

Abstract
Background: Mucopolysaccharidosis type II (MPSII), also known as Hunter syndrome, is an X-linked disorder caused by mutations in the iduronate 2 sulfatase (IDS) gene. This enzyme catalyzes the initial step in the catabolism of heparan sulfate and dermatan sulfate; thus, its deficiency leads to the accumulation of these glycosaminoglycans. MPS II has significant allelic heterogeneity, making the establishment of genotype-phenotype correlations difficult. This study assessed clinical features in combination with deep genotyping of a group of Colombian patients with MPS II and attempted to establish a degree of genotype-phenotype correlation by employing bioinformatic tools.
Methods: Eighteen patients were included in this study, 11% of whom were non-neuronopathic, and the other 89% were neuronopathic. Samples were all analyzed using three molecular methodologies: MLPA, direct exon sequencing, and RFLP analysis.
Results: A total of 13 mutations were identified, 6 of which were novel (c.548_564dup16, c.477insT, c.595_607del12, c. 549_562del13, c.182delC, and a complete deletion of exon 7). The frequency of common mutations (R468Q, Q465X, K347Q, K236N, S71N, R88H, and a conversion phenomenon) was 53.85%. The S71N mutation was frequent among the attenuated phenotype, while private frameshift mutations and rearrangements were seen in patients with severe phenotypes. Molecular docking was performed on the wild-type and mutant IDS proteins, which revealed changes in the enzyme-substrate interaction for the mutant IDS.
Conclusion: The frequency of novel mutations (46.15%) is similar to what has been reported elsewhere. The use of bioinformatic tools showed differences in enzyme-substrate interactions. Studies with larger groups of patients are needed.
Electronic supplementary material
The online version of this chapter (doi:10.1007/8904_2014_376) contains supplementary material, which is available to authorized users.
Keywords: Bioinformatic analysis, Colombian patients, Iduronate sulfatase, Molecular docking, Mucopolysaccharidosis II
Introduction
Mucopolysaccharidosis type II (OMIM 309900), or Hunter syndrome, is the only MPS with a recessive X-linked inheritance pattern (Yatziv et al. 1977). The primary defect in this condition is the absence or defective activity of the lysosomal iduronate 2 sulfatase enzyme (IDS; EC 3.1.6.13) due to mutations in the IDS gene.
The severity with which this syndrome presents ranges from a neuronopathic (Hunter A) to a non-neuronopathic phenotype (Hunter B) (Neufeld and Muenzer 2001; Young et al. 1982; Martin et al. 2008). The neuronopathic form has an earlier onset and severe systemic and neurological involvement (Yatziv et al. 1977; Jones et al. 2009). The non-neuronopathic phenotype has minimal or no neurological deterioration, although patients manifest all the systemic complications (Young et al. 1982).
The reported incidence of MPSII is 1 in 170,000 male births (Martin et al. 2008; Nelson 1997; Poorthuis et al. 1999; Malm et al. 2008). In Japan, the incidence is estimated to be 1 in 90,000 (Ochiai et al. 2007). There are no studies regarding the incidence and prevalence of MPS II in Colombia; however, the combined frequency of all MPS cases calculated in the departments of Cundinamarca and Boyacá was 1.98 in 100,000 live births, and the estimated frequency of MPSII was 0.45 in 100,000 live births (Gómez et al. 2012).
The IDS gene is located at Xq27.3-q28, spans 24 Kb, and is composed of nine exons (Wilson et al. 1990). To date, over 350 mutations in this gene have been described (The Human Mutation Gene Database, http://www.hgmd.cf.ac.uk/ac/index.php), and phenotype-genotype correlations are difficult due to this abundant allelic heterogeneity (Rathmann et al. 1996; Vafiadaki et al. 1998; Lualdi et al. 2005; Lagerstedt et al. 2000; Froissart et al. 2007). A pseudogene is located approximately 25 Kb telomeric to the functional gene, with homologous regions to IDS exons 2 and 3 and intron 2 and with 96% homology with intron 7, explaining the susceptibility to complex recombination events (Rathmann et al. 1995; Bondeson et al. 1995; Bunge et al. 1998; Lualdi et al. 2005).
IDS is a 550-amino acid, monomeric lysosomal enzyme (Wilson et al. 1990) and is part of the sulfatase superfamily, with domains highly conserved (Diez-Roux and Ballabio 2005). It has been hypothesized that Asp45, Asp46, Cys84, Asp334, and His335 comprise the active site of the enzyme, based on homology with the active sites of arylsulfatases A and B (Dierks et al. 1999; Waldow et al. 1999). The Cys84 residue is conserved among all eukaryotic sulfatases (Dierks et al. 1999). Because complete characterization of this enzyme is ongoing and because its structure has not yet been elucidated by X-ray crystallography, most of the information on its structure and function has been inferred from homology with other better characterized sulfatases (Miech et al. 1998).
This study focuses on the clinical characterization and mutational profiling of Colombian patients with MPS II. This is achieved by deep genotyping of the IDS gene and assessment of the consequences in an IDS 3D model using a bioinformatic approach.
