

Abstract
An air- and moisture-stable fluoroiodane in the presence of AgBF4 is suitable for selective geminal difluorination of styrenes under mild reaction conditions. One of the C=F bonds is formed by transfer of electrophilic fluorine from the hypervalent iodine reagent, while the other one arises from the tetrafluoroborate counterion of silver. Deuterium-isotope-labelling experiments and rearrangement of methyl styrene substrates suggest that the reaction proceeds through a phenonium ion intermediate.
Keywords: fluorine, hypervalent compounds, reaction mechanism, rearrangement, silver
Organofluorines are very important substances in pharmaceutical and agrochemical industries and have also had an increasing role in medicinal research and diagnostics.[1] As organofluorine compounds are specific regulator substances, inhibitors, or biomarkers, constant development of new methodologies for their selective synthesis is a very important task in synthetic chemistry. The appearance of new stable, electrophilic fluorinating reagents and methodologies allowed the extension of the new methodologies for selective synthesis of a large variety of organofluorine compounds.[2] In recent years creation of single C=F bonds, introduction of CF2, and trifluoromethylation reactions have received a lot of attention. Of these major areas the introduction of a CF2 group is probably the least developed.[3] Nevertheless, the difluoromethyl group proved to be an important motif in enzyme inhibitors.[4] This property is probably a result of the ability of the CF2H group to donate hydrogen bonds,[5] and, thus serve as a bioisoster for hydroxy and thiol moieties.[6] Furthermore, very recently CF2 groups have found new applications in medicinal diagnostics as well. A new trend in the synthesis of trifluoromethyl-group-based PET tracers is substitution of suitable sp3 difluoromethyl groups with 18F to obtain 18FCF2 functionalities.[7]
The early methods for introducing difluoromethyl groups to organic molecules were mainly based on the application of highly reactive inorganic fluorinating reagents, such as DAST,[8] Deoxofluor,[9] and XeF2.[10] However, application of these reagents may lead to problems in functional-group tolerance and selectivity, and causes hazardous HF development upon contact with water. The recently reported methodologies are usually based on metal-catalyzed cross-coupling of various CF2 carriers with aryl halides and boronates. For example the groups of Amii,[11] Hartwig,[12] Prakash and Olah,[13] and Qing[14] used a copper catalyst (or mediator), while the group of Zhang[15] used palladium catalysis for the introduction of a CF2 group to organic substrates. Baran and co-workers have published a series of papers on C=H difluoromethylation by CF2H radicals.[16] Introduction of the difluoromethyl group with consecutive difluorination reactions is a less common approach compared to the above-mentioned cross-coupling of CF2 units with the organic substrate. Tang and co-workers[17] have shown that consecutive geminal difluorination of aromatic benzyl groups can be achieved by using Selectfluor as a fluorine source in combination with a silver catalyst under oxidative conditions. A similar reaction has been reported by Chen and co-workers[18] using visible-light-promoted reactions under metal-free conditions. For both reactions a radical mechanism was postulated.
Recently, development of new fluorination reactions by application of hypervalent iodine reagents has attracted considerable attention.[2a,b,19] As a part of our synthetic fluorochemistry program,[20] we have studied the potential use of the hypervalent fluoroiodane 1[19f,21] as an electrophilic organofluorinating reagent [see Eq. (1)]. The fluoroiodane 1 is an air- and moisture-stable crystalline compound and structural analogue of the Togni reagent,[2a,22] which has been one of the most successful electrophilic trifluoromethylating reagents in organic synthesis. Another attractive property is that 1 can be obtained from its chloro analogue by addition of KF.[21b] Thus, synthesis of 1 using KF involves a simple umpolung method by changing a nucleophilic fluorine into an electrophilic one. This simple possibility for umpolung of the fluorine atom can be a useful feature for the development of new tracers for medicinal diagnostics.[23] The first report by Legault and Prévost[21a] on the attempts to use 1 for C=F bond-formation reactions was disappointing. In contrast to its bromo analogue, 1 was not suitable for the electrophilic halogenation of anisole. According to Stuart and co-workers[19f] 1 reacts with 1,3-diketoesters and 1,3-diketones in the presence of TREAT-HF to give mono- and difluoro products, respectively. As far as we know, this paper by Stuart and co-workers[19f] has been the only report on the application of 1 for creating C=F bonds.
