

Abstract
Objective
We have previously demonstrated that activation of toll-like receptor 4 (TLR4) in skeletal muscle results in an increased reliance on glucose as an energy source and a concomitant decrease in fatty acid oxidation under basal conditions. Herein, we examined the effects of lipopolysaccharide (LPS), the primary ligand for TLR4, on mitochondrial oxygen consumption in skeletal muscle cell culture and isolated mitochondria.
Materials/ methods
Skeletal muscle cell cultures were exposed to LPS and oxygen consumption was assessed using a Seahorse Bioscience extracellular flux analyzer. Mice were also exposed to LPS and oxygen consumption was assessed in mitochondria isolated from skeletal muscle.
Results
Acute LPS exposure resulted in significant reductions in cyanide 4-(trifluoromethoxy) phenylhydrazone (FCCP)-stimulated maximal respiration (state 3u) and increased oligomycin induced state 4 (state 4O) respiration in C2C12 and human primary myotubes. These findings were observed in conjunction with increased mRNA of uncoupling protein 3 (UCP3), superoxide dismutase 2 (SOD2), and pyruvate dehydrogenase activity. The LPS-mediated changes in substrate oxidation and maximal mitochondrial respiration were prevented in the presence of the antioxidants N-acetylcysteine and catalase, suggesting a potential role of reactive oxygen species in mediating these effects. Mitochondria isolated from red gastrocnemius and quadriceps femoris muscle from mice injected with LPS also demonstrated reduced respiratory control ratio (RCR), and ADP- and FCCP-stimulated respiration.
Conclusion
LPS exposure in skeletal muscle alters mitochondrial oxygen consumption and substrate preference, which is absent when antioxidants are present.
Keywords: Skeletal muscle, mitochondrial oxygen consumption, reactive oxygen species, respiration, C2C12 myotubes, human primary myotubes
Introduction
Toll-like receptor 4 (TLR4) is a trans-membrane protein integral to innate immunity [1,2]. Upon activation, TLR4 initiates an inflammatory response through the activation of a variety of pro- and anti-inflammatory pathways [3]. TLR4 has been implicated in a number of diseases including type 2 diabetes (T2DM) and cardiovascular disease [4–11]. Mutations resulting in nonfunctional TLR4 render protection from obesity, insulin resistance, and cardiovascular disease in rodent models [12,13]. In humans, polymorphisms in the TLR4 gene are associated with reduced incidence of heart disease and diabetes [14,15]. We, and others have observed increased expression of TLR4 in skeletal muscle from obese and T2DM humans relative to non-obese, healthy controls [16–18].
There has been growing interest in the role of circulating Lipopolysaccharide (LPS), also known as endotoxin, in the pathology of metabolic derangements associated with obesity and T2DM [19–25]. Rodent studies have revealed associations between elevated circulating LPS and metabolic dysregulation, both at the whole body and tissue level [19]. Specifically, high fat feeding in rodents elicits modest elevations in circulating LPS, a phenomenon that is termed metabolic endotoxemia [20]. In humans, a single high fat meal acutely increases circulating LPS concentrations, while obesity and T2DM are associated with metabolic endotoxemia [23,24,26,27]. In fact, it appears that blood LPS levels follow a circadian pattern peaking a couple of hours following a meal [23,26,28]. However with obesity or following chronic periods of high fat feeding this rhythm may be lost, resulting in subclinically elevated LPS levels [19,24].
Our group has recently demonstrated that acute activation of TLR4, in skeletal muscle cells, by LPS results in increased reliance on glucose as an oxidative substrate and a simultaneous decrease in fatty acid oxidation under non-insulin-stimulated conditions [17]. The underlying mechanism(s) of TLR4-induced effects on skeletal muscle substrate metabolism are not completely understood. However, the preferential utilization of glucose relative to fatty acids under LPS-stimulated conditions warrants an investigation into mitochondrial oxygen consumption under the same conditions. Additionally, examining the effects of acute versus chronic LPS administration on mitochondrial oxygen consumption may provide a better understanding of how LPS plays a role in the development of metabolic diseases. The purpose of the current study was to determine the effects of acute and chronic low dose LPS administration (similar to what is observed with metabolic endotoxemia) on mitochondrial oxygen consumption in skeletal muscle.
Materials and Methods
Cell culture
C2C12 murine myoblasts were purchased from the American Type Culture Collection (Manassas, VA) and cultured as previously described [17]. Cultures of human primary skeletal muscle cells from non-obese donors (Caucasian, healthy, males, BMI<25kg/m2, no evidence of metabolic disease, no family history of diabetes or cardiovascular disease) were performed as previously described [29]. All samples were obtained from subjects who provided written informed consent under an approved protocol by the Virginia Polytechnic Institute and State University Institutional Review Board. Experiments were conducted on day 4 of differentiation for C2C12 cells and day 7 of differentiation from human primary cells (first passage), unless otherwise noted. All experiments were conducted three times.
LPS treatment in cell culture studies
C2C12 skeletal muscle cells were treated with 50 pg/mL of LPS from Escherichia coli 0111:B4 (Sigma-Aldrich, St. Louis, MO) for 2, 12, or 24 hours, as described below. We have previously shown that both low (50pg/mL) and high dose (500ng/mL) concentrations of LPS result in a substrate preference for the oxidation of glucose over that of fatty acids following the above specified treatment time course in C2C12 cells [17]. The low dose was selected for the current work since it is synonymous with LPS levels reported in a state of metabolic endotoxemia [19].
Antioxidant treatment in cell culture
C2C12 cells were co-treated with either 50pg/mL of LPS with or without the cell-permeable antioxidants, N-acetyl-L-cysteine (NAC) or Catalase [30–33]. Cells were treated with either 20 mM of NAC (A7250; Sigma-Aldrich, St. Louis MO) or 25 U/mL of Catalase (C1345; Sigma-Aldrich, St. Louis MO) for 2 hours. Cells were pretreated with Catalase for 30 minutes before the addition of LPS.
Mitochondrial respiration in cell culture
Assessment of mitochondrial respiration in C2C12 skeletal muscle cells was performed using an XF24 extracellular flux analyzer (Seahorse Bioscience, North Billerica, MA) as described by Gerencser et. al., with modifications [34]. Briefly, cells were seeded into 20 wells of a XF24 V7 microplate at a density of 5,000 cells per well. Cells were grown to 80% confluence and differentiated into myotubes. Experiments were conducted in serum free media containing 25mmol glucose, 110mg/L pyruvate, and 4mM glutamate. Experiments consisted of 3-minute mixing, 2-minute wait, and 3-minute measurement cycles. Oxygen consumption was measured as previously described under basal conditions, in the presence of the ATP synthase inhibitor, oligomycin (state 4O; 0.5 μM, O4876; Sigma-Aldrich, St. Louis, MO), and in the presence of Carbonyl cyanide 4-(trifluoromethoxy) phenylhydrazone (state 3u; FCCP; 300 μM, C2920; Sigma-Aldrich, St. Louis, MO) a mitochondrial uncoupler, to assess maximal respiration [34,35]. Cellular respiratory control ratio (cRCR) was calculated as the ratio of FCCP stimulated maximal respiratory rate over oligomycin induced state 4 respiration. Data are expressed as percent change from baseline values to account for potential differences in cell number across wells. All experiments were performed at 37 °C.
