

Abstract
Canavan’s disease (CD) is a fatal autosomal recessive pediatric leukodystrophy in which patients show severe neurodegeneration and typically die by the age of 10, though life expectancy in patients can be highly variable. Currently, there is no effective treatment for CD; however, gene therapy seems to be a feasible approach to combat the disease. Being a monogenic defect, the disease provides an excellent model system to develop gene therapy approaches that can be extended to other monogenic leukodystrophies and neurodegenerative diseases. CD results from mutations in a single gene aspartoacylase which hydrolyses N-acetyl aspartic acid (NAA) which accumulates in its absences. Since CD is one of the few diseases that show high NAA levels, it can also be used to study the enigmatic biological role of NAA. The disease was first described in 1931, and this review traces the progress made in the past 8 decades to understand the disease by enumerating current hypotheses and ongoing palliative measures to alleviate patient symptoms in the context of the latest advances in the field.
Clinical Description
Canavan’s disease (CD) is a fatal pediatric leukodystrophy in which the central nervous system (CNS) white matter shows progressive spongy degeneration (Matalon et al. 1993). It results from deficiency of aspartoacylase (N-acetyl-l-aspartate amidohydrolase; EC 3.5.1.15) (Matalon et al. 1988), an enzyme required for catabolism of N-acetylaspartate (NAA), the second most abundant amino acid derivative in the CNS. The disease was thought to be ethnically confined to Ashkenazi Jews; however, a growing number of non-Jewish patients have been identified worldwide (Elpeleg and Shaag 1999).
Patients show vacuolization in the subcortical white matter (which becomes gelatinous), edema and demyelination, and thin delicate meninges. Swollen cortical protoplasmic astrocytes containing membrane-bound cytoplasmic vacuoles, elongated mitochondria with distorted cristae, degraded axonal medullary sheaths, and a prominent increase in protoplasmic astrocytes characterize the diseased brain. There also seems to be a significant loss of proteolipid protein and total lipids in the white matter (Beaudet 2001).
Clinical manifestations include atonia of neck muscles, hypotonia, severe psychomotor and mental retardation, seizures, NAAduria, blindness, and megalencephaly. Previously, the disease was classified based on onset and severity of symptoms into congenital, infantile, and juvenile forms, congenital being the most severe with symptom onset in the first few weeks of life (Adachi et al. 1973).
Aspartoacylase: Biochemistry and Genetics
CD was described as an enzyme deficiency disease (Matalon et al. 1988) when NAA-rich patient plasma and urine samples on incubation with extracts of normal fibroblasts increased aspartate levels suggesting a block in NAA catabolism. Aspartoacylase was detected by immunohistochemistry in oligodendrocytes and nonreactive microglia but not neurons (Madhavarao et al. 2004). However large reticular and motor neurons in the brainstem and spinal cord showed moderate staining for aspartoacylase (Klugmann et al. 2003).
Intensive genetic studies identified human aspartoacylase cDNA from kidney based on sequence information from bovine aspartoacylase. The gene was localized to 17p13-ter, comprises six exons encoding a 313 amino acid long polypeptide chain with a molecular mass of 36 kd (Kaul et al. 1993). Aspartoacylase deacetylates NAA (Birnbaum et al. 1952) with a Km of 8.5 × 10−4 mol/L and Vmax of 43,000 nmol/min per mg of protein (Kaul et al. 1991). Isolation of the aspartoacylase cDNA sequence and hence identification of causative mutations for CD was a landmark in the field as it made molecular diagnosis and gene therapy of CD possible (Kaul et al. 1994). Currently, >54 mutations are associated with Canavan’s disease (Hershfield et al. 2007) with new mutations being identified on a regular basis. Several mutations have been identified to result in a loss of aspartoacylase activity as well as lack of expression (Sommer and Sass 2012) though all mutations have not been completely characterized; hence, DNA analysis limits prenatal diagnosis to carrier couples in populations with known mutations. The previous method of measuring AspA activity in the chorionic villi or amniocytes from fetuses was deemed unreliable (Matalon and Michals-Matalon 1999a). Currently quantification of NAA in the amniotic fluid by chromatography coupled with mass spectrometry (Jakobs et al. 1991; Bennett et al. 1993; Al-Dirbashi et al. 2009) is considered to be a reliable alternative for diagnostic testing. Patients, however, follow a similar course of disease irrespective of the residual aspartoacylase activity, and as life expectancy is highly variable in CD patients even with the same genotype, hence there are no defined correlations between the disease phenotype and mutations (Matalon and Michals-Matalon 1999b).
Characterization of the Substrate
NAA is an abundant (5–10 mM) amino acid derivative in the vertebrate CNS with a molecular mass of 175.1 Da (Birken and Oldendorf 1989). Most neuropsychiatric disorders like schizophrenia show a decrease in NAA levels (Mondino and Saoud 2013); however, CD is a rare example along with sickle cell disease (Steen 2005), tardive dyskinesia (Tsai et al. 1998), and multiple sclerosis (Tortorella et al. 2011) that shows elevated NAA levels. NAAduria is a reliable and specific biochemical index for CD (Matalon et al. 1988). Additionally CD patients show very high amounts of NAA in their blood and CSF (Matalon et al. 1988).
NAA serves as a marker of neuronal integrity as high levels indicate brain injury and disease (Birken and Oldendorf 1989); however, the biological function of NAA remains enigmatic. It is metabolically compartmentalized, being exclusively produced in neuronal mitochondria by l-aspartate N-acetyltransferase (AspNAT) [EC 2.3.1.17] (Ariyannur et al. 2010), and metabolized in oligodendrocytes (Madhavarao et al. 2003) by aspartoacylase.
Theories Behind the Molecular Etiology of CD
A comprehensive investigation of aspartoacylase regulation in oligodendrocytes is essential to understand CD pathogenesis because the specific connection between aspartoacylase deficiency and the failure of proper CNS development and myelination remains unclear. Listed here are existing theories that attempt to offer an explanation.
Molecular Water Pump (MWP) and Osmolyte Imbalance Theory
The adult brain is a major source of metabolic water and uses about 20 % of the daily caloric intake. Additionally, two major CD symptoms – increased CSF pressure and intramyelinic edema – are hallmarks of profound fluid imbalance suggesting the existence of an efficient MWP (Baslow 1999).
The theory suggests that NAA accumulation could result in osmolytic imbalance in the brain since it is similar in nature to taurine (Taylor et al. 1995), an important CNS osmolyte. Furthermore, NAA has two juxtaposed anabolic (neurons) and catabolic (oligodendrocytes) compartments that suggest a mechanical framework for a MWP in the brain.
A MWP involves the synthesis and facilitated diffusion of a hydrated intracellular osmolyte (NAA with its ion-dipole and dipole–dipole associated water) down its gradient. At maturity, intraneuronal NAA concentration is ~10 ± 14 mM, while the interstitial concentration is only 80–100 μm (Sager and Hansen 1997), indicating a large outward-directed transport gradient. To maintain tissue–ECF osmotic balance (in the periaxonal space), the osmolyte (NAA) is rapidly hydrolyzed (by aspartoacylase to form aspartate and acetate) to maintain the concentration gradient. The metabolic products are dehydrated as they are once again taken up by an active transport mechanism to complete the cycle (Baslow 1999). Presumably, the acetate is taken up by astroglial processes at axon internodes and synapses (Tsacopoulos 1996) recycling the hydrolyzed products to produce more NAA (Fig. 1).
Fig. 1.
