

DDX6 is an abundant DEAD-box helicase associated with various complexes involved in mRNA decay and repression. Its interactome in human cells was analyzed to identify its most prominent partners. Among them, three proteins were essential for P-body assembly in all tested conditions: DDX6, 4E-T, and LSM14A.
Abstract
P-bodies are cytoplasmic ribonucleoprotein granules involved in posttranscriptional regulation. DDX6 is a key component of their assembly in human cells. This DEAD-box RNA helicase is known to be associated with various complexes, including the decapping complex, the CPEB repression complex, RISC, and the CCR4/NOT complex. To understand which DDX6 complexes are required for P-body assembly, we analyzed the DDX6 interactome using the tandem-affinity purification methodology coupled to mass spectrometry. Three complexes were prominent: the decapping complex, a CPEB-like complex, and an Ataxin2/Ataxin2L complex. The exon junction complex was also found, suggesting DDX6 binding to newly exported mRNAs. Finally, some DDX6 was associated with polysomes, as previously reported in yeast. Despite its high enrichment in P-bodies, most DDX6 is localized out of P-bodies. Of the three complexes, only the decapping and CPEB-like complexes were recruited into P-bodies. Investigation of P-body assembly in various conditions allowed us to distinguish required proteins from those that are dispensable or participate only in specific conditions. Three proteins were required in all tested conditions: DDX6, 4E-T, and LSM14A. These results reveal the variety of pathways of P-body assembly, which all nevertheless share three key factors connecting P-body assembly to repression.
INTRODUCTION
Protein production in the cytoplasm of eukaryotic cells depends on the balance between translation, storage, and degradation of mRNAs. Although it has only been recognized lately, posttranscriptional regulation appears critical for the control of gene expression. Strikingly, the first genome-scale prediction of synthesis rates of mRNAs and proteins in mammalian cells showed that the abundance of proteins is predominantly controlled at the level of translation (Schwanhäusser et al., 2011). Studies of mature oocytes and fertilized eggs, which rely on translational regulation because their transcription is shut down, led to the identification of several important factors, including the well-characterized CPEB complex (Minshall et al., 2007). Other cells also control expression posttranscriptionally, as illustrated by local translation in dendrites and axons, which involves some of the factors identified in early development, as well as neuron-specific ones (Holt and Schuman, 2013). More generally, the recently discovered RNA interference (RNAi) pathway is a major pathway that controls mRNA translation and stability by a mechanism involving the microRNA (miRNA)-associated RISC complex. Remarkably, many factors involved in these various processes accumulate in cytoplasmic ribonucleoprotein (RNP) granules, called P-bodies in somatic cells, along with the 5′ to 3′ mRNA degradation machinery (Buchan, 2014). P-bodies, which are conserved in all eukaryotes, vegetal and animal, are thought to play a role in mRNA degradation and/or storage, though overall their function remains elusive.
Several P-body proteins were shown to influence P-body number in mammalian cells, as their silencing led to the reduction or the disappearance of P-bodies. This includes GW182 (Yang et al., 2004), LSM1, DDX6, 4E-T (Andrei et al., 2005), EDC4 (Yu et al., 2005), LSM14A (Yang et al., 2006), CPEB1 (Wilczynska et al., 2005), and PAT1B (Marnef et al., 2010; Ozgur et al., 2010). This suggested that each of these proteins is required for P-body assembly. Regarding GW182 and CPEB1, we showed that P-bodies could nevertheless be robustly re-induced in their absence, by incubating cells briefly with arsenite, indicating that P-body assembly is a more complex process than anticipated (Serman et al., 2007). DDX6 was unique, in that arsenite could not induce any P-bodies in DDX6-depleted cells. We therefore focused on this RNA helicase as a key protein in the assembly process.
DDX6 (called RCK/p54 in humans, Me31B in Drosophila, CGH1 in Caenorhabditis elegans, Dhh1 in yeast) was initially described in two main types of complexes: the decapping complex and the CPEB1 translation-repression complex. Its involvement in 5′ cap removal has been mostly studied in yeast, where DDX6 coimmunoprecipitated with several decapping proteins, including DCP1, LSM1, and PAT1 (Coller et al., 2001). Moreover, DDX6 deletion led to the increased half-life of reporter mRNAs and to the accumulation of full-length deadenylated mRNAs. Similar interactions were then reported in Drosophila cells (Haas et al., 2010). Finally, a direct interaction of DDX6 with PAT1B and EDC3 was demonstrated by cocrystallization (Tritschler et al., 2009; Sharif et al., 2013).
