

Abstract
Background: Lysinuric protein intolerance (LPI; MIM# 222700) is a rare metabolic disorder caused by a defective cationic amino acids (CAA) membrane transport leading to decreased circulating plasma CAA levels and resulting in dysfunction of the urea cycle. Short stature is commonly observed in children with LPI and has been associated with protein malnutrition. A correlation between LPI and growth hormone deficiency (GHD) has also been postulated because of the known interaction between the AA arginine, ornithine, and lysine and growth hormone (GH) secretion. Our report describes a case of GHD in an LPI patient, who has not presented a significant increase in growth velocity with recombinant-human GH (rhGH) therapy, suggesting some possible pathogenic mechanisms of growth failure.
Case Presentation: The proband was a 6-year-old boy, diagnosed as suffering from LPI, erythrophagocytosis (HP) in bone marrow, and short stature. Two GH provocative tests revealed GHD. The patient started rhGH therapy and a controlled-protein diet initially with supplementation of oral arginine and then of citrulline. At 3-year follow-up, no significant increase in growth velocity and in insulin-like growth factor-1 (IGF-1) levels was observed. Inadequate nutrition and low plasmatic levels of arginine, ornithine, lysine, and HP may have contributed to his poor growth.
Conclusion: Our case suggests that growth failure in patients with GHD and LPI treated with rhGH could have a complex and multifactorial pathogenesis. Persistently low plasmatic levels of lysine, arginine, and ornithine, associated with dietary protein and caloric restriction and systemic inflammation, could determine a defect in coupling GH to IGF-1 production explaining why GH replacement therapy is not able to significantly improve growth impairment. We hypothesize that a better understanding of growth failure pathophysiology in these patients could lead to the development of more rational strategies to treat short stature in patients with LPI.
Keywords: Arginine, Growth hormone deficiency, Lysine, Lysinuric protein intolerance, Ornithine, Protein malnutrition
Introduction
Lysinuric protein intolerance (LPI; MIM 222700) is a rare autosomal recessive metabolic disorder caused by a defective CAA lysine, arginine, and ornithine membrane transport. Urinary excretion of CAA is increased and CAA are poorly adsorbed from the intestine leading to decreased circulating plasma CAA levels (Borsani et al. 1999; Torrents et al. 1999). This defect results in dysfunction of the urea cycle, causing episodes of postprandial hyperammonemia and spontaneous protein aversion (Ogier de Baulny et al. 2012; Sebastio et al. 2011; Palacin et al. 2004). The SLC7A7 gene, which encodes the y+L amino acid transporter-1 (y+LAT-1), is the only gene in which mutations are currently known to cause LPI (Torrents et al. 1999; Ogier de Baulny et al. 2012; Sebastio et al. 2011).
Typically, patients with LPI are asymptomatic while breast-feeding and develop symptoms later in life after weaning. Recurrent vomiting and episodes of diarrhea are observed as acute symptoms after protein ingestion; however, the diagnosis of LPI may be missed during infancy unless the presence of neurologic involvement triggers a diagnostic laboratory evaluation. Chronic symptoms of LPI are short stature, enlarged liver and spleen, osteoporosis, muscle hypotonia, and sometimes moderate mental retardation. Renal insufficiency, pulmonary alveolar proteinosis, and hematologic anomalies including HP are recurrent clinical features and are suggestive of multisystem involvement (Ogier de Baulny et al. 2012; Sebastio et al. 2011).
Current long-term treatment of LPI consists of controlled-protein diet and supplementation with oral l-citrulline, l-lysine, or l-arginine, in order to replenish the urea cycle and prevent hyperammonemia and orotic aciduria (Palacin et al. 2004; Awrich et al. 1975; Mizutani et al. 1984; Brosnan and Brosnan 2007).
Failure to thrive and short stature are commonly observed in children with LPI and have been associated with protein malnutrition (Sebastio and Nunes 2006). A correlation between LPI and GHD was also postulated because of the known interaction between the amino acids (AA) arginine, ornithine, and lysine and GH secretion (Alba-Roth et al. 1988; Knopf et al. 1965; van Vught et al. 2008; Isidori et al. 1981).