Cell Culture Substrate Oxidation and Pyruvate Deydrogenase Activity
Fatty acid oxidation was assessed in cell culture by measuring and summing 14CO2 production and 14C-labeled acid-soluble metabolites from the oxidation of [1-14C]-palmitic acid (Perkin Elmer, Waltham, MA), respectively, as previously described [17,36]. Briefly, cells were serum starved for 12 hours and then incubated in 0.5 uCi/ml of [1-14C]-palmitic acid for 3 hours. Media was then removed and exposed to 45% perchloric acid for 1 hour to liberate 14CO2, which was trapped in a tube containing 1M NaOH. The NaOH was then placed into a scintillation vial, 5ml scintillation fluid added, and then placed on a scintillation counter (LS 4500, Beckman Coulter) and counted for the presence of 14C. Acid soluble metabolites were determined by collecting the acidified media and measuring 14C content. Glucose oxidation was assessed by measuring 14CO2 production from the oxidation of 1uCi/ml [U-14C]-glucose (Perkin Elmer, Waltham, MA) in a manner similar to fatty acid oxidation with the exception that glucose was substituted for BSA-bound palmitic acid. [1-14C]-pyruvate oxidation was assessed in a similar manner to glucose oxidation with the exception that pyruvate was substituted for [U-14C]-glucose. Pyruvate oxidation was used to assess the activity of pyruvate dehydrogenase (PDH), the enzyme that catalyzes the oxidation of pyruvate resulting in the provision of glucose-derived acetyl CoA to the TCA cycle [37].
Total RNA extraction and qRT-PCR
Total cellular RNA was extracted using an RNeasy Mini Kit (Qiagen) and DNase I treatment (Qiagen, Valencia, CA), according to the manufacturer’s instructions. Target gene expression was normalized to β-actin (C2C12 cells) or Cyclophilin B (human primary cells) rRNA levels, which were assayed by multiplexing with the manufactures 5#VIC-labeled, primer-limited β-actin or cyclophilin B endogenous control premix. Primers and 5# FAM-labeled Taqman probes were purchased as pre-validated assays and qRT-PCR was performed using an ABI 7900HT (Applied Biosystems, Carlsbad, CA). Relative quantification of target genes was calculated using the 2Δ – CT method. Derivation of the 2 –ΔCT equation has been described in Applied Biosystems User Bulletin No. 2 (P/N 4303859).
Mitochondrial DNA copy number
Mitochondrial copy number was assessed according to the method of He et. al., [38]. C2C12 cells were exposed to 50 pg/ml of LPS for 2 hours. Immediately following LPS exposure, cells were collected and DNA was extracted using a commercially available kit (Invitrogen, Carlsbad, CA). TaqMan primers and probes were designed using Primer Express version 2.1 and real-time PCR was carried out in an ABI PRISM 7900 sequence detector (Applied Biosystems, Carlsbad, CA). Murine skeletal muscle genomic DNA copy number was measured at the Uncoupling Protein 2 (UCP2) gene and mitochondrial DNA (mtDNA) copy number was measured from the cytochrome C oxidase II (COX II) mtDNA gene. mtDNA copy number was calculated by the absolute value of ΔCt values between groups (control vs. LPS). Because amplification occurs exponentially (increasing twofold with each cycle of PCR), log base 2 of ΔCt is the copy number for each sample.
Animal studies
Mouse studies were performed under an approved protocol by the Institutional Animal Care and Use Committee at Virginia Tech. Mice for all studies were maintained on a normal chow diet and a 12:12-hr light-dark cycle. Two studies were performed in male C57BL/6J mice. In the first study, 8-wk-old male C57BL/6J mice were injected in the peritoneum with saline (control, n=6) or LPS (LPS, n=6; 0.01 μg/kg body weight) in the morning following an overnight fast. LPS from Escherichia coli 0111:B4 was used for all studies (L2630; Sigma-Aldrich, St. Louis, MO). Animals were sacrificed via CO2 asphyxiation/ cervical dislocation 4-hours post injection and respiration was measured in mitochondria isolated from the red portion of gastrocnemius muscle. Each mitochondrial sample was run in triplicate. In the second study, four C57BL/6J mice were sacrificed in the morning as described above and mitochondria were isolated from the red portion of gastrocnemius muscle. The mitochondria (5μg each well) from each animal were then directly exposed to either saline or LPS (50 pg/mL) for 1 hour while mitochondrial respiration was assessed. Each mitochondrial sample from each animal was run in triplicate (n=4*3=12, control; n=4*3=12 LPS).
Mitochondrial isolation from red gastrocnemius muscle
Mitochondria were isolated from red gastrocnemius muscle as previously described with modifications [39]. Freshly dissected muscle was placed in ice-cold buffer 1 for mitochondrial isolation (IBM1) containing 67mM of sucrose, 50mM Tris/ HCl, 50mM KCl, 10mM EDTA/ Tris, and 0.2% BSA. The fat and connective tissue was removed and the muscle was minced into very small pieces (<10mg). The tissue was transferred to a cell strainer, rinsed with PBS/ EDTA, and placed in IMB1/ 0.05% trypsin for digestion for 30 minutes. The sample was then centrifuged at 200g for 3 minutes at 4°C, the supernatant removed and the pellet was resuspended in IBM1. The sample was homogenized using a Potter Ehlvejhem glass/ teflon homogenizer (Thomas Scientific, Swedesboro, NJ), centrifuged at 700g for 10 minutes at 4°C. The supernatant was transferred to a polypropylene tube and was centrifuged at 8000g for 10 minutes at 4°C. The supernatant was carefully removed and the pellet suspended in buffer 2 for mitochondrial isolation (IBM2) containing 250mM Sucrose, 3mM EGTA/ Tris, 10mMTris HCl) and then centrifuged again at 8000g for 10 minutes at 4°C. Finally, the supernatant was carefully discarded and the pellet was resuspended in 200μL of IBM2. Protein concentration was determined using the bicinchoninic acid (BCA) assay.