The CPEB1 complex was characterized in detail in Xenopus, where it is required for the repression of maternal mRNAs. In oocytes, DDX6 coimmunoprecipitated and copurified in gel-filtration fractions with CPEB1 and associated proteins, including 4E-T, Pat1a, Lsm14B, and eIF4E1b, as well as ePAB (Minshall et al., 2007, 2009). Moreover, tethering DDX6 to reporter mRNAs repressed their translation (Minshall et al., 2009). In yeast, where CPEB is absent, DDX6 deletion prevented the general translational repression that is observed after glucose deprivation (Holmes et al., 2004). On the other hand, DDX6 overexpression led to general translational repression even in rich medium (Coller and Parker, 2005). Similarly, in human cells, DDX6 silencing stimulated cellular translation (Chu and Rana, 2006). Furthermore, DDX6 interacted with proteins of the RISC complex, including AGO1 and AGO2. Specific reporters of small interfering RNA (siRNA)- and miRNA-driven silencing suggested that DDX6 participated in the miRNA but not the siRNA pathway (Chu and Rana, 2006). Recently DDX6 was shown to interact directly with the CCR4/NOT complex, which deadenylates mRNAs before decapping. Moreover, a NOT1 peptide activated the ATPase activity of DDX6, which otherwise is undetectable (Mathys et al., 2014), and mutations abolishing DDX6 NOT1 binding prevented repression of miRNA reporters (Chen et al., 2014; Mathys et al., 2014; Rouya et al., 2014).
While DDX6 is essential for human P-body assembly (Serman et al., 2007; Minshall et al., 2009), there is so far no insight into which of its numerous functions is involved. The relative abundance of the various DDX6 complexes described above is not even known. The first question was therefore which of them, or another yet to be identified, was predominant in human epithelial cells. To address this, we undertook the identification of the DDX6 interactome in human cells using a tandem-affinity purification (TAP-tag) strategy coupled to mass spectrometry analysis. The top partners included the decapping complex, a CPEB-like complex, and an Ataxin2/Ataxin2L (ATXN2/ATXN2L) complex. On the basis of silencing experiments, we showed that translational repression complexes were central to P-body assembly, rather than decay or ATXN2/ATXN2L complexes.
RESULTS
Identification of the main DDX6 complexes
To identify the main complexes containing DDX6 in human somatic cells, we purified them from the epithelial cell line HEK293 by TAP-tag. Briefly, a plasmid encoding DDX6 fused to FLAG and hemagglutinin (HA) tags was transiently transfected into these cells. After 48 h, the tagged protein was expressed at similar levels and with a similar P-body localization and enrichment as the endogenous one, as indicated by Western blot and immunofluorescence analysis, respectively (Figure 1, A–C). It is important to note, as reported previously, that although DDX6 is highly enriched in P-bodies, most DDX6 protein is nevertheless localized out of P-bodies. Indeed, an accurate quantification of DDX6 distribution in HeLa cell thin sections in immunoelectron microscopy previously demonstrated that endogenous DDX6 is enriched 170-fold in P-bodies with respect to the surrounding cytoplasm (Ernoult-Lange et al., 2012). Taking into account their size, this means that, in a cell with five P-bodies, 94% of DDX6 is excluded from P-bodies. Although we did not perform such extensive quantification in HEK293 cells, our estimate is of the same order. Indeed, DDX6 is similarly concentrated in P-bodies, as observed in immunoelectron microscopy (Figure 1D), and P-bodies are often larger (650-nm vs. 500-nm diameter) but less numerous (two per cell vs. five) than in HeLa cells.
FIGURE 1:

Purification of DDX6 complexes by TAP-tag. (A) HEK293 cells were transfected with a FLAG-DDX6-HA or an empty plasmid as a control. After 48 h, proteins were analyzed by Western blotting with anti-DDX6 antibodies. (B) In parallel, transfected cells were stained with anti-FLAG (red) and anti-EDC4 (green) antibodies to detect the exogenous DDX6 protein and P-bodies, respectively. For comparison, untransfected cells were stained with anti-DDX6 and anti-EDC4 antibodies. Owing to the round shape of the cells, the maximal projection of a z-series of images is shown to better visualize P-bodies. Scale bar: 10 μm. (C) DDX6 enrichment in P-bodies with respect to the surrounding cytoplasm (n = 94 and 90 for transfected and untransfected samples, respectively) was quantified from single-plane images. (D) Endogenous DDX6 was detected in untransfected HEK293 cells by immunoelectron microscopy, as previously described (Souquere et al., 2009). One P-body highly enriched in DDX6 is shown. (E) Cells transfected with a FLAG-DDX6-HA or an empty plasmid were lysed in the presence of RNase inhibitor or RNase, and DDX6 complexes were purified by TAP-tag. Proteins were migrated on a denaturing gel along with a molecular weight marker (MW) and silver stained.
Cytoplasmic lysates were prepared from 2 × 108 cells, and DDX6 RNP complexes were successively purified on FLAG and HA affinity resins. As a control, the same procedure was applied to cells transfected with an empty vector. An aliquot of the resulting proteins was separated in denaturing SDS–PAGE and stained with silver to visualize the quality of the purification (Figure 1E, left panel). The tagged DDX6 specifically copurified with a large number of proteins, as expected for a protein involved in a variety of RNP complexes. The experiment was repeated after treatment of the cytoplasmic lysate with RNase A to identify proteins that interact with DDX6 independently of RNA. The silver staining pattern remained highly complex after RNA digestion (Figure 1E, right panel). Complexes purified from both untreated and RNase-treated lysates and from control lysates were then analyzed by mass spectrometry to identify their protein content.
