

Abstract
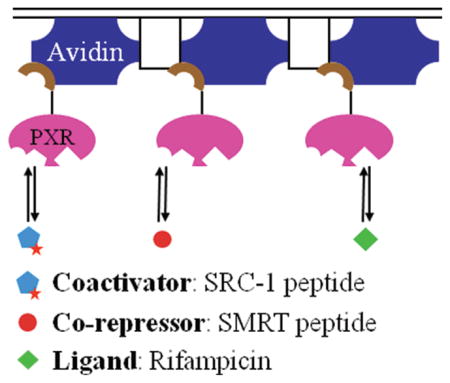
The pregnane X receptor (PXR), a member of the nuclear receptor superfamily, regulates the expression of drug-metabolizing enzymes in a ligand-dependent manner. The conventional view of nuclear receptor action is that ligand binding enhances the receptor’s affinity for coactivator proteins, while decreasing its affinity for corepressors. To date, however, no known rigorous biophysical studies have been conducted to investigate the interaction among PXR, its coregulators, and ligands. In this work, steady-state total internal reflection fluorescence microscopy (TIRFM) and total internal reflection with fluorescence recovery after photobleaching were used to measure the thermodynamics and kinetics of the interaction between the PXR ligand binding domain and a peptide fragment of the steroid receptor coactivator-1 (SRC-1) in the presence and absence of the established PXR agonist, rifampicin. Equilibrium dissociation and dissociation rate constants of ~5 μM and ~2 s−1, respectively, were obtained in the presence and absence of rifampicin, indicating that the ligand does not enhance the affinity of the PXR and SRC-1 fragments. Additionally, TIRFM was used to examine the interaction between PXR and a peptide fragment of the corepressor protein, the silencing mediator for retinoid and thyroid receptors (SMRT). An equilibrium dissociation constant of ~70 μM was obtained for SMRT in the presence and absence of rifampicin. These results strongly suggest that the mechanism of ligand-dependent activation in PXR differs significantly from that seen in many other nuclear receptors.
The nuclear receptor superfamily consists of structurally related proteins that regulate the transcription of target genes in a ligand-dependent manner. Nuclear receptors, which include the estrogen, androgen, thyroid, and vitamin D receptors, regulate a variety of biological processes, including reproduction, development, metabolism, and energy homeostasis, in response to various hydrophobic ligands. The pregnane X receptor (PXR), a member of the nuclear receptor superfamily, protects the body from potentially toxic compounds by regulating the expression of proteins that metabolize and excrete these compounds from cells.1
PXR binds promiscuously to a wide variety of compounds, including naturally occurring steroids, hormones, and bile acids, as well as exogenous ligands like insecticides, herbal extracts, and pharmaceutical products.1 PXR has been implicated in adverse drug–drug interactions, whereupon being activated by a drug, PXR promotes the expression of enzymes that metabolize the activating drug, as well as other co-administered therapeutics. Such adverse effects have been observed with hyperforin, a constituent in the herbal product St. John’s Wort, and rifampicin, an antibiotic, both of which have been shown to bind and activate PXR.2–7
PXR works in concert with its heterodimerization partner, the retinoid X receptor (RXR), to bind promoter regions of target genes and coactivators like the steroid receptor coactivator-1 (SRC-1). The PXR–RXR–coactivator–DNA complex then recruits and directs downstream members of the transcription machinery.5,6 Crystal structures of several nuclear receptor ligand binding domains (LBDs) in the apo and ligand-bound states indicate that in the presence of an agonist, an α-helix at the C-terminus called activation function-2 (AF-2) undergoes a conformational change that allows nuclear receptors to bind coactivators.8–11 Specifically, in the active conformation, nuclear receptors form a critical charge clamp with conserved LXXLL motifs (where X is any amino acid) found in coactivators.11,12
In the absence of ligand, the transcriptional activity of nuclear receptors is, in part, downregulated by the action of corepressor proteins. The nuclear receptor corepressor known as the silencing mediator of retinoid and thyroid receptors (SMRT) has been shown to repress both the basal and ligand-induced transcriptional activity of PXR.13–15 Corepressors bind nuclear receptors via receptor interaction domains containing conserved LXXXIXXXL motifs and recruit proteins, including histone deacetylases, that suppress transcription. A crystal structure of a nuclear receptor (peroxisome proliferator-activated receptor-α) LBD in complex with a peptide fragment of SMRT shows that AF-2 is displaced from its active conformation upon corepressor binding.16 Corepressors thereby inhibit the transcriptional activity of nuclear receptors by preventing the recruitment of coactivators.
This paper provides a biophysical assessment of the interaction between PXR-LBD and peptides derived from the coactivator, SRC-1, and the corepressor, SMRT, in the presence and absence of the well-established PXR agonist, rifampicin. For the first time, we report equilibrium constants for PXR-LBD–coregulator interactions and dissociation rate constants for the PXR-LBD–SRC-1 interaction measured by steady-state total internal reflection fluorescence microscopy (TIRFM)17–20 and total internal reflection with fluorescence recovery after photobleaching (TIR-FRAP),21–23 respectively. This quantitative information strongly suggests that the mode of ligand-dependent activation of PXR differs from that of most other nuclear receptors studied to date.
MATERIALS AND METHODS
PXR-LBD Cloning, Expression, Purification, and Labeling
The LBD of human PXR (residues 130–434) was expressed as a fusion protein with an amino-terminal AviTag and His6 tag (for purification). The AviTag allows for the specific biotinylation of the tagged protein by the Escherichia coli enzyme BirA. A codon-optimized version of the PXR-LBD gene (GenScript USA, Piscataway, NJ) was cloned into pET21c(+) between NdeI and HindIII, along with an N-terminal AviTag sequence (Avidity, Denver, CO) followed by a His6 sequence. With this insert, two stop codons were introduced upstream of the C-terminal His6 tag normally found in pET21c(+). PXR-LBD was coexpressed with an 88-amino acid fragment of SRC-1 to enhance PXR stability. The SRC-1 fragment (residues 623–710), along with the T7 promoter, had been previously inserted into the pACYC184 vector at the HindIII and BamHI sites.24,25 The pET21c-AviTag-His6-PXR-LBD and pACYC184-SRC-1 plasmids were cotransformed into the BL21 DE3 Gold E. coli strain.
Terrific Broth (3 L) supplemented with ampicillin (100 μg/mL) and chloramphenicol (35 μg/mL) was inoculated with an overnight culture (0.5% innoculant). Cells were grown at 37 °C to an OD600 of ~2.7 and induced with 0.1 mM isopropyl β-D-thiogalactopyranoside at 18 °C for ~16 h. Cells were harvested by centrifugation (30 min at 3500g and 4 °C) and stored at −80 °C. Cell pellets (~25 g) were resuspended in 125 mL of buffer A [20 mM Tris (pH 7.9), 250 mM NaCl, 5% glycerol, 20 mM imidazole, and 0.1 mM tris(2-carboxyethyl)phosphine (TCEP) (Soltec Ventures, Beverly, MA)] supplemented with three protease inhibitor tablets (complete, EDTA-free; Roche Diagnostics, Mannheim, Germany) and DNase (50 μg/mL; Worthington Biochemical Corp., Lakewood, NJ) and then subjected to Dounce homogenization. Cell homogenates were tip sonicated on ice, and the cell lysates were then clarified by centrifugation (45 min at 27000g and 4 °C). The clarified cell lysates were incubated with 750 μL of His-Select Ni resin (Sigma-Aldrich, St. Louis, MO) equilibrated in buffer A for 1 h at 4 °C. The resin was subsequently washed with 125 mL of buffer B [20 mM Tris (pH 7.9), 500 mM NaCl, 5% glycerol, and 20 mM imidazole]. PXR-LBD was eluted using six aliquots of 1 mL each of buffer C [20 mM Tris (pH 7.9), 250 mM NaCl, 5% glycerol, 2 mM TCEP, and 300 mM imidazole]. Protein fractions were combined and dialyzed against buffer D [20 mM Tris (pH 7.9), 250 mM NaCl, 5% glycerol, and 2 mM TCEP] at 4 °C. Thereafter, PXR-LBD was quickly frozen in liquid N2 and stored at −80 °C in 1 mL aliquots at a concentration of 0.1 mg/mL.
The AviTag-His6-PXR-LBD fusion protein, henceforth termed PXR-LBD, has a molecular mass of 38750 Da with an extinction coefficient at 280 nm of 34080 M−1 cm−1.26 Protein concentrations were measured by using both the absorbance at 280 nm and the Bradford assay (Bio-Rad Laboratories, Hercules, CA). Sodium dodecyl sulfate–polyacrylamide gel electrophoresis with Coomassie Brilliant Blue and silver stains indicated that the primary band was at the molecular mass expected for PXR-LBD and impurities were negligible. Silver-stained gels showed no bands close to or at 10 kDa, suggesting that the coexpressed, 9.8 kDa SRC-1 fragment was removed during PXR purification. Western blots with anti-biotin antibodies conjugated to horseradish peroxidase (Cell Signaling Technology, Beverly, MA) were used to confirm in vivo biotinylation. The molar ratio of biotin to PXR-LBD, as estimated by using Pierce Biotin Quantitation Kit (Thermo Scientific, Rockford, IL), ranged from 0.7 to 1.3, indicating that a majority of the protein was biotinylated.
For some control measurements, PXR-LBD was fluorescently labeled with an amine reactive dye, Alexa Fluor 488 5-TFP (Invitrogen, Carlsbad, CA). Briefly, PXR-LBD that eluted from the Ni affinity column during purification was concentrated to ~1.5 mg/mL and incubated with a 10–15-fold molar excess of the dye for 2 h at 4 °C with continuous, gentle stirring. Free dye was removed by passing the solution through an anion exchange column (4 mL) constructed from Dowex 1X8 resin (Acros Organics, Morris Plains, NJ) and equilibrated in 10 mM sodium phosphate and 5% glycerol (pH 5.3). Protein was immediately dialyzed against buffer D. The molar ratio of dye to protein, as determined by the relative absorptivities at 280 and 494 nm, was approximately 0.3. As before, protein aliquots were frozen in liquid N2 and stored at −80 °C.
