

Abstract
β-Ureidopropionase deficiency (OMIM #613161) is a rare autosomal recessive inborn error of metabolism due to mutations in the UPB1 gene, which encodes the third enzyme involved in the pyrimidine degradation pathway. A total of 28 cases have been reported, mainly presenting with seizures, microcephaly, and intellectual disabilities. However, 11 of them were asymptomatic cases (Nakajima et al., J Inherit Metab Dis 37(5):801–812, 2014). We report on a 9-year-old female presenting with intractable epilepsy, microcephaly, and global developmental delay. She was homozygous for p.R326Q (c.977G>A) and heterozygous for p.G31S (c.91G>A) in the UPB1 gene, detected by targeted next-generation sequencing test and subsequently confirmed by biochemical analysis of urine, plasma, and cerebrospinal fluid (CSF) using reversed-phase HPLC, combined with electrospray tandem mass spectrometry. We report a first Korean female case with β-ureidopropionase deficiency.
Keywords: Epilepsy, Korean, N-carbamyl-β-amino acids, Pyrimidine, ß-Ureidopropionase
Introduction
β-Ureidopropionase (β-UP), the enzyme involved in pyrimidine degradation pathway, catalyzes the conversion of N-carbamyl-β-alanine and N-carbamyl-β-aminoisobutyric acid to β-alanine and β-aminoisobutyric acid, respectively (van Kuilenburg et al. 2012; Yaplito-Lee et al. 2008). The prevalence of β-UP deficiency in Japan has been estimated at 1 in 6,000 (Kuhara et al. 2009). Molecular findings of 13 Japanese β-UP-deficient patients revealed three novel missense mutations (p.G31S, p.E271K, and p.I286T), with all 13 patients showing a p.R326Q mutation and 8 patients being homozygous. The p.R326Q mutation was previously described to abolish the enzyme activity (Nakajima et al. 2014). Two Chinese patients have been reported, both carried the p.R326Q mutation, but ß-UP deficiency has not been reported in Korea up to date (van Kuilenburg et al. 2012; Nakajima et al. 2014).
We report a first Korean case of ß-ureidopropionase deficiency who was homozygous for p.R326Q (c.977G>A) and heterozygous for p.G31S (c.91G>A) in the UPB1 gene detected by Illumina next-generation sequencer in panel gene test for microcephaly and lissencephaly and Sanger sequencing analysis and the diagnosis was subsequently confirmed by biochemical analysis of the pyrimidine pathway.
Case Report
A 3-month-old female visited the outpatient department after a generalized tonic-clonic seizure without fever. Her family history was nonspecific, and her medical history included a normal vaginal delivery at 41 weeks with a birth weight of 3,140 g (40th percentile), height of 51 cm (70th percentile), and head circumference of 32.5 cm (15th percentile) from non-consanguineous Korean parents. We performed an epilepsy workup with electroencephalography (EEG), brain magnetic resonance image (MRI), electrolyte, thyroid function test, and neonatal tandem mass screening, which were all nonspecific findings. A recurrent seizure was stopped after administration of phenobarbital medication. Development has been nearly normal with mild gross motor delay until the age of 12 months when recurring seizures began and multiple antiepileptic drug medications such as valproic acid, carbamazepine, and clobazam (Sentil®) were administered. Seizures were not well controlled, which required reevaluation and workups that included EEG, brain MRI, and tandem mass screening, but the results did not reveal any specific findings. At the age of 17 months, recurrent seizures continued with poor control, which lead to changes in her medication to valproic acid, topiramate, and pyridoxine. By that time, she showed microcephaly and global developmental delay. At the age of 27 months, we examined the patient using the Korean Bayley Scale of Infant Development-II (K-BSID-II) for developmental delay. Results showed a mental performance age of 4 months and a motor performance age of 6 months. At the age of 3 years, she had an episode of generalized status epilepticus. Further diagnostic workups including an MeCP2 gene test for Rett syndrome, methylation test for Prader-Willi/Angelman syndrome, and muscle biopsy for mitochondrial disease were all normal. At the age of 4 years, the patient had another episode of intractable epilepsy and subsequently started ketogenic diet with multiple antiepileptic drugs such as topiramate, lamotrigine, and levetiracetam. Seizures were aggravated by a minor upper respiratory infection or gastrointestinal infection. At 8 years of age, neurological development had deteriorated to generalized hypotonia, with inability to speak or sit upright by herself. At that time, she had still intractable epilepsy, but her brain MRI and EEG were nonspecific.