Without considering potential alternative isoforms, 367 and 323 proteins were specifically identified in untreated and RNase-treated DDX6 complexes compared with control lysates (Figure 2A, left panel, and Supplemental Table 1), and the two lists were limited to 299 and 233 proteins represented by at least two peptides, respectively. Based on Gene Ontology (GO) annotation, the vast majority of these were involved in RNA metabolism (GO 0003723): 226 and 112 proteins in untreated and RNase-treated lysates, respectively (GO p values = 10−197 and 10−64). Based on the literature, proteins were then manually annotated as involved in RNA or DNA metabolism, cytoskeleton, or mitochondria, and ranked by Mascot scores (Figure 2A, right panel). The low level of cytoskeletal and mitochondrial proteins confirmed the quality of the purification. Manual annotation and GO analysis indicated that even proteins with the lowest Mascot scores should be considered, as they were highly enriched for proteins involved in RNA metabolism (GO p values = 10−33 and 10−11, for untreated and RNase-treated lysates, respectively; Figure 2B). Compared with untreated lysates, RNase treatment severely decreased the number of proteins with high and middle Mascot scores (Figure 2A, left panel).
FIGURE 2:

Functional description of the proteins identified by mass spectrometry. (A) Proteins purified from lysates treated with RNase inhibitor (− RNase) or RNase (+ RNase) were ranked by Mascot score and arbitrarily divided into three groups of high, medium, and low scores, as shown in the left panel. Their distribution into functional categories is based on the literature, and shown in the right panel. (B) RNA-binding proteins were searched using gene ontology. The table presents for each group (all, high, medium, low Mascot scores), the number of proteins annotated in the GO database (GO annotated), the number of RNA-binding annotated proteins (GO 0003723), and the p value associated with their enrichment. (C) Proteins categorized in RNA metabolism in A were subcategorized into indicated functions based on the literature.
To further refine the functional description of DDX6 partners, we subclassified proteins involved in RNA metabolism into categories of mRNA decay, mRNA repression or localization, cap or poly(A)-binding proteins, ribosomal proteins, translation initiation or elongation factors, and splicing, based on the literature (Figure 2C). Even though arbitrary choices had to be made for proteins which were involved in several pathways, in particular to distinguish decapping activators and translational repressors as detailed in the introduction, it indicated three major classes of partners in terms of enrichment: decay factors, repression/localisation factors, and ribosomal proteins. Among the three, ribosomal proteins were the most affected by RNase treatment.
DDX6 in mRNA-decay complexes
Overall 28 DDX6 partners were classified in mRNA decay: 27 were present in untreated lysates (21 with scores > 160), and 17 in RNase-treated lysates (12 with scores > 160). While Mascot scores do not faithfully reflect protein abundance, we considered that the top list was likely to contain at least some components of the most abundant DDX6 complexes. In fact, four proteins of the mRNA decapping complex were present in the top 20 partners: EDC4, EDC3, DCP1A, and DCP1B (Table 1). Furthermore, all other components were present further down the list: XRN1, DCP2, and LSM1-7 (except LSM5; Table 2). XRN2 was also present, though with a lower score than XRN1. In contrast, proteins of the 3′ to 5′ mRNA degradation pathway were almost absent, whether components of the exosome or of the CCR4-NOT complex (Table 2). Similarly, apart from UPF1, which was abundantly present in untreated lysates, proteins of the nonsense-mediated decay pathway were almost absent (Table 2).
TABLE 1:
Top DDX6 partners.
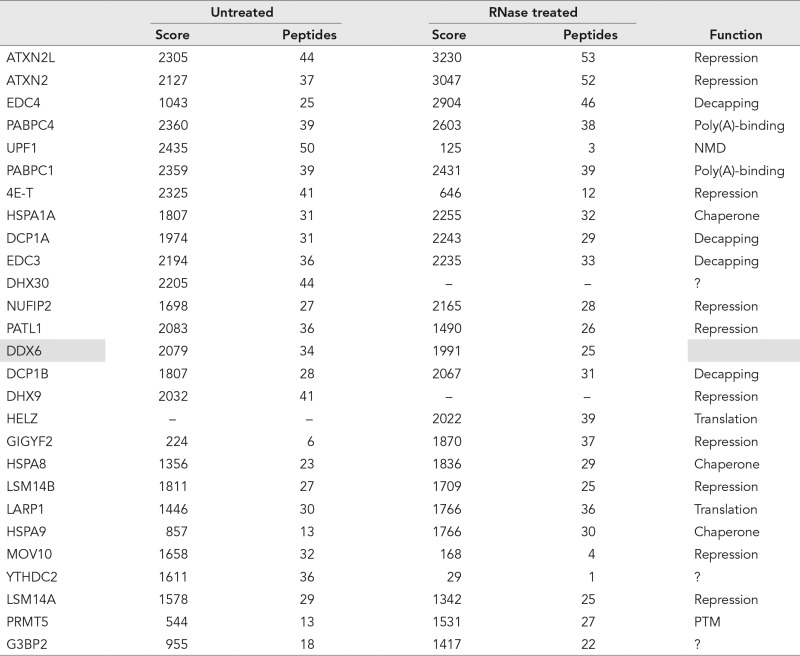 |
The 20 proteins with highest Mascot scores in either untreated or RNase-treated lysates are listed. The indicated function is based on the literature. Note that some of the proteins classified in translational repression are also decapping activators. NMD: Nonsense-mediated decay; PTM: posttranslational modification.