Methods
Phenotypic Analysis
This is a descriptive study without a hypothesis to prove. Diagnoses of MPS II were established clinically and confirmed by measurement of IDS activity. Eighteen patients (representing 15 families) were assessed by pediatric neurologists and clinical geneticists. The clinical assessment was performed following the criteria developed by Holt et al. for establishing the phenotype based on a severity score (Holt et al. 2011). This score takes into account 7 early clinical markers: sleep disturbance, increased activity, behavior difficulties, seizure-like behavior, perseverative chewing, and inability to achieve bowel training and bladder training. For each of these markers, a patient is assigned a 0 (zero) for “never” or a 1 for “ever.” Patients with a total score ≥3 have a high likelihood of developing CNS disease (Holt et al. 2011). The main phenotypic variables are described; the frequencies and percentages among patients are estimated for discrete variables. For continuous variables, the same procedure was performed and expressed in terms of central tendency and dispersion measurements.
A Kolmogorov-Smirnov nonparametric test was performed on a single sample to assess whether enzymatic activity values were normally distributed (Ho). The significance level required to reject Ho was 0.05 (IC 95). Once this was completed, a chi-squared (χ2) test with 9 degrees of freedom was performed to assess variation among the obtained data, with Ho being the existence of statistically significant differences when p < 0.05. These calculations were performed using the IBM SPSS free trial software v.21 (Chicago IL, USA).
Genotyping
Peripheral blood samples were obtained after written, informed consent was given. Genomic DNA extraction from 300 μL of blood was performed using the UltraClean Blood DNA Isolation Kit® (MOBIO Laboratories, USA), following the manufacturer’s instructions. Obtained DNA was quantified by spectrophotometry using a Nanodrop2000®, a Thermo Scientific® instrument. For genotyping, the following three steps were performed:
-
Step 1. Detection of large deletions and duplications using MLPA
MLPA (multiplex ligation-dependent probe amplification) (Schouten et al. 2002) was performed on all subjects, because it has been reported from several patient series that up to 20% of patients will have complex rearrangements (Froissart et al. 2007). SALSA® MLPA® P164 IDS (MRC-Holland) was used, and the protocol was followed according to the manufacturer’s specifications. Briefly, a minimum of 5 patients and 3 control DNA samples were used in each experiment, with the same amount of DNA (10 ng) per reaction. Capillary electrophoresis was performed using an ABI Prism 310 Genetic Analyzer. Genemapper® software was used to process raw data. Analysis of fragments was performed using the Coffalyser version 8 software.
-
Step 2. Sequencing
Direct sequencing of the 9 IDS exons was performed using primers modified from the initial reports by Froissart et al. (1998) and Lin et al. (2006) (Alves et al. 2006; Lau and Lam 2008) to amplify gene segments and intron-exon boundaries (Table 1, supplementary material). We did not analyze 5′UTR-3′UTR segments or epigenetic alterations. Conventional PCR and sequencing details of this work are available in Box 1 in supplementary material. Reaction products were analyzed on an ABI Prism 3500 sequencer (Applied Biosystems), and the obtained sequences were carefully revised using BioEdit® Sequence Alignment Editor. The BLASTn tool was employed for the identification of mutations, and the amino acid sequences were obtained using the Translate Tool from Proteomics Tools (ExPASy).
-
Step 3. Recombinant analysis by RFLPs
For subjects whose MLPA and sequencing results were negative, a rapid PCR and RFLP method for detection of the 4 most common types of IDS/IDSP1 rearrangements, as described by Lualdi and collaborators, was used (Lualdi et al. 2005). This method consists of two PCRs separated by digestion with restriction enzymes (HinfI, SacI, and Eco57I-AcuI) which recognize the breakage sites where these recombinations occur.
Table 1.
Phenotypic characteristics of the study sample
| Patient | Age (years) | Family history of MPSII | Age at diagnosis | IDS activitya | Initial symptom | Severity score (Holt et al. 2011) | PHENOTYPE |
|---|---|---|---|---|---|---|---|
| MPSII001 | 5 | No | 1 | 0.71 | Speech delay | 4 | Neuronopathic |
| MPSII002 | 18 | No | 4 | 0.24 | Speech delay | 7 | Neuronopathic |
| MPSII003 | 8 | No | 5 | 0.32 | Macrocephaly/speech delay | 5 | Neuronopathic |
| MPSII004 | 14 | Maternal uncle | 8 | 0.19 | Speech delay/coarse features | 5 | Neuronopathic |
| MPSII005 | 2 | No | 2 | 1.10 | Speech delay | 3 | Suggestive of. Neuronopathic |
| MPSII006 | 17 | Maternal uncle, cousins | 10 | 0.40 | Speech delay | 7 | Neuronopathic |
| MPSII007 | 6 | No | 5 | 0.00 | Pneumonia/visceromegaly/umbilical hernia | 0 | Non Neuronopathic |
| MPSII008 | 10 | Brother | 6 | 1.6 *(dried blood spot) | Speech regression/coarse features | 5 | Neuronopathic |
| MPSII009 | 14 | No | 8 | 1.8b | Speech delay | 5 | Neuronopathic |
| MPSII010 | 13 | No | 3 | 0.47 | Pneumonia/joint stiffness | 5 | Neuronopathic |
| MPSII011 | 13 | Maternal Uncle | 6 | 0.85 | Speech delay/joint stiffness. | 3 | Neuronopathic |
| MPSII012 | 8 | Maternal uncle, cousin | 2 | 26** (dried blood spot) | Macrocephaly/speech delay | 3 | Neuronopathic |
| MPSII013 | 7 | No | 3 | 1.34 | Hearing loss/joint stiffness | 3 | Neuronopathic |
| MPSII014 | 1 | No | 1 | 0.47 | Pneumonia | 3 | Suggestive of Neuronopathic |
| MPSII015 | 31 | Nephew | 9 | 9.46*** (dried blood spot) | Joint stiffness/umbilical hernia | 0 | Non Neuronopathic |
| MPSII016 | 16 | Uncle, cousins | 9 | UN | Speech delay | 6 | Neuronopathic |
| MPSII017 | 6 | Uncle, cousins | 2 | UN | Speech delay | 5 | Neuronopathic |
| MPSII018 | 16 | No | 10 | 22.27** (dried blood spot) | Speech delay/joint stiffness | 5 | Neuronopathic |
Reference values: a7.5–55.1 nmol/mg protein/h. b48–118 nmol/mg protein/h. IDS activity in dried blood spot (μmol/L h): *ref 12–23, **ref 57–149, ***ref 71–187
UN unavailable
Bioinformatic Analysis
Bioinformatic analysis of the effects of mutations on the IDS 3D protein structure was performed as described in Galvis et al. (2014). The first step was tridimensional modeling of the IDS protein. Measurement of the root mean square deviation (RMSD) was performed by superimposing the model and a template to evaluate the model’s precision; total RMSD values (the average distance between atoms in two overlapping proteins) of 1.0 or close to this value represent molecules with high structural similarity. Molecular docking of wild-type IDS was then performed, followed by modeling and docking for mutants, for which RMSD and ASA measurements were also calculated (Galvis et al. 2014).