A ligand-bound mimic of PXR-LBD was made by mutating two residues in the ligand binding pocket to tryptophans: S247W and C284W.27 The mutations were made sequentially using the QuikChange-II Site-Directed Mutagenesis Kit (Stratagene, La Jolla, CA) according to the manufacturer’s protocol. The following primers (with mutated nucleotides underlined) were used for the S247W mutation: forward, 5′-gcacatggcagatatgTGGacctatatgttcaaaggc-3′; and reverse, 5′-gcctttgaacatataggtCCAcatatctgccatgtgc-3′. For the C284W mutation, the following primers were used: forward, 5′-gcagcgtttgaactgTTGcagctgcgtttcaac-3′; and reverse, 5′-gttgaaacgcagctgCCAcagttcaaacgctgc-3′. Mutations were generated using the pET21c-AviTag-His6-PXR-LBD plasmid as a template and confirmed by sequencing. PXR-LBD (S247W/C284W) was expressed and purified as described above for wild-type (WT) PXR-LBD.
Coregulator Peptide Synthesis and Fluorescence Labeling
A 25-amino acid fragment of SRC-1 (676-CPSSWSSLTERHKILHRLLQEGSPS-700) was synthesized at the University of North Carolina Microprotein Sequencing and Peptide Synthesis Facility. Residue 680 was mutated from histidine to tryptophan (H680W) to facilitate peptide quantification. The average and monoisotopic molecular masses of the peptide are 2849.18 and 2847.44 Da, respectively. The molar absorptivity of the peptide at 280 nm is 5810 M−1 cm−1. The N-terminal cysteine residue was fluorescently labeled with a thiol reactive dye, fluorescein C5 maleimide (AnaSpec, Fremont, CA). Briefly, peptide (5 mg, 1.8 μM), a 2-fold molar excess of TCEP, and a 4-fold molar excess of dye were combined in 50 mM sodium phosphate (pH 7.6). The reaction mixture was stirred under argon for ~5 h at room temperature and for an additional ~19 h at 4 °C. Free dye was removed by passing the mixture through an anion exchange column (4 mL) constructed from Dowex 1X8 resin, both equilibrated and washed with 50 mM sodium acetate (pH 5.0). The peptide was further purified using high-performance liquid chromatography (HPLC) to remove salts. Briefly, a 0 to 40% gradient of solvent B (95% acetonitrile, 5% water, and 0.1% trifluoroacetic acid) in solvent A (95% water, 5% acetonitrile, and 0.1% trifluoroacetic acid) was generated on an Atlantis dC18 (10 mm × 100 mm) column and Waters HPLC with Delta 600 pumps (Waters Corp., Milford, MA) at a flow rate of 4 mL/min. Peptide fractions were combined, lyophilized, and stored at −20 °C. Peptide labeling was confirmed by mass spectrometry. The molar ratio of fluorescein to peptide was determined by using the absorptivities at 280 and 494 nm. Labeling ratios ranged from 0.35 to 0.50. The fluorescently labeled SRC-1 peptide is henceforth denoted as F-SRC-1. Labeled and unlabeled peptides were mixed to yield an overall F-SRC-1 labeling ratio of 0.10, unless otherwise indicated.
Peptide fragments of SMRT (2337-TNMGLEAIIRKALMGKYDQWEE-2358) and of a second corepressor protein, called the nuclear receptor corepressor (NCoR, 2251-GHSFADPASNLGLEDIIRKALMGSF-2275), were synthesized at the University of North Carolina Microprotein Sequencing and Peptide Synthesis Facility. The concentration of SMRT was determined spectrophotometrically using the peptide’s extinction coefficient at 280 nm, 6970 M−1 cm−1. The concentration of NCoR was determined by measuring the mass of the lyophilized peptide.
Other Reagents
D-Biotin (Acros Organics, Morris Plains, NJ), unreactive fluorescein reference standard (Invitrogen, Eugene, OR), NeutrAvidin (Thermo Scientific, Rockford, IL), ovalbumin (Sigma-Aldrich), and rifampicin (Fisher Scientific, Fairlawn, NJ) were obtained commercially. Concentrations for the last four reagents were determined spectrophotometrically by using the following extinction coefficients: fluorescein, 68000 M−1 cm−1 at 494 nm; NeutrAvidin, 99600 M−1 cm−1 at 280 nm; ovalbumin, 31500 M−1 cm−1 at 280 nm; and rifampicin, 26400 M−1 cm−1 at 334 nm. For some control measurements, NeutrAvidin was fluorescently labeled with Alexa Fluor 488 5-TFP (Invitrogen). NeutrAvidin (2 mg/mL) and a 5-fold molar excess of dye were dissolved in 100 mM sodium phosphate (pH 8.0). The mixture was incubated at 25 °C for 1 h with continuous, gentle stirring. Thereafter, free dye was removed by passing the mixture through an anion exchange column (4 mL) constructed from Dowex 1X8 resin and equilibrated in 100 mM sodium acetate with 500 mM NaCl (pH 4.0). Labeled protein was dialyzed against buffer D. The molar ratio of dye to protein, determined spectrophotometrically, was approximately 0.7.
Sample Preparation
Immediately before use, all protein samples were centrifuged (~100000g for 30 min; Airfuge; Beckman-Coulter, Fullerton, CA) to remove possible aggregates. NeutrAvidin and ovalbumin were filtered (0.1 μm, 13 mm, Anatop; Whatman, GE Healthcare, Piscataway, NJ) following centrifugation. Microscope (3 in. × 1 in. × 1 mm; Gold Seal Products, Porstmouth, NH) and fused silica (0.25 in. × 1 in. × 1 mm; Quartz Scientific, Fairport Harbor, OH) slides were cleaned by being boiled in ICN detergent (MP Biomedicals, Solon, OH) diluted in water, sonicated in a bath, rinsed extensively in deionized water, and dried at 120 °C. Immediately prior to the collection of data, the substrates were further cleaned in an argon ion plasma cleaner (PDC-3XG, Harrick Scientific, Ossining, NY) for 15 min at room temperature. Fused silica slides were mounted on microscope slides using double-sided, 0.13 mm thick, tape (part no. 021200-64988, 3M Corp., St. Paul, MN). A NeutrAvidin/ovalbumin mixture (0.5 and 0.1 mg/mL, respectively, in buffer D, 60 μL) was applied to the space between the fused silica and microscope slides. Slides were incubated at room temperature for 1 h, allowing the NeutrAvidin and ovalbumin to coat the surfaces of the substrates. Excess protein was removed by washing the inner sample chambers with buffer D (10 × 200 μL). NeutrAvidin- and ovalbumin-coated sample chambers were treated with PXR-LBD (0.1 mg/mL in buffer D, 200 μL) for 5 min at room temperature and washed with buffer D (10 × 200 μL) to remove excess PXR. For steady-state TIRFM and TIR-FRAP measurements to yield F-SRC-1–PXR-LBD equilibrium and dissociation rate constants, solutions (200 μL) containing F-SRC-1 and ligand in buffer D at the indicated concentrations were applied to the sample chambers. Immediately thereafter, samples were mounted onto the microscope for data collection. For competition curves to yield SMRT–PXR-LBD equilibrium constants, solutions (200 μL) containing 5 μM F-SRC-1, rifampicin, and SMRT in buffer D at the indicated concentrations were applied to the sample chambers. SMRT was replaced with a peptide fragment of the corepressor, NCoR, in some measurements.
Fluorescence Microscopy
Steady-state TIRFM and TIR-FRAP were conducted using the equipment described below. A through-prism TIRFM apparatus was used to generate evanescent illumination with an elliptically Gaussian spatial profile having the following 1/e2 radii: wx = 22.4 ± 0.5 μm, and wy = 65.0 ± 0.4 μm, respectively.28 Measurements were conducted using an argon ion laser (Innova 90-3, Coherent, Palo Alto, CA), an inverted microscope (Zeiss Axiovert 35, Carl Zeiss Inc., Thornwood, NY) with a 40×, 0.55 numerical aperture, long working distance objective (Nikon Instruments Inc., Nelville, NY), and an avalanche photodiode (SPCM-AQ-151, EG&G Optoelectronics, Quebec City, QC) detector. An in-house LabVIEW program and DAQ board (PCI-MIO-16XE-50, National Instruments, Austin, TX) were used to control the instruments. Fluorescence was excited at 488 nm and detected through a dichroic mirror and barrier filter, at room temperature. All data were fitted in SigmaPlot 11.0 (Systat Software Inc., San Jose, CA).
Steady-State Total Internal Reflection Fluorescence Microscopy
Steady-state TIRFM17–19 was used to measure equilibrium dissociation constants for the interaction between the F-SRC-1–PXR-LBD complex and the SMRT–PXR-LBD complex under different conditions, including different rifampicin concentrations. All samples were evanescently illuminated, and the surface-associated fluorescence was measured using a personal computer-based correlator board (ALV-5000/E, ALV, Langen, Germany). Fluorescence intensities, averaged over 10 s, were measured at eight distinct sites on a given sample.
To characterize the F-SRC-1–PXR-LBD interaction, the surface-associated fluorescence of samples was measured as a function of the F-SRC-1 concentration in solution. The measured surface-associated fluorescence was assumed to be proportional to the average density of fluorescent molecules bound to surface binding sites (PXR-LBD), as well as the average density of such molecules diffusing in the evanescent wave. To obtain the fluorescence associated with F-SRC-1–PXR-LBD complexes alone, fluorescence measured in the absence of PXR-LBD was subtracted from the total fluorescence. This background-corrected fluorescence was plotted as a function of the F-SRC-1 concentration and fit to a model of single-site binding to yield an apparent equilibrium dissociation constant.
Competition curves were used to determine the equilibrium dissociation constant for the interaction between PXR-LBD and corepressor peptides. TIRFM was used to measure the surface-associated fluorescence arising from 5 μM F-SRC-1 reversibly interacting with surface-immobilized PXR-LBD in the presence of an increasing concentration of the corepressor peptide. The corepressor competed with F-SRC-1 to bind PXR-LBD, and a corresponding decrease in the surface-associated fluorescence was observed. The background, arising from fluorescence due to the diffusion of free F-SRC-1 in the evanescent wave, was subtracted, and the data were fit to an appropriate model (see Results) to obtain the equilibrium dissociation constant for the interaction between the PXR and corepressor fragments.