TABLE 2:
Functional classification of DDX6 partners.
| Untreated | RNase treated | |||||
|---|---|---|---|---|---|---|
| Score | Peptides | Score | Peptides | Pathway/complex | ||
| mRNA decay | DCP1A | 1974 | 31 | 2243 | 29 | 5′, 3′ mRNA decay |
| DCP1B | 1807 | 28 | 2067 | 31 | ||
| EDC3 | 2194 | 36 | 2235 | 33 | ||
| EDC4 | 1043 | 25 | 2904 | 46 | ||
| XRN1 | 851 | 22 | 316 | 8 | ||
| XRN2 | 119 | 4 | – | – | ||
| DCP2 | 82 | 3 | 298 | 7 | ||
| LSM1 | 244 | 4 | 153 | 3 | ||
| LSM2 | 389 | 7 | 325 | 5 | ||
| LSM3 | 58 | 2 | 34 | 1 | ||
| LSM4 | 294 | 5 | 214 | 5 | ||
| LSM6 | 207 | 5 | 158 | 4 | ||
| LSM7 | 101 | 2 | 111 | 2 | ||
| CNOT6L | – | – | 30 | 1 | 3′, 5′ mRNA decay | |
| UPF1 | 2435 | 50 | 125 | 3 | NMD | |
| UPF3B | 39 | 1 | – | – | ||
| SMG6 | 149 | 4 | – | – | ||
| eIF4A3 | 592 | 14 | – | – | EJC core | |
| MAGOH | 193 | 5 | – | – | ||
| MLN51 | 187 | 4 | – | – | ||
| Y14 | 77 | 2 | – | – | ||
| Translational repressors | 4E-T | 2325 | 41 | 646 | 12 | CPEB complex |
| PAT1B | 2083 | 36 | 1490 | 26 | ||
| LSM14B | 1811 | 27 | 1709 | 25 | ||
| LSM14A | 1578 | 29 | 1342 | 25 | ||
| ATXN2L | 2305 | 44 | 3230 | 53 | miRNA pathway | |
| ATXN2 | 2127 | 37 | 3047 | 52 | ||
| MOV10 | 1658 | 32 | 168 | 4 | ||
| AGO2 | 104 | 3 | – | – | ||
| GIGYF2 | 224 | 6 | 1870 | 37 | eIF4E2-BP | |
| NUFIP2 | 1698 | 27 | 2165 | 28 | FMRP complex | |
| FMRP | 432 | 8 | – | – | ||
| FXR2 | 618 | 11 | – | – | ||
| FXR1 | 403 | 8 | – | – | ||
| Miscellaneous | PABPC4 | 2360 | 39 | 2603 | 38 | Poly(A)- binding |
| PABPC1 | 2359 | 39 | 2431 | 39 | ||
| PABPC3 | 1131 | 20 | 1206 | 21 | ||
| PABPN1 | – | – | 78 | 2 | ||
| LSM12 | 430 | 7 | 591 | 7 | ATXN2 associated | |
| DHX30 | 2205 | 44 | – | – | Helicases | |
| DHX9 | 2032 | 41 | – | – | ||
| HELZ | – | – | 2022 | 39 | ||
| YTHDC2 | 1611 | 36 | 29 | 1 | ||
| HSPA1A | 1807 | 31 | 2255 | 32 | Chaperone and regulators | |
| HSPA8 | 1356 | 23 | 1836 | 29 | ||
| HSPA9 | 857 | 13 | 1766 | 30 | ||
| HSPA5 | 1234 | 20 | 1312 | 21 | ||
| HSPH1 | – | – | 161 | 4 | ||
| BAG4 | 257 | 5 | 406 | 7 | ||
| BAG2 | – | – | 75 | 2 | ||
| G3BP2 | 955 | 18 | 1417 | 22 | G3BP complex | |
| G3BP1 | 548 | 10 | 809 | 15 | ||
| CAPRIN1 | 185 | 3 | – | – | ||
| LARP1 | 1446 | 30 | 1766 | 36 | TOP mRNA translation | |
| PRMT5 | 544 | 13 | 1531 | 27 | PTM | |
| NCBP1 | 1011 | 17 | – | – | Nuclear CBC and associated proteins | |
| NCBP2 | 237 | 4 | – | – | ||
| DDX3 | 106 | 3 | – | – | ||
| ZC3H18 | 192 | 6 | – | – | ||
| ARS2 | 100 | 4 | – | – | ||
| eIF4E2 | 112 | 2 | 268 | 7 | eIF4F | |
| eIF4E1 | 181 | 5 | 189 | 3 | ||
| eIF4G | 43 | 2 | 376 | 11 | ||
| eIF4B | 38 | 1 | 45 | 1 | ||
| eIF4A1 | – | – | 56 | 2 | ||
The 20 proteins with highest Mascot scores are in bold.