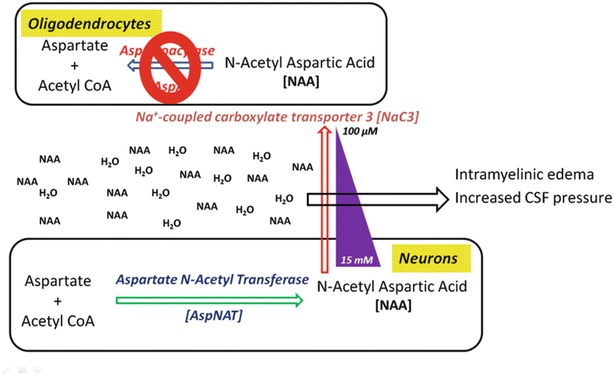
Molecular water pump theory: accumulation of the l-enantiomer of NAA in interstitial space draws out water from surrounding cells causing edema in the brain as well as shrinking nearby cells to give rise to vacuoles
Paranodal seals connecting oligodendrocytes to axons could serve as the site for the NAA inter-compartmental bidirectional cycle (Baslow 1999). In CD, aspartoacylase deficiency would lead to accumulation of NAA and water and result in increased hydrostatic pressure that could loosen the tight junctional seals separating interlamellar spaces from the extracellular periaxonal and parenchymatous spaces resulting in intramyelinic edema (Hirano 1981). Subsequent demyelination could create vacant spaces within the white matter leading to the spongy brain phenotype.
Dysmyelination Theory
NAA-derived acetyl groups were shown to be involved in fatty acid synthesis as acetyl-labeled NAA injection into rats resulted in maximum fatty acid incorporation just before and during myelination (D'Adamo Jr et al. 1968). Later studies indicated that NAA was transferred from the axon to myelin, and NAA-derived acetate was incorporated into myelin lipids (Chakraborty et al. 2001).
This theory proposes that deficiency of NAA-derived acetate decreases the synthesis of myelin-associated lipids in CD leading to dysmyelination (Madhavarao et al. 2005) (Fig. 2). Additionally, temporal correlations have been shown between developmental increases in aspartoacylase activity and myelination (Bhakoo et al. 2001). Similar to human CD patients, brain acetate levels are reduced by ~80% in aspartoacylase knockout (ASPAKO) mice during peak postnatal myelination, while myelin lipids such as cerebrosides and sulfatides are reduced (Madhavarao et al. 2005; Ahmed et al. 2013). These data speculate that NAA-derived acetate is essential during postnatal myelination to supply substrate for some proportion of the lipids that make up myelin sheaths in the developing brain.
Fig. 2.
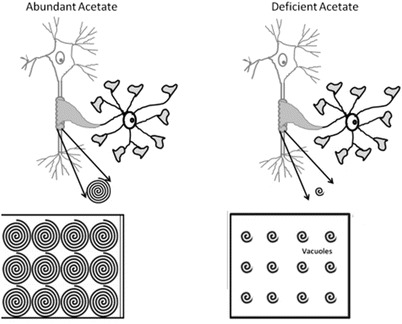
Demyelination/dysmyelination theory: abundant acetate allows for proper formation of myelin bilayers, while in acetate deficiency, the layers may not be completely and tightly bound giving rise to vacuoles in interstitial space
Studies on the Nur7 KO mouse (Traka et al. 2008) suggest that spongy degeneration is not dependent on disrupted myelin synthesis. Even though Nur7 mice are heterozygous for a null allele of a galactolipid-synthesizing enzyme which could further reduce brain cerebroside content, they do not show more severe myelin pathology implicating additional mechanisms in the pathophysiology of aspartoacylase deficiency (Madhavarao et al. 2009). Animal models lacking functional aspartoacylase show significant albeit structurally abnormal myelination probably because parallel pathways for myelination exist during initial stages of myelinogenesis (Wang et al. 2009).
Deficiency of AspA-Derived Acetate Compromises Oligodendrocyte Differentiation
Studies on oligodendrocyte maturation have highlighted the importance of epigenetic control in differentiation (Copray et al. 2009). Since neurons transfer NAA to oligodendrocytes (Chakraborty et al. 2001), NAA-derived acetate is probably important for histone acetylation reactions that regulate chromatin structure and gene transcription in these cells. Dramatic reduction of acetate resulting from aspartoacylase deficiency could impact histone reactions required for epigenetic gene regulation preventing normal differentiation leading to oligodendrocyte cell death and neuronal injury possibly contributing to vacuole formation.
Protein Folding and Stabilization Theory
Cells like oligodendrocytes, which have active protein secretory pathways, are sensitive to disorders of protein misfolding. Acetyl CoA is an important substrate for acetylation and deacetylation of nascent polypeptide chains in the endoplasmic reticulum (ER) required for stabilization and correct folding of proteins (Spange et al. 2009). Reduced acetyl CoA availability due to aspartoacylase deficiency could negatively impact protein folding and stabilization, targeting proteins for degradation. Oligodendrocytes are highly susceptible to ER stress associated with disruptions in protein synthesis and trafficking (Lin 2009). In the ASPAKO mouse, a severe loss of myelin basic protein and PLP/DM20 proteolipid proteins has been observed, combined with a decrease in myelinated fibers (Kumar et al. 2009).
Oxidative Stress Theory
Intracerebroventricular administration of NAA induces seizures in normal rats, probably by neuronal overexcitation (Akimitsu et al. 2000); further, tardive dyskinesia patients also have significantly higher CSF concentrations of NAA like CD patients (Tsai et al. 1998). Animal studies show that epileptic seizures result in free radical production and oxidative damage to cellular proteins, lipids, and DNA (Bruce 1995) implicating oxidative stress as one of the possible causes of neurological impairment. Recent work suggests that chronic mitochondrial oxidative stress and resultant dysfunction can render the brain more susceptible to epileptic seizures (Patel 2004). Hence it seems that there is a role for oxidative stress both as a cause and a consequence of epileptic seizures.
NAA probably promotes oxidative stress by decreasing nonenzymatic antioxidant defenses and stimulating oxidative damage to both lipids and proteins by enhancing reactive species in cerebral cortex (Fig. 3). To define the role of oxidative stress in NAA neurotoxicity, its effects on the antioxidant enzymes including catalase, superoxide dismutase (SOD) ,and glutathione peroxidase (GPX) were studied. NAA inhibited the functions of catalase and GPX enzymes indicating impaired detoxification of hydrogen peroxide, but it had no effect on SOD. Acute administration of NAA also enhanced levels of hydrogen peroxide in vitro which could possibly be involved in the progression of the characteristic neurodegeneration in CD (Pederzolli et al. 2007). Though these results could not be extrapolated to humans, they do reveal probable mechanisms since the oxidative stress parameters occurred with concentrations of NAA (~4fold higher) observed in the plasma and cerebrospinal fluid of patients affected by CD (Tsai and Coyle 1995). Based on the supposition that NAA may promote oxidative stress in vitro and in vivo both by enhancing reactive species and diminishing antioxidant defenses, administration of antioxidants, especially vitamins E and C, could be considered as a potential adjuvant therapy for patients affected by CD (Pederzolli et al. 2007).
Fig. 3.
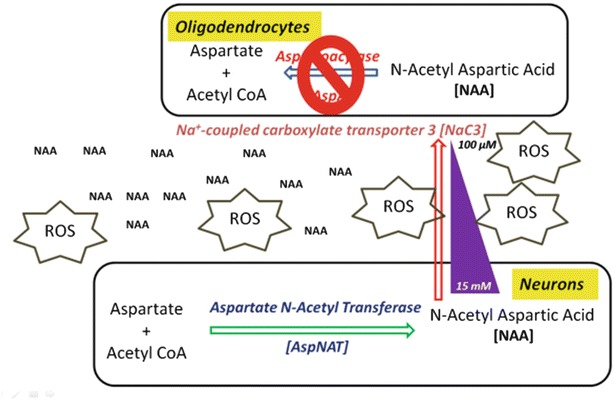
Oxidative stress theory: accumulation of excess l-enantiomer of NAA could lead to generation of reactive oxygen species that could potentially generate vacuoles in brain interstitium
A previous study linked NAA accumulation to nitric oxide (NO) toxicity as it upregulates inducible nitric oxide synthase (iNOS) and stimulates neuronal and endothelial nitric oxide synthase (Surendran 2009). Increased NO levels lead to disturbances in DNA structure and enhance protein interaction (Lee 2007). Discrepancies in the molecular weight of aspartoacylase when prepared using different methods (Kaul et al. 1993) suggest that it may dimerize based on its concentration. It is possible that upregulation of NO synthase as a direct result of NAA accumulation may nitrosylate AspA causing it to dimerize. This hypothesis proposes that NAA upregulation and aspartoacylase malfunction could be cyclically linked.