We have now found [Eq. (1)] that mixing of equimolar amounts of the styrene 2 a, 1, and AgBF4 (3) results in the difluoro compound 4 a under mild reaction conditions (40 °C). The reaction was very clean and the yield of the isolated product was over 50 %, thus indicating that in the geminally difluorinated product one of the fluorine atoms was derived from 1, while the other one originated from the BF4− counterion of silver. Thus, under mild reaction conditions, using air- and moisture-stable starting materials a formal F2 addition to 2 a could be performed.
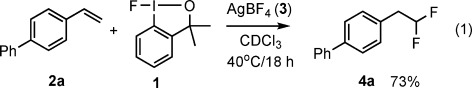 |
(1) |
Deviation from the above mentioned [Eq. (1)] optimal reaction conditions led to either lower yields of isolated 4 a or no reaction of 2 a (Table 1). Application of catalytic amounts of AgBF4, instead of 1 equivalent, led to a significant drop in yield from 73 to 36 % (entry 1). Using AgSbF6 as a mediator and F source led to formation of 4 a (entry 2) but the reaction was much slower than that with AgBF4, as a lot of unreacted starting material (2 a) remained. When AgOAc, AgCN, or AgF was applied instead of 3 the styrene 2 a remained intact (entry 3). A combination of catalytic amounts of AgOAc and 1 equivalent Bu4NBF4 was inefficient (entry 4), but upon treatment with (tBu)3PHBF4, instead of Bu4NBF4, 4 a was isolated with 44 % yield (entry 5). This result and the above experiment (entry 1) with a catalytic amount of 3 indicate that the geminal difluorination reaction can be achieved with a catalytic amount of a silver salt. Thus, the source of the second fluoride [Eq. (1)] is the BF4−, which must not necessarily be added as part of a silver salt. However, the yields of 4 are much higher with stoichiometric amounts of AgBF4. Therefore, we employed one equivalent of AgBF4 as a mediator and secondary fluorine source in additional applications (see also Table 2). When we replaced 1 with Selectfluor, fluorination of 2 a was not observed (entry 6). In the case of using Zn(BF4)2 or Cu(MeCN)4BF4, instead of AgBF4, we could observe the difluorination reaction and isolate 4 a in the corresponding yields of 46 and 32 %. However, particularly with a copper salt, several other byproducts were formed (entries 7 and 8).
Table 1.
Variation of the yield of the isolated product resulting from changes to the reaction conditions [see Eq. (1)].
| Entry | Deviation from the reaction conditions given in Equation (1) | Yield [%] |
|---|---|---|
| 1 | 10 mol % AgBF4 | 36 |
| 2 | 1 equiv AgSbF6 | 18 |
| 3 | 1 equiv AgOAc, AgCN and AgF | <5 |
| 4 | 10 mol % AgOAc, 1 equiv Bu4NBF4 | <5 |
| 5 | 10 mol % AgOAc, 1 equiv (tBu)3PHBF4 | 44 |
| 6 | 1 equiv AgBF4, 1 equiv Selectfluor instead of 1 | <5 |
| 7 | 1 equiv Zn(BF4)2 | 46 |
| 8 | 1 equiv Cu(MeCN)4BF4 | 32 |
| 9 | 1 equiv ZnF2, CuF2 | <5 |
| 10 | MeOH as solvent | <5 |
| 11 | toluene as solvent | trace |
Table 2.