To circumvent potential artificial interactions resulting from transgene expression, we then performed DDX6 immunoprecipitation experiments using untransfected cells (Figure 3A). Anti-DDX6 antibodies coimmunoprecipitated EDC3, EDC4, DCP1A, and XRN1 efficiently in the presence or absence of RNase, though to various extents. The data were then quantitated to estimate the fraction of these proteins associated with DDX6. While weak coimmunoprecipitations are inconclusive, as the interactions could be lost during purification, efficient ones should reveal proteins that are mostly bound to DDX6. Strikingly, a major fraction of the EDC3 and DCP1A pools, but little EDC4, was associated with endogenous DDX6 in an RNA-independent manner. RNA digestion reproducibly enhanced EDC4 interaction, possibly due to an increased accessibility of the DDX6 epitope in the absence of RNA. Only the interaction with XRN1 was partly mediated by RNA. In agreement with previous reports, these four proteins were strongly enriched in P-bodies, with nevertheless some signal in the cytosol, as for DDX6 (Supplemental Figure 1A).
FIGURE 3:
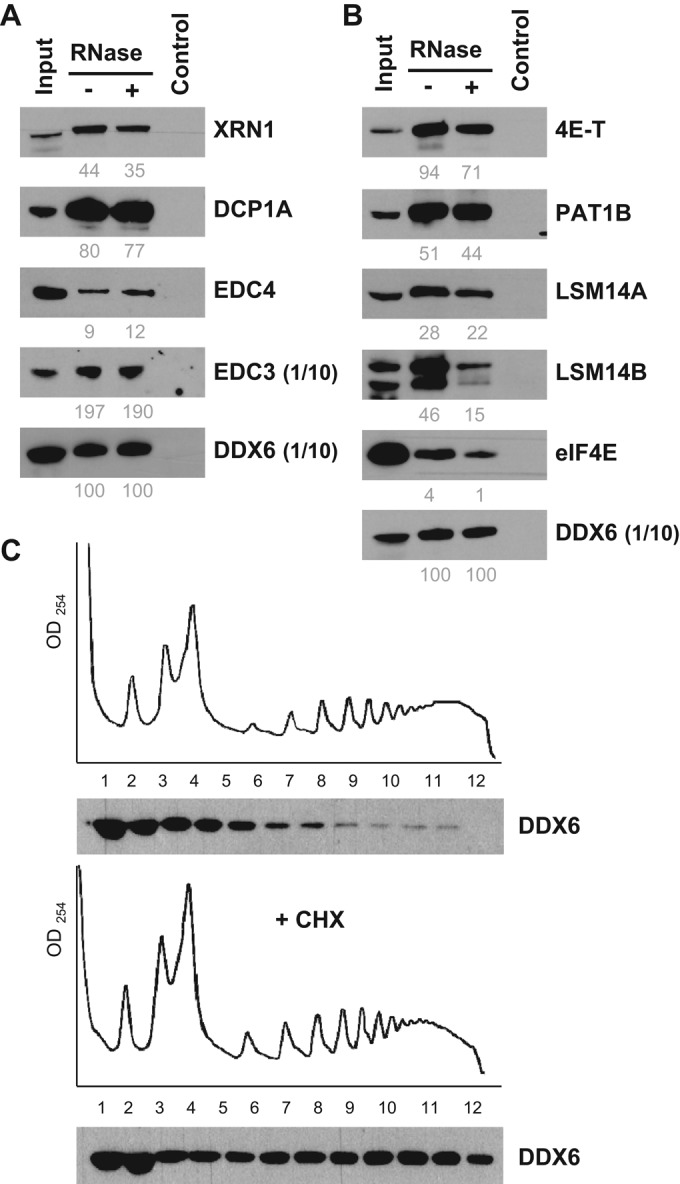
Characterization of decay and repression complexes in untransfected cells. (A and B) HEK293 cytoplasmic lysates were immunoprecipitated with anti-DDX6 antibodies and analyzed by Western blotting for components of (A) decapping and (B) repression complexes, as indicated. For DDX6 and EDC3, only one-tenth of the immunoprecipitate was loaded on the gel to avoid saturation. For facilitation of comparison, the immunoprecipitation efficiency was estimated from scanned images, as described in Materials and Methods, and indicated below as percent values. (C) Cytoplasmic lysates from HEK293 incubated or not with cycloheximide (+ CHX) were separated by centrifugation on sucrose gradients and analyzed by optical densitometry (top panels). Proteins from collected fractions were analyzed by Western blotting with anti-DDX6 antibodies (bottom panels).