There are only a few cases in the literature dealing with LPI patients in whom the GH/IGF-1 axis was investigated (Awrich et al. 1975; Goto et al. 1984; Esposito et al. 2006; Niinikoski et al. 2011), and a true GHD was observed in only one case (Esposito et al. 2006). In that case the patient was a 12-year-old severely stunted girl with renal tubular disease as well; she responded well to GH replacement therapy, but she had an age close to puberty and the follow-up period was only 1 year.
Our report describes a case of GHD in an LPI patient, who has not presented a significant increase in growth velocity with recombinant-human GH (rhGH) therapy, suggesting some possible pathogenic mechanisms of growth failure.
Case Presentation
Case Report
Our patient was a 6-year-9-month-old boy born at 38 weeks after an uneventful pregnancy from two unrelated healthy parents. His birth weight was 2,750 g (between 10th and 25th percentile on the Italian Neonatal Study growth charts (INeS) www.inescharts.com) and length was 47 cm (between 10th and 25th percentile on the INeS growth charts). He underwent normal weaning but subsequently showed recurrent vomiting, diarrhea, and an aversion to protein-rich foods that his parents gradually eliminated from his diet. In the first years of life, the boy suffered from failure to thrive and many upper respiratory infections. While assessing his problems, laboratory tests revealed high serum ferritin (244 ng/mL) and lactic dehydrogenase (LDH) values (1017 U/L).
He was brought to our attention for short stature. He was in good general conditions and his clinical examination was normal. His weight was 16.8 kg (<3rd percentile on the growth charts of the Center for Disease Control, CDC, and Prevention www.cdc.gov/growthcharts) and his height was 107 cm (<3rd percentile on CDC growth charts) (Fig. 1). The height velocity during the year before our evaluation was of 5 cm/year. Tanner stages were of G1, PH1. Midparental height (MPH) was used as an indicator of genetic growth potential, and the genetic target height of the patient was 172.5 cm (between 25th and 50th percentile on CDC growth charts) (Fig. 1). His bone age was delayed, being 4 years and 6 months according to Greulich and Pyle Method.
Fig. 1.
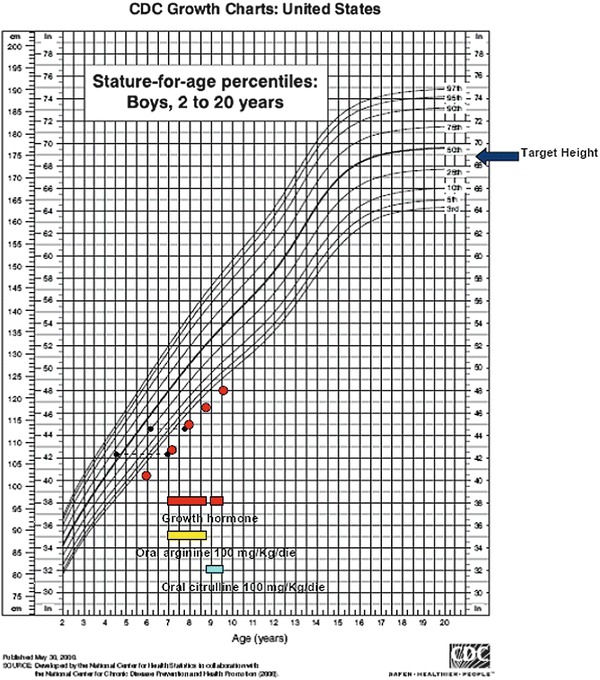
Growth chart of the patient, plotted by CDC charts percentiles. The time when GH treatment and oral supplementation with arginine and citrulline were started are indicated
Two GH provocative tests were performed to measure GH peak serum levels, revealing in both cases GH values well below 10 μg/L; in particular, the arginine stimulation test induced a GH peak level of 1.1 μg/L and the insulin stimulation test of 6.8 μg/L. IGF-1 value was very low (3 nmol/L, reference range 9–30 nmol/L). Abdominal ultrasonography revealed only a mild hepatosplenomegaly. Blood cell and platelet count, renal and liver function tests, and urine analysis were all normal.