Treatment Strategies
CD was described as the only neurological disease which manifests increased NAA levels in the brain; most other neurological diseases like schizophrenia show low NAA levels (Mondino and Saoud 2013). Since an increasing number of substances can affect NAA levels in the brain (Baslow and Resnik 1997), slowing down the anabolic portion of the NAA cycle using pharmaceuticals may allow the existing, albeit enzyme-deficient oligodendrocytes to produce a stable myelin sheath and restore neurological function. Surprisingly the kidney has high levels of aspartoacylase; however, since NAA levels in the kidney are very low, aspartoacylase may have additional catalytic functions. Thus, alternate approaches may be needed to deal with the pathological consequences of loss in aspartoacylase activity in such peripheral tissues.
A large number of preclinical proof-of-concept studies have been performed on various CD animal models including two rodent models – a naturally existing Tremor rat (Kitada et al. 2000) and an artificially engineered ASPAKO mouse (Matalon et al. 2000). Newer studies have created a knock-in model (Mersmann et al. 2011) and a model with a single-point mutation in Nur7 (Traka et al. 2008) making the development of therapeutic modalities much easier. Though currently the patients are mostly supported by palliative measures, existing treatment modules focus on different aspects of the disease phenotypes in CD and are described below.
Palliative Measures
Most of the palliative measures for patients of Canavan’s disease follow the care provided for patients of other pediatric neurodegenerative diseases (Hunt and Burne 1995). Current palliative measures for respiratory issues in patients with Canavan’s disease include suction and cough assist machines to clear secretions and mucous from the mouth, throat, nose, and upper lobes of the lungs, the Vesta system for airway clearance and oxygen concentrators to provide a continuous flow of oxygen for easy breathing. Nebulizers also help to administer medication as aerosols for these patients.
Hypotonia being a major Canavan symptom requires positioning equipment which may include foam supports, feeder seats, specialized strollers, and bath chairs to help patients with their positioning needs. Feeding pumps are also used to assist in dispensing liquid nutrients at predetermined rates for feeding.
Symptomatic Treatment of Disease
The earliest human clinical trials on CD patients used acetazolamide to reduce water concentration and NAA levels in white matter for a period of 5 months. The drug reduced the intracranial pressure, but did not reduce water content or NAA levels (Bluml et al. 1998). A ketogenic diet increased the levels of β-hydroxybutyrate in the brain but did not affect the elevated levels of NAA (Novotny Jr et al. 1999). These treatment strategies were primarily targeted to alleviate edema; however, there was little to no benefit for the patients.
In recent years, intraperitoneal injections of lipoic acid [which can cross the blood brain barrier (Samuel et al. 2005)] have also been tried in preclinical studies using tremor rats, the naturally occurring animal model for CD (Pederzolli et al. 2007) based on the fact that NAA induces oxidative stress in the CNS. The encouraging results suggest that this might be a good therapeutic approach for symptomatic treatment. With seizures being a hallmark of the disease, CD patients have canonically been targeted using anticonvulsive drugs like acetazolamide, clonazepam, and oxcarbazepine (Leone et al. 2012).
Addressing Elevated Substrate in the Context of Deficient Aspartoacylase
Dietary supplementation of acetate in newborn patients was proposed as therapeutic for CD since the primary pathogenesis is postnatal and deficiency of aspartoacylase and acetate are concurrent. This group reported phenotypic improvements in myelin galactocerebroside content and brain vacuolation and also showed a partial reversion in motor dysfunction in GTA-treated tremor rats over a course of 4 months (Madhavarao et al. 2009). In conclusion, dietary supplementation may not totally alleviate an inherent genetic metabolic disorder but may definitely offer partial symptomatic alleviation. Clinical translation of GTA to human infants showed no significant side effects or toxicity but showed no motor improvement (Segel et al. 2011).
A recent study proposed dietary triheptanoin supplementation in Nur7 mice to support fatty acid synthesis and TCA cycles and hence improve the redox status in diseased animals. It showed phenotypic improvements suggesting that the underlying pathological mechanism of CD may be a combination of several factors (Francis et al. 2013).
Neuroprotective strategies may be needed to counteract neurological damage caused by oxidative stress. Pharmacologically, lithium has been neuroprotective for dementia patients (Kessing et al. 2008) possibly by reducing expression of proapoptotic proteins (Chen 1999) and increasing the levels of antiapoptotic proteins (Chang et al. 2009). Intraperitoneal lithium administration caused a significant drop in brain NAA levels in wild-type rats (O'Donnell et al. 2000) and tremor rats (Baslow et al. 2002). After 1 year of treatment in patients, NAA levels in both urine and brain were decreased. Patients showed improved alertness and visual tracking; however axial hypotonia and spastic dysplegia were unaffected (Solsona et al. 2012).
Controlling brain NAA levels for CD (Assadi et al. 2010) may not be a highly effective therapeutic strategy since the NAA system is not universally present in the neurons (Baslow 1997). A complete inhibition or even a significant reduction in NAA production may thus be a viable option in controlling demyelination and improving the quality of life in CD patients.
Addressing the Deficiency of the Enzyme Aspartoacylase
Using enzyme replacement as a therapy for neurological disorders has been difficult because of the challenging blood–brain barrier. Surface lysyl groups of human aspartoacylase were modified through PEGylation to decrease immune response and increase circulation half-life with the intention of treating CD patients (Zano et al. 2011).
Most of these modalities as CD therapeutics mostly showed good tolerance however did not cause any significant improvement in the quality of life in the patients. However as CD is a monogenic defect with pathology being most evidently localized in the CNS, it presents an attractive target for gene therapy.
Gene Therapy Using Gene Replacement Strategy
Gene therapy is the supplementation or alteration of DNA as a therapy in organisms to alleviate disease. The most common gene therapy approach involves replacement of a mutant gene that causes a genetic disease by a fully functional gene. The therapeutic DNA is packaged into a “vector” that delivers the DNA in cells after which the cellular machinery takes over and produces the deficient protein the absence of which resulted in the disease.
Nonviral gene delivery systems are one of the conventional approaches for gene therapy; however they are limited by efficiency of gene transfer. Direct injections of plasmids (naked or in complexes) are also inefficient because of limited uptake due to aggregate formation, low rates of diffusion, endotoxin contamination and transient expression necessitating improvements in design. The first clinical trial for CD used a nonviral gene transfer technique with intraventricular injections of a non-aggregating lipid plasmid formulation LPD, composed of a recombinant plasmid with a condensing agent [poly-l-lysine or protamine sulfate] and a liposomal formulation [DC-CHOL/DOPE] on two Canavan patients (Leone et al. 2000). The study established that the gene transfer technique worked and was safe but differences in the responses of the patients made it difficult to conclude if the gene therapy was successful.
One of the popular approaches in viral gene delivery is using adeno-associated viruses (AAVs). These are one of the smallest nonpathogenic mammalian parvoviruses that are replication incompetent, are almost completely nontoxic after CNS delivery in non-preexposed mammals (Samulski et al. 1999), and have a high level of sustained gene expression (Samulski et al. 1999) in nondividing cells. The most common serotype (AAV2) was the first to be successfully used for gene transfer (Hermonat 1984); however its use for CNS disorders was limited due to the presence of the blood–brain barrier (BBB) (which physically excludes foreign molecules and microorganisms based on size, charge, and lipid solubility from the blood to the brain) efficiently blocking rAAV diffusion into the CNS (Zlokovic 2008) rendering it incapable of marked therapeutic benefit for global neurological disorders like CD which have no cure to date.
Preclinical viral gene therapy studies involved stereotactical adenovirus-mediated delivery of aspartoacylase in tremor rats that showed reduction of seizures (Seki et al. 2002) and AAV2 mediated delivery that showed significant improvement in motor abilities and elevation of aspartoacylase expression (McPhee et al. 2005).