DDX6 in translational repression complexes
Overall 50 DDX6 partners were classified in mRNA repression and/or localization: 48 were present in untreated lysates (41 with scores > 160) and 20 in RNase-treated lysates (17 with scores > 160). Four proteins characteristic of the CPEB1 complex were present in the top 20 list: 4E-T, PAT1B, LSM14A, and LSM14B (Table 1). Nevertheless, CPEB1 itself was not detected, possibly reflecting its relatively low abundance in HEK293 cells. We will therefore refer to this complex as the CPEB-like complex hereafter. The top list also included NUFIP2, which binds FMRP. Besides FMRP itself, other FMRP-associated proteins were present lower down the list, including FXR1 and FXR2 (Table 2). Finally, there were two eIF4E-binding proteins, 4E-T (also in the CPEB1 complex; Kamenska et al., 2014) which interacts with both eIF4E1 and eIF4E2, and GIGYF2 specific to eIF4E2 (Morita et al., 2012). Consistent with DDX6 association with repressed mRNAs, most translation initiation factors were absent. The only exceptions were the eIF4F components, albeit with medium to low scores. Immunoprecipitation experiments using untransfected cells confirmed the efficient copurification of the major fraction of 4E-T, PAT1B, LSM14A, and LSM14B with DDX6. Contrary to decay factors, all interactions were partly sensitive to RNase treatment (Figure 3B). In immunofluorescence, the five proteins were strongly enriched in P-bodies (Supplemental Figure 1B). In contrast, only a minor fraction of eIF4E1 coprecipitated with DDX6, and eIF4E1 was modestly enriched in P-bodies, which is consistent with its main role in active translation initiation.
Surprisingly, a large number of ribosomal proteins were also specifically present in DDX6 complexes (Figure 2C). Although ribosomal proteins are common contaminants of purified complexes, they were unexpected at such levels. Indeed, 58 proteins of the 40S and 60S subunits were present, including 32 with Mascot scores above 160. RNase treatment strongly decreased both their number and abundance: 36 proteins were left, including only six with scores above 160, without bias toward 40S or 60S subunits. We therefore analyzed the distribution of endogenous DDX6 in polysomes using sucrose gradients (Figure 3C, top panel). While a large part of the protein was present in subpolysomal fractions, some was spread across the entire gradient. For ensuring that these heavy complexes were polysomes, cells were incubated with cycloheximide for 30 min before lysis to specifically accumulate polysomes (Figure 3C, bottom panel). The amount of DDX6 increased in the heavy fractions, confirming that a fraction of DDX6 was associated with polysomes.
DDX6 in ATXN2 and ATXN2L complexes
Interestingly, both ATXN2 and ATXN2L were in the top 20 proteins interacting with DDX6 (Table 1). These related proteins contain an LSm domain. They were shown to interact together and with PABP, and ATXN2 also interacts with LSM12 (Nonhoff et al., 2007; Kaehler et al., 2012). In fact, PABPC1 and PABPC4 were also in the top list, while PABPC3 and LSM12 were present further down the list (Table 2). ATXN2 is functionally required for miRNA function in Drosophila cells (McCann et al., 2011). A second regulator of the miRNA pathway, MOV10, was also in the top 20 list (Table 1; Liu et al., 2012). Nevertheless, with the exception of a small amount of AGO2, core proteins of the RISC complex were absent, including TNRC6A, 6B, and 6C and AGO1, -3, and -4 proteins. DDX6 interaction with ATXN2, ATXN2L, PABPC1, and LSM12 was confirmed in untransfected cells by coimmunoprecipitation (Figure 4A). It was fully resistant to RNase, except for LSM12 which was partly RNA-mediated. RNA digestion reproducibly enhanced PABPC1 interaction, as observed for EDC4. In contrast to proteins of the decapping and CPEB1 complexes, none of these proteins localized to P-bodies (Supplemental Figure 1C). Therefore DDX6 participates in at least three major complexes: the decapping complex and the repression complex, both of which can be recruited into P-bodies, and an ATXN2/ATXN2L complex, which is excluded from P-bodies.
FIGURE 4:

Characterization of the ATXN2/2L complex and EJC in untransfected cells. (A) Components of the ATXN2/2L complex were analyzed as in Figure 3, A and B. (B) HeLa cells were transfected with indicated siRNAs. After 48 h, cells were stressed with arsenite for 30 min. Localization of ATXN2, ATXN2L, and DDX6 in stress granules was analyzed by immunofluorescence, using TIA1 as a stress granule marker. Scale bar: 10 μm. (C) Components of the EJC were analyzed as in A.
In stress conditions, ATXN2 and ATXN2L are recruited with repressed mRNAs into a distinct type of granule, called stress granules (Nonhoff et al., 2007; Kaehler et al., 2012). As some DDX6 also relocates to stress granules, we investigated whether their recruitment could be coordinated. For this experiment, we used HeLa cells, which are flatter than HEK293 cells and thus more appropriate for cell imaging. Cells were transfected with siRNAs targeting ATXN2 or ATXN2L, and after 48 h, stress granules were induced with arsenite. The silencing was efficient, as verified by immunostaining of ATXN2 and ATXN2L (unpublished data). In control cells, ATXN2 and ATXN2L were recruited in stress granules, as expected, while DDX6 was found in both P-bodies and stress granules (Figure 4B). Similar results were observed following ATXN2 silencing. However, when ATXN2L was depleted, DDX6 was exclusively localized in P-bodies. These results supported the DDX6-ATXN2L interaction in vivo and unexpectedly showed that ATXN2L is required for the recruitment of DDX6 to stress granules.