After the first laboratory tests, we found again high ferritin (770 ng/mL) and LDH levels (1,007 U/L). Therefore, a bone marrow aspiration was performed, evidencing HP. Given the history of poor growth associated with aversion to protein-rich food, HP in bone marrow, and hepatosplenomegaly, we assessed plasma and urinary AA as well. We found lower than normal plasmatic levels of lysine (62 μmol/L; normal values 115–278 μmol/L), ornithine (13.9 μmol/L; n.v. 25–159 μmol/L), and arginine (22.1 μmol/L; n.v. 38–135 μmol/L) and higher than normal urinary levels of lysine (247 mmol/mol of creatinine; n.v. 10–56 mmol/mol of creatinine), arginine (89.5 mmol/mol of creatinine; n.v. 0–6 mmol/mol of creatinine), and ornithine (20.5 mmol/mol of creatinine; n.v. 0–6 mmol/mol of creatinine) (Tables 1 and 2). Based on these results, we diagnosed LPI with HP in bone marrow and GHD.
Table 1.
Results of plasma cationic amino acids analyses at LPI diagnosis and after 24 and 36 months of therapy
| Amino acids | Proband: at diagnosis (μmol/L) | Proband: after 24 months of therapy (μmol/L) | Proband: after 36 months of therapy (μmol/L) | Reference range (μmol/L) |
|---|---|---|---|---|
| Lysine | 62 | 73.81 | 36.96 | 115–278 |
| Arginine | 22.1 | 53.70 | 18.96 | 38–135 |
| Ornithine | 13.9 | 16.21 | 17.45 | 25–159 |
| Glutamine | 1,025 | 1,041 | 1,061 | 399–823 |
Reference range is also indicated
Table 2.
Results of urinary amino acids analyses at LPI diagnosis
| Amino acids | Proband: at diagnosis (mmol/mol of creatinine) | Reference range (mmol/mol creatinine) |
|---|---|---|
| Lysine | 247 | 10–56 |
| Arginine | 89.5 | 0–6 |
| Ornithine | 20.5 | 0–6 |
Reference range is also indicated
After PCR-amplified fragments sequencing, the patient was reported as a homozygote for a known missense mutation 1001T>G in the SLC7A7 gene, leading to a premature stop codon in exon 8. His parents were heterozygous carriers of the same mutation.
At 7 years (height 108 cm), the patient started a controlled-protein diet (1 g/kg/day) and an oral supplementation with l-arginine (100 mg/kg/die) and vitamin D (800 IU/day). Following magnetic resonance imaging to rule out abnormalities of the hypothalamo-pituitary axis, the patient began daily subcutaneous rhGH therapy at a dose of 30 μg/kg/day for 6 days a week.
After 12 months from the institution of the rhGH therapy, despite good compliance with therapy and diet, the patient showed only a mild acceleration of growth velocity (6.5 cm/year). GH therapy promoted appetite, but his protein, in gram per kilogram per day, and his caloric intake, in Kcal per kilogram per day, showed no clear differences. IGF-1 level increased only from 3 to 10 nmol/L and bone age was of 6 years.
After 12 months, GHD was retested; l-dopa stimulation test revealed a GH peak level of 5.6 μg/L, confirming GHD. Because of the gastrointestinal symptoms experienced by the patient, in particular intestinal bloating, we stopped oral l-arginine and started oral supplementation with l-citrulline (100 mg/kg/day). Lysine supplement was not given because of his experienced gastrointestinal symptoms.
At 10 years, after 36 months of good compliance with rhGH therapy and diet, his height was 125.4 cm, still under 3rd percentile on CDC growth charts, and his growth velocity was of 5.4 cm/year.
Therapy was monitored on the basis of plasma ammonia, plasma AA, and urinary orotic acid excretion. Postprandial plasma ammonia levels and orotic aciduria were always in the normal range. Plasmatic levels of arginine, ornithine, and lysine remained low. Glutamine levels remained around 1,000 μmol/L (Table 1).
Although a gradual GH dosage adjustment, IGF-1 levels persisted low (11.7 nmol/L).
During the whole follow-up period of 3 years, the patient maintained good general conditions and no acute episode of metabolic derangement was reported. His renal and pulmonary function was normal.