Total Internal Reflection with Fluorescence Recovery after Photobleaching
TIR-FRAP21,22,29,30 was used to measure the apparent dissociation rate constant for the F-SRC-1–PXR-LBD interaction. In this technique, an evanescently illuminated area is photobleached, and the subsequent fluorescence recovery is observed as a function of time. Fluorescence recovery, which occurs when photobleached molecules on the surface are replaced by unbleached molecules from solution, is proportional to the intrinsic rate at which photobleached molecules dissociate from surface binding sites, in the absence of surface rebinding. Recovery curves were fit to an appropriate exponential model to obtain the off rate for the F-SRC-1–PXR-LBD interaction (see Results for more detail). TIR-FRAP measurements were taken using bleach pulses with intensities of 100–300 mW and associated bleach times of 50–100 ms. Fluorescence recovery was monitored for 30 s after photobleaching.
RESULTS
Overview
TIRFM and related techniques specifically allow one to probe the behavior of fluorescent species close to or at interfaces. Therefore, we immobilized biotinylated PXR-LBD on NeutrAvidin-coated microscope slides via the biotin–avidin linkage. TIRFM-based techniques were used to examine coactivator and corepressor peptides in solution reversibly interacting with surface-bound PXR-LBD, in the presence and absence of rifampicin.
Control Measurements
It was necessary, first, to confirm that NeutrAvidin and PXR-LBD were irreversibly bound to the surface. To probe the surface lifetime of NeutrAvidin, fluorescently labeled NeutrAvidin (0.5 mg/mL) along with ovalbumin (0.1 mg/mL) was immobilized on microscope slides; the slides were then washed, and the surface-associated fluorescence was monitored for 90 min at 30 min intervals using TIRFM (data not shown). Although the fluorescence decreased 13 ± 3% over the first 30 min period, no further decrease was observed. To probe the surface lifetime of PXR-LBD, slides were coated with a mixture of NeutrAvidin (0.5 mg/mL) and ovalbumin (0.1 mg/mL), washed, treated with fluorescently labeled PXR-LBD (0.1 mg/mL), and washed, and then the surface-associated fluorescence was monitored for 90 min at 30 min intervals using TIRFM (data not shown). Although the fluorescence decreased 13 ± 3% over the 90 min period, the decrease was only 3 ± 3% after 30 min. As all TIRFM equilibrium and TIR-FRAP kinetic measurements, for individual samples, were obtained within at least 30 min of the final wash, the calibrated surface residency times for NeutrAvidin and PXR were judged to be sufficient.
PXR-LBD was biotinylated at its N-terminus, away from the ligand binding pocket, the SRC-1 peptide binding site, the SMRT binding site, and AF-2,7,25 to minimize the possibility that biotinylation and subsequent immobilization would affect the interaction with ligands and coregulators. To determine whether PXR-LBD specifically bound NeutrAvidin during surface immobilization, fluorescently labeled PXR-LBD (F-PXR-LBD) was applied to NeutrAvidin-coated substrates in the presence and absence of excess (100 μM) D-biotin. Figure 1 shows that in the presence of excess D-biotin the surface-associated fluorescence decreased markedly, indicating that F-PXR-LBD specifically bound NeutrAvidin. All subsequent measurements were conducted at a PXR-LBD concentration of 0.1 mg/mL.
Figure 1.

Specificity of PXR-LBD immobilization. The surface-associated fluorescence of F-PXR-LBD bound to immobilized NeutrAvidin was measured, after surfaces had been treated in the presence (▼) and absence (●) of 100 μM D-biotin. The molar ratios of D-biotin to PXR-LBD ranged from approximately 400 (0.01 mg/mL PXR-LBD) to 40 (0.1 mg/mL PXR-LBD). Fluorescence was measured after incubation of F-PXR-LBD with surface-bound NeutrAvidin for 5 min and washing with 2 mL of buffer D. Mean values from three separate samples (obtained from eight points per sample) were averaged to generate the curves. Uncertainties are standard deviations associated with the 3-fold averages. All subsequent measurements were taken with 0.1 mg/mL PXR-LBD.
PXR has the ability to bind many types of ligands, and in the measurements reported here, the SRC-1 peptide was labeled with fluorescein maleimide. To ensure that fluorescein is not a PXR ligand, the surface-associated fluorescence as a function of the concentration of the unreactive fluorescein reference standard in solution was examined in the presence and absence of surface-bound PXR-LBD (data not shown). Data were obtained for fluorescein concentrations ranging up to 9 μM, above the maximum concentration of labeled F-SRC-1 used in subsequent measurements (usually 3.5 μM). The evanescently excited fluorescence intensities measured in the presence and absence of PXR-LBD were identical and linear with the fluorescein concentration, indicating that the fluorescence arose solely from fluorescein in solution and that, therefore, fluorescein is not a PXR ligand.
F-SRC-1–PXR-LBD Equilibrium Dissociation Constants Measured by Steady-State TIRFM
Steady-state TIRFM was used to examine the thermodynamics of the interaction between F-SRC-1 and PXR-LBD at different rifampicin concentrations. The surface-associated fluorescence arising from both F-SRC-1 bound to immobilized PXR-LBD and free F-SRC-1 in solution that were close enough to the surface to be excited by the evanescent field, denoted by F(+), was measured as a function of the F-SRC-1 concentration in solution. Also measured was the fluorescence solely from F-SRC-1 in solution, denoted by F(−), obtained from samples not treated with PXR-LBD. The difference, which gives a measure of the density of specifically bound F-SRC-1, is denoted by F(+)–F(−). These three quantities are, in the simplest case, given by
| (1) |
where Q is a proportionality constant, S is the surface density of PXR-LBD, A is the solution concentration of F-SRC-1 (only 10 or 30% of which is actually labeled), Kd is the equilibrium dissociation constant describing the reversible association of F-SRC-1 with PXR-LBD, d is the depth of the evanescent field, and b is a constant background signal.
For a given, matched set of measured F(+) and F(−) intensities (see Figure 2A), the following procedure was used to find the best-fit value of the equilibrium dissociation constant, Kd. The experimentally determined values of F(−) were fit to the second expression in eq 1 with A as the abscissa and Qd and b as free parameters. The best-fit values of Qd and b were used to calculate theoretical values for F(−) for the values of A at which F(+) intensities had been obtained. The theoretical values of F(−) were subtracted from the experimental values of F(+) (see Figure 2B). The fluorescence differences were then fit to the third expression in eq 1 with QS and Kd as free parameters. A representative binding curve from a single trial is shown in Figure 2.
Figure 2.

Representative F-SRC-1–PXR-LBD binding isotherm. These plots, obtained from a single trial, show the surface-associated fluorescence of F-SRC-1 interacting with PXR-LBD in the presence of 1 μM rifampicin. (A) Representative values of F(+) (●) and F(−) (○). (B) Background-subtracted data, F(+) – F(−), curve-fit to the third expression in eq 1, which yields a Kd of 4.5 ± 1.4 μM, where the error is that associated with the fit. The PXR-LBD surface site density is (2.0 ± 0.2) × 103 molecules/μm2.
Equilibrium dissociation constants pertaining to the F-SRC-1–PXR-LBD interaction, obtained at rifampicin concentrations that ranged from 0 to 100 μM, are listed in Table 1. As the reported equilibrium dissociation constants for rifampicin and PXR-LBD are ≤10 μM,31,32 at 100 μM rifampicin one would expect most (≥91%) of the surface-immobilized PXR-LBD to be in the ligand-bound state. The measured dissociation constants are within error of each other, indicating that rifampicin does not affect the affinity of the receptor for F-SRC-1. To verify these results, binding curves for which the surface-associated fluorescence was measured as a function of the rifampicin concentration, ranging from 0 to 100 μM, were obtained (data not shown). In these measurements, the F-SRC-1 concentration was kept constant at 2.5 μM. The surface-associated fluorescence did not change, within experimental uncertainty, with rifampicin concentration, supporting our previous finding that rifampicin does not affect the interaction between PXR-LBD and the SRC-1 fragment. A PXR-LBD mutant (S247W/C284W), which was made to mimic a ligand-bound state of the receptor via the introduction of bulky tryptophan residues into the ligand binding pocket,27 served as a control. Many such mutants have been shown to constitutively recruit the coactivator and promote the transcription of reporter genes in a ligand-independent manner.7,27 These measurements indicate that the F-SRC-1 peptide interacts with PXR-LBD as though it is ligand-bound, even in the absence of rifampicin (Table 1). Additional measurements were taken with 5 mM TCEP or F-SRC-1 with 30% labeling, as opposed to 2 mM TCEP and 10% labeling for all other curves (Table 1). Equilibrium constants obtained at 5 mM TCEP were within error of the Kd values obtained at 2 mM TCEP (10% labeling). This result indicates that 2 mM TCEP was sufficient to prevent significant dimerization of unlabeled SRC-1 molecules via the formation of disulfide bonds and to maintain cysteine residues in PXR-LBD in their reduced state, as they would be in vivo. Equilibrium constants obtained using F-SRC-1 with 30% labeling were within error of those obtained using coactivator with 10% labeling (2 mM TCEP), indicating that the fluorescein tag does not significantly interfere with PXR-LBD–SRC-1 binding.
Table 1.
F-SRC-1–PXR-LBD Equilibrium Dissociation Constants Measured by Steady-State TIRFMa
| PXR-LBD | [rifampicin] (μM) | Kd (μM) |
|---|---|---|
| WT | 0 | 4 ± 2 |
| WT | 0.1 | 5 ± 3 |
| WT | 1 | 4 ± 2 |
| WT | 10 | 5 ± 3 |
| WT | 100 | 4 ± 3 |
| S247W/C284W | 0 | 4 ± 2 |
| WT & S247W/C284W (5 mM TCEP) | 0 | 4 ± 2 |
| WT (30% labeled F-SRC-1) | 0 | 5 ± 2 |
Reported Kd values are averages of values obtained from two binding isotherms. Controls were conducted with a ligand-bound mimic of the receptor, PXR-LBD (S247W/C284W); 5 mM TCEP, as opposed to 2 mM; and F-SRC-1 with 30% labeling, instead of 10%. For the 5 mM TCEP control, one curve was measured for WT PXR-LBD and the other for the double tryptophan mutant. Uncertainties are propagated from the errors associated with each of the two fits. WT PXR-LBD refers to the AviTag-His6-PXR-LBD (130–434) fusion protein, with no other modifications.