DDX6 complexes in oocytes
To evaluate cell-specific variations of DDX6 complexes, we conducted a similar experiment in Xenopus oocytes. Several hundred stage VI oocytes were injected with in vitro transcribed RNA encoding FLAG-MS2-tagged DDX6 or tags alone. After 48 h, DDX6 RNP complexes were immunoprecipitated with anti-FLAG antibodies, and proteins were identified by mass spectrometry (Supplemental Table 2). Among the 27 proteins specifically present in oocytes, 19 were directly related to mRNA metabolism. From these, 14 were also found in HEK293 cells, either identically or in their somatic version. Among the five absent proteins, some were in fact oocyte specific (Zar1, Zar2, CPEB1). In a second experiment, oocytes were matured by treatment with progesterone before DDX6 RNP purification (Supplemental Table 2). Of the 24 proteins identified, 20 were related to mRNA metabolism, and 16 of these were also found in HEK293 cells.
In the two experiments, the highest Mascot scores were found for proteins involved in translational repression, while proteins involved in decapping came second. The top repressive proteins were components of the CPEB1 complex, including Lsm14B, Pat1a, and 4E-T. In oocytes, the decapping complex was only partially represented, with most proteins of the Lsm1-7 complex, but neither Dcp1a, Dcp1b, Dcp2 proteins nor Edc4. Edc3 was only detected after progesterone treatment. Instead, a distinct decapping enzyme, Nudt16, was present (Ghosh et al., 2004). As quantitative proteomic analysis indicates that Dcp1a, Dcp1b, and Edc4 are nevertheless expressed in Xenopus eggs as in human epithelial cells (Supplemental Table 2; Nagaraj et al., 2011; Wühr et al., 2014), these results raise the possibility that DDX6 participates in a different decapping complex in oocytes compared with epithelial cells. Finally, there was no evidence of any Atxn2 or Atxn2L complex, and Pabps were also absent (Supplemental Table 2). Again, these proteins are nevertheless expressed, at least in eggs. Thus, while DDX6 partners have similar types of function in oocytes and in epithelial cells, there are important cell-specific variations, which include in particular the nature of the decapping complex and the ATXN2/2L complex.
DDX6 complexes and EJC
DDX6 is a cytoplasmic protein, and the TAP-tag experiment was performed with cytoplasmic extracts of HEK293 cells. Surprisingly, the four core proteins of the EJC (exon junction complex) were found: eIF4A3, MLN51, MAGOH, and Y14 (Table 2). The EJC is thought to be removed in the cytoplasm at the first round of translation, upon ribosome scanning. A second concurrent event is the replacement of the nuclear poly(A)-binding protein PABPN1 by its cytoplasmic counterparts PABPC1-4, and the replacement of the nuclear cap-binding complex NCBP1/NCBP2 by its cytoplasmic counterpart eIF4E1 or eIF4E2. Strikingly, while a small amount of PABPN1 was detected, both NCBP1 and NCBP2 were present with scores that were even higher than the eIF4E1 and eIF4E2 proteins.
The interactions with EJC proteins, PABPN1, and NCBP1 were confirmed by coimmunoprecipitation in untransfected cells (Figure 4C). The interactions with the EJC and NCBP1 were fully RNA mediated, while PABPN1 was partly RNA independent. These results raised the possibility that DDX6 binds mRNAs not only following translational repression but at the nuclear exit, before any translation. Nevertheless, in contrast to the repressors above, none of these proteins localized to P-bodies, except for a small amount of MLN51 (Supplemental Figure 1D).
Role of DDX6 partners in P-body maintenance
To rank DDX6 partners in terms of requirement for P-body maintenance, we compared the effect of silencing DDX6 and some of its main partners on P-bodies. HeLa cells were silenced for 48 h with siRNA targeting DDX6 partners, including EDC3 and EDC4, PAT1B, 4E-T, LSM14A and LSM14B, and ATXN2 and ATXN2L. For each transfection, the silencing efficiency was confirmed by Western blot analysis (Figure 5A). In parallel, P-bodies were detected by immunostaining of EDC4 or DDX6, depending on the silenced protein and its antibody host species (Figure 5B). Silencing 4E-T or LSM14A suppressed P-bodies as efficiently as did silencing DDX6, in agreement with previous reports (Ferraiuolo et al., 2005; Chu and Rana, 2006; Yang et al., 2006). Depleting PAT1B led to a 50% decrease of P-body number (Figure 5C), as previously reported (Marnef et al., 2010). The decrease after silencing EDC4 was only 24%, indicating a minor contribution of EDC4 to P-body assembly. This is in contradiction to a previous report (Yu et al., 2005) and may be related to the use of different P-body markers. ATXN2L silencing reproducibly led to a minor decrease, which was lower than previously reported (Kaehler et al., 2012), possibly due to the shorter silencing duration. Finally, silencing EDC3 and ATXN2 did not impact P-bodies, while depleting LSM14B even increased their number. The absence or decrease of P-bodies was confirmed by immunostaining of a second P-body marker, XRN1 (Supplemental Figure 2A). These results pointed to a major role of 4E-T and LSM14A along with DDX6 in the maintenance of P-bodies, and to an accessory role of PAT1B and EDC4.