Discussion
Among the several metabolic-endocrine disorders causing short stature, especially in cases of aversion to protein-rich foods, LPI must always be considered. The clinical onset of the disease is usually delayed by breast-feeding or by the use of milk-based infant formula due to their relatively low protein content (Ogier de Baulny et al. 2012; Sebastio et al. 2011; Palacin et al. 2004), but in the case of our patient, the diagnosis was further delayed because his parents gradually eliminated protein-rich foods from his diet. Moreover, during the first 6 years of his life, failure to thrive and the gastrointestinal symptoms misled the diagnosis toward gastrointestinal disorders.
The growth failure frequently observed in children with LPI appears to be a postnatal phenomenon, because intrauterine development is usually normal (Bröer 2007), as observed in our patient, and poor growth is generally attributed to protein malnutrition (Sebastio and Nunes 2006). Nevertheless, Sperandeo et al. (Sperandeo et al. 2007) reported on the generation of a slc7a7−/− mice that experienced intrauterine growth restriction (IUGR) by downregulating IGF-1 expression in fetal liver.
In humans with LPI, there are only a few cases in whom the GH/IGF-1 axis was investigated (Awrich et al. 1975; Goto et al. 1984; Esposito et al. 2006; Niinikoski et al. 2011), and a true GHD was documented in only one of these patients (Esposito et al. 2006). Esposito et al. in 2006 described the first case of a 12-year-old LPI patient with documented GHD that was tested with arginine (GH peak 6.4 μg/L) and clonidine stimulation test (GH peak 2.7 μg/L). After 1 year of GH replacement therapy (35 μg/kg/day) with oral citrulline supplementation (150 mg/kg/day) and protein-restricted diet (1.2 g/kg/day), the authors observed a significant acceleration of growth velocity (from 2 to 8 cm/year) associated with a significant increase in IGF-1 levels (from 95 to 379 μg/L). Nevertheless, the follow-up period of this patient was only 1 year and the patient had an age close to puberty and a renal tubular disease as well.
Some observations suggest a possible link between pathophysiology of LPI and alterations in GH/IGF-1 axis. Niinikoski et al. (2011) observed in four LPI children very low IGF-1 concentrations (5–13 nmol/L), despite arginine-insulin GH provocative test revealed an abnormal GH peak level (< 10 ug/L) in only one of the patients. Nevertheless, a normal GH response to provocative tests in patients with auxology suggestive of GHD does not definitively rule out GHD because they may have a low spontaneous GH secretion over 24 h, likely reflecting GH neurosecretory dysfunction (Gasco et al. 2008). Moreover, today, there is no gold standard for estimating GH secretion in LPI patients. In our case a very low GH peak was obtained with the arginine intravenous (IV) stimulation test; it is likely that this test may not be an appropriated test for GHD evaluation in patients with LPI because of their increased renal clearance of arginine.
Some studies suggest that GH secretion in LPI patients could be altered due to specific lysine, ornithine, and arginine deficiencies (Awrich et al. 1975; Goto et al. 1984). Goto et al. (1984) observed in a 14-year-old LPI boy a better response to the same GH stimulation test after treatment with arginine. Awrich et al. (1975) observed in a 9-year-old girl with LPI – but without GHD – an improvement of growth after oral supplement of arginine, lysine, and citrulline.
Although studies on slc7a7−/− mice exclude CAA depletion as a primary cause of IUGR (Sperandeo et al. 2007), it is not possible to exclude its role in postnatal growth delay. In fact, it is well known that GH secretion can be promoted by intravenous administration of various AA, and the stimulatory effect of some AA on GH is clinically used as a method to assess the responsiveness of the GH secretory system. In particular, it is known that IV arginine increases GH release by suppressing endogenous somatostatin secretion (Alba-Roth et al. 1988), and infusion of lysine also promotes relatively large increases (~8- to 22-fold) in circulating GH levels (Knopf et al. 1965). IV administration of ornithine stimulates GH release as well (Evain-Brion et al. 1982). The stimulatory effect of these AA on GH secretion is also evident upon oral administration (Chromiak and Antonio 2002), suggesting a role of dietary AA in the regulation of plasma GH concentrations (van Vught et al. 2008; Isidori et al. 1981).