Results
Clinical Characterization of Patients
A total of 18 Colombian patients were included in this study, and the average age of the patients was 10.8 years (SD ±7.8 years). Following the criteria of Holt et al. for determining the phenotype, a severity score was calculated for each patient (Table 1). Those patients with a score ≥3 were classified as neuronopathic, or as suggestive of a neuronopathic phenotype if they were younger than 3 years old. The severity score was 0 in only two patients, both of whom were above 5 years of age, which classified them as non-neuronopathic. All remaining patients had a score of 3 or higher.
It was found that neuronopathic Hunter syndrome is the most common, accounting for 89% of cases, while the non-neuronopathic type was seen in 11% of cases. The mean age of symptom onset for all patients was 2.5 years (SD ±1.55 years), but among those with the neuronopathic phenotype was 2.7 years (SD ±1.63 years).
The mean age at diagnosis was 6 years (SD ±4.38 years) for the whole group, 5 years (SD ±3.21) for the neuronopathic group, and 7 years (SD ± 2.13) for non-neuronopathic group. Initial symptoms commonly reported in the neuronopathic phenotype group were speech delay (81.2%) and joint rigidity (18.8%). For the non-neuronopathic group, initial manifestations included recurrent pneumonia, visceromegaly, and joint rigidity (Table 1).
Enzyme activity levels were analyzed by a nonparametric Kolmogorov-Smirnov test and Chi-squared (χ2) test. No statistically significant differences in enzyme activity were found between individuals for the whole group.
Genotyping of MPSII Patients
Using MLPA, it was possible to detect two types of mutations in two patients. In patient MPSII001, a gain in exon 4 and in the pseudogene was identified. Direct sequencing confirmed that this gain consisted of a 1 nucleotide insertion (Table 2). For patient MPSII012, an absence of signal for exon 7 was detected, indicating a complete deletion of said exon (Fig. 1). A search of the X chromosome gene database® and Human Gene Mutation Database® from Biobase confirmed that deletion of exon 7 is a novel mutation.
Table 2.
Genotypes of Colombian MPSII patients
| Code | Mutation (cDNA) | Mutation (protein) | Phenotype | Reporta |
|---|---|---|---|---|
| MPSII001 | c.477 insT | P160S fsX4 | N | This study |
| MPSII002 | c.548_564dupTTGCCCTGTGGATGTG | D190P fsX13 | N | This study |
| MPSII003 | c.1403G>A | R468Q | N | Whitley (1993); Villani (2000); Sukegawa-Hayasaka et al. (2006) |
| MPSII004 | c.263G>A | R88H | N | Rathmann et al. (1996) |
| MPSII005 | c.708G>C | K236N | Suggest. N | Gucev (2011) |
| MPSII006 | c.1393C>T | Q465X | N | Li (1996) |
| MPSII007 | c. 212G>A (polymorphism c.438 C>T) | S71N | No N | Froissart et al. (1998) |
| MPSII008 | c.1403G>A | R468Q | N | Whitley (1993), Villani (2000) |
| MPSII009 | c.1039A>C | K347Q | N | Lissens (1997) |
| MPSII010 | c.595_607delACAGAGCACTGA | Del.Q200_E2003 (QSTE) | N | This study |
| MPSII011 | c. 549_562delTGCCCTGTGGATG | P185WfsX23 | N | This study |
| MPSII012 | Deletion of exon 7 | R294GfsX2 | N | This study |
| MPSII013 | Conversion involving intron 3 | – | N | Lualdi et al. (2005) |
| MPSII014 | c.263G>A | R88H | Suggest. N | Rathmann et al. (1996) |
| MPSII015 | c. 212G>A (polymorphism c.438 C>T) | S71N | Non N | Froissart et al. (1998) |
| MPSII016 | c.1393C>T | Q465X | N | Li (1996) |
| MPSII017 | c.1393C>T | Q465X | N | Li (1996) |
| MPSII018 | c.182 delC | P62QfsX67 | N | This study |
aThese mutations were verified against the X Chromosome gene database ® and Human Gene Mutation Database ® from Biobase, confirming whether these were novel or previously reported mutations
Fig. 1.