The density, S, of PXR-LBD at the surface was experimentally determined by using the best-fit values of QS (the saturating fluorescence in the binding isotherm) and Qd (the slope of the background), and an estimated d value of ≈85 nm for our system.33 The density of PXR-LBD at the surface served as an internal control, as the values of S should be on the same order of magnitude for all binding curves and within error of each other for measurements conducted with a single batch of PXR. The densities ranged from 1 to 7 × 103 molecules/μm2, with an average of (4 ± 2) × 103 molecules/μm2. For measurements taken using the same batch of PXR, the S values, as returned by the fitting program, were always within error of each other.
F-SRC-1–PXR-LBD Dissociation Rate Constants Measured by TIR-FRAP
TIR-FRAP was employed to examine the kinetics of the interaction between F-SRC-1 and PXR-LBD at 0, 10, and 100 μM rifampicin. In conducting these measurements, we monitored the surface-associated fluorescence generated by TIR illumination before and after a short, intense bleach pulse. Once an evanescently illuminated area is photobleached, the fluorescence recovers as surface-bound, photobleached molecules dissociate and are replaced by unbleached molecules in solution. The rate of this recovery reflects the rate at which photobleached molecules dissociate from surface binding sites.22,29 In the absence of surface rebinding, the temporal shape of the recovery curve is predicted to be a single exponential with a rate equal to the intrinsic off rate.34
Figure 3 shows two representative fluorescence recovery curves for 1.25 μM (Figure 3A) and 20 μM (Figure 3B) F-SRC-1 interacting with PXR-LBD in the absence of rifampicin. The curves were obtained using a 300 mW, 50 ms bleach pulse, and fluorescence was measured for 30 s after photobleaching. As shown, fluorescence recovery was rapid and essentially complete after 30 s.
Figure 3.

Representative fluorescence recovery curves. These plots show typical fluorescence recovery curves for (A) 1.25 μM and (B) 20 μM F-SRC-1 interacting with PXR-LBD in the absence of ligand, obtained using a 300 mW, 50 ms bleach pulse. Fluorescence values have been normalized to average prebleach fluorescence values and then fit to one-exponential (green curves; f2 = 0 in eq 2) and two-exponential (red curves; f2 ≠ 0 in eq 2) models. It is evident, especially at the early recovery times, that the two-exponential model is a better fit for the data. All reported values are derived from two-exponential fits. For the curves shown here, the values of f1, k1, f2, k2, and μ were (A) 8.0 ± 0.4 kHz, 0.76 ± 0.08 s−1, 4.3 ± 0.4 kHz, 0.126 ± 0.012 s−1, and 0.88, respectively, and (B) 15.0 ± 0.8 kHz, 1.56 ± 0.13 s−1, 2.5 ± 0.3 kHz, 0.15 ± 0.02 s−1, and 0.83, respectively. The weighted average of the two rate constants, koff, was (A) 0.54 or (B) 1.36 s−1.
To confirm that F-SRC-1 was indeed highly reversibly interacting with immobilized PXR-LBD, we measured the surface-associated fluorescence following the application of F-SRC-1 and immediately after washing with 2 mL of buffer D. The surface-associated fluorescence was at background levels after washing, indicating that F-SRC-1 was completely washed away and interacts only reversibly with PXR-LBD. This observation also informs one that the larger fragment of SRC-1, with which PXR-LBD is co-expressed, is most likely removed during the washing step following the application of PXR-LBD to the substrate, if not before during PXR-LBD purification [as indicated by gel electrophoresis (see Materials and Methods)].
In a system such as ours where F-SRC-1 reversibly interacts with surface-bound PXR-LBD, one would at first expect a single intrinsic dissociation rate constant. Hence, fluorescence recovery curves were expected to be monoexponential, with the single exponent reflecting the one rate constant. However, as illustrated in Figure 3 and as previously observed for a variety of other systems,19,22,29 the curves are not monoexponential but are in fact better described by the sum of two exponentials
| (2) |
where f0 is the fluorescence at time zero, defined as the center of the bleach pulse. The weighted average of the two exponential factors, k1 and k2, can be used to determine an average off rate, as follows:
| (3) |
The fractions of the prebleach fluorescence, fp, that were bleached (β) and then recovered (μ) were calculated as follows:
| (4) |
In the TIR-FRAP measurements, the monitoring excitation intensity was sufficiently low that in the absence of a bleach pulse, the evanescently excited fluorescence was constant with no measurable decrease during the typical postbleach monitoring time of 30 s. Potential photoinduced artifacts arising from the intense photobleaching pulse were ruled out by three different types of control measurements. Here, quantitative analysis of recovery curves indicated no significant difference in the best-fit values of koff for (a) a 3-fold increase in the bleaching intensity, (b) a 2-fold increase in the bleaching time, or (c) recovery curves obtained following sequential bleaching of identical regions after previous fluorescence recovery.
TIR-FRAP recovery curves were obtained for a range of F-SRC-1 concentrations, at three different rifampicin concentrations. As shown in Figure 4, fluorescence recovery was significantly slower at lower F-SRC-1 concentrations. This observation is almost certainly due to the fact that at lower F-SRC-1 concentrations there is an increased density of free PXR-LBD on the surface. Consequently, bleached molecules that dissociate from surface binding sites rebind free PXR-LBD at a higher frequency, reducing the overall rate of fluorescence recovery and thereby the observed rate constants. The intrinsic dissociation rate constant was taken to be the value at which the weight-averaged off rate, koff, was maximized and independent of the F-SRC-1 concentration. Toward this end, average off rates were plotted as a function of the F-SRC-1 concentration and fit to the model shown in eq 529 to determine the limit of the off rate as the F-SRC-1 concentration approached infinity, given by :
| (5) |
where koff(A) is the measured average off rate for a given value of A and C is an arbitrary constant. The free parameters were and C. Intrinsic, average dissociation rate constants, , were obtained for the F-SRC-1–PXR-LBD interaction at 0, 10, and 100 μM rifampicin and found to be 2.0 ± 0.1, 2.4 ± 0.3, and 2.4 ± 0.1 s−1, respectively (Figure 4A). As with the thermodynamics, rifampicin does not affect the kinetics of the interaction between F-SRC-1 and PXR-LBD.
Figure 4.

F-SRC-1–PXR-LBD dissociation rate constants measured by TIR-FRAP and theoretical probabilities of rebinding. (A) Four to six recovery curves were measured for each of four (0 μM rifampicin) or three (10 and 100 μM rifampicin) independently prepared samples, for each F-SRC-1 concentration. These measurements were taken using 300 mW, 50 ms; 200 mW, 50 ms; and 100 mW, 100 ms bleach pulses. Each recovery curve was fit to eq 2, and the best-fit values of f1, k1, f2, and k2 were used to calculate koff according to eq 3. The off rates (koff) obtained from the recovery curves pertaining to a single sample were averaged. The points shown in the plot are the averages of three or four of these mean koff values for each F-SRC-1 and rifampicin concentration, with the associated standard deviation. These values of koff as a function of F-SRC-1 concentration, in the absence (○) and presence [10 μM (●) or 100 μM (▼)] of rifampicin, were fit to the model in eq 5. The best-fit values of the intrinsic dissociation rates, , were 2.0 ± 0.1, 2.4 ± 0.3, and 2.4 ± 0.1 s−1 at 0, 10, and 100 μM rifampicin, respectively. (B) Probabilities of rebinding computed by numerically integrating eq 6 from time zero (the center of the bleach pulse) to 30 s (after photobleaching), using the following values: Kd = 4 μM, koff = 2 s−1, S = 3000 molecules/μm2, D = 100 μm2/s, wx = 22.4 μm, and wy = 65.0 μm.
As previously shown,35 the probability that a bleached molecule that dissociates from the origin at time zero has rebound at least once within the illuminated and observed area after the duration of the postbleach observation is
| (6) |
where tf is the duration of the observation time after photobleaching, wx and wy are the 1/e2 radii of the elliptically Gaussian illuminated area, D is the diffusion coefficient of F-SRC-1 in solution, and η describes the propensity for rebinding. The parameter η is given by
| (7) |
where kon is the association rate constant, obtained by computing the quotient of the measured dissociation rate constant and equilibrium dissociation constant. Figure 4B shows the values of eq 6 numerically evaluated for the following values: tf = 30 s, Kd = 4 μM, koff = 2 s−1, S = 3000 molecules/μm2, D = 100 μm2/s, wx = 22.4 μm, and wy = 65.0 μm. As shown, P is significant at the lower F-SRC-1 concentrations but becomes negligibly small for the higher F-SRC-1 concentrations. This information is consistent with the interpretation that the measured fluorescence recovery curves are significantly affected by surface rebinding at low F-SRC-1 concentrations but that, at the higher F-SRC-1 concentrations, the effects of surface rebinding are negligible and the recovery curves accurately report the intrinsic surface dissociation kinetics.