CAA depletion could also compromise IGF-1 production. In fact, it has been observed that in humans, lysine sufficiency increased IGF-1 concentrations (Jansen 1962), and in animals low lysine intakes decreased muscle protein accretion and significantly downregulated hepatic IGFBP-1, as well as muscle GH-R and IGF-II (Ishida et al. 2011; Hevrǿy et al. 2007). Furthermore, arginine has been demonstrated to stimulate IGF-1 production (Chevalley et al. 1998).
It is interesting to note that, although a gradual rhGH dosage adjustment, in our patient, IGF-1 levels remained very low, suggesting a defect in coupling GH to IGF-1 production.
Inadequate nutrition, in particular protein malnutrition and low caloric intake, may well contribute to the poor growth, because the functioning of GH/IGF-1 axis appears optimal only with adequate dietary intake. In fact, infants and young children having higher energy demands are especially vulnerable to nutritional imbalances. In particular, nutritional status is a critical element in regulating the GH/IGF-1 system, modulating the responsiveness of the liver to GH (Zamboni et al. 1996; Trobec et al. 2011; Hintz et al. 1978) and influencing IGF-1 gene expression also in extra-hepatic tissues (Naranjo et al. 2002). Moreover, malnutrition and protein deprivation are associated to high levels of GH accompanied by marked decrease in circulating IGF-1 levels (Zamboni et al. 1996; Trobec et al. 2011), and in specifically protein-malnourished children, it has been observed that serum IGF-1 bioactivity is low (Hintz et al. 1978). Furthermore, caloric intake is positively correlated with the increment in growth velocity during GH therapy (Zadik et al. 2005).
These points suggest that it is very important to optimize nutrition to improve growth in LPI patients, even more in GHD patients treated with GH. In fact, because GH therapy can potentially induce a rapid acceleration in growth, it should be suspected that a child who does not increase accordingly could experience compromised nutritional intake or a deficiency in some essential nutrients.
Systemic disorder-related manifestations of LPI (e.g., hematological abnormalities, immune dysfunction, pulmonary and renal involvement) could also exacerbate growth failure in these patients.
In fact, chronic inflammatory diseases have been associated with growth retardation in children (Pozzo and Kemp 2012). Moreover, the increased energy expenditure observed in chronic inflammatory disorders could impair linear growth, and inflammatory cytokines can adversely affect a number of components of growth plate chondrogenesis (De Benedetti 2009; Ahmed and Sävendahl 2009) and can also disrupt the GH/IGF-1 axis (Savage 2013).
A role of an unbalanced metabolism of intracellular arginine, which causes intracellular arginine accumulation and increased nitric oxide (NO) production, has been hypothesized to explain especially the inflammatory complications affecting lung and immune and hematologic systems in LPI patients (Sebastio et al. 2011), but currently, it is not clear if this mechanism could play a role in the pathophysiology of alterations in GH/IGF-1 axis and growth failure.
A good and precocious treatment of both GHD and LPI is also very important. Several studies have demonstrated that growth response on GH treatment is positively influenced by a young age at the start of treatment (Ranke et al. 2007). Moreover, optimal long-term outcome, such as stature, in inherited disorders depends on early diagnosis and good metabolic control (MaCdonald et al. 2012). Our patient was treated by 7 years of age, and chronic dysmetabolism and delayed rhGH therapy could be responsible for growth impairment.
Nevertheless, delayed puberty and prolonged growth due to delayed bone age, frequently observed in LPI patients, may explain why the final height in treated LPI patients is anyhow usually subnormal or low normal (Palacin et al. 2004).
In conclusion, our case suggests that growth failure in patients with GHD and LPI treated with rhGH has a complex and multifactorial pathogenesis. Persistently low IGF-1 levels, observed in our patient although a gradual rhGH dosage adjustment, suggest that CAA deficiency, associated with dietary protein and caloric restriction and systemic inflammation, could determine a defect in coupling GH to IGF-1 production explaining why GH replacement therapy is not able to significantly improve growth impairment. CAA deficiency could be responsible also for reduced spontaneous GH secretion.