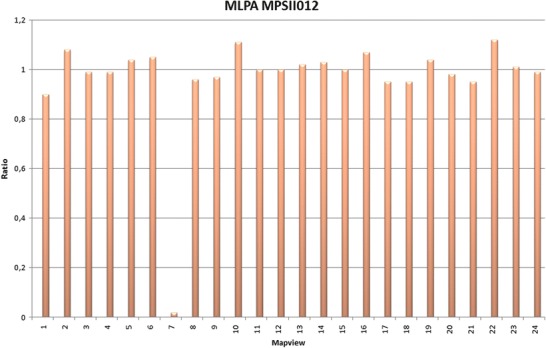
Complete deletion of exon 7 in patient MPSII012. The X-axis of this bar diagram displays specific fragments, and the Y-axis shows normalized ratios. The fragment for exon 7 (7) yielded no signal (0)
Direct sequencing of the nine gene segments and sequence comparison to the reference genome allowed identification of point mutations (Table 2).
Neither MLPA nor direct sequencing allowed for the establishment of a molecular diagnosis in patient MPSII013. For this patient, we used the PCR method described by Lualdi et al. (Lualdi et al. 2005). The product of a first PCR was digested with Hinfl and Sacl to identify recombination sites on intron 7, with negative results. Finally, an AcuI digestion of the second PCR product led to the identification of a conversion event in intron 3, where thymine 292879 is the breakage site and is replaced by a cytosine from the chimeric 3–7 introns (Fig. 2).
Fig. 2.
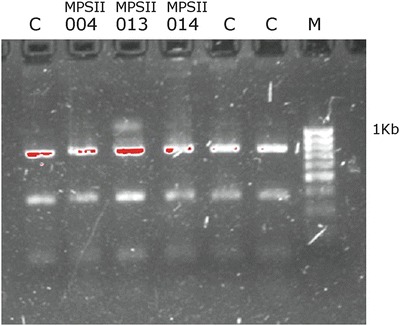
PCR 2 products after digestion with AcuI. C control. Note the 1,174 bp product on well 3, corresponding to MPSII013. M molecular weight
Employing these three molecular approaches, gross rearrangements were identified in 15.4% of patients, whereas small indels and point mutations were identified in 84.6% of patients. Novel mutations accounted for 46.15% of all the mutations found (Table 2).
Bioinformatic Results
All the results are shown in the supplementary material. The wild-type hIDS model superposed against its main template yielded an RMSD of 0.97 Å, indicating the model had high accuracy (Fig. 1, supplementary material). Wild-type hIDS docking interactions involved the O-2 sulfate of glucuronic acid (GlcA) with Cys84, Tyr165, Leu244, and Arg297 (Fig. 2, supplementary material) (Galvis et al. 2014). RMSD, ASA, and docking results for the IDS mutants are shown in the supplementary material (Tables 2 and 3 and Fig. 3) (Galvis et al. 2014).
Discussion
This is the first study in South America to perform a molecular analysis of MPS II with three available laboratory methodologies and in silico simulations with the identified mutations.
Clinical Characterization
Even though Holt et al. included only patients over 3 years of age in their work, the first five clinical markers are documented to have onset ages before even 2 years of age, and the first three have onset ages in the first months of life (Holt et al. 2011). We employed their methodology with all the patients of this study, but those younger than 2 years of age with scores ≥3 were classified as being “suggestive of neuronopathic phenotype.”
According to the literature, two thirds of patients have a neuronopathic phenotype (Wraith et al. 2008b), which is similar to the neuronopathic phenotype occurrence observed in our population, which was 89%. In a Brazilian study, initial symptoms for the severe phenotype were macrocephaly, speech delay, and behavior abnormalities, with symptom onset at 1.5 years (6–24 months) (Schwartz et al. 2007). The same age at onset is reported in HOS (Wraith et al. 2008a, b). For the neuronopathic phenotype, the initial symptoms more commonly reported in our study were speech delay (81%) and joint stiffness (18.8%), but age at symptom onset was 2.7 years on average, which differs from the reported result due to an atypical age at onset (5 years old) for patient 008, as the parents recalled.
Reported first manifestations of the non-neuronopathic phenotype were skeletal abnormalities, joint stiffness, and hearing loss (Schwartz et al. 2007). We observed recurrent pneumonia, umbilical hernia, visceromegaly, and joint stiffness (Table 1).
The Hunter Outcome Survey reported a median age at diagnosis of Hunter syndrome of 3.5 years (Wraith et al. 2008a). In a group of 77 patients in Brazil, a mean age at diagnosis of 6 years was found (Schwartz et al. 2007), which is more similar to our results (6 ± 4.38 years). This is most likely a consequence of a delay in recognition of early signs, and/or difficulties in accessing health services in some geographical, cultural, or social contexts.
In Latin America, patients with an attenuated phenotype are reported to be diagnosed at age 14 on average, while those with a severe phenotype are diagnosed at 7 years old (Schwartz et al. 2007). In our study, patients with the non-neuronopathic phenotype were diagnosed at 7 years of age and those with the neuronopathic phenotype (or with symptoms suggestive of it) at 5 years of age, on average.
IDS quantity and activity do not correlate with phenotype (Parkinson et al. 2004), and the enzyme is equally deficient in both forms of the disease (Scarpa et al. 2011). Here we found no differences in IDS activity among the whole group of patients.