SMRT–PXR-LBD Equilibrium Dissociation Constants Measured by Steady-State TIRFM
SMRT–PXR-LBD equilibrium constants were obtained from competition curves in which SMRT was titrated against a fixed concentration of F-SRC-1 (5 μM). Steady-state TIRFM was used to measure the decrease in surface-associated fluorescence as increasing amounts of SMRT competed with a fixed concentration of F-SRC-1 to bind surface-bound PXR-LBD. Competition curves were obtained at rifampicin concentrations of 0, 10, and 100 μM. Background-subtracted data were fit to eq 8 to obtain the equilibrium dissociation constant, Kd′, for the interaction between the corepressor and PXR-LBD.
| (8) |
where Q is a proportionality constant, S is the surface density of PXR-LBD, Kd is the F-SRC-1–PXR-LBD dissociation constant, A is the concentration of F-SRC-1, and B is the concentration of the corepressor. QS and Kd′ were free parameters, while Kd and A were fixed at 5 μM. Figure 5 shows the background-subtracted, SMRT competition curve obtained in the absence of ligand. Fitting the data in Figure 5 to eq 8 yielded an equilibrium dissociation constant of 65 ± 13 μM for the interaction between SMRT and PXR-LBD. An equilibrium dissociation constant of 51 ± 14 μM was obtained at 100 μM rifampicin (see Table 2). The fact that these values are within error of each other indicates that rifampicin does not alter the affinity of SMRT and PXR fragments. Competition curves were also obtained using a peptide fragment of another corepressor called the nuclear receptor corepressor (NCoR). NCoR, which has been reported not to interact with PXR,13 competed even more weakly with F-SRC-1 to bind surface-bound PXR-LBD [see Figure 5]. Fitting these data to eq 8 yielded an equilibrium constant of 200 ± 50 μM for the NCoR–PXR-LBD interaction in the absence of ligand. This value was within error of those obtained at 10 and 100 μM rifampicin as shown in Table 2.
Figure 5.

Representative corepressor competition data. This plot shows the background-subtracted and normalized fluorescence as a function of SMRT (●) and NCoR (○) concentrations obtained at 5 μM F-SRC-1, in the absence of ligand. Background fluorescence, measured in the absence of surface-immobilized PXR-LBD, was subtracted, and the data were fit to eq 8 with QS and Kd′ as free parameters and using the following fixed parameters: A = 5 μM, and Kd = 5 μM. Equilibrium dissociation constants of 65 ± 13 and 200 ± 50 μM were obtained for the SMRT–PXR-LBD (●) and NCoR–PXR-LBD (○) interactions, respectively. These data were normalized to fluorescence counts determined using the value of the fit parameter QS, A = 5 μM, Kd = 5 μM, and B = 0 μM (no corepressor). The data are averages of two trials, and the errors are the corresponding standard deviations.
Table 2.
Corepressor–PXR-LBD Equilibrium Dissociation Constants Measured by Steady-State TIRFMa
| [rifampicin] (μM) |
Kd′ (μM
|
|
|---|---|---|
| SMRT | NCoR | |
| 0 | 65 ± 13 | 200 ± 50 |
| 10 | 82 ± 11 | 160 ± 40 |
| 100 | 51 ± 14 | 160 ± 40 |
Reported Kd′ values are averages obtained from two competition curves. Uncertainties are propagated from the errors associated with each of the two fits.
DISCUSSION
The nuclear receptor PXR plays an important role in the metabolism of endobiotic and xenobiotic compounds, including many pharmaceutical products, by regulating the expression of drug-metabolizing enzymes. The conventional view of nuclear receptor action is that upon binding agonists, nuclear receptors preferentially associate with coactivators, which in turn recruit downstream members of the transcription machinery.36 In this study, we investigated the interaction between the ligand binding domain of PXR and a relevant peptide derived from the coactivator, SRC-1, in the presence and absence of the PXR agonist, rifampicin. Specifically, TIRFM and TIR-FRAP were employed to examine the thermodynamics and kinetics of PXR-LBD interacting with a fluorescently labeled SRC-1 peptide at different rifampicin concentrations. The equilibrium and dissociation rate constants for the PXR-LBD–F-SRC-1 interaction were unchanged in the presence of rifampicin. In the absence of ligand, the basal transcriptional activity of PXR was reported to be significantly reduced by the corepressor SMRT.13 Thus, the thermodynamics of the interaction between PXR-LBD and a peptide fragment of SMRT was also measured using TIRFM. Again, rifampicin had no effect on the PXR-LBD–SMRT interaction. These results indicate that the agonist rifampicin does not affect PXR’s affinity for (at least these two) coregulators.
While no known rigorous biophysical studies have been previously conducted to quantitatively characterize the interaction among PXR, coactivators, and ligands, the thermodynamics and kinetics of other nuclear receptors, particularly the steroid hormone receptors, interacting with coregulators and ligands have been measured. As reported above, apparent equilibrium and dissociation rate constants of 5 μM and 2 s−1, respectively, were obtained for the interaction between PXR-LBD and SRC-1 (676–700) in the presence and absence of rifampicin. This measured apparent equilibrium dissociation constant is at the weak end of the spectrum of affinities measured for nuclear receptors, many of which fall in the nanomolar range. Fluorescence polarization assays of full-length SRC-1 interacting with the full-length estrogen receptor (ER) and ER-LBD in the presence of estrogen yielded a Kd of ~30 nM on both occasions.37 In the absence of ligand, no interaction was observed. Equilibrium constants of full-length ER and another member of the steroid receptor coactivator family, SRC-2, interacting in live cells have been estimated using fluorescence cross-correlation spectroscopy. Estimated Kd values of ~200 nM, <6 nM, and >3 μM were obtained for ER/SRC-2 in the apo, agonist-bound, and antagonist-bound states, respectively.38 Equilibrium constants of ~160 nM were obtained for the thyroid receptor LBD interacting with a SRC-2 fragment in the presence of thyroxin.39,40 Surface plasmon resonance yielded an apparent dissociation rate constant on the order of 1 × 10−2 s−1 for this interaction.40 However, a recent study showed that the glucocorticoid receptor (GR), like PXR, binds many coregulator peptides with micromolar affinity in the presence of the GR agonist, dexamethasone.41 Further studies have shown that another receptor, the peroxisome proliferator-activated receptor-γ (PPARγ), is able to bind SRC-1 in the absence of ligand with an equilibrium dissociation constant of 34.2 μM.42 Addition of a ligand did increase the affinity of PPARγ and SRC-1, and a Kd of 0.96 μM was obtained. The ability of both PXR and PPARγ to bind coactivators in the absence of ligand may account for their relatively high level of basal activity. FRET measurements have been used to examine the interaction between full-length, human PXR and the same 25-amino acid fragment of SRC-1 used in this study, as a function of the rifampicin concentration.43 While the assay did not yield equilibrium dissociation constants for PXR and SRC-1, it did show, contrary to our findings, that rifampicin slightly, but within experimental uncertainty, increased the extent of coactivator recruitment. We cannot yet account for this discrepancy.
The structures of LBDs are conserved among the various members of the nuclear receptor superfamily.44,45 Briefly, nuclear receptor LBDs consist of approximately 12 α-helices arranged into three layers and two to five β-strands that line a side of the ligand binding pocket. The ligand binding pocket exists as a cavity on one side of the LBD and is lined primarily by hydrophobic residues from several α-helices and β-strands. The coactivator binding site consists of a hydrophobic groove on the surface of the LBD, created by α-helices 3, 4, and 12 in PXR.
Crystal structures of nuclear receptors in the apo and agonist-bound states have contributed to an existing molecular model of ligand-mediated interaction between nuclear receptors and coactivators. A crystal structure of the retinoid acid receptor in the apo state showed helix 12 (H12), or activation function-2 (AF-2), extended away from the main body of the LBD,9 whereas subsequent structures of ligand-bound nuclear receptor LBDs showed H12 folded against the body of the LBD.10,11 In this folded conformation, LBDs can bind conserved LXXLL motifs in coactivators via a charge clamp.11 Therefore, it is thought that H12 serves as the molecular switch that is modulated by ligands to promote interaction of the nuclear receptor with coactivators. PXR-LBD was cocrystallized with the same 25-amino acid fragment of SRC-1 (residues 676–700) used in this study, and the agonist SR12813.12 The SRC-1 fragment formed a kinked α-helix and bound to a groove on the surface of PXR-LBD created by helices 3 (H3), 4 (H4) and 12. Two polar contacts between PXR-LBD and SRC-1 [K259 (H3), carbonyl oxygen of L694; E427 (H12), amine nitrogen of I689] constituted the charge clamp that has also been observed in other nuclear receptor–coactivator complexes. In addition, a hydrogen bond was formed between K227 (H4) and H687. This lysine residue is also conserved in other nuclear receptors, including the constitutive androstane, liver X, farnesoid X, and vitamin D receptors.
Crystal structures of ligand-bound nuclear receptors have helped elucidate the manner in which many endogenous ligands stabilize H12 in the folded or active conformation. It appears that a canonical π–cation interaction stabilizes H12 in the folded state in the steroid hormone receptors,46 which encompass the glucocorticoid, mineralcorticoid, progesterone, androgen, and estrogen receptors, as well as in other nuclear receptors, including the vitamin D, thyroid hormone, farnesoid X, and liver X receptors.47 In the non-steroid hormone receptors, an oxygen atom from each receptor’s endogenous ligand forms an electrostatic interaction with a conserved histidine in H10 or H11, which in turn makes π–cation interactions with a conserved tryptophan or phenylalanine in H12. In this way, ligands of many nuclear receptors indirectly stabilize H12 in the active conformation. However, it is not immediately clear from crystal structures of ligand-bound PXR-LBD how agonists might stabilize PXR’s H12 in the folded state. PXR ligands are structurally diverse and appear to lack a common chemical feature that could stabilize the active conformation. Furthermore, attempts to design an antagonist directed at the PXR ligand binding pocket have thus far failed, with many of the proposed compounds instead serving as agonists.48 It appears that any compound that binds PXR’s ligand binding pocket serves to activate the receptor. (Compounds that antagonize PXR by competing with coactivators to bind the receptor have been described previously.27,31) Therefore, it is likely that PXR ligands do not directly stabilize the active conformation and increase the receptors’s affinity for coactivators but instead work through an alternate mechanism to upregulate PXR activity.