A deoxyribonucleic acid (DNA) sample was sent to the Seattle Children’s Hospital Research Institute for analysis by target panel test after IRB approval. Targeted massive parallel sequencing was performed using GAIIx instrument (Illumina, San Diego, CA). The exons of a total of 1,461 genes were targeted for various neurological conditions, including seizures, brain development, intellectual disability, and movement disorders (SureSelect, Agilent). Sequence was aligned to hg19 using BWA 0.5.7, and single-nucleotide variants and indels were called using GATK (version 1.6.5). Mean read depth of the targeted exome was calculated with the GATK DepthOfCoverage walker. Annotation of variants was performed with SeattleSeq Annotation 134. Common variants were identified by filtering against the NHLBI Exome Variant Server. Variants identified as possibly disease causing were confirmed by standard polymerase-chain-reaction (PCR) combined with bidirectional Sanger sequencing using standard methods.
The UPB1 gene for β-UP deficiency was one of the genes included in this panel, which revealed that she was homozygous for p.R326Q (c.977G>A) and heterozygous for p.G31S (c.91G>A) in the UPB1 gene (confirmed by Sanger sequencing analysis, NM_016327). Her family was subsequently subjected to sequencing analysis, which revealed heterozygosity for p.R326Q in the father and heterozygosity for both p.R326Q and p.G31S in the mother. Her two asymptomatic siblings were heterozygous for p.R326Q (Fig. 1). Biochemical analysis of pyrimidine metabolites in patient samples (serum, urine, and CSF) revealed highly elevated N-carbamyl-β-amino acids and moderately elevated levels of dihydrothymine in CSF and plasma, supporting the biochemical diagnosis of β-UP deficiency (Table 1).
Fig. 1.
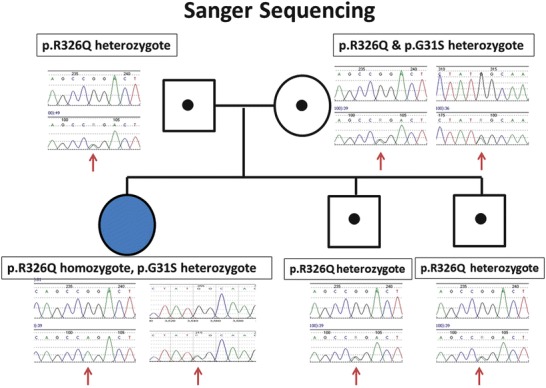
Sanger sequencing analysis for the UPB1 gene. Proband was homozygous for p.R326Q (c.977G>A) and heterozygous for p.G31S (c.91G>A) in the UPB1 gene. All family members were found to be carriers, with heterozygosity for p.R326Q in the father and heterozygosity for both p.R326Q and G31S in the mother and heterozygosity for p.R326Q in two siblings
Table 1.
Pyrimidine metabolite levels in urine, plasma, and cerebrospinal fluid (CSF) of patient
| Urine (μmol/mmol creatinine) | Plasma (μM) | Cerebrospinal fluid (μM) | ||||
|---|---|---|---|---|---|---|
| Pyrimidine metabolites | Patient | Reference | Patient | Reference | Patient | Reference |
| N-carbamyl-β-alanine | 248 | 11.0 ± 9.2 | 6.81 | 0.2 ± 0.3 | 0.90 | 0.1 ± 0.3 |
| N-carbamyl-β-aminoisobutyric acid | 186 | 1.8 ± 2.3 | 25.15 | 0.1 ± 0.2 | 1.60 | 0.01 ± 0.04 |
| Dihydrouracil | 16 | 6.3 ± 5.3 | 0.81 | 1.3 ± 0.8 | 1.60 | 2.1 ± 1.0 |
| Dihydrothymine | 47 | 3.1 ± 2.1 | 3.58 | 0.9 ± 0.3 | 4.30 | 1.1 ± 0.3 |
| Uracil | 4 | 11.8 ± 9.1 | <0.6 | 0.2 ± 0.4 | 0.20 | 0.1 ± 0.2 |
| Thymine | 1 | 0.5 ± 0.6 | <0.08 | 0.05 ± 0.03 | <0.1 | <0.1 |
The reference values are depicted as mean ± SD
At the age of 8 years and 10 months, the patient showed failure to thrive, global developmental delay, and microcephaly. She had body weight of 11.2 kg (<1 percentile), height of 103 cm (<1 percentile), and a head circumference of 44.5 cm (<1 percentile). At 9 years and 1 month of age, she started a restricted purine and pyrimidine diet, which helped reduce her frequency of drop attack seizure and generalized tonic-clonic seizure from 20–30 attacks per day to 1–3 attacks per day. Now, her age is 9 years and 8 months of age; she is still receiving multiple antiepileptic drug medications such as valproic acid, clobazam (Sentil®), zonisamide (Excegran®), and recently added rufinamide (Inovelon®).
Her two siblings, age 5 years and 21 months, show development in the normal range and have no seizure history.