Mutational Profile
Using MLPA, genotyping, and RFLPs, we were able to confirm the molecular diagnosis in 100% of patients in this study. In a series of 155 European patients, it was reported that up to 64% of patients have private mutations (Froissart et al. 2007). However, novel mutations in Colombian patients were detected with a frequency of 46.15%, similar to what was observed in 38 patients from China (Zhang et al. 2011).
The most commonly reported mutation (in 3.2–11% of patients) is R468Q (Isogai et al. 1998; Vafiadaki et al. 1998; Lin et al. 2006; Froissart et al. 2007; Zhang et al. 2011), which was found in 15.4% of our study population. R88H is also pan-ethnic (2–5.4% of patients) (Rathmann et al. 1996; Froissart et al. 2007; Zhang et al. 2011; Sohn et al. 2012), with a frequency in our study of 15.4%.
Two mutations were complex rearrangements: a conversion at intron 3 detected by RFLPs (Lualdi et al. 2005) and a deletion of exon 7 detected by MLPA, together accounting for 15.4% of all cases. This is similar to previously reported results, which have calculated that recombination phenomena account for 20% of cases (Steen-Bondeson et al. 1992; Bunge et al. 1998; Lualdi et al. 2005; Froissart et al. 2007).
A recent study of 103 unrelated South American patients reported that 29% of observed mutations were novel (Brusius-Facchin et al. 2014), which differs from the findings in our study. The same authors (Brusius-Facchin et al. 2014) found alterations such as inversions/disruptions and partial/total deletions of the IDS gene in 20/103 (19%) patients. Small (<22 bp) insertions/deletions/indels and point mutations were identified in 83/103 (88%) patients, which is very similar to our findings; in our Colombian group, we found small indels and point mutations in 84.6% of patients.
Bioinformatic Analysis and Effect of Mutations on the Tridimensional Structure of hIDS
Regarding mutant IDS, modeling revealed that even point mutations can alter the complete 3D structure, as reflected in the RMSD values (Galvis et al. 2014).
In 2005, Kato et al. performed tridimensional modeling of IDS and reported that the residues at the catalytic site are Asp45, Asn46, Cys84, Arg88, Lys135, His138, Asp334, His335, and Lys347 (Kato et al. 2005). By contrast, our docking simulation found that besides Cys84, the residues Arg297, Leu244, Tyr165, and Asn106 are part of the catalytic site as well (Galvis et al. 2014). It is likely that the cationic metal union required for the catalytic activity induces changes in the amino acids of the catalytic pocket, but these types of simulations are outside the scope of this study (Galvis et al. 2014).
In IDS mutants, docking simulations performed here showed differences with regard to interactions with substrates. Kato et al. hypothesized that a nonconservative mutation in R468Q should affect the electrostatic field for substrate entrance into the active site (Kato et al. 2005). Furthermore, Western blot analysis showed only primary precursors (Sukegawa-Hayasaka et al. 2006). In the mutation Q465X, an early stop codon causes the loss of the two potential glycosylation sites N513 and N537, with potential consequences for subcellular transport (Millat et al. 1997). For the S71N mutation, we propose that new hydrogen bonds seen within the catalytic site hinder the resolution of the ester-sulfate ester, but further experimental studies are needed. For example, mutation correction at a translational level has been found, especially in cases of attenuated presentation (Bunge et al. 1993; Lualdi et al. 2010).
The new mutations Del.Q200_E203 and R294GfsX2 (Del. exon 7) showed markedly distorted interactions within the pocket (Galvis et al. 2014).
Conclusions
This is the first study in South America which performs a molecular analysis of MPSII with three available laboratory methodologies and performs in silico simulations with the identified mutations. In our population, the observed age at diagnosis was similar to what has been reported in the literature. The genotyping study based on 18 samples achieved a sensitivity of 100% when employing three methodologies, but the result of the study will possibly be different if a larger number of samples are included. The frequency of novel mutations (46.15%) is similar to what has been reported elsewhere. The use of bioinformatic tools was of utility and molecular docking allowed for identification of changes at the level of the catalytic site of each mutant. However, given the small sample size, a phenotype-genotype correlation was not feasible; studies with larger groups of patients are needed.
Electronic Supplementary Material
Acknowledgments
To patients and their families, MRC-Holland for the donation of the MLPA kit for this research, ACOPEL, and Doctors Gabriel Sierra, Luz Norella Correa, Gustavo Contreras, Sandra Mansilla, and Juan Carlos Prieto. To professors Mauricio Rey, Rita Baldrich, Blanca Schroeder, Henry J Rodríguez, and other members of immunogenetics/biology group at Universidad Nacional de Colombia for their support relating to laboratory work. To Tatiana Vinasco, David Serrano, and Andres Gutierrez for contribution of literature and counseling.
One-Sentence Take-Home Message
Deep genotyping and bioinformatic approaches can lead to a better understanding of Hunter syndrome (MPSII).
Compliance with Ethics Guidelines
All procedures followed were in accordance with the ethical standards of the responsible committees on human experimentation (institutional and national) and with the Helsinki Declaration of 1975, as revised in 2000. Informed consent for study participation was obtained from all patients included in the study.
This study included 18 Colombian patients with a diagnosis of MPS II whose familial or legal guardians gave informed consent. The informed consent was previously evaluated and approved by the Ethics Committee of the School of Medicine of the National University of Colombia (Comité de Etica de la Facultad de Medicina de la Universidad Nacional de Colombia). By Act Number 16 of November 5, 2010, the Ethics Committee approved the study. After a careful review, the Committee determined that the project meets the ethical requirements and complies with Approbatory Concept. Signed: Prof. CARLOS ARTURO GUERRERO, President of the Ethics Committee.