Respiration in isolated mitochondria
Respirometry measures of isolated mitochondria were performed using an XF24 extracellular flux analyzer (Seahorse Bioscience, North Billerica, MA). Immediately following mitochondrial isolation, protein was quantified using a Pierce bicinchoninic acid assay (Thermo Scientific, Rockford, IL) and mitochondria were plated on Seahorse cell culture plates at a concentration of 5 μg/ well in the presence of 10 mM pyruvate (P5280; Sigma-Aldrich, St. Louis, MO) and 5 mM malate (P5280; Sigma-Aldrich, St. Louis, MO). Experiments consisted of 25 second mixing and 4–7 minute measurement cycles. Oxygen consumption was measured as previously described under state 2 respiration (in the presence of pyruvate and malate), ADP (5 mM, Sigma-Aldrich, St. Louis, MO) state 3 stimulated respiration (State 3), oligomycin (2μM) induced state 4 respiration (State 4O), and uncoupled, maximal respiration in the presence of FCCP (3 μM) to assess respiratory capacity (State 3u) [35,40]. Respiratory control ratio (RCR) was calculated as the ratio of ADP stimulated state 3 and oligomycin induced state 4 respiration. Oligomycin induced state 4 respiration was used to account for any contaminating ATPase activity that may prevent the restoration of a low respiration [35,40]. Data are expressed as pmol/min. All experiments were performed at 37 °C.
Statistics
Results were analyzed with Graph Pad Prism Software (Version 6). All data was tested for normality using the Shapiro-Wilk normality test. Data were analyzed using a 2-tailed Student’s T-test or a 2-way ANOVA with a Tukey post-hoc test for multiple comparisons in the case of normally distributed data. If it was determined that the data was not normally distributed, a Mann-Whitney test was conducted in place of a T-test and a Kruskal-Wallis was used in place of the ANOVA. Results are presented as mean ± SD. The level of significance was set a priori at P<0.05.
Results
Low levels of LPS alter mitochondrial oxygen consumption in C2C12 cells
Two hours of exposure to 50pg/ml of LPS resulted in a significant decline in cRCR in C2C12 muscle cells (Figure 1A). These changes were a direct result of higher rates of respiration in the presence of oligomycin (inhibitor of ATP synthesis) and lower rates of FCCP-stimulated (mitochondrial uncoupler) maximal respiration (Figure 1, B and C). Chronic treatment (12 and 24 hours) with LPS also resulted in a significant decline in FCCP-stimulated maximal respiration (Figure 2, A and B), which resulted in a trending decline in cRCR (Figure 2, C; p=0.1 and D; p=0.08). However, chronic LPS treatment had no effect on respiration in the presence of oligomycin (Figure 2, E and F).
Figure 1. Acute TLR4 activation alters mitochondrial oxygen consumption in C2C12 skeletal muscle cells.
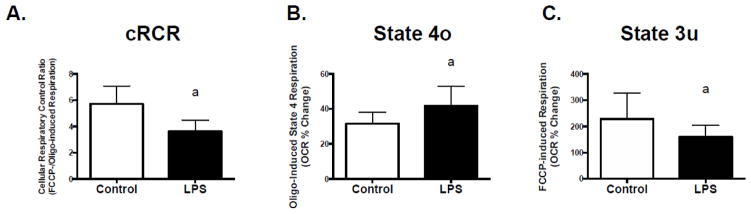
C2C12 myotubes were exposed to 50 pg/mL LPS for 2 hours followed by assessment of mitochondrial respiration using a Seahorse XF24 extracellular flux analyzer. Experiments were conducted in serum free DMEM with glucose and pyruvate as the substrates under basal conditions, in the presence of the ATP synthase inhibitor oligomycin (0.5 μM, State 4o), and in the presence of the mitochondrial uncoupler, FCCP (300 μM, State 3u) to assess maximal respiration. Cellular respiratory control ratio (cRCR) was calculated as FCCP-stimulated maximal respiration rate/oligomycin-induced state 4 respiration rate. (A) cRCR, (B) State 4o, and (C) State 3u. Data are presented as means ± SD. a, significantly different from control, p< 0.05.
Figure 2. Chronic TLR4 activation impairs mitochondrial respiratory capacity in C2C12 cells.

Cells were exposed to 50 pg/mL LPS for 12 or 24 hours followed by assessment of mitochondrial respiration using a Seahorse XF24 extracellular flux analyzer. Experiments were conducted as described in figure one (A) 12-hour State 3u, (B) 24-hour State 3u, (C) 12-hour cRCR, (D) 24-hour cRCR, (E) 12-hour State 4o, (F) 24-hour State 4o. Data are presented as means ± SD. a, significantly different from control, p< 0.05.
Pyruvate dehydrogenase activity is increased in C2C12 cells in response to LPS
The activity of pyruvate dehydrogenase was assessed via pyruvate oxidation [37]. Following a 2 hour treatment with 50 pg/mL of LPS, PDH activity was significantly increased compared to controls (Figure 3).
Figure 3. Pyruvate dehydrogenase activity is increased in C2C12 cells following TLR4 activation.

14CO2 production from the oxidation of [1–14C]-pyruvate was used to assess PDH activity. C2C12 myotubes were exposed to 50 pg/mL LPS for 2 hours. Immediately following exposure, pyruvate dehydrogenase activity was assessed. Data are presented as means ± SD. a, significantly different from control, p< 0.05.
The LPS-induced effects in C2C12 cells are independent of changes to mitochondrial DNA copy number
Low dose LPS treatment for 2 hours had no effect on mtDNA copy number in C2C12 cells (Control, 126.9±6.9 au vs. LPS, 127.1±2.9 au, p=0.9).
LPS treatment results in the up-regulation of antioxidant gene expression
Two-hour LPS treatment at 50pg/ml significantly increased uncoupling protein 3 (UCP3) and mitochondrial/ manganese superoxide dismutase 2 (SOD2) mRNA levels in C2C12 (Figure 4, A and B, respectively). Additionally, these effects were either attenuated or completely blocked in the presence of NAC.
Figure 4. Antioxidant mRNA is up-regulated following LPS exposure and blocked by the antioxidant NAC in C2C12 cells.
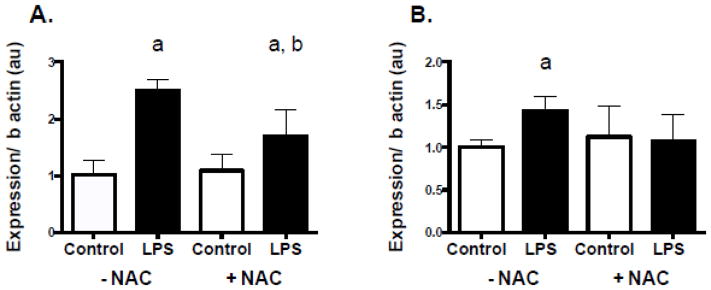
C2C12 cells were treated with 50 pg/mL of LPS with and without 20mM NAC for 2 hours. (A) Uncoupling protein 3 (UCP3) mRNA following LPS exposure in the presence and absence of NAC. (B) Mitochondrial/ manganese superoxide dismutase 2 (SOD2) mRNA levels following LPS exposure in the presence and absence of NAC. Data are presented as means ± SD. a, significantly different from control –NAC, p<0.05. b, significantly different LPS –NAC, p<0.05.