Discussion
Pyrimidine nucleotides are essential for a number of biological processes, such as the synthesis of ribonucleic acid (RNA), DNA, phospholipids, and glycogen and the sialylation and glycosylation of proteins. Metabolic changes affecting the levels of pyrimidines may lead to abnormal neurological activity, as they play an important role in the regulation of the central nervous system (CNS) (van Kuilenburg et al. 2004).
Metabolism of pyrimidine nucleotides is divided into three pathways: biosynthetic, catabolic, and salvage. In the catabolic pathway of pyrimidine nucleosides, uracil and thymine are degraded in three steps: dihydropyrimidine dehydrogenase, dihydropyrimidinase, and β-UP (Saudubray et al. 2012; Kuhara et al. 2009). β-UP is the last enzyme involved in the pyrimidine degradation pathway, catalyzing the conversion of N-carbamyl-β-alanine and N-carbamyl-β-aminoisobutyric acid to β-alanine and β-aminoisobutyric acid, respectively (van Kuilenburg et al. 2012; Yaplito-Lee et al. 2008).
Clinical presentation of β-UP-deficient patients varies from asymptomatic to severe neurological deterioration (Nakajima et al. 2014). A number of symptoms have been reported, including muscular hypotonia, dystonic movement (Assmann et al. 1998), febrile status epilepticus (Assmann et al. 2006b), congenital anomalies of the urogenital and colorectal systems (Yaplito-Lee et al. 2008), growth retardation, microcephaly, intellectual disability, autism, progressive mood change, abnormal facial morphology such as prominent metopic suture, dolichocephaly, recurrent episodes of desaturation unrelated to seizure activity, recurrent attacks of vomiting and severe dehydration, bilateral microphthalmia, nephrotic syndrome, transient cerebral blindness, abnormal eye movement, abnormal retina pigmentation, severe speech delay, and mild optic atrophy (van Kuilenburg et al. 2012; Nakajima et al. 2014). Our patient’s clinical presentations were comparable with other severe cases previously reported such as microcephaly, intractable epilepsy, status epilepticus, and global developmental delay.
Diagnosis of β-UP deficiency is challenging as neurological manifestations and MRI abnormalities such as cortical dysplasia, atrophia cerebri, delayed myelination, vermis hypoplasia, brain stem hypoplasia, callosal body hypoplasia, and subdural hematoma are overall nonspecific. Biochemical analysis for pyrimidine metabolites shows highly elevated levels of N-carbamyl-β-amino acids, moderately elevated levels of the dihydrouracil and dihydrothymine, but normal levels of uracil and thymine in urine, plasma, and CSF (van Kuilenburg et al. 2012).
Mutations of the UPB1 gene can cause β-UP deficiency. Eight missense and three splice site mutations in UPB1 were reported (van Kuilenburg et al. 2012), and three novel missense mutation and one p.R326Q mutation were identified in 13 patients from 12 unrelated Japanese families (Nakajima et al. 2014). No genotype-phenotype correlation was detected. In Japan, 1.8% of the Japanese population is heterozygous for the p.R326Q mutation and an estimated 1 in 12,500 individuals is homozygous for the p.R326Q mutation. Transient expression of mutant β-UP enzymes in HEK293 cells revealed that the p.R326Q mutation decreased residual activities to less than 1.3%. However, the p.G31S mutation showed a residual activity of 50% (Nakajima et al. 2014). Of note, four asymptomatic neonates were detected during a pilot study of newborns, and the prevalence of a β-UP deficiency in Japan has been estimated to be 1 in 6,000 (Kuhara et al. 2009). In a more recent study, Nakajima et al. reported 13 Japanese β-UP-deficient patients, 4 of which were symptomatic with seizures, motor retardation, mental retardation, and autism, but nine were asymptomatic and remained asymptomatic up to 10 years (Nakajima et al. 2014). Many of these asymptomatic cases were homozygous for the p.R326Q mutation.
Treatment of the ß-UP-deficient patient is symptomatic. Treatment with ß-alanine for more than 1.5 years revealed no clinical improvement (Assmann et al. 2006a). ß-Aminoisobutyric acid supplementation and ß-UP administration for removal of N-carbamyl-ß-alanine and N-carbamyl-ß-aminoisobutyric acid have not yet been attempted in ß-UP-deficient patients. A pyrimidine-restrictive diet could be an option for treating ß-UP-deficient patients and may help to reduce the seizure frequency as observed in our patient.
In conclusion, ß-UP deficiency should be considered for a possible diagnosis in patients with unexplained symptoms such as intractable epilepsy, progressive neurologic degeneration, and microcephaly; p.R326Q variation in UPB1 gene seems a prevalent one in Far Eastern Asians.
Acknowledgment
We would like to thank the Green Cross Laboratories for delivering samples to the Netherlands.