In another study, intraparenchymal delivery of rAAV2 to the brains of ASPAKO mice showed increased aspartoacylase activity and reduced NAA levels (Matalon et al. 2003). Vacuolation near the injection site was improved; however distant sites such as cerebellum were unaffected implying that the requirement for multisite injections to get optimal enzyme activity in the entire brain.
A second clinical trial (Janson et al. 2006) involved intracranial injections of rAAV2 vectors into the parietal, occipital, and frontal lobes in the brain of a large group of CD patients [FDA-IND#9119; NIH (RAC) #0001-381]. Clinical changes in most participants were not pronounced, relatively transient, and thought to be due to inadequacies of the vector or delivery system. Moreover, in CD patients, elevated NAA levels appear to cause white matter pathology but rAAV2 was conclusively demonstrated to transduce only neurons (McCown 2005). In a follow-up study on 13 of the 28 patients enrolled in this trial, the long-term safety, dosing parameters, and efficacy of the treatment were evaluated (Leone et al. 2012). From the study, it is evident that rAAV-mediated gene therapy is the most promising safe therapeutic modality for CD to date. The treatment stabilized the atrophy and even slowed progression of the disease in some patients, but there was a lack of a uniform response across cohorts. Standardized neurological exams and motor function tests post-gene therapy showed improvements in younger patients though the raw scores still indicated spastic quadriplegia. This result underlines the importance of defining a therapeutic window that will help improve the quality of life for the patients. This study clearly established the long-term safety of rAAVs as gene therapy vectors and indicated the need for serotypes and delivery strategies that lead to widespread and efficient transduction of the brain.
Various neurological diseases are subject of several clinical trials using rAAV (Asokan et al. 2012) and the available results indicate that the vectors mediate stable gene expression in the human brain (Muramatsu et al. 2010; Hwu et al. 2012) and are safe (Leone et al. 2012). rAAV2 vectors were the first vectors used for CNS gene transfer in animal models as well as in humans, and incidentally Canavan’s disease was one of the first CNS diseases to go for clinical trials using rAAV (Leone et al. 2000; Janson et al. 2002). It was not until the discovery of a large family of novel primate-derived AAVs (Gao et al. 2002) that gene delivery to the CNS made great strides. Encouraging results with stereotactic delivery of rAAV2 to the brain led to intraparenchymal testing of other more efficient rAAV serotypes like 1,5,7,8,9, and rh.10 (Burger et al. 2004; Cearley and Wolfe 2006; Cearley et al. 2008) for CNS gene delivery. Direct infusion of the vectors in the brain parenchyma using stereotactic equipment guided needle placement is the most effective means for structure-specific gene delivery and proved to be beneficial for diseases with localized pathology.
Although intraparenchymal infusion is a great option for localized corrections of pathology, it is not suitable for widespread CNS transduction due to localized delivery and limitations in diffusion. Treatment of neurological disorders caused by single-gene defects requires global CNS transduction, intravenous injections would be extremely beneficial as the brain is rich in blood capillaries, allowing a wider distribution of the therapeutic vector concurrently being less invasive than intracranial delivery. A breakthrough in the field occurred when AAV9 was found to cross the BBB (Foust et al. 2009) and intravascular administration of rAAV9 in mice resulted in a widespread transduction of the CNS. Discovery of more serotypes that can cross the BBB (Zhang et al. 2011; Yang et al. 2014) paves the way for more efficient CNS gene transfer using the rAAV vectors. It is noteworthy that the rAAV vectors appear to be relatively consistent in their CNS gene transfer properties even across animal models like cats, dogs, and monkeys (Duque et al. 2009; Gray et al. 2013; Swain et al. 2013).
An exciting study recently demonstrated that single intravenous injection of rAAVs that can cross the BBB achieved sustained therapeutic benefit in ASPAKO mice even when vector was administered as late as P21 (Ahmed et al. 2013). This is extremely significant because untreated animals died by the age of 28 days. One of the most relevant issues in clinical application of gene therapy is the age of the patient. Though genetic screening allows for detection of inherited diseases in newborns, some diseases may manifest when the patient is well advanced in adulthood. In both cases gene therapy methods would need to be tailored to the specific patient type since the dosage and route of administration may not be equally therapeutic in both cases. Preclinical studies using rAAV9 also showed neuronal transduction accompanied by dramatic improvement in the diseased mice (Ahmed et al. 2013). Taken together, these observations suggest that de novo expression of aspartoacylase, regardless of the enzyme’s physical location, can reduce NAA levels indicating that gene correction of every cell in the brain is not necessary (Matalon et al. 2003). The study addressed a couple of key issues, one of which was the timing of vector administration. Intravenous rAAV9 injections were performed at different ages of mouse pups to primarily document survival and motor functions. They found that although therapeutic benefits were indeed conferred upon all the treated animals, an earlier intervention was more beneficial presumably because certain developmental stages are more responsive to the therapeutics based on the stage of myelination.
It is evident that systemic delivery of rAAV9 at early neonatal stages achieves an extensive and robust transduction of neural cell types in CD that can be extended to developing therapeutics for other neurodegenerative diseases like Parkinson’s disease using neurotrophic growth factors. Supraphysiological aspartoacylase levels or for that matter, growth factors, in other peripheral tissues have not been documented, may be potentially harmful and even lead to deleterious immune responses. To address this issue, the same study harnessed the endogenous miRNA machinery to limit transduction of unintended target tissues opening up newer possibilities of treatment. miRNA binding sites of specific miRNAs that are highly enriched in tissues like the liver, heart, and skeletal muscle were used to construct vectors that would detarget aspartoacylase expression from these tissues. The study showed successful alleviation of symptoms even when aspartoacylase expression was detargeted documenting the practical applications of miRNA-based therapeutics in leukodystrophies that can be extended for other diseases as well.
Although CD is defined as a leukodystrophy, it is yet to be established if the major event in this disease is demyelination or dysmyelination. Several therapeutic attempts, more notably the metabolic correction approach, seem to indicate an occurrence of demyelination. In this case, supplementation of substrates required for myelination would markedly increase the longevity as well as the quality of life of patients. However, most metabolic supplementation studies do not show any dramatic reversal of symptoms indicating that the molecular etiology might be more complicated. Thin layer chromatography (TLC) and X-ray diffraction studies conducted on ASPAKO mice indicate abnormal myelin composition in untreated animals (Ahmed et al. 2013) indicating that it would be worth exploring if the diseased oligodendrocytes are unable to synthesize normal myelin or are incapable of myelination itself. It would be appropriate to indicate here that systemic delivery of AAV9 resulted in a primarily neuronal transduction. As neurons do not divide, they would act as in situ factories that constantly produce the therapeutic protein. Additionally, since neurons are the site of NAA synthesis, expression of the metabolizing enzyme would reduce the load of NAA to a significant extent, especially when it appears that most of the CD symptoms like hydrocephaly, vacuolation, and NAAduria arise from accumulation of excess NAA. Targeting oligodendrocytes specifically would prove to be beneficial since aspartoacylase is localized in these cells, however the chances of transduced cells dividing and diluting out the therapeutic benefits seem imminent and the approach would be fraught by the danger of reduced therapeutic benefit with the progression of time.
Perspectives and Future Directions
It has been more than eight decades since CD was first described in 1931 (Canavan 1931), and there was little progress in understanding its pathogenesis until it was identified as an aspartoacylase deficiency almost five decades later (Matalon et al. 1988). Description of the human aspartoacylase cDNA sequence led to a huge leap in molecular diagnosis for CD patients (Kaul et al. 1993); however the molecular etiology has remained controversial. To resolve this, strong efforts in modeling CD in animals significantly broadened avenues to investigate pathological mechanisms. Preclinical studies began with the naturally existing tremor rats and then expanded to include the ASPAKO mouse and later the Nur7 and the LacZ knock-in mouse models. Currently the field seems poised for more revolutionary progress with the advent of newer serotypes and advances in capsid evolution for rAAVs that are specific for different neural cell types and can cross the BBB.