Conflict of Interest
Johanna Galvis, Jannet González, Alfredo Uribe declare that they have no conflict of interest. Harvy Velasco declares that he received an educative grant from Shire.
Details of the Contribution of Individual Authors
Johanna Galvis: Planning of the work; patients’ recruitment; clinical data extraction and analysis; genotyping steps at laboratory; bioinformatic simulations; reporting of the results; writing and revision of manuscript. Guarantor 1
Jannet Gonzalez: Bioinformatic simulations. Revision of bioinformatic results
Alfredo Uribe: IDS Enzyme assays results, additional clinical data of patients
Harvy Velasco: Planning of the work; patients’ recruitment; clinical data extraction and analysis. Revision of manuscript. Guarantor 2
Footnotes
Competing interests: None declared
Contributor Information
Johanna Galvis, Email: djgalvisr@unal.edu.co, Email: juana7@gmail.com.
Collaborators: Johannes Zschocke
References
- Alves S, Mangas M, Prata MJ, et al. Molecular characterization of Portuguese patients with mucopolysaccharidosis type II shows evidence that the IDS gene is prone to splicing mutations. J Inherit Metab Dis. 2006;29:743–754. doi: 10.1007/s10545-006-0403-z. [DOI] [PubMed] [Google Scholar]
- Bondeson ML, Dahl N, Malmgren H, et al. Inversion of the IDS gene resulting from recombination with IDS-related sequences is a common cause of the Hunter syndrome. Hum Mol Genet. 1995;4:615–621. doi: 10.1093/hmg/4.4.615. [DOI] [PubMed] [Google Scholar]
- Brusius-Facchin AC, Schwartz IV, Zimmer C, et al. Mucopolysaccharidosis type II: identification of 30 novel mutations among Latin American patients. Mol Genet Metab. 2014;111:133–138. doi: 10.1016/j.ymgme.2013.08.011. [DOI] [PubMed] [Google Scholar]
- Bunge S, Steglich C, Zuther C, et al. Iduronate-2-sulfatase gene mutations in 16 patients with mucopolysaccharidosis type II (Hunter syndrome) Hum Mol Genet. 1993;2:1871–1875. doi: 10.1093/hmg/2.11.1871. [DOI] [PubMed] [Google Scholar]
- Bunge S, Rathmann M, Steglich C, et al. Homologous nonallelic recombinations between the iduronate-sulfatase gene and pseudogene cause various intragenic deletions and inversions in patients with mucopolysaccharidosis type II. Eur J Hum Genet. 1998;6:492–500. doi: 10.1038/sj.ejhg.5200213. [DOI] [PubMed] [Google Scholar]
- Dierks T, Lecca MR, Schlotterhose P, Schmidt B, von Figura K. Sequence determinants directing conversion of cysteine to formylglycine in eukaryotic sulfatases. EMBO J. 1999;18:2084–2091. doi: 10.1093/emboj/18.8.2084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diez-Roux G, Ballabio A. Sulfatases and human disease. Annu Rev Genomics Hum Genet. 2005;6:355–379. doi: 10.1146/annurev.genom.6.080604.162334. [DOI] [PubMed] [Google Scholar]
- Froissart R, Maire I, Millat G, et al. Identification of iduronate sulfatase gene alterations in 70 unrelated Hunter patients. Clin Genet. 1998;53:362–368. doi: 10.1111/j.1399-0004.1998.tb02746.x. [DOI] [PubMed] [Google Scholar]
- Froissart R, Da Silva IM, Maire I. Mucopolysaccharidosis type II: an update on mutation spectrum. Acta Paediatr Suppl. 2007;96:71–77. doi: 10.1111/j.1651-2227.2007.00213.x. [DOI] [PubMed] [Google Scholar]
- Galvis J, Gonzalez J, Torrente D, Velasco H, Barreto G. In silico analysis of iduronate 2 sulfatase mutations in Colombian patients with hunter syndrome (MPSII) in: advances in computational biology. Adv Intel Syst Comput. 2014;232(2014):205–212. doi: 10.1007/978-3-319-01568-2_30. [DOI] [Google Scholar]
- Gómez A, García-Robles R, Suárez-Obando F. Estimación de las frecuencias de las mucopolisacaridosis y análisis de agrupamiento espacial en los departamentos de Cundinamarca y Boyacá. Biomedica. 2012;32:602–609. doi: 10.7705/biomedica.v32i4.574. [DOI] [PubMed] [Google Scholar]
- Gucev ZS, Tasic V, Sinigerska I, Kremensky I et al (2011) Hunter syndrome (Mucopolysacharidosis Type II) in Macedonia and Bulgaria. Prilozi 32(2):187–198 [PubMed]
- Holt J, Poe MD, Escolar ML. Early clinical markers of central nervous system involvement in mucopolysaccharidosis type II. J Pediatr. 2011;159(320–326):e322. doi: 10.1016/j.jpeds.2011.03.019. [DOI] [PubMed] [Google Scholar]
- Isogai K, Sukegawa K, Tomatsu S, et al. Mutation analysis in the iduronate-2-sulphatase gene in 43 Japanese patients with mucopolysaccharidosis type II (Hunter disease) J Inherit Metab Dis. 1998;21:60–70. doi: 10.1023/A:1005363414792. [DOI] [PubMed] [Google Scholar]