The LPS effect on metabolism in C2C12 cells is blocked by antioxidant treatment
To determine if LPS-mediated effects on oxygen consumption could be blocked by antioxidant treatment, FCCP-stimulated maximal respiration was assessed in C2C12 cells following 2 hours of exposure to 50 pg/mL of LPS in the presence and absence of NAC and Catalase. The presence of NAC and Catalase prevented the LPS-induced decline in FCCP-stimulated maximal respiration (Figure 5, A and B), however had no effects on respiration in the presence of oligomycin (data not shown). To determine whether the LPS-mediated effects on substrate metabolism could also be blocked by antioxidant treatment, glucose and fatty acid oxidation were assessed following 2 hours of exposure to 50 pg/mL of LPS with and without NAC. The LPS-induced changes in glucose oxidation and fatty acid oxidation were also blocked in the presence of NAC (Figure 5, C and D).
Figure 5. The TLR4 effect on metabolism in C2C12 cells is blocked by antioxidant treatment.

C2C12 cells were treated with 50 pg/mL of LPS with and without 20mM NAC or 25 U/mL Catalase for 2 hours. (A) FCCP-stimulated maximal respiration following acute LPS exposure with and without NAC. (B) FCCP-stimulated maximal respiration following acute LPS exposure with and without Catalase. (C) Glucose oxidation following acute LPS exposure with and without NAC, and (D) fatty acid oxidation following acute LPS exposure with and without NAC. Data are presented as means ± SD. a, significantly different from control –NAC, p < 0.05. b, significantly different from LPS –NAC, p <0.05.
LPS alters mitochondrial oxygen consumption in human primary skeletal muscle cells
Based on the effects of low levels of LPS on mitochondrial function in C2C12 cells and mitochondria isolated from mice, we assessed mitochondrial function in human primary skeletal muscle cells under identical conditions. Two hours of LPS treatment resulted in lower cRCR, higher rates of respiration in the presence of oligomycin (inhibitor of ATP synthesis) and lower rates of FCCP-stimulated (mitochondrial uncoupler) maximal respiration (Figure 6, A–C). Chronic treatment (12 and 24 hours) with LPS also resulted in a significant decline in FCCP-stimulated maximal respiration (Figure 7, A and B), which also resulted in a trend for a decline in cRCR (Figure 7, C; p=0.1 and D; p=0.06). However, there were no significant effects on respiration in the presence of oligomycin (Figure 7, E and F).
Figure 6. Acute TLR4 activation alters mitochondrial function in human primary skeletal muscle cells.
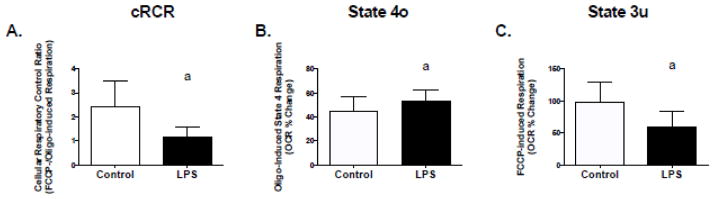
Human primary myotubes were exposed to 50 pg/mL LPS for 2 hours followed by assessment of mitochondrial respiration using a Seahorse XF24 extracellular flux analyzer. Experiments were conducted as described in figure one. (A) cRCR, (B) oligomycin-induced state 4 respiration rate, and (C) FCCP-stimulated maximal respiration. Data are presented as means ± SD. a, significantly different from control, p < 0.05.
Figure 7. Chronic TLR4 activation of impairs mitochondrial respiratory capacity in human primary myotubes.

Cells were exposed to 50 pg/mL LPS for 12 or 24 hours followed by assessment of mitochondrial respiration using a Seahorse XF24 extracellular flux analyzer. Experiments were conducted as describe in figure one. (A) 12-hour State 3u, (B) 24-hour State 3u, (C) 12-hour cRCR, (D) 24-hour cRCR, (E) 12-hour State 4o, (F) 24-hour State 4o. Data are presented as means ± SD. a, significantly different from control, p < 0.05.
Consistent with C2C12 cell experiments under the same conditions, significant increases in mRNA levels of UCP3 and SOD2 were also observed in human primary muscle cells (Figure 8, A and B, respectively), and were blocked in the presence of a NAC. Collectively, these data support our previous work [17], showing that low levels of LPS also affect skeletal muscle metabolism in human skeletal muscle.
Figure 8. Antioxidant mRNA is up-regulated following LPS exposure and blocked by the antioxidant NAC in human primary myotubes.
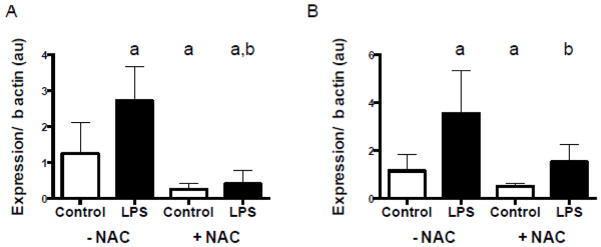
Human primary myotubes were treated with 50 pg/mL of LPS with and without NAC for 2 hours. (A) Uncoupling protein 3 (UCP3) mRNA following LPS exposure in the presence and absence of NAC. (B) Mitochondrial/ manganese superoxide dismutase 2 (SOD2) mRNA levels following LPS exposure in the presence and absence of NAC. Data are presented as means ± SD. a indicates significantly different from control –NAC, p < 0.05. b, significantly different from LPS –NAC, p <0.05.
Low levels of LPS alter oxygen consumption in mitochondria isolated from skeletal muscle
As a follow-up to the C2C12 studies, male C57BL/6J mice were injected i.p., with either saline or LPS (~0.025μg per mouse), euthanized 4 hours post injection, and mitochondrial respiration was assessed in mitochondria isolated from red gastrocnemius skeletal muscle. Compared to saline controls, LPS resulted in significant declines in RCR, ADP-stimulated state 3 respiration, and FCCP-stimulated maximal respiration (Figure 9, A–C). No effects on state 2 or state 4o respiration were observed (Figure 6, D and E). Previous research has demonstrated that direct LPS exposure to isolated mitochondria results in significant reductions in state 3 respiration [41,42]. However, these earlier studies were conducted using very high doses (>100 μg/mL) of LPS. To test whether LPS directly altered mitochondrial respiration at doses relevant to metabolic endotoxemia, mitochondria were isolated from red gastrocnemius skeletal obtained from male C57BL/6J mice and treated with 50 pg/mL of LPS. LPS had no effects on any aspects of mitochondrial oxygen consumption in isolated mitochondria (Basal, Control, 25.5 ± 7.3; N=5; vs. LPS, 17.5 ± 7.3pmol/min, p=0.34; State 3, Control, 772.5 ± 44.47 vs. LPS 626.0 ± 143.3 pmol/min, p= 0.32; State 4O, Control, 115.3 ± 10.34 vs. LPS, 104.0 ± 12.32 pmol/min, p=0.50; State 3u, Control, 558.8 ± 46.82 vs. LPS, 510.0 ± 153.3 pmol/min, p=0.75; RCR, Control, 6.850 ± 0.3971 vs. LPS, 5.704 ± 0.7555, p=0.19).