Pathogenesis of growth failure in LPI is still partially understood. We hypothesize that patients with LPI and GHD could better elucidate the biochemical causes of growth failure pathophysiology leading to the development of more rational strategies to treat short stature in LPI.
Acknowledgements
The Authors are grateful to CEINGE Laboratories of University of Naples Federico II (Italy) for testing SLC7A7 gene, and to Alice Dianin for dietetic assessment of the patient to Ms. Judyth Dillon for editing the English version of the manuscript.
Abbreviations
- AA
Amino acids
- CAA
Cationic amino acids
- GH
Growth hormone
- GHD
Growth hormone deficiency
- HP
Erythrophagocytosis
- IGF-1
Insulin-like growth factor-1
- IUGR
Intrauterine growth restriction
- LDH
Lactic dehydrogenase
- LPI
Lysinuric protein intolerance
- MPH
Midparental height
- NO
Nitric oxide
Conflict of Interest
Maines Evelina, Morandi Grazia, Olivieri Francesca, Camilot Marta, Cavarzere Paolo, Gaudino Rossella, Antoniazzi Franco, and Andrea Bordugo declare that they have no conflict of interest.
Informed Consent
All procedures followed were in accordance with the ethical standards of the responsible committee on human experimentation (institutional and national) and with the Helsinki Declaration of 1975, as revised in 2000.
Written informed consent was obtained from the parents of the patient for publication of this case report. A copy of the written consent is available for review by the Editor-in-Chief of this journal.
Animal Rights
This article does not contain any studies with human or animal subjects performed by the any of the authors.
Author’s Contributions
Dr. Gaudino and Dr. Cavarzere conceived and designed the study and ensured the accuracy of the data and analysis. Dr. Olivieri collected the data and revised the manuscript critically for important intellectual content. Dr. Maines and Dr. Morandi ensured the accuracy of the data and analysis, wrote the initial draft, and critically revised the manuscript for important intellectual content. Dr. Camilot ensured the accuracy of the draft and edited the English version of the manuscript. Dr. Antoniazzi conceived and designed the study, ensured the accuracy of the data analysis, and critically revised the manuscript for important intellectual content. Dr. Bordugo revised the manuscript critically for important intellectual content.
Footnotes
Competing interests: None declared
An erratum to this chapter is available at 10.1007/8904_2014_362
Contributor Information
Maines Evelina, Email: evelina.maines@gmail.com.
Collaborators: Johannes Zschocke
References
- Ahmed SF, Sävendahl L. Promoting growth in chronic inflammatory disease: lessons from studies of the growth plate. Horm Res. 2009;72(Suppl 1):42–47. doi: 10.1159/000229763. [DOI] [PubMed] [Google Scholar]
- Alba-Roth J, Muller OA, Schopohl J, et al. Arginine stimulates growth hormone secretion by suppressing endogenous somatostatin secretion. J Clin Endocrinol Metab. 1988;67:1186–1189. doi: 10.1210/jcem-67-6-1186. [DOI] [PubMed] [Google Scholar]
- Awrich AE, Stackhouse WJ, Cantrell JE, et al. Hyperdibasicaminoaciduria, hyperammonemia, and growth retardation: treatment with arginine, lysine and citrulline. J Pediatr. 1975;87:731–738. doi: 10.1016/S0022-3476(75)80296-4. [DOI] [PubMed] [Google Scholar]
- Borsani G, Bassi MT, Sperandeo MP, et al. SLC7A7, encoding a putative permease-related protein, is mutated in patients with lysinuric protein intolerance. Nat Genet. 1999;21:297–301. doi: 10.1038/6815. [DOI] [PubMed] [Google Scholar]