Take-Home Message
We report a first Korean case of ß-ureidopropionase deficiency presenting with intractable seizure, intellectual disability, and microcephaly who was homozygous for p.R326Q and heterozygous for p.G31S in the UPB1 gene, presenting with typical biochemical analysis results in pyrimidine pathways.
Contribution of Individual Authors
Si Houn Hahn and Valeria Vasta performed genetic study interpret and analyze the results. André B.P. van Kuilenburg and N. G. G. M. Abeling performed biochemical analysis. Si Houn Hahn and André B.P. van Kuilenburg participated in drafting the manuscript. Jun Hwa Lee drafted the manuscript and was the treating pediatricians.
Guarantor for the Article
Jun Hwa Lee
Details of Funding
None
Details of Ethics Approval
The study was approved by the Institutional Review Board of Samsung Changwon Hospital (IRB study #2013-SCMC-058-00).
Conflicting Interests
Jun Hwa Lee declares that he has no conflict of interest.
André B.P. van Kuilenburg declares that he has no conflict of interest.
Valeria Vasta declares that she has no conflict of interest.
N. G. G.M. Abeling declares that he has no conflict of interest.
Si Houn Hahn declares that he has no conflict of interest.
Informed Consent
All procedures followed were in accordance with the ethical standards of the responsible committee on human experimentation (Samsung Changwon Hospital, Republic of Korea) and with the Helsinki Declaration of 1975, as revised in 2000 (5). Informed consent was obtained from the parents.
Footnotes
Competing interests: None declared
Contributor Information
Jun Hwa Lee, Email: ljh3643@hanmail.net, Email: ljh3643@skku.edu.
Collaborators: Johannes Zschocke
References
- Assmann B, Gohlich-Ratmann G, Brautigam C, et al. Presumptive ureidopropionase deficiency as a new defect in pyrimidine catabolism found with in vitro H-NMR spectroscopy. J Inherit Metab Dis. 1998;21(suppl.2):1. [Google Scholar]
- Assmann B, Gohlich G, Baethmann M, et al. Clinical findings and a therapeutic trial in the first patient with beta-ureidopropionase deficiency. Neuropediatrics. 2006;37:20–25. doi: 10.1055/s-2006-923933. [DOI] [PubMed] [Google Scholar]
- Assmann BE, Van Kuilenburg AB, Distelmaier F, et al. Beta-ureidopropionase deficiency presenting with febrile status epilepticus. Epilepsia. 2006;47:215–217. doi: 10.1111/j.1528-1167.2006.00391.x. [DOI] [PubMed] [Google Scholar]
- Kuhara T, Ohse M, Inoue Y, Shinka T. Five cases of beta-ureidopropionase deficiency detected by GC/MS analysis of urine metabolome. J Mass Spectrom. 2009;44:214–221. doi: 10.1002/jms.1500. [DOI] [PubMed] [Google Scholar]
- Nakajima Y, Meijer J, Dobritzsch D, et al. Clinical, biochemical and molecular analysis of 13 Japanese patients with beta-ureidopropionase deficiency demonstrates high prevalence of the p. 977G>A (p.R326Q) mutation. J Inherit Metab Dis. 2014;37(5):801–812. doi: 10.1007/s10545-014-9682-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saudubray JM, van Berghe G, Walter JH. Disorders of purine and pyrimidine metabolism. In: van Berghe G, Vincent MF, Marie S, editors. Inborn metabolic disease. 5. Heidelberg: Springer; 2012. pp. 511–515. [Google Scholar]
- van Kuilenburg AB, van Lenthe H, Ratmann GG, et al. Confirmation of the enzyme defect in the first case of beta-ureidopropionase deficiency. Beta-alanine deficiency. Adv Exp Med Biol. 2000;486:243–246. doi: 10.1007/0-306-46843-3_47. [DOI] [PubMed] [Google Scholar]
- van Kuilenburg AB, Meinsma R, Beke E, et al. beta-Ureidopropionase deficiency: an inborn error of pyrimidine degradation associated with neurological abnormalities. Hum Mol Genet. 2004;13:2793–2801. doi: 10.1093/hmg/ddh303. [DOI] [PubMed] [Google Scholar]
- van Kuilenburg AB, Dobritzsch D, Meijer J, et al. ß-ureidopropionase deficiency: phenotype, genotype and protein structural consequences in 16 patients. Biochim Biophys Acta. 2012;1822:1096–1108. doi: 10.1016/j.bbadis.2012.04.001. [DOI] [PubMed] [Google Scholar]
- Yaplito-Lee J, Pitt J, Meijer J, Zoetekouw L, Meinsma R, van Kuilenburg AB. Beta-ureidopropionase deficiency presenting with congenital anomalies of the urogenital and colorectal systems. Mol Genet Metab. 2008;93:190–194. doi: 10.1016/j.ymgme.2007.09.009. [DOI] [PubMed] [Google Scholar]