Figure 9. TLR4 activation causes mitochondrial dysfunction in skeletal muscle from C57BL/6J mice.

C57BL/6J mice were injected with LPS at a dose of 1 μg/kg body mass. Four hours post injection, red gastrocnemius muscle mitochondria were isolated, and oxygen consumption was measured. Respirometry measures of isolated mitochondria were performed using a Seahorse XF24 extracellular flux analyzer. Oxygen consumption was measured under basal conditions, ADP (5 mM) stimulated state 3 respiration, oligomycin (2μM) induced state 4 respiration, and uncoupled respiration in the presence of FCCP (0.3 μM) to assess maximal respiration. Respiratory control ratio was calculated as the ratio of state 3 respiration to oligomycin induced state 4 respiration. (A) RCR, (B) ADP-stimulated state 3 respiration, (C) FCCP-stimulated maximal respiration, (D) basal respiration, and (E) oligomycin respiration rate. Data are presented as means ± SD. a, significantly different from control, p < 0.05.
Discussion
The goal of the current study was to gain a better understanding of the effects of acute and chronic exposure of low dose LPS administration on mitochondrial oxygen consumption in skeletal muscle. Our group has previously reported that acute LPS exposure in skeletal muscle results in the preferential oxidation of glucose over that of fatty acids in the basal, non-insulin stimulated state [17]. Furthermore, recent work highlighting metabolic endotoxemia in the development of metabolic disease warrants the need to understand the role of acute versus chronic LPS exposure on mitochondrial function in skeletal muscle [19–25]. The important observations reported herein are that acute LPS exposure in both C2C12 and human primary myotubes results in increased state 4O respiration and reduced state 3u respiration. Moreover, these effects occur in the context of increased reliance on glucose as an oxidative substrate for the mitochondria [43]. However, chronic exposure to LPS (12–24 hours) results in reduced state 3u respiration only.
Earlier studies demonstrated that LPS administered to isolated mitochondrial preparations resulted in inhibition of respiration and declines in respiratory control [41,42,44,45]. More recent studies conducted in animal models also showed that LPS administration in vivo results in a decline in mitochondrial respiration and respiratory control as well as declines in the activity of Complexes I and III of the electron transport chain [2,46,47]. However, these earlier studies were conducted with very high doses of LPS (>100ug) and in some conditions, direct treatment of mitochondrial preparations, which may have been toxic to the mitochondria [2]. The current studies demonstrate that much lower doses of LPS, similar to what is observed in obesity, T2DM, and metabolic endotoxemia, also results in changes in respiration [19,25].
The mechanisms responsible for the effects of LPS on mitochondrial respiration in our study are not known; however a number of possibilities exist. First, the acute effects of LPS exposure may not be mitochondrial dysfunction per se, but rather mitochondria responding to physiological cues leading to mitochondrial uncoupling, subsequent increases in respiration, relieved inhibition of PDH, and ultimately a switch to a more rapidly metabolized substrate for ATP production (e.g., glucose) [37,48–50]. For example, increased reactive oxygen species production could result in increased mitochondrial proton leak and uncoupling through either UCP3 dependent or independent mechanisms [51,52]. Conversely, the LPS induced increase in state 4O respiration could also be increased in response to an increase in substrate kinetics, as evidenced by the increase in PDH activity; however the contribution of substrate oxidation to oligomycin resistant respiration compared to mitochondrial proton leak is low [35]. Future studies are warranted to elucidate the mechanism(s) underlying the effects of LPS on mitochondrial respiration.
The mechanism(s) for the LPS-mediated increased PDH activity is unknown, however a few possibilities exist. It is well established that acetyl-CoA, NADH and pyruvate dehydrogenase kinase 4 (PDK4) are negative regulators of PDH, and that protein phosphatases (1 and 2) are positive regulators of PDH [37,48–50,53]. The LPS induced -increased electron transport flux via uncoupling could result in reduced concentrations of acetyl-CoA and NADH, thus relieving inhibition of PDH. On the other hand, LPS may be altering the enzyme kinetics of PDK4 or protein phosphatases to increase PDH activity [53]. Future studies will be necessary to discern the precise mechanism(s) by which LPS exposure increases PDH activity.
It has also been well established in non-muscle cells, that activation of TLR4 via LPS increases ROS production [30,32,54–57]. Sandstrom et al. reported that contraction-induced increases in glucose uptake in isolated mouse extensor digitorum longus (EDL) muscle was reduced by 50% in the presence of the antioxidants NAC and ebselen [33]. We recently reported that 2 hours of LPS exposure caused a significant increase in basal (non-insulin stimulated) glucose uptake and lactate production in skeletal muscle cells, suggesting an increase in glycolytic flux [17]. Herein, we show that the LPS-mediated increase in glucose oxidation (as well as the reduction in fat oxidation and FCCP stimulated respiration) is blocked in the presence of the antioxidant NAC. In the same context, increases in SOD2 and UCP3 gene expression are also blocked in the presence of NAC. It is important to point out that NAC significantly decreased basal mRNA expression of SOD2 and UCP3 and this decrease may be mediated, at least in part, by NFκB for several reasons. NAC is a precursor of glutathione and has been shown to down regulate NFκB activity [58,59]. In turn, NFκB has been shown to regulate SOD2 and UCP3 expression either directly or through TNFa [60,61]. Therefore the reduction in basal gene expression may be in response to a NAC induced reduction in NFκB. It is not evident in the current work if the effects of NAC on glucose or pyruvate oxidation are also observed with changes in glucose uptake and lactate production as demonstrated previously [17]. Nonetheless, based on these observations, it is plausible that LPS-mediated ROS production in skeletal muscle is the mechanism by which low levels of LPS modulate mitochondrial substrate preference.
We also present that a more chronic exposure to LPS (12–24 hours) results in impaired mitochondrial respiratory capacity with no evidence of mitochondrial proton leak. Under maximally stimulated conditions, respiration is dependent on multiple factors such as substrate supply and coordination between the TCA cycle and the electron transport chain [35,40]. As such, reduced respiration in response to FCCP in the absence of increased state 4O respiration, is more indicative of impaired mitochondrial respiratory capacity and not only mild mitochondrial uncoupling [35]. This is supported by data from the isolated mitochondrial experiments demonstrating significant reductions in RCR, ADP-stimulated, and FCCP-stimulated respiration in the presence of LPS. These data suggest that prolonged exposure to LPS is disruptive to mitochondrial function, whereas short-term exposure may serve a modulatory role in substrate metabolism.
There are some limitations to the current work. Studies were conducted in cell culture and animal models and therefore it is unknown if similar effects are observed in humans. Additionally, the underlying mechanisms for the effects of LPS on mitochondrial oxygen consumption are not entirely understood, although it is suggested that ROS may be involved. The current data does highlight a novel link between inflammatory pathways and oxygen consumption in skeletal muscle and may therefore provide additional insight into the development of metabolic dysfunction in the face of chronic low-grade inflammation as observed in obesity. Human studies are warranted to better understand the relationship between feeding and circulating LPS concentrations, and how these fluctuations may pertain to substrate preference and oxidation.