Difluorination of styrene derivatives with the iodiane 1 and AgBF4.[a]
 |
[a] Styrene 2 (0.1 mmol), 1 (0.1 mmol), and AgBF4 (0.1 mmol) in chloroform (0.5 mL) was stirred at 40 °C for 18 h. [b] Unless otherwise stated yield is that of isolated product. [c] Yield determined by NMR spectroscopy.
The reaction with palladium salts was rather interesting. [Pd(MeCN)4(BF4)2] proved to be a rather efficient catalyst, thus affording 4 a with 50 % yield [Eq. (2)]. However, when the BF4− counter ion exchanged to Cl− the outcome of the reaction was completely different. When using [PdCl2(MeCN)2] as the catalyst 4 a did not form at all but the reaction gave the iodofluorinated product 5, which could be isolated with 43 % yield. Further studies indicated that in the geminal difluorination reaction with 1, AgF, ZnF2, or CuF2 were not able to serve as fluoride sources (Table 1, entries 3 and 9). We employed CDCl3 as the solvent, which allowed the careful analysis of the crude reaction mixtures by 1H and 19F NMR spectroscopy. Change of the solvent to MeOH or THF completely hindered any transformation of 2 a (entry 10), while using toluene led to formation of only traces of 4 a (entry 11).
 |
(2) |
Subsequently, we studied the synthetic scope of the reaction (Table 2). Both α- and β-naphtyl styrenes (2 b and 2 c) underwent geminal difluorination with high yields (entries 2 and 3). Despite the use of stoichiometric amounts of AgBF4 the bromo substituent in 2 d was tolerated and 4 d was formed in a clean difluorination process (entry 4). The compound 4 d can be a useful intermediate for modular synthesis of difluoromethyl compounds, as the bromo functionality can easily transformed by Suzuki–Miyaura coupling.[24] The difluorination reaction also tolerates oxygen-containing substituents, such as the ether in 2 e and carboxylate in 4 f (entries 5 and 6). However, for these substrates, we obtained somewhat lower yields than those for the hydrocarbon substrates 2 a–c. The parent styrene 2 g reacted readily with 1 but the isolation of 4 g was difficult because of its volatility. Therefore, we determined the yield of 4 g by 1H NMR spectroscopy. meta-Phenyl (2 h) and bromo (2 i) styrenes reacted readily to provide the corresponding difluorinated compounds 4 h and 4 i (entries 8 and 9). However, our attempts to obtain geminal difluorinated products from ortho-substituted styrenes remained fruitless. The reactions with the α-methyl styrenes 2 j–l gave surprising results. We still obtained the β-difluorinated products 4 j–l but the methyl group migrated from the α- to the β-position (entries 10–12). The rearrangement proceeds very cleanly, as we could not observe any other isomers of 4 j–l in the crude reaction mixture.
This finding and the fact that one of the fluorines arises from an electrophilic source (from 1) and the other from a nucleophilic reagent (from a BF4− anion) suggests a very interesting mechanism for the geminal difluorination process. To explore the mechanism, we prepared the deutero analogue of 2 a, [D2]-2 a. When [D2]-2 a was reacted with 1 in the presence of AgBF4 under the usual reaction conditions the compound [D2]-4 a was obtained in a very clean reaction [Eq. (3)]. This outcome was a surprising result, since the shift of one of the deuterium atoms of [D2]-2 a was expected. The double deuterium shift suggested a significant deuterium isotope effect. Therefore, we performed a competitive difluorination reaction between 2 a and [D2]-2 a [Eq. (4)]. However, according to these studies the deuterium isotope effect was found to be weak (about 1.3).
