Isolation and characterization of two binding proteins for advanced glycosylation end products from bovine lung which are present on the endothelial cell surface. J. Biol. Chem. 1992;267:14987–14997. [PubMed] [Google Scholar]
- 86.Hudson BI, Kalea AZ, Del Mar Arriero M, et al. Interaction of the RAGE cytoplasmic domain with diaphanous-1 is required for ligand-stimulated cellular migration through activation of Rac1 and Cdc42. J. Biol. Chem. 2008;283:34457–34468. doi: 10.1074/jbc.M801465200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Krebs A, Rothkegel M, Klar M, Jockusch BM. Characterization of functional domains of mDia1, a link between the small GTPase Rho and the actin cytoskeleton. J. Cell. Sci. 2001;114:3663–3672. doi: 10.1242/jcs.114.20.3663. [DOI] [PubMed] [Google Scholar]
- 88.Tominaga T, Sahai E, Chardin P, et al. Diaphanous-related formins bridge Rho GTPase and Src tyrosine kinase signaling. Mol. Cell. 2000;5:13–25. doi: 10.1016/s1097-2765(00)80399-8. [DOI] [PubMed] [Google Scholar]
- 89.Sakaguchi M, Murata H, Yamamoto K, et al. TIRAP, an adaptor protein for TLR2/4, transduces a signal from RAGE phosphorylated upon ligand binding. PLoS One. 2011;6:e23132. doi: 10.1371/journal.pone.0023132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Young KG, Copeland JW. Formins in cell signaling. Biochim. Biophys. Acta. 2010;1803:183–190. doi: 10.1016/j.bbamcr.2008.09.017. [DOI] [PubMed] [Google Scholar]
- 91.Geneste O, Copeland JW, Treisman R. LIM kinase and Diaphanous cooperate to regulate serum response factor and actin dynamics. J. Cell Biol. 2002;157:831–838. doi: 10.1083/jcb.200203126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Copeland JW, Treisman R. The diaphanous-related formin mDia1 controls serum response factor activity through its effects on actin polymerization. Mol. Biol. Cell. 2002;13:4088–4099. doi: 10.1091/mbc.02-06-0092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Xu Y, Toure F, Qu W, et al. Advanced glycation end product (AGE)-receptor for AGE (RAGE) signaling and up-regulation of Egr-1 in hypoxic macrophages. J. Biol. Chem. 2010;285:23233–23240. doi: 10.1074/jbc.M110.117457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Balasubbu S, Sundaresan P, Rajendran A, et al. Association analysis of nine candidate gene polymorphisms in Indian patients with type 2 diabetic retinopathy. BMC Med. Genet. 2010;11:158. doi: 10.1186/1471-2350-11-158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Zong H, Ward M, Madden A, et al. Hyperglycaemia-induced pro-inflammatory responses by retinal Muller glia are regulated by the receptor for advanced glycation end-products (RAGE) Diabetologia. 2010;53:2656–2666. doi: 10.1007/s00125-010-1900-z. [DOI] [PubMed] [Google Scholar]
- 96.Zhang HM, Chen LL, Wang L, et al. Association of 1704G/T and G82S polymorphisms in the receptor for advanced glycation end products gene with diabetic retinopathy in Chinese population. J. Endocrinol. Invest. 2009;32:258–262. doi: 10.1007/BF03346463. [DOI] [PubMed] [Google Scholar]
- 97.Barile GR, Schmidt AM. RAGE and its ligands in retinal disease. Curr. Mol. Med. 2007;7:758–765. doi: 10.2174/156652407783220778. [DOI] [PubMed] [Google Scholar]
- 98.Kaji Y, Usui T, Ishida S, et al. Inhibition of diabetic leukostasis and blood-retinal barrier breakdown with a soluble form of a receptor for advanced glycation end products. Invest. Ophthalmol. Vis. Sci. 2007;48:858–865. doi: 10.1167/iovs.06-0495. [DOI] [PubMed] [Google Scholar]
- 99.Pachydaki SI, Tari SR, Lee SE, et al. Upregulation of RAGE and its ligands in proliferative retinal disease. Exp. Eye Res. 2006;82:807–815. doi: 10.1016/j.exer.2005.09.022. [DOI] [PubMed] [Google Scholar]
- 100.Barile GR, Pachydaki SI, Tari SR, et al. The RAGE axis in early diabetic retinopathy. Invest. Ophthalmol. Vis. Sci. 2005;46:2916–2924. doi: 10.1167/iovs.04-1409. [DOI] [PubMed] [Google Scholar]
- 101.Brussee V, Guo G, Dong Y, et al. Distal degenerative sensory neuropathy in a long-term type 2 diabetes rat model. Diabetes. 2008;57:1664–1673. doi: 10.2337/db07-1737. [DOI] [PubMed] [Google Scholar]
- 102.Toth C, Rong LL, Yang C, et al. Receptor for advanced glycation end products (RAGEs) and experimental diabetic neuropathy. Diabetes. 2008;57:1002–1017. doi: 10.2337/db07-0339. [DOI] [PubMed] [Google Scholar]
- 103.Haslbeck KM, Schleicher E, Bierhaus A, et al. The AGE/RAGE/NF-(kappa)B pathway may contribute to the pathogenesis of polyneuropathy in impaired glucose tolerance (IGT) Exp. Clin. Endocrinol. Diabetes. 2005;113:288–291. doi: 10.1055/s-2005-865600. [DOI] [PubMed] [Google Scholar]
- 104.Bierhaus A, Haslbeck KM, Humpert PM, et al. Loss of pain perception in diabetes is dependent on a receptor of the immunoglobulin superfamily. J. Clin. Invest. 2004;114:1741–1751. doi: 10.1172/JCI18058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Berlanga J, Cibrian D, Guillen I, et al. Methylglyoxal administration induces diabetes-like microvascular changes and perturbs the healing process of cutaneous wounds. Clin. Sci. 2005;109:83–95. doi: 10.1042/CS20050026. [DOI] [PubMed] [Google Scholar]
- 106.Wear-Maggitti K, Lee J, Conejero A, et al. Use of topical sRAGE in diabetic wounds increases neovascularization and granulation tissue formation. Ann. Plast. Surg. 2004;52:519–521. doi: 10.1097/01.sap.0000122857.49274.8c. Discussion 522. [DOI] [PubMed] [Google Scholar]
- 107.Santana RB, Xu L, Chase HB, et al. A role for advanced glycation end products in diminished bone healing in type 1 diabetes. Diabetes. 2003;52:1502–1510. doi: 10.2337/diabetes.52.6.1502. [DOI] [PubMed] [Google Scholar]
- 108.Goova MT, Li J, Kislinger T, et al. Blockade of receptor for advanced glycation end-products restores effective wound healing in diabetic mice. Am. J. Pathol. 2001;159:513–525. doi: 10.1016/S0002-9440(10)61723-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Ishihara K, Tsutsumi K, Kawane S, et al. The receptor for advanced glycation end-products (RAGE) directly binds to ERK by a D-domain-like docking site. FEBS Lett. 2003;550:107–113. doi: 10.1016/s0014-5793(03)00846-9. [DOI] [PubMed] [Google Scholar]