- Bröer S. Lysinuric protein intolerance: one gene, many problems. Am J Physiol Cell Physiol. 2007;293:C540–C541. doi: 10.1152/ajpcell.00166.2007. [DOI] [PubMed] [Google Scholar]
- Brosnan ME, Brosnan JT. Orotic acid excretion and arginine metabolism. J Nutr. 2007;137:1656–1661. doi: 10.1093/jn/137.6.1656S. [DOI] [PubMed] [Google Scholar]
- Chevalley T, Rizzoli R, Manen D, et al. Arginine increases insulin-like growth factor-1 production and collagen synthesis in osteoblast-like cells. Bone. 1998;23:103–109. doi: 10.1016/S8756-3282(98)00081-7. [DOI] [PubMed] [Google Scholar]
- Chromiak JA, Antonio J. Use of amino acids as growth-hormone-releasing agents by athletes. Nutrition. 2002;18:657–661. doi: 10.1016/S0899-9007(02)00807-9. [DOI] [PubMed] [Google Scholar]
- De Benedetti F. The impact of chronic inflammation on the growing skeleton: lessons from interleukin-6 transenic mice. Horm Res. 2009;72(Suppl 1):26–29. doi: 10.1159/000229760. [DOI] [PubMed] [Google Scholar]
- Esposito V, Lettiero T, Fecarotta S, et al. Growth hormone deficiency in a patient with lysinuric protein intolerance. Eur J Pediatr. 2006;165:763–766. doi: 10.1007/s00431-006-0170-8. [DOI] [PubMed] [Google Scholar]
- Evain-Brion D, Donnadie M, Roger M, Job JC. Simultaneous study of somatotropic and corticotropic pituitary secretions during ornithine infusion test. Clin Endocrinol. 1982;17:119. doi: 10.1111/j.1365-2265.1982.tb01571.x. [DOI] [PubMed] [Google Scholar]
- Gasco V, Corneli G, Beccuti G, et al. Retesting the childhood-onset GH-deficient patient. Eur J Endocrinol. 2008;159(Suppl 1):S45–S52. doi: 10.1530/EJE-08-0293. [DOI] [PubMed] [Google Scholar]
- Goto I, Yoshimura T, Kuroiwa Y. Growth hormone studies in lysinuric protein intolerance. Eur J Pediatr. 1984;141:240–242. doi: 10.1007/BF00572769. [DOI] [PubMed] [Google Scholar]
- Hevrø´y EM, El-Mowafi A, Taylor RG et al (2007) Lysine intake affects gene expression of anabolic hormones in Atlantic salmon, Salmo salar. Gen Comp Endocrinol 152:39–46 [DOI] [PubMed]
- Hintz RL, Suskind R, Amatayakul K, et al. Plasma somatomedin and growth hormone values in children with protein-calorie malnutrition. J Pediatr. 1978;92:153–156. doi: 10.1016/S0022-3476(78)80099-7. [DOI] [PubMed] [Google Scholar]
- Ishida A, Kyoya T, Nakashima K, et al. Muscle protein metabolism during compensatory growth with changing dietary lysine levels from deficient to sufficient in growing rats. J Nutr Sci Vitaminol. 2011;57:401–408. doi: 10.3177/jnsv.57.401. [DOI] [PubMed] [Google Scholar]
- Isidori A, Lo Monaco A, Cappa M. A study of growth hormone release in man after oral administration of amino acids. Curr Med Res Opinion. 1981;7:475. doi: 10.1185/03007998109114287. [DOI] [PubMed] [Google Scholar]
- Jansen GR. Lysine in human nutrition. J Nutr. 1962;76:1–35. doi: 10.1093/jn/76.2_Suppl.1. [DOI] [PubMed] [Google Scholar]
- Knopf RF, Conn JW, Fajanss SS, Floyd JC et al (1965) Plasma growth hormone response to intravenous administration of amino acids. J Clin Endocrinol Metab 25:1140–1144 [DOI] [PubMed]
- MaCdonald A, van Rijn M, Feillet F, et al. Adherence issues in inherited metabolic disorders treated by low natural protein diets. Ann Nutr Metab. 2012;61:289–295. doi: 10.1159/000342256. [DOI] [PubMed] [Google Scholar]
- Mizutani N, Kato T, Maehara M, et al. Oral administration of arginine and citrulline in the treatment of Lysinuric protein intolerance. Tohoku J Exp Med. 1984;142:15–24. doi: 10.1620/tjem.142.15. [DOI] [PubMed] [Google Scholar]