Currently CD falls in a category of diseases, the treatment of which could have wide implications in developing therapeutics for a vast group of leukodystrophies and neurodegenerative diseases. Being a monogenic defect, CD is a perfect target for gene therapy attempts which could be utilized as tools to tease out the underlying pathology of leukodystrophies and develop efficient therapeutics for them. Mouse models that show a less severe phenotype resembling infantile and juvenile CD patients could be used to determine disease progression. Additionally, the unique symptom of elevated NAA levels in CD also makes it a perfect disease model to study functions of NAA. An important element for translating observations from ASPAKO mice would be to couple in vivo physiology and imaging in the mouse to functional neuroimaging in patients to help identify conserved neural circuit phenotypes and pave way to improve upon therapeutics.
In addition to existing therapeutic modalities, alternative means of treatment for CD could include partial silencing of the NAA biosynthetic enzyme AspNAT (Madhavarao et al. 2003) to control the NAA metabolism cycle. Additionally inhibition of NAA export from the neuronal mitochondria could markedly decrease NAA accumulation in interstitial spaces that probably causes osmotic dysregulation in the CNS. Pharmacological protection of oligodendrocytes against damage in demyelinating diseases could also be a promising avenue of treatment (Waksman 1999). Intravenous delivery of rAAV9 (Ahmed et al. 2013) in the ASPAKO model suggests that lowering NAA levels in the brain probably led to alleviation of NAAduria as well as a decrease in edema indicating that the MWP theory could partially explain pathogenesis in CD.
Inclusion of newer CD animal models, continuously expanding viral vector repertoire and less invasive delivery methods for pan CNS delivery, makes it likely that gene therapy for CD and other such diseases will advance rapidly in the near future. It should be noted, however, in spite of newer serotypes, that there will still remain the issue of half-life of the transduced cells. All of these factors increasingly indicate the potential for combinatorial therapy strategies that could be translated to human patients.
An important issue would be to determine how far along in disease progression can the treatment be administered to patients to achieve substantial therapeutic benefits. Neurometabolic diseases like globoid cell leukodystrophy show favorable outcomes for presymptomatic intervention but not later indicating the existence of a restricted therapeutic time window (Escolar et al. 2005). Similar outcomes were seen in preclinical studies (Ahmed et al. 2013) and follow-up study on CD clinical trials (Leone et al. 2012) indicating that future gene therapy interventions should begin before irreversible neuro-structural changes occur underlining the importance of rodent studies. However, an important issue in the translation of the intravenous rAAV therapeutic dose from mice to humans is the huge vector manufacturing burden-making treatment costs prohibitive. Tweaking the expression cassette by codon optimization would increase expression efficiency and require lower therapeutic doses. Moreover the delivery of vector to the CSF directly would entail more diffusion in the CNS and less spread in peripheral organs.
The core clinical features of CD are very distinct and provide good targets for therapeutics. Long-term studies on rAAV gene therapy-treated patients have documented that the therapy is safe and without risks and it is only a matter of time until the inefficiencies are improved. Efficient translation of the comprehensive data-driven validation of effective therapy in mouse models to humans would push forward the successful development of therapeutics like rAAVs and help to alleviate the sufferings of the hitherto helpless patients.
Acknowledgment
We would like to acknowledge the grant support from Jacob’s Cure, NTSAD Foundation, Canavan Foundation, and Public Health Service grants 1R01NS076991, P01 HL59407-11, P01 AI100263-01 from National Institutes of Health to GG.
One Sentence Synopsis
In this review, we follow the evolution of research on Canavan’s disease and discuss current understanding of molecular pathogenesis as well as developing potential therapeutics for the disease.
Compliance with Ethics Guidelines
Conflict of Interest
Seemin Seher Ahmed declares that she has no conflict of interest.
Guangping Gao is a founder of Voyager Therapeutics and holds equity in the company. He is also an inventor on patents with potential royalties licensed to Voyager Therapeutics. Voyager Therapeutics is a newly launched gene therapy company that focuses on using the recombinant adeno-associated virus platform technology for the development of gene therapeutics to treat a wide range of CNS disorders.
Informed Consent
This article directly does not contain any studies with human subjects performed by the authors.
Animal Rights
All institutional and national guidelines for the care and use of laboratory animals were followed in the work done in the authors’ laboratory.
Details of the Contributions of Individual Authors
Seemin Seher Ahmed planned and drafted the review.
Guangping Gao reviewed the drafts and provided critical inputs.
Footnotes
Competing interests: None declared
Contributor Information
Guangping Gao, Email: guangping.gao@umassmed.edu.
Collaborators: Johannes Zschocke
References
- Adachi M, Schneck L, Cara J, Volk BW. Spongy degeneration of the central nervous system (van Bogaert and Bertrand type; Canavan’s disease). A review. Hum Pathol. 1973;4(3):331–347. doi: 10.1016/S0046-8177(73)80098-X. [DOI] [PubMed] [Google Scholar]
- Ahmed SS, Li H, Cao CSE, Denninger AR, Su Q, Eaton S, Liso Navarro AA, Xie J, Szucs S, Zhang H, Moore C, Kirschner DA, Seyfried TN, Flotte TR, Matalon R, Gao G. A single intravenous rAAV injection as late as P20 achieves efficacious and sustained CNS gene therapy in Canavan mice. Mol Ther. 2013;12:2136–2147. doi: 10.1038/mt.2013.138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akimitsu TKK, Hanaya R, Iida K, Kiura Y, Arita K, Matsubayashi H, Ishihara K, Kitada K, Serikawa T, Sasa M. Epileptic seizures induced by N-acetyl-L-aspartate in rats: in vivo and in vitro studies. Brain Res. 2000;861(1):143–150. doi: 10.1016/S0006-8993(00)02028-X. [DOI] [PubMed] [Google Scholar]
- Al-Dirbashi OY, Kurdi W, Imtiaz F, et al. Reliable prenatal diagnosis of Canavan disease by measuring N-acetylaspartate in amniotic fluid using liquid chromatography tandem mass spectrometry. Prenat Diagn. 2009;29(5):477–480. doi: 10.1002/pd.2223. [DOI] [PubMed] [Google Scholar]
- Ariyannur PS, Moffett JR, Manickam P, et al. Methamphetamine-induced neuronal protein NAT8L is the NAA biosynthetic enzyme: implications for specialized acetyl coenzyme A metabolism in the CNS. Brain Res. 2010;1335:1–13. doi: 10.1016/j.brainres.2010.04.008. [DOI] [PubMed] [Google Scholar]