- Jones SA, Almassy Z, Beck M, et al. Mortality and cause of death in mucopolysaccharidosis type II-a historical review based on data from the Hunter Outcome Survey (HOS) J Inherit Metab Dis. 2009;32:534–543. doi: 10.1007/s10545-009-1119-7. [DOI] [PubMed] [Google Scholar]
- Kato T, Kato Z, Kuratsubo I, et al. Mutational and structural analysis of Japanese patients with mucopolysaccharidosis type II. J Hum Genet. 2005;50:395–402. doi: 10.1007/s10038-005-0266-4. [DOI] [PubMed] [Google Scholar]
- Lagerstedt K, Carlberg BM, Karimi-Nejad R, Kleijer WJ, Bondeson ML. Analysis of a 43.6 kb deletion in a patient with Hunter syndrome (MPSII): identification of a fusion transcript including sequences from the gene W and the IDS gene. Hum Mutat. 2000;15:324–331. doi: 10.1002/(SICI)1098-1004(200004)15:4<324::AID-HUMU4>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- Lau C, Lam CW. Molecular investigations of a novel iduronate-2-sulfatase mutant in a Chinese patient. Clin Chim Acta. 2008;392:8–10. doi: 10.1016/j.cca.2008.02.009. [DOI] [PubMed] [Google Scholar]
- Li P, Thompson JN. Detection of four novel mutations in the iduronate-2-sulphatase gene by single-strand conformation polymorphism analysis of genomic amplicons. J Inher Metab Dis. 1996;19(1):93–94. doi: 10.1007/BF01799358. [DOI] [PubMed] [Google Scholar]
- Lin SP, Chang JH, Lee-Chen GJ, Lin DS, Lin HY, Chuang CK. Detection of Hunter syndrome (mucopolysaccharidosis type II) in Taiwanese: biochemical and linkage studies of the iduronate-2-sulfatase gene defects in MPS II patients and carriers. Clin Chim Acta. 2006;369:29–34. doi: 10.1016/j.cca.2006.01.001. [DOI] [PubMed] [Google Scholar]
- Lissens W, Seneca S, Liebaers I. Molecular analysis in 23 Hunter disease families. J Inher Metab Dis. 1997;20:453–456. doi: 10.1023/A:1005335624386. [DOI] [PubMed] [Google Scholar]
- Lualdi S, Regis S, Di Rocco M, et al. Characterization of iduronate-2-sulfatase gene-pseudogene recombinations in eight patients with Mucopolysaccharidosis type II revealed by a rapid PCR-based method. Hum Mutat. 2005;25:491–497. doi: 10.1002/humu.20165. [DOI] [PubMed] [Google Scholar]
- Lualdi S, Tappino B, Di Duca M, et al. Enigmatic in vivo iduronate-2-sulfatase (IDS) mutant transcript correction to wild-type in Hunter syndrome. Hum Mutat. 2010;31:E1261–E1285. doi: 10.1002/humu.21208. [DOI] [PubMed] [Google Scholar]
- Malm G, Lund AM, Mansson JE, Heiberg A. Mucopolysaccharidoses in the Scandinavian countries: incidence and prevalence. Acta Paediatr. 2008;97:1577–1581. doi: 10.1111/j.1651-2227.2008.00965.x. [DOI] [PubMed] [Google Scholar]
- Martin R, Beck M, Eng C, et al. Recognition and diagnosis of mucopolysaccharidosis II (Hunter syndrome) Pediatrics. 2008;121:e377–e386. doi: 10.1542/peds.2007-1350. [DOI] [PubMed] [Google Scholar]
- Miech C, Dierks T, Selmer T, von Figura K, Schmidt B. Arylsulfatase from Klebsiella pneumoniae carries a formylglycine generated from a serine. J Biol Chem. 1998;273:4835–4837. doi: 10.1074/jbc.273.9.4835. [DOI] [PubMed] [Google Scholar]
- Millat G, Froissart R, Maire I, Bozon D. Characterization of iduronate sulphatase mutants affecting N-glycosylation sites and the cysteine-84 residue. Biochem J. 1997;326(Pt 1):243–247. doi: 10.1042/bj3260243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson J. Incidence of the mucopolysaccharidoses in Northern Ireland. Hum Genet. 1997;101:355–358. doi: 10.1007/s004390050641. [DOI] [PubMed] [Google Scholar]
- Neufeld EF, Muenzer J. The mucopolysaccharidoses. In: Scriver CR, editor. The metabolic and molecular bases of inherited disease. New York, NY: McGraw-Hill; 2001. [Google Scholar]
- Ochiai T, Suzuki Y, Kato T, et al. Natural history of extensive Mongolian spots in mucopolysaccharidosis type II (Hunter syndrome): a survey among 52 Japanese patients. J Eur Acad Dermatol Venereol. 2007;21:1082–1085. doi: 10.1111/j.1468-3083.2007.02203.x. [DOI] [PubMed] [Google Scholar]
- Parkinson EJ, Muller V, Hopwood JJ, Brooks DA. Iduronate-2-sulphatase protein detection in plasma from mucopolysaccharidosis type II patients. Mol Genet Metab. 2004;81:58–64. doi: 10.1016/j.ymgme.2003.11.002. [DOI] [PubMed] [Google Scholar]
- Poorthuis BJ, Wevers RA, Kleijer WJ, et al. The frequency of lysosomal storage diseases in The Netherlands. Hum Genet. 1999;105:151–156. doi: 10.1007/s004399900075. [DOI] [PubMed] [Google Scholar]