A crystal structure of apo-PXR-LBD shows H12 in the active conformation, indicating that the folded state may be favored even in the absence of ligand.25 If the active conformation is energetically favored in the apo state, that fact could explain our observation that PXR-LBD interacts with the SRC-1 fragment in a ligand-independent manner. Furthermore, PXR has a high level of basal activity relative to other nuclear receptors,49 indicating that the receptor may be able to adopt a stable active conformation in the absence of ligand. Supporting this conclusion, molecular dynamics simulations on PXR have shown that the AF-2 region of the receptor moves as a correlated unit in the absence of ligands.50
PXR is somewhat unique because it binds promiscuously to structurally diverse ligands. In fact, ligands that range in molecular weight from 232 (phenobarbitol) to 823 (rifampicin) have been shown to activate PXR. A 60-amino acid insert found in PXR-LBD that serves to create a five-stranded β-sheet lining one side of the ligand binding pocket, as opposed to the two- or three-stranded β sheet seen in other NRs, is thought to contribute to a large, flexible ligand binding pocket that allows PXR to accommodate diverse ligands.25 The ligand binding pocket can range in volume from 1150 Å3 in the apo state25 to ~1900 Å3 with rifampicin bound.7 Such a large cavity in the body of this globular protein may serve to destabilize apo-PXR. Ligand binding may increase the stability of PXR and thereby its lifetime in vivo. Ligand-dependent activation of PXR might then be a consequence of the increased stability of the ligand-bound receptor.
Nonetheless, studies of the interaction between PXR and SRC-1 in cells suggest that the interaction is enhanced in the presence of ligands. In vitro coprecipitation assays of bacterially expressed GST-tagged PXR-LBD and a 35S-labeled SRC-1 fragment, containing the coactivator’s receptor interaction domain (RID), showed a weakly enhanced interaction in the presence of various agonists, including rifampicin.5,51 It is worth noting that the corresponding interaction between a GST-tagged LBD of the estrogen receptor (ER) and radiolabeled SRC-1-RID was much more greatly enhanced in the presence of estradiol.51 Mammalian two-hybrid assays have also demonstrated a ligand-dependent interaction for SRC-1-RID and PXR, with similar results being obtained for both full-length PXR and PXR-LBD.31,52 Indeed, these studies were conducted with SRC-1-RID, which contains three LXXLL motifs, as opposed to the 25-amino acid fragment used in this study, which has only one LXXLL motif, albeit the most strongly interacting one.53 However, yeast two-hybrid assays have shown that coactivator fragments as small as eight amino acids, containing a single LXXLL motif, are able to interact with ER-LBD in a ligand-dependent manner.54 Therefore, it is unlikely that our observation that the interaction between PXR-LBD and the SRC-1 fragment is rifampicin-independent is due to the use of a 25-amino acid fragment of SRC-1, as opposed to the use of a larger fragment or the full-length protein that is 1441 residues long. It is possible that the observed ligand dependence in the in vivo biochemical assays is due to an increased stability of ligand-bound PXR.
Thermal denaturation studies using circular dichroism spectropolarimetry have shown that ligands do stabilize PXR-LBD. Melting temperatures of 43.0 ± 0.08,12 48.4 ± 0.05,12 and 46.5 ± 0.0555 °C were obtained for apo, SR12813-bound, and rifampicin-bound PXR-LBD, respectively. A complex of SRC-1 and PXR-LBD had a melting temperature of 48.2 ± 0.08 °C.12 These results show that agonists and the coactivator each serve to stabilize PXR-LBD. Ternary complexes of PXR-LBD and SRC-1 with either SR12813 or rifampicin were even more stable with melting temperatures of 52.5 ± 0.0512 and 52.6 ± 0.0355 °C, respectively. One might expect that this increase in stability upon addition of the third component would mean that the agonist and coactivator bind cooperatively, resulting in a ligand-dependent increase in affinity for the coactivator and vice versa. However, if the increased thermal stability is due to rifampicin and SRC-1 independently stabilizing different regions of PXR-LBD, binding of one may not increase the receptor’s affinity for the other.
Another possibility is that ligand-dependent activation of PXR occurs through a novel pathway that is as yet uncharacterized. A recent study demonstrated that the nuclear receptor peroxisome proliferator-activated receptor-γ (PPARγ) is activated by structurally diverse serotonin and fatty acid metabolites.42 PPARγ, like PXR, has a flexible ligand binding pocket that can accommodate many different endogenous and exogenous ligands. Crystal structures of PPARγ in complex with a mimic of serotonin metabolites, indomethacin (IDM), showed that it bound in a distinct region of the ligand binding pocket and made direct contact with H12, securing the helix in the active conformation. However, fatty acid metabolites bound away from H12 and failed to make any contact with the helix. Surface plasmon resonance studies showed IDM induced PPARγ to recruit SRC-1. In fact, equilibrium dissociation constants of 34.2 and 0.96 μM were obtained for PPARγ and SRC-1 in the absence and presence of IDM, respectively. While IDM precipitated a 30-fold increase in the affinity of PPARγ for SRC-1, the fatty acid metabolite, nitro-233, failed to enhance the interaction between the receptor and coactivator. Instead, it was proposed that nitro-233 may modulate heterodimerization with the retinoid X receptor. Given the structural diversity of PXR ligands, agonists like rifampicin may very well use different pathways to activate PXR.
Another aspect related to nuclear receptor function is the previous observation that many receptors have the capability of forming homodimers or heterodimers with other nuclear receptors. In the case of PXR, the physiologically relevant receptor dimer is thought to be one with RXR. Nonetheless, a previous work has shown that PXR-LBD homodimerizes in vitro with an equilibrium dissociation constant of 4.5 μM.26 A dimerization-null mutant was found to be incapable of binding the SRC-1 peptide either in the presence or in the absence of ligand. However, this result is not completely consistent with a separate report in which ligand-dependent interaction of the SRC-1 peptide with PXR-LBD was observed in vitro for a receptor concentration of 20 nM,43 well below the Kd for dimerization where one would predict that the PXR-LBD was present almost exclusively in monomeric form. One possibility is that the discrepancy arises in an indirect manner as a consequence of the mutations introduced to form the dimerization-null PXR-LBD. Regardless of these results, it is important to address the question of the state of dimerization of the PXR-LBD used in the work reported here. The solution concentration before application to the surface was 0.1 mg/mL (2.6 μM); thus, the previously measured dimerization Kd would imply that the solutions contained primarily monomeric PXR-LBD with a non-negligible fraction of dimeric PXR-LBD. It is not possible to determine, after application to the surfaces and washing, what fraction of the immobilized PXR-LBD was in the monomeric or dimeric form. Nonetheless, the results show clearly that the SRC-1 peptide does specifically and reversibly interact with at least a fraction of the immobilized PXR-LBD. Definitive conclusions about the interaction of SRC-1 peptides with monomeric and dimeric forms of PXR-LBD await further measurements.
The interaction between corepressors and nuclear receptors, particularly PXR, has not been as extensively studied as that between coactivators and nuclear receptors. Cell-based assays have been used to show that the SMRT-RID specifically interacts with PXR while that of NCoR does not.13 Hence, our finding that the peptide derived from SMRT binds PXR-LBD with greater affinity (Kd′ ~70 μM) than the NCoR fragment (Kd′ ~170 μM) is consistent with what has been reported in the literature. Only one of two interaction domains (LXXXIXXXL; ID1 and ID2), ID2, in the SMRT-RID was shown to actually bind PXR.14 In addition, of the two major SMRT splicing isoforms, α and τ, ID2 derived from SMRTα was found to bind preferentially to PXR.15 Compared to SMRTτ, SMRTα contains a 46-amino acid insert immediately downstream of ID2. For the purposes of this study, a peptide containing ID2 from SMRTα was used. Reporter assays showed that SMRT (α and τ) reduces both the basal and rifampicin-induced transcriptional activity of PXR on the CYP3A4 promoter.13,14 These results indicate that SMRT is able to compete with coactivators to bind PXR in the absence and presence of ligand and thereby reduce the receptor’s transcriptional activity.
In addition to the fact that SMRTα binds preferentially to PXR, it has been shown that SMRTα resists rifampicin-induced dissociation from PXR15 while SMRTτ does not.14 These results are consistent with our observation that the corepressor peptide derived from SMRTα binds PXR-LBD with equal affinity in the presence and absence of rifampicin. However, the measured equilibrium dissociation constant of SMRTα (2337–2358) and PXR-LBD, which averages to ~70 μM over the three rifampicin concentrations (see Table 2), is quite weak. In comparison, fluorescence polarization measurements of the thyroid hormone receptor-β LBD and SMRTα (2329–2358) yielded equilibrium dissociation constants of ~1 μM.16 It is unclear whether a larger fragment of SMRTα would have interacted with PXR-LBD with greater affinity. However, if the measured equilibrium constant is accurate, the interaction between SMRTα and PXR-LBD may not be physiologically relevant. It is possible that there are other corepressors that interact with PXR with a higher affinity and in a ligand-dependent manner.
Crystal structures of nuclear receptor LBDs and peptide fragments of SMRT have revealed the mode of corepressor binding and thereby elucidated the reason for the competition between corepressors and coactivators to bind nuclear receptors. A crystal structure of a PPAR isoform, PPARα, LBD in complex with an antagonist, GW490544, and the same peptide fragment of SMRTα (2337–2358) that was used in this work showed that the corepressor bound to a hydrophobic groove on the surface of the LBD that overlapped with the coactivator binding site.16 GW490544, like many nuclear receptor antagonists, had a portion that protruded out from the ligand binding pocket and prevented H12 (AF-2) from assuming its active conformation. This repositioning of H12 allowed the corepressor peptide to bind a groove formed by H3, H3′, H4, and H5. In the ternary complex, SMRT adopted a three-turn α-helix, unlike the two-turn α-helix formed by SRC-1. The additional helical turn in SMRT extended into the space that is normally occupied by H12 in the active conformation. The corepressor–PPARα complex was stabilized by polar contacts between the backbone carbonyls of A2348 and L2349 and the amine nitrogen of the same, conserved lysine residue in H3 that helps form the charge clamp with coactivators. Because the coactivator and corepressor binding sites overlap so greatly, one of these coregulators binding a nuclear receptor would necessarily prevent the other from binding. In the apo state, when H12 is allowed to freely sample active and inactive conformations, one would expect coactivators and corepressors to compete with each other to bind nuclear receptors. If in fact rifampicin fails to secure H12 in the active conformation, as is implied by existing structural data and our thermodynamic and kinetic data for SRC-1–PXR-LBD interactions, one would expect, as observed, that rifampicin does not affect the ability of corepressors to bind PXR-LBD.