- Asokan A, Schaffer DV, Samulski RJ. The AAV vector toolkit: poised at the clinical crossroads. Mol Ther. 2012;20(4):699–708. doi: 10.1038/mt.2011.287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Assadi M, Janson C, Wang DJ, et al. Lithium citrate reduces excessive intra-cerebral N-acetyl aspartate in Canavan disease. Eur J Paediatr Neurol. 2010;14(4):354–359. doi: 10.1016/j.ejpn.2009.11.006. [DOI] [PubMed] [Google Scholar]
- Baslow M. A review of phylogenetic and metabolic relationships between the acylamino acids, N-acetyl-L-aspartic acid and N-acetyl-L-histidine, in the vertebrate nervous system. J Neurochem. 1997;68(4):1335–1344. doi: 10.1046/j.1471-4159.1997.68041335.x. [DOI] [PubMed] [Google Scholar]
- Baslow MH. Molecular water pumps and the aetiology of Canavan disease: a case of the sorcerer’s apprentice. J Inherit Metab Dis. 1999;22(2):99–101. doi: 10.1023/A:1005437915117. [DOI] [PubMed] [Google Scholar]
- Baslow MH, Resnik TR. Canavan disease. Analysis of the nature of the metabolic lesions responsible for development of the observed clinical symptoms. J Mol Neurosci. 1997;9(2):109–125. doi: 10.1007/BF02736855. [DOI] [PubMed] [Google Scholar]
- Baslow MH, Kitada K, Suckow RF, Hungund BL, Serikawa T. The effects of lithium chloride and other substances on levels of brain N-acetyl-L-aspartic acid in Canavan disease-like rats. Neurochem Res. 2002;27(5):403–406. doi: 10.1023/A:1015504031229. [DOI] [PubMed] [Google Scholar]
- Beaudet A (2001) Aspartoacylase deficiency (Canavan disease) In: Scriver CR, Beaudet AL, Sly WS, Valle D (eds) The metabolic and molecular bases of inherited disease, McGraw-Hill Publishers, pp 5799–5805
- Bennett MJ, Gibson KM, Sherwood WG, et al. Reliable prenatal diagnosis of Canavan disease (aspartoacylase deficiency): comparison of enzymatic and metabolite analysis. J Inherit Metab Dis. 1993;16(5):831–836. doi: 10.1007/BF00714274. [DOI] [PubMed] [Google Scholar]
- Bhakoo KK, Craig TJ, Styles P. Developmental and regional distribution of aspartoacylase in rat brain tissue. J Neurochem. 2001;79(1):211–220. doi: 10.1046/j.1471-4159.2001.00561.x. [DOI] [PubMed] [Google Scholar]
- Birken DL, Oldendorf WH. N-acetyl-L-aspartic acid: a literature review of a compound prominent in 1H-NMR spectroscopic studies of brain. Neurosci Biobehav Rev. 1989;13(1):23–31. doi: 10.1016/S0149-7634(89)80048-X. [DOI] [PubMed] [Google Scholar]
- Birnbaum SM, Levinton L, Kingsley RB, Greenstein JP. Specificity of amino acid acylases. J Biol Chem. 1952;194:455–462. [PubMed] [Google Scholar]
- Bluml S, Seymour K, Philippart M, Matalon R, Ross B (1998) Elevated brain water in Canavan disease: impact of a diuretic therapy. In: Book elevated brain water in Canavan disease: impact of a diuretic therapy. p 171
- Bruce AJBM. Oxygen free radicals in rat limbic structures after kainate-induced seizures. Free Radic Biol Med. 1995;18(6):993–1002. doi: 10.1016/0891-5849(94)00218-9. [DOI] [PubMed] [Google Scholar]
- Burger C, Gorbatyuk OS, Velardo MJ, et al. Recombinant AAV viral vectors pseudotyped with viral capsids from serotypes 1, 2, and 5 display differential efficiency and cell tropism after delivery to different regions of the central nervous system. Mol Ther. 2004;10(2):302–317. doi: 10.1016/j.ymthe.2004.05.024. [DOI] [PubMed] [Google Scholar]
- Canavan MM (1931) Schilder’s encephalitis periaxialis diffusa. Arch Neurol Psychiat 25:299–308
- Cearley CN, Wolfe JH. Transduction characteristics of adeno-associated virus vectors expressing cap serotypes 7, 8, 9, and Rh10 in the mouse brain. Mol Ther. 2006;13(3):528–537. doi: 10.1016/j.ymthe.2005.11.015. [DOI] [PubMed] [Google Scholar]
- Cearley CN, Vandenberghe LH, Parente MK, Carnish ER, Wilson JM, Wolfe JH. Expanded repertoire of AAV vector serotypes mediate unique patterns of transduction in mouse brain. Mol Ther. 2008;16(10):1710–1718. doi: 10.1038/mt.2008.166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakraborty G, Mekala P, Yahya D, Wu G, Ledeen RW. Intraneuronal N-acetylaspartate supplies acetyl groups for myelin lipid synthesis: evidence for myelin-associated aspartoacylase. J Neurochem. 2001;78(4):736–745. doi: 10.1046/j.1471-4159.2001.00456.x. [DOI] [PubMed] [Google Scholar]
- Chang YC, Rapoport SI, Rao J. Chronic administration of mood stabilizers upregulates BDNF and bcl-2 expression levels in rat frontal cortex. Neurochem Res. 2009;34(3):536–541. doi: 10.1007/s11064-008-9817-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen RWCD. Long term lithium treatment suppresses p53 and Bax expression but increases Bcl-2 expression. A prominent role in neuroprotection against excitotoxicity. J Biol Chem. 1999;274(10):6039–6042. doi: 10.1074/jbc.274.10.6039. [DOI] [PubMed] [Google Scholar]
- Copray SHJ, Sher F, Casaccia-Bonnefil P, Boddeke E. Epigenetic mechanisms facilitating oligodendrocyte development, maturation, and aging. Glia. 2009;57(15):1579–1587. doi: 10.1002/glia.20881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Adamo AF Jr, Gidez LI, Yatsu FM (1968) Acetyl transport mechanisms. Involvement of N-acetyl aspartic acid in de novo fatty acid biosynthesis in the developing rat brain. Exp Brain Res 5(4):267–273 [DOI] [PubMed]
- Duque S, Joussemet B, Riviere C, et al. Intravenous administration of self-complementary AAV9 enables transgene delivery to adult motor neurons. Mol Ther. 2009;17(7):1187–1196. doi: 10.1038/mt.2009.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elpeleg ON, Shaag A. The spectrum of mutations of the aspartoacylase gene in Canavan disease in non-Jewish patients. J Inherit Metab Dis. 1999;22(4):531–534. doi: 10.1023/A:1005512524957. [DOI] [PubMed] [Google Scholar]
- Escolar MLPM, Provenzale JM, Richards KC, Allison J, Wood S, Wenger DA, Pietryga D, Wall D, Champagne M, Morse R, Krivit W, Kurtzberg J. Transplantation of umbilical-cord blood in babies with infantile Krabbe’s disease. N Engl J Med. 2005;352(20):2069–2081. doi: 10.1056/NEJMoa042604. [DOI] [PubMed] [Google Scholar]
- Foust KD, Nurre E, Montgomery CL, Hernandez A, Chan CM, Kaspar BK. Intravascular AAV9 preferentially targets neonatal neurons and adult astrocytes. Nat Biotechnol. 2009;27(1):59–65. doi: 10.1038/nbt.1515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francis J, Markov V, Leone P. Dietary triheptanoin rescues oligodendrocyte loss, dysmyelination and motor function in the nur7 mouse model of Canavan disease. J Inherit Metab Dis. 2013;37(3):369–381. doi: 10.1007/s10545-013-9663-6. [DOI] [PubMed] [Google Scholar]