 |
(3) |
 |
(4) |
The above results indicate that the hydrogen shift from the α- to the β-position of styrene is unlikely. We reasoned that the difluorination reaction probably proceeds by an α- to β-carbon atom exchange of the styrene derivative through a possible phenonium ion intermediate. Such types of intermediates were first suggested by Cram[25] and subsequently observed by Olah and co-workers.[26]
Accordingly, our plausible mechanism (Figure 1) involves silver-activated addition of the iodane to the double bond of [D2]-2 a to give the iodonium ion 6. Togni and co-workers[27] studied the activation of the CF3 analogue of 1 using zinc salts. A similar metal-mediated activation is conceivable for 1 as well. As mentioned above (Table 1, entry 7) Zn(BF4)2 is also a viable mediator of the difluorination process. Moreover, suitable copper and palladium salts [Table 1, entry 8 and Eq. (2)] may have a similar activating effect. The electron deficiency of iodine in 6 can be relieved by fluorine migration to give 7. The π donation of the aromatic ring in 7 may result in formation of the phenonium ion 8. In this process the iodoaryl group arising from 1 serves as a leaving group. The positive charge may delocalize over five carbon atoms (for sake of clarity only one of the resonance structures is given in Figure 1).
Figure 1.

Plausible mechanism for the geminal difluorination of styrenes.
By using the [PdCl2(MeCN)2] catalyst [Eq. (2)] the faith of 7 was probably different and it underwent C(sp2)=I bond fission to result in 5. The only possible source of iodine in 5 is 1. However, in the case of AgBF4 (or a few other catalysts or mediators) the fluorinated phenonium ion 8 forms (Figure 1), and may undergo a second fluorination by a nucleophilic fluorine arising from the BF4−. The BF4− anion is an unexpected but not unusual source of nucleophilic fluoride. Our recent studies[28] have shown the BF4− from [Pd(MeCN)4(BF4)2] [see Eq. (2)] may serve as a fluorine source in the activation of (SiMe3)2 for catalytic silylation of allyl alcohols. Gandon and co-workers[29] have recently shown that BF4− from AgBF4 is an excellent fluorinating reagent for organometallic compounds. The present study is probably one of the few examples demonstrating that a C=F bond can also be formed under mild reaction conditions using BF4− as a fluorine source. An additional remarkable feature of the above-presented reaction is that the opening of the cyclopropyl ring of 8 is highly regioselective. We could not observe formation of α-,β-difluorinated regioisomers of 4 a in the 1H and 19F NMR spectra of the crude reaction mixtures. Opening of the cyclopropane ring of the fluorinated carbon in 8 also easily explains the apparent migration of the methyl group in the difluorination of 2 j–l (entries 10–12 in Table 2). A further confirmation of the plausible mechanism in Figure 1 could be the analogue reactivity of phenyl iodonium acetate (PIDA) or phenyl iodonium trifluoroacetate (PIFA) with styrene derivatives. PIDA and PIFA can be considered structural analogues of 1. Tellitu and Domínguez,[30] and the group of Wirth[31] also postulated that the dioxo substitution of styrenes with PIDA and PIFA probably proceeds via phenonium intermediates.
In summary, we have shown that the hypervalent iodine 1 and AgBF4 (and a few other metal tetrafluoroborates) induce geminal difluorination of styrenes. These air- and moisture-stable reagents react under mild reaction conditions to perform a selective formal F2 addition. Using equimolecular amounts of 1 and styrene derivatives resulted in yields over 50 %, thus indicating that one of the C=F bonds is created by electrophilic fluorinating reagent 1, while the other one results from fluorine transfer from BF4−. The deuterium-labelling experiments indicate that the process probably proceeds via phenonium intermediates. The reaction is suitable for mild synthesis of β-difluoro aromatic compounds, which are bioisosters[4–6] of natural compounds with benzylalcohol and thiol motifs. In addition, our method extends the scope of the fluorination reactions, including a new application of the easily accessible, stable, and safe electrophilic fluoroidane reagent 1.
Experimental Section
The iodane 1 (28.0 mg, 0.1 mmol), AgBF4 3 (19.4 mg, 0.1 mmol), and the styrene 2 (0.1 mmol, 1 equiv) were mixed in CDCl3 (0.5 mL) and this mixture was stirred at 40 °C for 18 h. Then product 4 was isolated by chromatography.