Our thermodynamic and kinetic measurements indicate that rifampicin does not increase the affinity of PXR-LBD for the SRC-1 fragment or decrease the receptor’s affinity for SMRT. The vast structural diversity of PXR ligands makes it unlikely that all agonists would be able to form direct or indirect interactions with residues in H12, as seen with the π– cation interactions, to stabilize the active conformation. It is possible that the different PXR ligands work through an alternate mechanism, or even several mechanisms like in the case with PPARγ, to activate the receptor. One such possibility is that ligands, by filling the large cavity that is the ligand binding pocket, confer stability to PXR and increase the receptor’s in vivo lifetime. Other plausible ligand-dependent regulatory mechanisms include an interplay between (physiologically relevant) corepressors and coactivators, homodimerization or heterodimerization with RXR, phosphorylation, and the differential affinity of PXR for various chaperone proteins in the ligand-bound versus apo states. These possible mechanisms of action are all avenues for future study.
Acknowledgments
Funding
This work was supported by National Science Foundation Grant MCB-0641087 (N.L.T.) and a University of North Carolina Graduate School Dissertation Completion Fellowship (P.N.).
We thank Linda L. Spremulli, Brooke E. Christian, Valerie Hansen, Laurie Betts, Kaiulani Houston, Marcey Waters, Qunzhao Wang, David Lawrence, Yuan Cheng, and Xiang Wang for their assistance.
ABBREVIATIONS
- PXR
pregnane X receptor
- TIRFM
total internal reflection fluorescence microscopy
- SRC-1
steroid receptor coactivator-1
- SMRT
silencing mediator of retinoid and thyroid receptors
- RXR
retinoid X receptor
- AF-2
activation function-2
- LBD
ligand binding domain
- TIR-FRAP
total internal reflection combined with fluorescence recovery after photobleaching
- TCEP
tris(2-carboxyethyl)phosphine
- TFP
tetrafluorophenyl ester
- NCoR
nuclear receptor corepressor
- ER
estrogen receptor
- GR
glucocorticoid receptor
- PPAR
peroxisome proliferator-activated receptor
- GST
glutathione S-transferase
- RID
receptor interaction domain
- IDM
indomethacin
References
- 1.di Masi A, De Marinis E, Ascenzi P, Marino M. Nuclear receptors CAR and PXR: Molecular, functional, and biomedical aspects. Mol Aspects Med. 2009;30:297–343. doi: 10.1016/j.mam.2009.04.002. [DOI] [PubMed] [Google Scholar]
- 2.Moore LB, Goodwin B, Jones SA, Wisely GB, Serabjit-Singh CJ, Willson TM, Collins JL, Kliewer SA. St. John’s wort induces hepatic drug metabolism through activation of the pregnane X receptor. Proc Natl Acad Sci USA. 2000;97:7500–7502. doi: 10.1073/pnas.130155097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wentworth J, Agostini M, Love J, Schwabe J, Chatterjee V. St John’s wort, a herbal antidepressant, activates the steroid X receptor. J Endocrinol. 2000;166:R11–R16. doi: 10.1677/joe.0.166r011. [DOI] [PubMed] [Google Scholar]
- 4.Watkins RE, Maglich JM, Moore LB, Wisely GB, Noble SM, Davis-Searles PR, Lambert MH, Kliewer SA, Redinbo MR. 2.1 Å crystal structure of human PXR in complex with the St. John’s Wort compound hyperforin. Biochemistry. 2003;42:1430–1438. doi: 10.1021/bi0268753. [DOI] [PubMed] [Google Scholar]
- 5.Lehmann JM, McKee DD, Watson MA, Willson TM, Moore JT, Kliewer SA. The human orphan nuclear receptor PXR is activated by compounds that regulate CYP3A4 gene expression and cause drug interactions. J Clin Invest. 1998;102:1016–1023. doi: 10.1172/JCI3703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bertilsson G, Heidrich J, Svensson K, Åsman M, Jendeberg L, Sydow-Bäckman M, Ohlsson R, Postlind H, Blomquist P, Berkenstam A. Identification of a human nuclear receptor defines a new signaling pathway for CYP3A induction. Proc Natl Acad Sci USA. 1998;95:12208–12213. doi: 10.1073/pnas.95.21.12208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chrencik JE, Orans J, Moore LB, Xue Y, Peng L, Collins JL, Wisely GB, Lambert MH, Kliewer SA, Redinbo MR. Structural Disorder in the Complex of Human Pregnane X Receptor and the Macrolide Antibiotic Rifampicin. Mol Endocrinol. 2005;19:1125–1134. doi: 10.1210/me.2004-0346. [DOI] [PubMed] [Google Scholar]
- 8.Nagy L, Schwabe JWR. Mechanism of the nuclear receptor molecular switch. Trends Biochem Sci. 2004;29:317–324. doi: 10.1016/j.tibs.2004.04.006. [DOI] [PubMed] [Google Scholar]
- 9.Bourguet W, Ruff M, Chambon P, Gronemeyer H, Moras D. Crystal structure of the ligand-binding domain of the human nuclear receptor RXR-α. Nature. 1995;375:377–382. doi: 10.1038/375377a0. [DOI] [PubMed] [Google Scholar]
- 10.Renaud JP, Rochel N, Ruff M, Vivat V, Chambon P, Gronemeyer H, Moras D. Crystal structure of the RAR-γ ligand-binding domain bound to all-trans retinoic acid. Nature. 1995;378:681–689. doi: 10.1038/378681a0. [DOI] [PubMed] [Google Scholar]
- 11.Nolte RT, Wisely GB, Westin S, Cobb JE, Lambert MH, Kurokawa R, Rosenfeld MG, Willson TM, Glass CK, Milburn MV. Ligand binding and coactivator assembly of the peroxisome proliferator-activated receptor-γ. Nature. 1998;395:137–143. doi: 10.1038/25931. [DOI] [PubMed] [Google Scholar]
- 12.Watkins RE, Davis-Searles PR, Lambert MH, Redinbo MR. Coactivator binding promotes the specific interaction between ligand and the pregnane X receptor. J Mol Biol. 2003;331:815–828. doi: 10.1016/s0022-2836(03)00795-2. [DOI] [PubMed] [Google Scholar]
- 13.Takeshita A, Taguchi M, Koibuchi N, Ozawa Y. Putative Role of the Orphan Nuclear Receptor SXR (Steroid and Xenobiotic Receptor) in the Mechanism of CYP3A4 Inhibition by Xenobiotics. J Biol Chem. 2002;277:32453–32458. doi: 10.1074/jbc.M111245200. [DOI] [PubMed] [Google Scholar]
- 14.Johnson DR, Li CW, Chen LY, Ghosh JC, Chen JD. Regulation and Binding of Pregnane X Receptor by Nuclear Receptor Corepressor Silencing Mediator of Retinoid and Thyroid Hormone Receptors (SMRT) Mol Pharmacol. 2006;69:99–108. doi: 10.1124/mol.105.013375. [DOI] [PubMed] [Google Scholar]
- 15.Li CW, Dinh GK, Chen JD. Preferential Physical and Functional Interaction of Pregnane X Receptor with the SMRTα Isoform. Mol Pharmacol. 2009;75:363–373. doi: 10.1124/mol.108.047845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Xu HE, Stanley TB, Montana VG, Lambert MH, Shearer BG, Cobb JE, McKee DD, Galardi CM, Plunket KD, Nolte RT, Parks DJ, Moore JT, Kliewer SA, Willson TM, Stimmel JB. Structural basis for antagonist-mediated recruitment of nuclear co-repressors by PPARα. Nature. 2002;415:813–817. doi: 10.1038/415813a. [DOI] [PubMed] [Google Scholar]
- 17.Pisarchick ML, Thompson NL. Binding of a monoclonal antibody and its Fab fragment to supported phospholipid monolayers measured by total internal reflection fluorescence microscopy. Biophys J. 1990;58:1235–1249. doi: 10.1016/S0006-3495(90)82464-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hsieh HV, Poglitsch CL, Thompson NL. Direct measurement of the weak interactions between a moue Fc receptor (Fcgamma RII) and IgG1 in the absence and presence of hapten: A total internal reflection fluorescence microscopy study. Biochemistry. 1992;31:11562–11566. doi: 10.1021/bi00161a039. [DOI] [PubMed] [Google Scholar]
- 19.Gesty-Palmer D, Thompson NL. Binding of the soluble, truncated form of an Fc receptor (mouse FcγRII) to membrane-bound IgG as measured by total internal reflection fluorescence microscopy. J Mol Recognit. 1997;10:63–72. doi: 10.1002/(SICI)1099-1352(199703/04)10:2<63::AID-JMR342>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- 20.Jung H, Yang T, Lasagna MD, Shi J, Reinhart GD, Cremer PS. Impact of Hapten Presentation on Antibody Binding at Lipid Membrane Interfaces. Biophys J. 2008;94:3094–3103. doi: 10.1529/biophysj.107.115519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pisarchick ML, Gesty D, Thompson NL. Binding kinetics of an anti-dinitrophenyl monoclonal Fab on supported phospholipid monolayers measured by total internal reflection with fluorescence photobleaching recovery. Biophys J. 1992;63:215–223. doi: 10.1016/S0006-3495(92)81592-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pearce KH, Hiskey RG, Thompson NL. Surface binding kinetics of prothrombin fragment 1 on planar membranes measured by total internal reflection fluorescence microscopy. Biochemistry. 1992;31:5983–5995. doi: 10.1021/bi00141a005. [DOI] [PubMed] [Google Scholar]
- 23.Sund SE, Axelrod D. Actin Dynamics at the Living Cell Submembrane Imaged by Total Internal Reflection Fluorescence Photobleaching. Biophys J. 2000;79:1655–1669. doi: 10.1016/S0006-3495(00)76415-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jones SA, Moore LB, Shenk JL, Wisely GB, Hamilton GA, McKee DD, Tomkinson NCO, LeCluyse EL, Lambert MH, Willson TM, Kliewer SA, Moore JT. The pregnane X receptor: A promiscuous xenobiotic receptor that has diverged during evolution. Mol Endocrinol. 2000;14:27–39. doi: 10.1210/mend.14.1.0409. [DOI] [PubMed] [Google Scholar]