- Gao GP, Alvira MR, Wang L, Calcedo R, Johnston J, Wilson JM. Novel adeno-associated viruses from rhesus monkeys as vectors for human gene therapy. Proc Natl Acad Sci U S A. 2002;99(18):11854–11859. doi: 10.1073/pnas.182412299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray SJ, Nagabhushan Kalburgi S, McCown TJ, Samulski RJ. Global CNS gene delivery and evasion of anti-AAV-neutralizing antibodies by intrathecal AAV administration in non-human primates. Gene Ther. 2013;20(4):450–459. doi: 10.1038/gt.2012.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hermonat PLMN. Use of adeno-associated virus as a mammalian DNA cloning vector: transduction of neomycin resistance into mammalian tissue culture cells. Proc Natl Acad Sci U S A. 1984;81(20):6466–6470. doi: 10.1073/pnas.81.20.6466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hershfield JR, Pattabiraman N, Madhavarao CN, Namboodiri MA. Mutational analysis of aspartoacylase: implications for Canavan disease. Brain Res. 2007;1148:1–14. doi: 10.1016/j.brainres.2007.02.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirano A. Structure of normal central myelinated fibers. Adv Neurol. 1981;31:51–68. doi: 10.1212/WNL.31.1.51. [DOI] [PubMed] [Google Scholar]
- Hunt A, Burne R. Medical and nursing problems of children with neurodegenerative disease. Palliative Med. 1995;9(1):19–26. doi: 10.1177/026921639500900104. [DOI] [PubMed] [Google Scholar]
- Hwu WL, Muramatsu S, Tseng SH, et al. Gene therapy for aromatic L-amino acid decarboxylase deficiency. Sci Transl Med. 2012;4(134):134–161. doi: 10.1126/scitranslmed.3003640. [DOI] [PubMed] [Google Scholar]
- Jakobs C, ten Brink HJ, Langelaar SA, et al. Stable isotope dilution analysis of N-acetylaspartic acid in CSF, blood, urine and amniotic fluid: accurate postnatal diagnosis and the potential for prenatal diagnosis of Canavan disease. J Inherit Metab Dis. 1991;14(5):653–660. doi: 10.1007/BF01799929. [DOI] [PubMed] [Google Scholar]
- Janson C, McPhee S, Bilaniuk L, et al. Clinical protocol. Gene therapy of Canavan disease: AAV-2 vector for neurosurgical delivery of aspartoacylase gene (ASPA) to the human brain. Hum Gene Ther. 2002;13(11):1391–1412. doi: 10.1089/104303402760128612. [DOI] [PubMed] [Google Scholar]
- Janson CG, McPhee SW, Francis J, et al. Natural history of Canavan disease revealed by proton magnetic resonance spectroscopy (1H-MRS) and diffusion-weighted MRI. Neuropediatrics. 2006;37(4):209–221. doi: 10.1055/s-2006-924734. [DOI] [PubMed] [Google Scholar]
- Kaul R, Casanova J, Johnson AB, Tang P, Matalon R. Purification, characterization, and localization of aspartoacylase from bovine brain. J Neurochem. 1991;56(1):129–135. doi: 10.1111/j.1471-4159.1991.tb02571.x. [DOI] [PubMed] [Google Scholar]
- Kaul R, Gao GP, Balamurugan K, Matalon R. Cloning of the human aspartoacylase cDNA and a common missense mutation in Canavan disease. Nat Genet. 1993;5(2):118–123. doi: 10.1038/ng1093-118. [DOI] [PubMed] [Google Scholar]
- Kaul R, Gao GP, Aloya M, et al. Canavan disease: mutations among Jewish and non-Jewish patients. Am J Hum Genet. 1994;55(1):34–41. [PMC free article] [PubMed] [Google Scholar]
- Kessing LVSL, Forman JL, Andersen PK. Lithium treatment and risk of dementia. Arch Gen Psychiatry. 2008;65(11):1331–1335. doi: 10.1001/archpsyc.65.11.1331. [DOI] [PubMed] [Google Scholar]
- Kitada K, Akimitsu T, Shigematsu Y, et al. Accumulation of N-acetyl-L-aspartate in the brain of the tremor rat, a mutant exhibiting absence-like seizure and spongiform degeneration in the central nervous system. J Neurochem. 2000;74(6):2512–2519. doi: 10.1046/j.1471-4159.2000.0742512.x. [DOI] [PubMed] [Google Scholar]
- Klugmann M, Symes CW, Klaussner BK, et al. Identification and distribution of aspartoacylase in the postnatal rat brain. Neuroreport. 2003;14(14):1837–1840. doi: 10.1097/00001756-200310060-00016. [DOI] [PubMed] [Google Scholar]
- Kumar S, Biancotti JC, Matalon R, de Vellis J. Lack of aspartoacylase activity disrupts survival and differentiation of neural progenitors and oligodendrocytes in a mouse model of Canavan disease. J Neurosci Res. 2009;87(15):3415–3427. doi: 10.1002/jnr.22233. [DOI] [PubMed] [Google Scholar]
- Lee DHPG. Mutagenesis induced by the nitric oxide donor sodium nitroprusside in mouse cells. Mutagenesis. 2007;22(1):63–67. doi: 10.1093/mutage/gel051. [DOI] [PubMed] [Google Scholar]
- Leone P, Janson CG, Bilaniuk L, et al. Aspartoacylase gene transfer to the mammalian central nervous system with therapeutic implications for Canavan disease. Ann Neurol. 2000;48(1):27–38. doi: 10.1002/1531-8249(200007)48:1<27::AID-ANA6>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- Leone P, Shera D, McPhee SW et al (2012) Long-term follow-up after gene therapy for Canavan disease. Sci Transl Med 4(165):165ra163 [DOI] [PMC free article] [PubMed]
- Lin WPB. Endoplasmic reticulum stress in disorders of myelinating cells. Nat Neurosci. 2009;12(4):379–385. doi: 10.1038/nn.2273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madhavarao CN, Chinopoulos C, Chandrasekaran K, Namboodiri MA. Characterization of the N-acetylaspartate biosynthetic enzyme from rat brain. J Neurochem. 2003;86(4):824–835. doi: 10.1046/j.1471-4159.2003.01905.x. [DOI] [PubMed] [Google Scholar]
- Madhavarao CN, Moffett JR, Moore RA, Viola RE, Namboodiri MA, Jacobowitz DM. Immunohistochemical localization of aspartoacylase in the rat central nervous system. J Comp Neurol. 2004;472(3):318–329. doi: 10.1002/cne.20080. [DOI] [PubMed] [Google Scholar]
- Madhavarao CN, Arun P, Moffett JR, et al. Defective N-acetylaspartate catabolism reduces brain acetate levels and myelin lipid synthesis in Canavan’s disease. Proc Natl Acad Sci U S A. 2005;102:5221–5226. doi: 10.1073/pnas.0409184102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madhavarao CN, Arun P, Anikster Y, et al. Glyceryl triacetate for Canavan disease: a low-dose trial in infants and evaluation of a higher dose for toxicity in the tremor rat model. J Inherit Metab Dis. 2009;32(5):640–650. doi: 10.1007/s10545-009-1155-3. [DOI] [PubMed] [Google Scholar]
- Matalon R, Michals-Matalon K. Biochemistry and molecular biology of Canavan disease. Neurochem Res. 1999;24(4):507–513. doi: 10.1023/A:1022531829100. [DOI] [PubMed] [Google Scholar]
- Matalon R, Michals-Matalon K. Prenatal diagnosis of Canavan disease. Prenat Diagn. 1999;19(7):669–670. doi: 10.1002/(SICI)1097-0223(199907)19:7<669::AID-PD630>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- Matalon R, Michals K, Sebesta D, Deanching M, Gashkoff P, Casanova J. Aspartoacylase deficiency and N-acetylaspartic aciduria in patients with Canavan disease. Am J Med Genet. 1988;29(2):463–471. doi: 10.1002/ajmg.1320290234. [DOI] [PubMed] [Google Scholar]
- Matalon R, Kaul R, Michals K. Canavan disease: biochemical and molecular studies. J Inherit Metab Dis. 1993;16(4):744–752. doi: 10.1007/BF00711906. [DOI] [PubMed] [Google Scholar]
- Matalon R, Rady PL, Platt KA, et al. Knock-out mouse for Canavan disease: a model for gene transfer to the central nervous system. J Gene Med. 2000;2(3):165–175. doi: 10.1002/(SICI)1521-2254(200005/06)2:3<165::AID-JGM107>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- Matalon R, Surendran S, Rady PL, et al. Adeno-associated virus-mediated aspartoacylase gene transfer to the brain of knockout mouse for Canavan disease. Mol Ther. 2003;7(5 Pt 1):580–587. doi: 10.1016/S1525-0016(03)00066-2. [DOI] [PubMed] [Google Scholar]
- McCown T. Adeno-associated virus (AAV) vectors in the CNS. Curr Gene Ther. 2005;5(3):333–338. doi: 10.2174/1566523054064995. [DOI] [PubMed] [Google Scholar]