In conclusion, acute exposure of LPS at levels synonymous with metabolic endotoxemia cause skeletal muscle to preferentially rely on glucose as an oxidative substrate, which occurs in the context of uncoupled mitochondrial respiration. Moreover, these effects are absent in the presence of antioxidants, suggesting these effects are ROS dependent. Data were also presented to show that more chronic exposure to LPS (12–24h) caused impaired mitochondrial function in C2C12 cells, which may shed some light on a possible mechanism by which chronic metabolic endotoxemia causes whole body metabolic dysregulation, as reported by others [19–21,62,63]. Based on this data set and our previous work, we postulate that transient increases in LPS elicit physiological cues that modulate substrate metabolism, and that chronic elevations in LPS are disruptive to normal metabolic function [43].
Acknowledgments
This work was supported by the National Institutes of Health-NIDDK (RO1DK078765, MWH), The Obesity Society New Investigator’s Award (MIF), and The Fralin Life Science Research Institute at Virginia Tech (MWH and MIF).
Footnotes
Conflicts of Interest. There are no conflicts of interests
Disclosures
None
Author Contributions: MIF: Conception and design of research, performed experiments, analyzed data, interpreted results of experiments, prepared figures, drafted manuscript, edited and revised manuscript; YW: Performed experiments, analyzed data, interpreted results of experiments, edited and revised manuscript; RPM: Performed experiments, analyzed data, edited and revised manuscript; KAV: Performed experiments, analyzed data, edited and revised manuscript; KAW: Performed experiments; AA: Edited and revised manuscript; NB: Edited and revised manuscript; KR: Performed experiments; ER: Interpreted results of experiments; MWH: Conception and design of research, analyzed data, interpreted results of experiments, edited and revised manuscript, approved final version of manuscript.
Conflict of interest: The authors have no competing financial interests to declare.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Medzhitov R, Preston-Hurlburt P, Janeway CA., Jr A human homologue of the Drosophila Toll protein signals activation of adaptive immunity. Nature. 1997;388:394–397. doi: 10.1038/41131. [DOI] [PubMed] [Google Scholar]
- 2.Vanasco V, Cimolai MC, Evelson P, Alvarez S. The oxidative stress and the mitochondrial dysfunction caused by endotoxemia are prevented by alpha-lipoic acid. Free Radic Res. 2008;42:815–823. doi: 10.1080/10715760802438709. [DOI] [PubMed] [Google Scholar]
- 3.Lang CH, Silvis C, Deshpande N, Nystrom G, Frost RA. Endotoxin stimulates in vivo expression of inflammatory cytokines tumor necrosis factor alpha, interleukin-1beta, -6, and high-mobility-group protein-1 in skeletal muscle. Shock. 2003;19:538–546. doi: 10.1097/01.shk.0000055237.25446.80. [DOI] [PubMed] [Google Scholar]
- 4.Belforte FS, Coluccio Leskow F, Poskus E, Penas Steinhardt A. Toll-like receptor 4 D299G polymorphism in metabolic disorders: a meta-analysis. Mol Biol Rep. 2013;40:3015–3020. doi: 10.1007/s11033-012-2374-5. [DOI] [PubMed] [Google Scholar]
- 5.Bjorkbacka H. Multiple roles of Toll-like receptor signaling in atherosclerosis. Curr Opin Lipidol. 2006;17:527–533. doi: 10.1097/01.mol.0000245258.25387.ec. [DOI] [PubMed] [Google Scholar]
- 6.Fukata M, Abreu MT. TLR4 signalling in the intestine in health and disease. Biochem Soc Trans. 2007;35:1473–1478. doi: 10.1042/BST0351473. [DOI] [PubMed] [Google Scholar]
- 7.Jia SJ, Niu PP, Cong JZ, Zhang BK, Zhao M. TLR4 signaling: A potential therapeutic target in ischemic coronary artery disease. Int Immunopharmacol. 2014;23:54–59. doi: 10.1016/j.intimp.2014.08.011. [DOI] [PubMed] [Google Scholar]
- 8.Jialal I, Kaur H, Devaraj S. Toll-like receptor status in obesity and metabolic syndrome: a translational perspective. J Clin Endocrinol Metab. 2014;99:39–48. doi: 10.1210/jc.2013-3092. [DOI] [PubMed] [Google Scholar]
- 9.Miller YI. Toll-like receptors and atherosclerosis: oxidized LDL as an endogenous Toll-like receptor ligand. Future Cardiol. 2005;1:785–792. doi: 10.2217/14796678.1.6.785. [DOI] [PubMed] [Google Scholar]
- 10.Soares JB, Pimentel-Nunes P, Roncon-Albuquerque R, Leite-Moreira A. The role of lipopolysaccharide/toll-like receptor 4 signaling in chronic liver diseases. Hepatol Int. 2010;4:659–672. doi: 10.1007/s12072-010-9219-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Watanabe Y, Nagai Y, Takatsu K. Activation and regulation of the pattern recognition receptors in obesity-induced adipose tissue inflammation and insulin resistance. Nutrients. 2013;5:3757–3778. doi: 10.3390/nu5093757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shi H, Kokoeva MV, Inouye K, Tzameli I, Yin H, et al. TLR4 links innate immunity and fatty acid-induced insulin resistance. J Clin Invest. 2006;116:3015–3025. doi: 10.1172/JCI28898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tsukumo DM, Carvalho-Filho MA, Carvalheira JB, Prada PO, Hirabara SM, et al. Loss-of-function mutation in Toll-like receptor 4 prevents diet-induced obesity and insulin resistance. Diabetes. 2007;56:1986–1998. doi: 10.2337/db06-1595. [DOI] [PubMed] [Google Scholar]
- 14.Kolek MJ, Carlquist JF, Muhlestein JB, Whiting BM, Horne BD, et al. Toll-like receptor 4 gene Asp299Gly polymorphism is associated with reductions in vascular inflammation, angiographic coronary artery disease, and clinical diabetes. Am Heart J. 2004;148:1034–1040. doi: 10.1016/j.ahj.2004.05.049. [DOI] [PubMed] [Google Scholar]
- 15.Manolakis AC, Kapsoritakis AN, Tiaka EK, Sidiropoulos A, Gerovassili A, et al. TLR4 gene polymorphisms: evidence for protection against type 2 diabetes but not for diabetes-associated ischaemic heart disease. Eur J Endocrinol. 2011;165:261–267. doi: 10.1530/EJE-11-0280. [DOI] [PubMed] [Google Scholar]