Supporting information for this article is available on the WWW under http://dx.doi.org/10.1002/anie.201408812.
References
- [1a].Müller K, Faeh C, Diederich F. Science. 2007;317:1881. doi: 10.1126/science.1131943. [DOI] [PubMed] [Google Scholar]
- [1b].Purser S, Moore PR, Swallow S, Gouverneur V. Chem. Soc. Rev. 2008;37:320. doi: 10.1039/b610213c. [DOI] [PubMed] [Google Scholar]
- [1c].Tredwell M, Preshlock SM, Taylor NJ, Gruber S, Huiban M, Passchier J, Mercier J, Génicot C, Gouverneur V. Angew. Chem. Int. Ed. 2014;53:7751. doi: 10.1002/anie.201404436. [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014;126 [Google Scholar]
- [1d].Jeschke P. ChemBioChem. 2004;5:570. doi: 10.1002/cbic.200300833. [DOI] [PubMed] [Google Scholar]
- [1e].Wang J, Sánchez-Roselló M, Aceña JL, del Pozo C, Sorochinsky AE, Fustero S, Soloshonok VA, Liu H. Chem. Rev. 2014;114:2432. doi: 10.1021/cr4002879. [DOI] [PubMed] [Google Scholar]
- [2a].J. Charpentier, N. Früh, A. Togni, Chem. Rev2014. , DOI: [DOI] [PubMed]
- [2b].Liang T, Neumann CN, Ritter T. Angew. Chem. Int. Ed. 2013;52:8214. doi: 10.1002/anie.201206566. [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2013;125 [Google Scholar]
- [2c].Egami H, Sodeoka M. Angew. Chem. Int. Ed. 2014;53:8294. doi: 10.1002/anie.201309260. [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014;126 [Google Scholar]
- [2d].Chen P, Liu G. Synthesis. 2013:2919. [Google Scholar]
- [2e].Merino E, Nevado C. Chem. Soc. Rev. 2014;43:6598. doi: 10.1039/c4cs00025k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Hu J, Zhang W, Wang F. Chem. Commun. 2009:7465. doi: 10.1039/b916463d. [DOI] [PubMed] [Google Scholar]
- [4a].Aráoz R, Anhalt E, René L, Badet-Denisot M-A, Courvalin P, Badet B. Biochemistry. 2000;39:15971. doi: 10.1021/bi001408b. [DOI] [PubMed] [Google Scholar]
- [4b].Hope HR, Heuvelman D, Duffin K, Smith C, Zablocki J, Schilling R, Hegde S, Lee L, Witherbee B, Baganoff M, Bruce C, Tall AR, Krul E, Glenn K, Connolly DT. J. Lipid Res. 2000;41:1604. [PubMed] [Google Scholar]
- [4c].Narjes F, Koehler KF, Koch U, Gerlach B, Colarusso S, Steinkühler C, Brunetti M, Altamura S, De Francesco R, Matassa VG. Bioorg. Med. Chem. Lett. 2002;12:701. doi: 10.1016/s0960-894x(01)00842-3. [DOI] [PubMed] [Google Scholar]
- [4d].Xu Y, Qian L, Pontsler AV, McIntyre TM, Prestwich GD. Tetrahedron. 2004;60:43. [Google Scholar]
- [4e].Xu Y, Prestwich GD. J. Org. Chem. 2002;67:7158. doi: 10.1021/jo0203037. [DOI] [PubMed] [Google Scholar]
- [5].Erickson JA, McLoughlin JI. J. Org. Chem. 1995;60:1626. [Google Scholar]
- [6].Meanwell NA. J. Med. Chem. 2011;54:2529. doi: 10.1021/jm1013693. [DOI] [PubMed] [Google Scholar]
- [7a].Mizuta S, Stenhagen ISR, O’Duill M, Wolstenhulme J, Kirjavainen AK, Forsback SJ, Tredwell M, Sandford G, Moore PR, Huiban M, Luthra SK, Passchier J, Solin O, Gouverneur V. Org. Lett. 2013;15:2648. doi: 10.1021/ol4009377. [DOI] [PubMed] [Google Scholar]