- McPhee SW, Francis J, Janson CG, et al. Effects of AAV-2-mediated aspartoacylase gene transfer in the tremor rat model of Canavan disease. Brain Res Mol Brain Res. 2005;135(1–2):112–121. doi: 10.1016/j.molbrainres.2004.12.007. [DOI] [PubMed] [Google Scholar]
- Mersmann N, Tkachev D, Jelinek R, et al. Aspartoacylase-lacZ knockin mice: an engineered model of Canavan disease. PLoS One. 2011;6(5):e2033. doi: 10.1371/journal.pone.0020336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mondino M BJ, Saoud M. (2013) N-acetyl-aspartate level is decreased in the prefrontal cortex in subjects at-risk for schizophrenia. Front Psychiatry 4:Article 99 [DOI] [PMC free article] [PubMed]
- Muramatsu S, Fujimoto K, Kato S, et al. A phase I study of aromatic L-amino acid decarboxylase gene therapy for Parkinson’s disease. Mol Ther. 2010;18(9):1731–1735. doi: 10.1038/mt.2010.135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novotny EJ Jr, Hyder J, Rothman Dl (1999) Cerebral amino acids and metabolites in aminoacylase II deficiencies. J Mol Neurosci 12(3):174–175
- O'Donnell T, Rotzinger S, Nakashima TT, Hanstock CC, Ulrich M, Silverstone PH (2000) Chronic lithium and sodium valproate both decrease the concentration of myo-inositol and increase the concentration of inositol monophosphates in rat brain. Brain Res 880(1–2):84–91 [DOI] [PubMed]
- Patel M. Mitochondrial dysfunction and oxidative stress: cause and consequence of epileptic seizures. Free Radic Biol Med. 2004;37(12):1951–1962. doi: 10.1016/j.freeradbiomed.2004.08.021. [DOI] [PubMed] [Google Scholar]
- Pederzolli CDMC, Scapin F, Rockenbach FJ, Sgaravatti AM, Sgarbi MB, Wyse AT, Wannmacher CM, Wajner M, Dutra-Filho CS. N-acetylaspartic acid promotes oxidative stress in cerebral cortex of rats. Int J Dev Neurosci. 2007;25(5):317–324. doi: 10.1016/j.ijdevneu.2007.04.002. [DOI] [PubMed] [Google Scholar]
- Sager TNF-JA, Hansen AJ. Transient elevation of interstitial N-acetylaspartate in reversible global brain ischemia. J Neurochem. 1997;68(2):675–682. doi: 10.1046/j.1471-4159.1997.68020675.x. [DOI] [PubMed] [Google Scholar]
- Samuel SKR, Jayavelu T, Chinnakkannu P. Protein oxidative damage in arsenic induced rat brain: influence of DL-a-lipoic acid. Toxicol Lett. 2005;155:27–34. doi: 10.1016/j.toxlet.2004.08.001. [DOI] [PubMed] [Google Scholar]
- Samulski RJ, Sally M, Muzyczka N, TF eds (1999) Adeno associated viral vectors: the development of human gene therapy. Cold Spring Harbor Press, New York, pp 131–172
- Segel RAY, Zevin S, Steinberg A, Gahl WA, Fisher D, Staretz-Chacham O, Zimran A, Altarescu G. A safety trial of high dose glyceryl triacetate for Canavan disease. Mol Genet Metab. 2011;103(3):203–206. doi: 10.1016/j.ymgme.2011.03.012. [DOI] [PubMed] [Google Scholar]
- Seki T, Matsubayashi H, Amano T, et al. Adenoviral gene transfer of aspartoacylase into the tremor rat, a genetic model of epilepsy, as a trial of gene therapy for inherited epileptic disorder. Neurosci Lett. 2002;328(3):249–252. doi: 10.1016/S0304-3940(02)00522-0. [DOI] [PubMed] [Google Scholar]
- Solsona MDFL, Boquet EM, Andrés JL. Lithium citrate as treatment of Canavan disease. Clin Neuropharmacol. 2012;35(3):150–151. doi: 10.1097/WNF.0b013e3182515c9d. [DOI] [PubMed] [Google Scholar]
- Sommer A, Sass JO. Expression of aspartoacylase (ASPA) and Canavan disease. Gene. 2012;505(2):206–210. doi: 10.1016/j.gene.2012.06.036. [DOI] [PubMed] [Google Scholar]
- Spange SWT, Heinzel T, Krämer OH. Acetylation of non-histone proteins modulates cellular signalling at multiple levels. Int J Biochem Cell Biol. 2009;41(1):185–198. doi: 10.1016/j.biocel.2008.08.027. [DOI] [PubMed] [Google Scholar]
- Steen RGOR. Abnormally high levels of brain N-acetylaspartate in children with sickle cell disease. AJNR Am J Neuroradiol. 2005;26(3):463–468. [PMC free article] [PubMed] [Google Scholar]
- Surendran S. Upregulation of N-acetylaspartic acid alters inflammation, transcription and contractile associated protein levels in the stomach and smooth muscle contractility. Mol Biol Rep. 2009;36(1):201–206. doi: 10.1007/s11033-007-9167-2. [DOI] [PubMed] [Google Scholar]
- Swain GP, Prociuk M, Bagel JH, et al. Adeno-associated virus serotypes 9 and rh10 mediate strong neuronal transduction of the dog brain. Gene Ther. 2013;21(1):28–36. doi: 10.1038/gt.2013.54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor DLDS, Obrenovitch TP, Doheny MH, Patsalos PN, Clark JB, Symon L. Investigation into the role of N-acetylaspartate in cerebral osmoregulation. J Neurochem. 1995;65(1):275–281. doi: 10.1046/j.1471-4159.1995.65010275.x. [DOI] [PubMed] [Google Scholar]
- Tortorella C, Ruggieri M, Di Monte E, et al. Serum and CSF N-acetyl aspartate levels differ in multiple sclerosis and neuromyelitis optica. J Neurol Neurosurg Psychiatry. 2011;82:1355–1359. doi: 10.1136/jnnp.2011.241836. [DOI] [PubMed] [Google Scholar]
- Traka M, Wollmann RL, Cerda SR, Dugas J, Barres BA, Popko B. Nur7 is a nonsense mutation in the mouse aspartoacylase gene that causes spongy degeneration of the CNS. J Neurosci. 2008;28(45):11537–11549. doi: 10.1523/JNEUROSCI.1490-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsacopoulos MMP. Metabolic coupling between glia and neurons. J Neurosci. 1996;16(3):877–885. doi: 10.1523/JNEUROSCI.16-03-00877.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai G, Coyle J. N-acetylaspartate in neuropsychiatric disorders. Prog Neurobiol. 1995;46(5):531–540. doi: 10.1016/0301-0082(95)00014-M. [DOI] [PubMed] [Google Scholar]
- Tsai GGD, Chang RW, Flood J, Baer L, Coyle JT. Markers of glutamatergic neurotransmission and oxidative stress associated with tardive dyskinesia. Am J Psychiatry. 1998;155(9):1207–1213. doi: 10.1176/ajp.155.9.1207. [DOI] [PubMed] [Google Scholar]
- Waksman B. Demyelinating disease: evolution of a paradigm. Neurochem Res. 1999;24(4):491–495. doi: 10.1023/A:1022527628192. [DOI] [PubMed] [Google Scholar]
- Wang J, Leone P, Wu G, et al. Myelin lipid abnormalities in the aspartoacylase-deficient tremor rat. Neurochem Res. 2009;34(1):138–148. doi: 10.1007/s11064-008-9726-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang B, Li S, Wang H, et al. Global CNS transduction of adult mice by intravenously delivered rAAVrh.8 and rAAVrh.10 and nonhuman primates by rAAVrh.10. Mol Ther. 2014;22(7):1299–1309. doi: 10.1038/mt.2014.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zano S, Malik R, Szucs S, Matalon R, Viola RE. Modification of aspartoacylase for potential use in enzyme replacement therapy for the treatment of Canavan disease. Mol Genet Metab. 2011;102(2):176–180. doi: 10.1016/j.ymgme.2010.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Yang B, Mu X et al (2011) Several rAAV vectors efficiently cross the blood–brain barrier and transduce neurons and astrocytes in the neonatal mouse central nervous system. Mol Ther 8(19):1440–1448 [DOI] [PMC free article] [PubMed]
- Zlokovic B. The blood–brain barrier in health and chronic neurodegenerative disorders. Neuron. 2008;57:178–201. doi: 10.1016/j.neuron.2008.01.003. [DOI] [PubMed] [Google Scholar]