- 16.Dasu MR, Devaraj S, Park S, Jialal I. Increased toll-like receptor (TLR) activation and TLR ligands in recently diagnosed type 2 diabetic subjects. Diabetes Care. 2010;33:861–868. doi: 10.2337/dc09-1799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Frisard MI, McMillan RP, Marchand J, Wahlberg KA, Wu Y, et al. Toll-like receptor 4 modulates skeletal muscle substrate metabolism. Am J Physiol Endocrinol Metab. 2010;298:E988–998. doi: 10.1152/ajpendo.00307.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Reyna SM, Ghosh S, Tantiwong P, Meka CS, Eagan P, et al. Elevated toll-like receptor 4 expression and signaling in muscle from insulin-resistant subjects. Diabetes. 2008;57:2595–2602. doi: 10.2337/db08-0038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cani PD, Amar J, Iglesias MA, Poggi M, Knauf C, et al. Metabolic endotoxemia initiates obesity and insulin resistance. Diabetes. 2007;56:1761–1772. doi: 10.2337/db06-1491. [DOI] [PubMed] [Google Scholar]
- 20.Cani PD, Bibiloni R, Knauf C, Waget A, Neyrinck AM, et al. Changes in gut microbiota control metabolic endotoxemia-induced inflammation in high-fat diet-induced obesity and diabetes in mice. Diabetes. 2008;57:1470–1481. doi: 10.2337/db07-1403. [DOI] [PubMed] [Google Scholar]
- 21.Cani PD, Delzenne NM. The role of the gut microbiota in energy metabolism and metabolic disease. Curr Pharm Des. 2009;15:1546–1558. doi: 10.2174/138161209788168164. [DOI] [PubMed] [Google Scholar]
- 22.Creely SJ, McTernan PG, Kusminski CM, Fisher M, Da Silva NF, et al. Lipopolysaccharide activates an innate immune system response in human adipose tissue in obesity and type 2 diabetes. Am J Physiol Endocrinol Metab. 2007;292:E740–747. doi: 10.1152/ajpendo.00302.2006. [DOI] [PubMed] [Google Scholar]
- 23.Ghanim H, Abuaysheh S, Sia CL, Korzeniewski K, Chaudhuri A, et al. Increase in plasma endotoxin concentrations and the expression of Toll-like receptors and suppressor of cytokine signaling-3 in mononuclear cells after a high-fat, high-carbohydrate meal: implications for insulin resistance. Diabetes Care. 2009;32:2281–2287. doi: 10.2337/dc09-0979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Harte AL, Varma MC, Tripathi G, McGee KC, Al-Daghri NM, et al. High fat intake leads to acute postprandial exposure to circulating endotoxin in type 2 diabetic subjects. Diabetes Care. 35:375–382. doi: 10.2337/dc11-1593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Musso G, Gambino R, Cassader M. Interactions between gut microbiota and host metabolism predisposing to obesity and diabetes. Annu Rev Med. 2011;62:361–380. doi: 10.1146/annurev-med-012510-175505. [DOI] [PubMed] [Google Scholar]
- 26.Deopurkar R, Ghanim H, Friedman J, Abuaysheh S, Sia CL, et al. Differential effects of cream, glucose, and orange juice on inflammation, endotoxin, and the expression of Toll-like receptor-4 and suppressor of cytokine signaling-3. Diabetes Care. 2010;33:991–997. doi: 10.2337/dc09-1630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Erridge C, Attina T, Spickett CM, Webb DJ. A high-fat meal induces low-grade endotoxemia: evidence of a novel mechanism of postprandial inflammation. Am J Clin Nutr. 2007;86:1286–1292. doi: 10.1093/ajcn/86.5.1286. [DOI] [PubMed] [Google Scholar]
- 28.Ghanim H, Sia CL, Upadhyay M, Korzeniewski K, Viswanathan P, et al. Orange juice neutralizes the proinflammatory effect of a high-fat, high-carbohydrate meal and prevents endotoxin increase and Toll-like receptor expression. Am J Clin Nutr. 2010;91:940–949. doi: 10.3945/ajcn.2009.28584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hulver MW, Berggren JR, Carper MJ, Miyazaki M, Ntambi JM, et al. Elevated stearoyl-CoA desaturase-1 expression in skeletal muscle contributes to abnormal fatty acid partitioning in obese humans. Cell Metab. 2005;2:251–261. doi: 10.1016/j.cmet.2005.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Asehnoune K, Strassheim D, Mitra S, Kim JY, Abraham E. Involvement of reactive oxygen species in Toll-like receptor 4-dependent activation of NF-kappa B. J Immunol. 2004;172:2522–2529. doi: 10.4049/jimmunol.172.4.2522. [DOI] [PubMed] [Google Scholar]
- 31.Pi J, Bai Y, Zhang Q, Wong V, Floering LM, et al. Reactive oxygen species as a signal in glucose-stimulated insulin secretion. Diabetes. 2007;56:1783–1791. doi: 10.2337/db06-1601. [DOI] [PubMed] [Google Scholar]
- 32.Ryan KA, Smith MF, Jr, Sanders MK, Ernst PB. Reactive oxygen and nitrogen species differentially regulate Toll-like receptor 4-mediated activation of NF-kappa B and interleukin-8 expression. Infect Immun. 2004;72:2123–2130. doi: 10.1128/IAI.72.4.2123-2130.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sandstrom ME, Zhang SJ, Bruton J, Silva JP, Reid MB, et al. Role of reactive oxygen species in contraction-mediated glucose transport in mouse skeletal muscle. J Physiol. 2006;575:251–262. doi: 10.1113/jphysiol.2006.110601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gerencser AA, Neilson A, Choi SW, Edman U, Yadava N, et al. Quantitative microplate-based respirometry with correction for oxygen diffusion. Anal Chem. 2009;81:6868–6878. doi: 10.1021/ac900881z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Brand MD, Nicholls DG. Assessing mitochondrial dysfunction in cells. Biochem J. 2011;435:297–312. doi: 10.1042/BJ20110162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hulver MW, Berggren JR, Cortright RN, Dudek RW, Thompson RP, et al. Skeletal muscle lipid metabolism with obesity. Am J Physiol Endocrinol Metab. 2003;284:E741–747. doi: 10.1152/ajpendo.00514.2002. [DOI] [PubMed] [Google Scholar]
- 37.Zhang S, Hulver MW, McMillan RP, Cline MA, Gilbert ER. The pivotal role of pyruvate dehydrogenase kinases in metabolic flexibility. Nutr Metab (Lond) 2014;11:10. doi: 10.1186/1743-7075-11-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.He L, Chinnery PF, Durham SE, Blakely EL, Wardell TM, et al. Detection and quantification of mitochondrial DNA deletions in individual cells by real-time PCR. Nucleic Acids Res. 2002;30:e68. doi: 10.1093/nar/gnf067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Frezza C, Cipolat S, Scorrano L. Organelle isolation: functional mitochondria from mouse liver, muscle and cultured fibroblasts. Nat Protoc. 2007;2:287–295. doi: 10.1038/nprot.2006.478. [DOI] [PubMed] [Google Scholar]