- [7b].Riss PJ, Ruehl T, Rafique W, Lien VT. Chem. Commun. 2014;50:0. doi: 10.1039/c4cc01641f. [DOI] [PubMed] [Google Scholar]
- [7c].van der Born D, Sewing C, Herscheid JDM, Windhorst AD, Orru RVA, Vugts DJ. Angew. Chem. Int. Ed. 2014;53:11046. doi: 10.1002/anie.201406221. [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014;126 [Google Scholar]
- [8a].Singh RP, Shreeve JnM. Synthesis. 2002:2561. [Google Scholar]
- [8b].Dolbier WR, Xie P, Zhang L, Xu W, Chang Y, Abboud KA. J. Org. Chem. 2008;73:2469. doi: 10.1021/jo7026849. [DOI] [PubMed] [Google Scholar]
- [9].Middleton WJ. J. Org. Chem. 1975;40:574. [Google Scholar]
- [10].Patrick TB, Qian S. Org. Lett. 2000;2:3359. doi: 10.1021/ol006450d. [DOI] [PubMed] [Google Scholar]
- [11].Fujikawa K, Fujioka Y, Kobayashi A, Amii H. Org. Lett. 2011;13:5560. doi: 10.1021/ol202289z. [DOI] [PubMed] [Google Scholar]
- [12].Fier PS, Hartwig JF. J. Am. Chem. Soc. 2012;134:5524. doi: 10.1021/ja301013h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Prakash GKS, Ganesh SK, Jones J-P, Kulkarni A, Masood K, Swabeck JK, Olah GA. Angew. Chem. Int. Ed. 2012;51:12090. doi: 10.1002/anie.201205850. [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2012;124 [Google Scholar]
- [14].Jiang X-L, Chen Z-H, Xu X-H, Qing F-L. Org. Chem. Frontiers. 2014;1:774. [Google Scholar]
- [15].Feng Z, Min Q-Q, Xiao Y-L, Zhang B, Zhang X. Angew. Chem. Int. Ed. 2014;53:1669. doi: 10.1002/anie.201309535. [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014;126 [Google Scholar]
- [16a].Fujiwara Y, Dixon JA, O’Hara F, Funder ED, Dixon DD, Rodriguez RA, Baxter RD, Herle B, Sach N, Collins MR, Ishihara Y, Baran PS. Nature. 2012;492:95. doi: 10.1038/nature11680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16b].Fujiwara Y, Dixon JA, Rodriguez RA, Baxter RD, Dixon DD, Collins MR, Blackmond DG, Baran PS. J. Am. Chem. Soc. 2012;134:1494. doi: 10.1021/ja211422g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16c].Zhou Q, Ruffoni A, Gianatassio R, Fujiwara Y, Sella E, Shabat D, Baran PS. Angew. Chem. Int. Ed. 2013;52:3949. doi: 10.1002/anie.201300763. [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2013;125 [Google Scholar]
- [17].Xu P, Guo S, Wang L, Tang P. Angew. Chem. Int. Ed. 2014;53:5955. doi: 10.1002/anie.201400225. [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014;126 [Google Scholar]
- [18].Xia J-B, Zhu C, Chen C. J. Am. Chem. Soc. 2013;135:17494. doi: 10.1021/ja410815u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19a].Tredwell M, Gouverneur V. Angew. Chem. Int. Ed. 2012;51:11426. doi: 10.1002/anie.201204687. [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2012;124 [Google Scholar]
- [19b].Suzuki S, Kamo T, Fukushi K, Hiramatsu T, Tokunaga E, Dohi T, Kita Y, Shibata N. Chem. Sci. 2014;5:2754. [Google Scholar]