- 25.Watkins RE, Wisely GB, Moore LB, Collins JL, Lambert MH, Williams SP, Willson TM, Kliewer SA, Redinbo MR. The human nuclear xenobiotic receptor PXR: Structural determinants of directed promiscuity. Science. 2001;292:2329–2333. doi: 10.1126/science.1060762. [DOI] [PubMed] [Google Scholar]
- 26.Noble SM, Carnahan VE, Moore LB, Luntz T, Wang H, Ittoop OR, Stimmel JB, Davis-Searles PR, Watkins RE, Wisely GB, Lecluyse E, Tripathy A, McDonnell DP, Redinbo MR. Human PXR forms a tryptophan zipper-mediated homodimer. Biochemistry. 2006;45:8579–8589. doi: 10.1021/bi0602821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang H, Li H, Moore LB, Johnson MD, Maglich JM, Goodwin B, Ittoop OR, Wisely B, Creech K, Parks DJ, Collins JL, Wilson TM, Kalpana GV, Venkatesh M, Xie W, Cho SY, Roboz J, Redinbo M, Moore JT, Mani S. The phytoestrogen coumestrol is a naturally occurring antagonist of the human pregnane X receptor. Mol Endocrinol. 2008;22:838–857. doi: 10.1210/me.2007-0218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Slade KM, Steele BL, Pielak GJ, Thompson NL. Quantifying green fluorescent protein diffusion in Escherichia coli by using continuous photobleaching with evanescent illumination. J Phys Chem B. 2009;113:4837–4845. doi: 10.1021/jp810642d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lagerholm BC, Starr TE, Volovyk ZN, Thompson NL. Rebinding of IgE Fabs at haptenated planar membranes: Measurement by total internal reflection with fluorescence photobleaching recovery. Biochemistry. 2000;39:2042–2051. doi: 10.1021/bi9917434. [DOI] [PubMed] [Google Scholar]
- 30.Hsieh HV, Thompson NL. Dissociation kinetics between a mouse FC receptor (FCGAMMA RII) and IgG: Measurement by total internal reflection with fluorescence photobleaching recovery. Biochemistry. 1995;34:12481–12488. doi: 10.1021/bi00038a047. [DOI] [PubMed] [Google Scholar]
- 31.Huang H, Wang H, Sinz M, Zoeckler M, Staudinger J, Redinbo MR, Teotico DG, Locker J, Kalpana GV, Mani S. Inhibition of drug metabolism by blocking the activation of nuclear receptors by ketoconazole. Oncogene. 2007;26:258–268. doi: 10.1038/sj.onc.1209788. [DOI] [PubMed] [Google Scholar]
- 32.Xiao L, Nickbarg E, Wang W, Thomas A, Ziebell M, Prosise WW, Lesburg CA, Taremi SS, Gerlach VL, Le HV, Cheng KC. Evaluation of in vitro PXR-based assays and in silico modeling approaches for understanding the binding of a structurally diverse set of drugs to PXR. Biochem Pharmacol. 2011;81:669–679. doi: 10.1016/j.bcp.2010.12.003. [DOI] [PubMed] [Google Scholar]
- 33.Pero JK, Haas EM, Thompson NL. Size dependence of protein diffusion very close to membrane surfaces: Measurement by total internal reflection with fluorescence correlation spectroscopy. J Phys Chem B. 2006;110:10910–10918. doi: 10.1021/jp056990y. [DOI] [PubMed] [Google Scholar]
- 34.Thompson NL, Burghardt TP, Axelrod D. Measuring surface dynamics of biomolecules by total internal reflection fluorescence with photobleaching recovery or correlation spectroscopy. Biophys J. 1981;33:435–454. doi: 10.1016/S0006-3495(81)84905-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Thompson NL, Navaratnarajah P, Wang X. Measuring surface binding thermodynamics and kinetics by using total internal reflection with fluorescence correlation spectroscopy: Practical considerations. J Phys Chem B. 2011;115:120–131. doi: 10.1021/jp1069708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Carnahan VE, Redinbo MR. Structure and Function of the Human Nuclear Xenobiotic Receptor PXR. Curr Drug Metab. 2005;6:357–367. doi: 10.2174/1389200054633844. [DOI] [PubMed] [Google Scholar]
- 37.Margeat E, Poujol N, Boulahtouf A, Chen Y, Müller JD, Gratton E, Cavailles V, Royer CA. The human estrogen receptor α dimer binds a single SRC-1 coactivator molecule with an affinity dictated by agonist structure. J Mol Biol. 2001;306:433–442. doi: 10.1006/jmbi.2000.4418. [DOI] [PubMed] [Google Scholar]
- 38.Savatier J, Jalaguier S, Ferguson ML, Cavaillès V, Royer CA. Estrogen receptor interactions and dynamics monitored in live cells by fluorescence cross-correlation spectroscopy. Biochemistry. 2010;49:772–781. doi: 10.1021/bi9013006. [DOI] [PubMed] [Google Scholar]
- 39.Arnold LA, Estébanez-Perpiñá E, Togashi M, Jouravel N, Shelat A, McReynolds AC, Mar E, Nguyen P, Baxter JD, Fletterick RJ, Webb P, Guy RK. Discovery of Small Molecule Inhibitors of the Interaction of the Thyroid Hormone Receptor with Transcriptional Coregulators. J Biol Chem. 2005;280:43048–43055. doi: 10.1074/jbc.M506693200. [DOI] [PubMed] [Google Scholar]
- 40.Valadares NF, Polikarpov I, Garratt RC. Ligand induced interaction of thyroid hormone receptor β with its coregulators. J Steroid Biochem Mol Biol. 2008;112:205–212. doi: 10.1016/j.jsbmb.2008.10.006. [DOI] [PubMed] [Google Scholar]
- 41.Pfaff SJ, Fletterick RJ. Hormone and coregulator binding to the glucocorticoid receptor are allosterically coupled. J Biol Chem. 2010;285:15256–15267. doi: 10.1074/jbc.M110.108118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Waku T, Shiraki T, Oyama T, Maebara K, Nakamori R, Morikawa K. The nuclear receptor PPARγ individually responds to serotonin- and fatty acid-metabolites. EMBO J. 2010;29:3395–3407. doi: 10.1038/emboj.2010.197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mitro N, Vargas L, Romeo R, Koder A, Saez E. T0901317 is a potent PXR ligand: Implications for the biology ascribed to LXR. FEBS Lett. 2007;581:1721–1726. doi: 10.1016/j.febslet.2007.03.047. [DOI] [PubMed] [Google Scholar]
- 44.Ingraham HA, Redinbo MR. Orphan nuclear receptors adopted by crystallography. Curr Opin Struct Biol. 2005;15:708–715. doi: 10.1016/j.sbi.2005.10.009. [DOI] [PubMed] [Google Scholar]
- 45.Moore JT, Collins JL, Pearce KH. The Nuclear Receptor Superfamily and Drug Discovery. Chem Med Chem. 2006;1:504–523. doi: 10.1002/cmdc.200600006. [DOI] [PubMed] [Google Scholar]
- 46.Queralt-Rosinach N, Mestres J. A canonical cation-π interaction stabilizes the agonist conformation of estrogen-like nuclear receptors. Eur Biophys J. 2010;39:1471–1475. doi: 10.1007/s00249-010-0602-2. [DOI] [PubMed] [Google Scholar]
- 47.Huang P, Chandra V, Rastinejad F. Structural overview of the nuclear receptor superfamily: Insights into physiology and therapeutics. Annu Rev Physiol. 2010;72:247–272. doi: 10.1146/annurev-physiol-021909-135917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Xue Y, Chao E, Zuercher WJ, Willson TM, Collins JL, Redinbo MR. Crystal structure of the PXR-T1317 complex provides a scaffold to examine the potential for receptor antagonism. Bioorg Med Chem. 2007;15:2156–2166. doi: 10.1016/j.bmc.2006.12.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Goodwin B, Redinbo MR, Kliewer SA. Regulation of CYP3A gene transcription by the pregnane X receptor. Annu Rev Pharmacol Toxicol. 2002;42:1–23. doi: 10.1146/annurev.pharmtox.42.111901.111051. [DOI] [PubMed] [Google Scholar]
- 50.Teotico DG, Frazier ML, Ding F, Dokholyan NV, Temple BRS, Redinbo MR. Active Nuclear Receptors Exhibit Highly Correlated AF-2 Domain Motions. PLoS Comput Biol. 2008;4:e1000111. doi: 10.1371/journal.pcbi.1000111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kliewer SA, Moore JT, Wade L, Staudinger JL, Watson MA, Jones SA, McKee DD, Oliver BB, Willson TM, Zetterström RH, Perlmann T, Lehmann JM. An orphan nuclear receptor activated by pregnanes defines a novel steroid signaling pathway. Cell. 1998;92:73–82. doi: 10.1016/s0092-8674(00)80900-9. [DOI] [PubMed] [Google Scholar]
- 52.Wang H, Huang H, Li H, Teotico DG, Sinz M, Baker SD, Staudinger J, Kalpana G, Redinbo MR, Mani S. Activated pregnenolone X receptor is a target for ketoconazole and its analogs. Clin Cancer Res. 2007;13:2488–2495. doi: 10.1158/1078-0432.CCR-06-1592. [DOI] [PubMed] [Google Scholar]
- 53.Darimont BD, Wagner RL, Apriletti JW, Stallcup MR, Kushner PJ, Baxter JD, Fletterick RJ, Yamamoto KR. Structure and specificity of nuclear receptor–coactivator interactions. Genes Dev. 1998;12:3343–3356. doi: 10.1101/gad.12.21.3343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Heery DM, Kalkhoven E, Hoare S, Parker MG. A signature motif in transcriptional coactivators mediates binding to nuclear receptors. Nature. 1997;387:733–736. doi: 10.1038/42750. [DOI] [PubMed] [Google Scholar]
- 55.Carnahan VE. MS Thesis. University of North Carolina; Chapel Hill, NC: 2007. Characterizing the pregnane X receptor’s interactions and biophysical properties. [Google Scholar]