- 40.Brand MD. Top-down elasticity analysis and its application to energy metabolism in isolated mitochondria and intact cells. Mol Cell Biochem. 1998;184:13–20. [PubMed] [Google Scholar]
- 41.Kato M. Site of action of lipid A on mitochondria. J Bacteriol. 1972;112:268–275. doi: 10.1128/jb.112.1.268-275.1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.McGivney A, Bradley SG. Action of bacterial lipopolysaccharide on the respiration of mouse liver mitochondria. Infect Immun. 1980;27:102–106. doi: 10.1128/iai.27.1.102-106.1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Frisard MI, McMillan RP, Marchand J, Wahlberg KA, Wu Y, et al. Toll-like receptor 4 modulates skeletal muscle substrate metabolism. Am J Physiol Endocrinol Metab. 298:E988–998. doi: 10.1152/ajpendo.00307.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Greer GG, Milazzo FH. Pseudomonas aeruginosa lipopolysaccharide: an uncoupler of mitochondrial oxidative phosphorylation. Can J Microbiol. 1975;21:877–883. doi: 10.1139/m75-130. [DOI] [PubMed] [Google Scholar]
- 45.McGivney A, Bradley SG. Action of bacterial endotoxin and lipid A on mitochondrial enzyme activities of cells in culture and subcellular fractions. Infect Immun. 1979;25:664–671. doi: 10.1128/iai.25.2.664-671.1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Joshi MS, Crouser ED, Julian MW, Schanbacher BL, Bauer JA. Digital imaging analysis for the study of endotoxin-induced mitochondrial ultrastructure injury. Anal Cell Pathol. 2000;21:41–48. doi: 10.1155/2000/201406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Trumbeckaite S, Opalka JR, Neuhof C, Zierz S, Gellerich FN. Different sensitivity of rabbit heart and skeletal muscle to endotoxin-induced impairment of mitochondrial function. Eur J Biochem. 2001;268:1422–1429. doi: 10.1046/j.1432-1327.2001.02012.x. [DOI] [PubMed] [Google Scholar]
- 48.Holness MJ, Sugden MC. Regulation of pyruvate dehydrogenase complex activity by reversible phosphorylation. Biochem Soc Trans. 2003;31:1143–1151. doi: 10.1042/bst0311143. [DOI] [PubMed] [Google Scholar]
- 49.Patel MS, Nemeria NS, Furey W, Jordan F. The pyruvate dehydrogenase complexes: structure-based function and regulation. J Biol Chem. 2014;289:16615–16623. doi: 10.1074/jbc.R114.563148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Spriet LL, Heigenhauser GJ. Regulation of pyruvate dehydrogenase (PDH) activity in human skeletal muscle during exercise. Exerc Sport Sci Rev. 2002;30:91–95. doi: 10.1097/00003677-200204000-00009. [DOI] [PubMed] [Google Scholar]
- 51.Cannon B, Shabalina IG, Kramarova TV, Petrovic N, Nedergaard J. Uncoupling proteins: a role in protection against reactive oxygen species--or not? Biochim Biophys Acta. 2006;1757:449–458. doi: 10.1016/j.bbabio.2006.05.016. [DOI] [PubMed] [Google Scholar]
- 52.Mailloux RJ, Harper ME. Uncoupling proteins and the control of mitochondrial reactive oxygen species production. Free Radic Biol Med. 2011;51:1106–1115. doi: 10.1016/j.freeradbiomed.2011.06.022. [DOI] [PubMed] [Google Scholar]
- 53.Peters SJ. Regulation of PDH activity and isoform expression: diet and exercise. Biochem Soc Trans. 2003;31:1274–1280. doi: 10.1042/bst0311274. [DOI] [PubMed] [Google Scholar]
- 54.Park HS, Jung HY, Park EY, Kim J, Lee WJ, et al. Cutting edge: direct interaction of TLR4 with NAD(P)H oxidase 4 isozyme is essential for lipopolysaccharide-induced production of reactive oxygen species and activation of NF-kappa B. J Immunol. 2004;173:3589–3593. doi: 10.4049/jimmunol.173.6.3589. [DOI] [PubMed] [Google Scholar]
- 55.Pi Y, Zhang LL, Li BH, Guo L, Cao XJ, et al. Inhibition of reactive oxygen species generation attenuates TLR4-mediated proinflammatory and proliferative phenotype of vascular smooth muscle cells. Lab Invest. 2013;93:880–887. doi: 10.1038/labinvest.2013.79. [DOI] [PubMed] [Google Scholar]
- 56.Qin L, Li G, Qian X, Liu Y, Wu X, et al. Interactive role of the toll-like receptor 4 and reactive oxygen species in LPS-induced microglia activation. Glia. 2005;52:78–84. doi: 10.1002/glia.20225. [DOI] [PubMed] [Google Scholar]
- 57.Xiang M, Fan J, Fan J. Association of Toll-like receptor signaling and reactive oxygen species: a potential therapeutic target for posttrauma acute lung injury. Mediators Inflamm. 2010;2010 doi: 10.1155/2010/916425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Chen G, Shi J, Hu Z, Hang C. Inhibitory effect on cerebral inflammatory response following traumatic brain injury in rats: a potential neuroprotective mechanism of N-acetylcysteine. Mediators Inflamm. 2008;2008:716458. doi: 10.1155/2008/716458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kim H, Seo JY, Roh KH, Lim JW, Kim KH. Suppression of NF-kappaB activation and cytokine production by N-acetylcysteine in pancreatic acinar cells. Free Radic Biol Med. 2000;29:674–683. doi: 10.1016/s0891-5849(00)00368-3. [DOI] [PubMed] [Google Scholar]
- 60.Peristeris P, Clark BD, Gatti S, Faggioni R, Mantovani A, et al. N-acetylcysteine and glutathione as inhibitors of tumor necrosis factor production. Cell Immunol. 1992;140:390–399. doi: 10.1016/0008-8749(92)90205-4. [DOI] [PubMed] [Google Scholar]
- 61.Zelko IN, Mariani TJ, Folz RJ. Superoxide dismutase multigene family: a comparison of the CuZn-SOD (SOD1), Mn-SOD (SOD2), and EC-SOD (SOD3) gene structures, evolution, and expression. Free Radic Biol Med. 2002;33:337–349. doi: 10.1016/s0891-5849(02)00905-x. [DOI] [PubMed] [Google Scholar]
- 62.Clemente-Postigo M, Queipo-Ortuno MI, Murri M, Boto-Ordonez M, Perez-Martinez P, et al. Endotoxin increase after fat overload is related to postprandial hypertriglyceridemia in morbidly obese patients. J Lipid Res. 2012;53:973–978. doi: 10.1194/jlr.P020909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Osto M, Zini E, Franchini M, Wolfrum C, Guscetti F, et al. Subacute endotoxemia induces adipose inflammation and changes in lipid and lipoprotein metabolism in cats. Endocrinology. 2011;152:804–815. doi: 10.1210/en.2010-0999. [DOI] [PubMed] [Google Scholar]