- [19c].Kong W, Feige P, de Haro T, Nevado C. Angew. Chem. Int. Ed. 2013;52:2469. doi: 10.1002/anie.201208471. [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2013;125 [Google Scholar]
- [19d].Rotstein BH, Stephenson NA, Vasdev N, Liang SH. Nat. Commun. 2014;5:4365. doi: 10.1038/ncomms5365. [DOI] [PubMed] [Google Scholar]
- [19e].Kitamura T, Kuriki S, Morshed MH, Hori Y. Org. Lett. 2011;13:2392. doi: 10.1021/ol200632d. [DOI] [PubMed] [Google Scholar]
- [19f].Geary GC, Hope EG, Singh K, Stuart AM. Chem. Commun. 2013;49:9263. doi: 10.1039/c3cc44792h. [DOI] [PubMed] [Google Scholar]
- [20a].Janson PG, Ghoneim I, Ilchenko NO, Szabó KJ. Org. Lett. 2012;14:2882. doi: 10.1021/ol3011419. [DOI] [PubMed] [Google Scholar]
- [20b].Ilchenko NO, Janson PG, Szabó KJ. Chem. Commun. 2013;49:6614. doi: 10.1039/c3cc43357a. [DOI] [PubMed] [Google Scholar]
- [20c].Ilchenko NO, Janson PG, Szabo KJ. J. Org. Chem. 2013;78:11087. doi: 10.1021/jo401831t. [DOI] [PubMed] [Google Scholar]
- [20d].Larsson JM, Pathipati SR, Szabo KJ. J. Org. Chem. 2013;78:7330. doi: 10.1021/jo4010074. [DOI] [PubMed] [Google Scholar]
- [21a].Legault CY, Prévost J. Acta Crystallogr. Sect. E. 2012;68:1238. doi: 10.1107/S1600536812012822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21b].Matoušek V, Pietrasiak E, Schwenk R, Togni A. J. Org. Chem. 2013;78:6763. doi: 10.1021/jo400774u. [DOI] [PubMed] [Google Scholar]
- [22].Eisenberger P, Gischig S, Togni A. Chem. Eur. J. 2006;12:2579. doi: 10.1002/chem.200501052. [DOI] [PubMed] [Google Scholar]
- [23a].Lee E, Kamlet AS, Powers DC, Neumann CN, Boursalian GB, Furuya T, Choi DC, Hooker JM, Ritter T. Science. 2011;334:639. doi: 10.1126/science.1212625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23b].Brandt JR, Lee E, Boursalian GB, Ritter T. Chem. Sci. 2014;5:169. doi: 10.1039/C3SC52367E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24a].Miyaura N, Suzuki A. Chem. Rev. 1995;95:2457. [Google Scholar]
- [24b].Miyaura N. Top. Curr. Chem. 2002;219:11. [Google Scholar]
- [25].Cram DJ. J. Am. Chem. Soc. 1949;71:3863. [Google Scholar]
- [26a].Olah GA, Porter RD. J. Am. Chem. Soc. 1970;92:7627. [Google Scholar]
- [26b].Olah GA, Porter RD. J. Am. Chem. Soc. 1971;93:6877. [Google Scholar]
- [27].Koller R, Stanek K, Stolz D, Aardoom R, Niedermann K, Togni A. Angew. Chem. Int. Ed. 2009;48:4332. doi: 10.1002/anie.200900974. [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2009;121 [Google Scholar]
- [28].Larsson JM, Szabó KJ. J. Am. Chem. Soc. 2013;135:443. doi: 10.1021/ja309860h. [DOI] [PubMed] [Google Scholar]
- [29].Bour C, Monot J, Tang S, Guillot R, Farjon J, Gandon V. Organometallics. 2014;33:594. [Google Scholar]
- [30].Tellitu I, Domínguez E. Tetrahedron. 2008;64:2465. [Google Scholar]
- [31].Boye AC, Meyer D, Ingison CK, French AN, Wirth T. Org. Lett. 2003;5:2157. doi: 10.1021/ol034616f. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.