- Naranjo WM, Yakar S, Sanchez-Gomez M, et al. Protein calorie restriction affects nonhepatic IGF-a production and the lymphoid system: studies using the liver-specific IGF-1 gene-deleted mouse model. Endocrinology. 2002;143:2233–2241. doi: 10.1210/endo.143.6.8852. [DOI] [PubMed] [Google Scholar]
- Niinikoski H, Lapatto R, Nuutinen M, et al. Growth hormone therapy is safe and effective in patients with lysinuric protein intolerance. JIMD Reports. 2011;1:43–47. doi: 10.1007/8904_2011_15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogier de Baulny H, Schiff M, et al. Lysinuric protein intolerance (LPI): a multi organ disease by far more complex than a classic urea cycle disorder. Mol Genet Metab. 2012;106:12–17. doi: 10.1016/j.ymgme.2012.02.010. [DOI] [PubMed] [Google Scholar]
- Palacin M, Bertran J, Chillaron J, et al. Lysinuric protein intolerance: mechanisms of pathophysiology. Mol Gen Metab. 2004;81:S27–S37. doi: 10.1016/j.ymgme.2003.11.015. [DOI] [PubMed] [Google Scholar]
- Pozzo AM, Kemp SF. Growth and growth hormone treatment in children with chronic diseases. Endocrinol Metab Clin North Am. 2012;41:747–759. doi: 10.1016/j.ecl.2012.07.001. [DOI] [PubMed] [Google Scholar]
- Ranke MB, Lindberg A, Price DA, et al. Age at growth hormone therapy start and first-year responsiveness to growth hormone are major determinants of height outcome in idiopathic short stature. Horm Res. 2007;68:53–62. doi: 10.1159/000098707. [DOI] [PubMed] [Google Scholar]
- Savage MO. Insulin-like growth factors, nutrition and growth. World Rev Nutr Diet. 2013;106:52–59. doi: 10.1159/000342577. [DOI] [PubMed] [Google Scholar]
- Sebastio G, Nunes V (2006) Lysinuric protein intolerance. In: Pagon RA, Adam MP, Ardinger HH et al (eds) Gene reviews™ [Internet]. University of Washington, Seattle, 1993–2014 [PubMed]
- Sebastio G, Sperandeo M, Andria G. Lysinuric Protein Intolerance: reviewing concepts on a multisystem disease. Am J Med Genet Part C. 2011;157:54–62. doi: 10.1002/ajmg.c.30287. [DOI] [PubMed] [Google Scholar]
- Sperandeo MP, Anunziata P, Bozzato A, et al. Slc7a7 disruption causes fetal growth retardation by downregulating Igf1 in the mouse model of lysinuric protein intolerance. Am J Physiol Cell Physiol. 2007;293:C191–C198. doi: 10.1152/ajpcell.00583.2006. [DOI] [PubMed] [Google Scholar]
- Torrents D, Mykkanen J, Pineda M, et al. Identification of SLC7A7, encoding y + LAT-1, as the lysinuric protein intolerance gene. Nat Genet. 1999;21:293–296. doi: 10.1038/6809. [DOI] [PubMed] [Google Scholar]
- Trobec K, von Hachling S, Anker SD, et al. Growth hormone, insulin-like growth factor 1, and insulin signalling-a pharmacological target in body wasting and cachexia. J Cachexia Sarcopenia Muscle. 2011;2:191–200. doi: 10.1007/s13539-011-0043-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Vught AJ, Nieuwenhuizen AG, Brummer RJM, et al. Effects of oral ingestion of amino acids and proteins on the somatotropic axis. J Clin Endocrinol Metab. 2008;93:584–590. doi: 10.1210/jc.2007-1784. [DOI] [PubMed] [Google Scholar]
- Zadik Z, Sinai T, Zung A, Reifen R. Effect of nutrition on growth in short stature before and during growth-hormone therapy. Pediatrics. 2005;116:68–72. doi: 10.1542/peds.2004-1129. [DOI] [PubMed] [Google Scholar]
- Zamboni G, Dufillot D, Antoniazzi F, et al. Growth hormone-binding proteins and insulin-like growth factor-binding proteins in protein-energy malnutrition, before and after nutritional rehabilitation. Pediatr Res. 1996;39:410–414. doi: 10.1203/00006450-199603000-00006. [DOI] [PubMed] [Google Scholar]