- Rathmann M, Bunge S, Steglich C, Schwinger E, Gal A. Evidence for an iduronate-sulfatase pseudogene near the functional Hunter syndrome gene in Xq27.3-q28. Hum Genet. 1995;95:34–38. doi: 10.1007/BF00225070. [DOI] [PubMed] [Google Scholar]
- Rathmann M, Bunge S, Beck M, Kresse H, Tylki-Szymanska A, Gal A. Mucopolysaccharidosis type II (Hunter syndrome): mutation “hot spots” in the iduronate-2-sulfatase gene. Am J Hum Genet. 1996;59:1202–1209. [PMC free article] [PubMed] [Google Scholar]
- Scarpa M, Almassy Z, Beck M, et al. Mucopolysaccharidosis type II: European recommendations for the diagnosis and multidisciplinary management of a rare disease. Orphanet J Rare Dis. 2011;6:72. doi: 10.1186/1750-1172-6-72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schouten JP, McElgunn CJ, Waaijer R, Zwijnenburg D, Diepvens F, Pals G. Relative quantification of 40 nucleic acid sequences by multiplex ligation-dependent probe amplification. Nucleic Acids Res. 2002;30:e57. doi: 10.1093/nar/gnf056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz IV, Ribeiro MG, Mota JG, et al. A clinical study of 77 patients with mucopolysaccharidosis type II. Acta Paediatr Suppl. 2007;96:63–70. doi: 10.1111/j.1651-2227.2007.00212.x. [DOI] [PubMed] [Google Scholar]
- Sohn YB, Ki CS, Kim CH, et al. Identification of 11 novel mutations in 49 Korean patients with mucopolysaccharidosis type II. Clin Genet. 2012;81:185–190. doi: 10.1111/j.1399-0004.2011.01641.x. [DOI] [PubMed] [Google Scholar]
- Steen-Bondeson ML, Dahl N, Tonnesen T, et al. Molecular analysis of patients with Hunter syndrome: implication of a region prone to structural alterations within the IDS gene. Hum Mol Genet. 1992;1:195–198. doi: 10.1093/hmg/1.3.195. [DOI] [PubMed] [Google Scholar]
- Sukegawa-Hayasaka K, Kato Z, Nakamura H, et al. Effect of Hunter disease (mucopolysaccharidosis type II) mutations on molecular phenotypes of iduronate-2-sulfatase: enzymatic activity, protein processing and structural analysis. J Inherit Metab Dis. 2006;29:755–761. doi: 10.1007/s10545-006-0440-7. [DOI] [PubMed] [Google Scholar]
- Vafiadaki E, Cooper A, Heptinstall LE, Hatton CE, Thornley M, Wraith JE. Mutation analysis in 57 unrelated patients with MPS II (Hunter’s disease) Arch Dis Child. 1998;79:237–241. doi: 10.1136/adc.79.3.237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villani GR, Daniele A, Balzano N, Di Natale P (2000) Expression of five iduronate-2-sulfatase site-directed mutations. Biochim Biophys Acta 1501(2–3):71–80 [DOI] [PubMed]
- Waldow A, Schmidt B, Dierks T, von Bulow R, von Figura K. Amino acid residues forming the active site of arylsulfatase A. Role in catalytic activity and substrate binding. J Biol Chem. 1999;274:12284–12288. doi: 10.1074/jbc.274.18.12284. [DOI] [PubMed] [Google Scholar]
- Whitley CB, Anderson RA, Aronovich EL et al (1993) Caveat to genotype-phenotype correlation in mucopolysaccharidosis type II: discordant clinical severity of R468W and R468Q mutations of the iduronate-2-sulfatase gene. Hum Mutat 2(3):235–23 [DOI] [PubMed]
- Wilson PJ, Morris CP, Anson DS, et al. Hunter syndrome: isolation of an iduronate-2-sulfatase cDNA clone and analysis of patient DNA. Proc Natl Acad Sci U S A. 1990;87:8531–8535. doi: 10.1073/pnas.87.21.8531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wraith JE, Beck M, Giugliani R, Clarke J, Martin R, Muenzer J. Initial report from the hunter outcome survey. Genet Med. 2008;10:508–516. doi: 10.1097/GIM.0b013e31817701e6. [DOI] [PubMed] [Google Scholar]
- Wraith JE, Scarpa M, Bec M, et al. Mucopolysaccharidosis type II (Hunter syndrome): a clinical review and recommendations for treatment in the era of enzyme replacement therapy. Eur J Pediatr. 2008;167:267–277. doi: 10.1007/s00431-007-0635-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yatziv S, Erickson RP, Epstein CJ. Mild and severe Hunter syndrome (MPS II) within the same sibships. Clin Genet. 1977;11:319–326. doi: 10.1111/j.1399-0004.1977.tb01323.x. [DOI] [PubMed] [Google Scholar]
- Young ID, Harper PS, Newcombe RG, Archer IM. A clinical and genetic study of Hunter’s syndrome. 2. Differences between the mild and severe forms. J Med Genet. 1982;19:408–411. doi: 10.1136/jmg.19.6.408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Li J, Zhang X et al (2011) Analysis of the IDS gene in 38 patients with Hunter syndrome: the c.879G > A (p.Gln293Gln) synonymous variation in a female create exonic splicing. PLoS One 6:e22951 [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.