FIGURE 5:

Role of P-body proteins in the maintenance of P-bodies. (A–C) HeLa cells were transfected with indicated siRNAs. After 48 h, (A) proteins were analyzed by Western blotting with indicated antibodies, and (B) P-bodies were analyzed by immunofluorescence with anti-EDC4 or DDX6 antibodies, along with antibodies directed against the silenced protein to check the silencing at the individual cell level (unpublished data). Scale bar: 10 μm. (C) P-bodies were counted in three independent experiments, and their average number per cell was plotted. **, p < 0.005. (D and E) After transfection of siLSM14A and siLSM14B, proteins were analyzed by Western blotting using indicated antibodies (D). Signals were quantified in three independent experiments and plotted (E).
In view of their strong homology and their similar localization in P-bodies, the opposite effect of silencing LSM14A and LSM14B was unexpected. When analyzing the efficiency of LSM14A and LSM14B silencing by Western blot, we observed a cross-regulation of the two proteins (Figure 5D). Silencing LSM14B led to an almost twofold increase of LSM14A protein, and conversely, depleting LSM14A enhanced expression of LSM14B (Figure 5E). This regulation did not extend to other P-body proteins such as DDX6 (Figure 5D). Thus the induction of P-bodies observed after LSM14B silencing could be the result of increasing LSM14A abundance.
LSM14A interacts directly with DDX6 through its FDF motif, and this interaction is prevented by a mutation of the FDF-binding pocket (Mut1; Tritschler et al., 2009). To determine whether this pocket was involved in P-body assembly, we used a complementation assay previously set up to demonstrate the requirement for DDX6 helicase motifs (Minshall et al., 2009). In the presence of endogenous DDX6, Mut1 DDX6 localized to P-bodies similar to the wild-type protein (Supplemental Figure 3A). When P-bodies were suppressed by siDDX6 transfection before plasmid transfection, Mut1 DDX6 was two times less potent than wild-type DDX6 at assembling P-bodies (Supplemental Figure 3B). Thus the Mut1 mutation partially impairs the capacity of DDX6 to assemble P-bodies. The FDF-binding pocket of DDX6 binds several ligands, including LSM14A, but also PAT1B and EDC3 (Tritschler et al., 2009; Sharif et al., 2013). As EDC3 was dispensable for P-bodies (Figure 5C), these results suggested that a direct interaction between DDX6 and LSM14A or PAT1B facilitated P-body assembly.
Role of DDX6 partners in de novo P-body assembly
We have shown previously that, besides being required for the maintenance of pre-existing P-bodies, DDX6 is also required for the induction of new P-bodies following arsenite treatment (Serman et al., 2007; Minshall et al., 2009). To address the same question for its partners, we transfected HeLa cells with various siRNAs and treated them with arsenite for 30 min to induce new P-bodies. The action of arsenite was controlled by immunostaining of ATXN2 (Supplemental Figure 2B). The silencing was efficiently maintained during arsenite treatment, as controlled by Western blotting (Supplemental Figure 2D). P-bodies were detected by immunostaining of EDC4 or LSM14A, depending on the silenced protein (Figure 6A). In control cells and in cells transfected with siRNAs targeting EDC3, arsenite treatment led to the doubling of P-body number (Figure 6, A and B, compared with Figure 5, B and C). The same was observed after PAT1B silencing, despite the decrease of P-bodies observed before arsenite addition. Thus PAT1B was dispensable for de novo P-body assembly. In contrast, silencing 4E-T prevented P-body reassembly, as did DDX6 depletion. Silencing EDC4 and LSM14A was partially inhibitory but with distinct outcomes. Normal-sized P-bodies still formed in the absence of EDC4 but not in the absence of LSM14A, as evidenced when quantitation was restricted to large P-bodies (Figure 6B). Overall these results indicated that DDX6, 4E-T, and, to a lesser extent, LSM14A were required for P-body assembly in response to arsenite, whereas PAT1B and EDC3 were dispensable, and EDC4 only facilitated assembly.
FIGURE 6:

Role of P-body proteins in P-body assembly after arsenite treatment. HeLa cells were transfected with indicated siRNAs and analyzed as in Figure 5, except that cells were treated with arsenite for 30 min before fixation. P-bodies were immunostained (A) and counted (B) as in Figure 5 (all), except that LSM14A antibodies were used in place of DDX6, as this accumulated in both stress granules and P-bodies. A second quantitation restricted to P-bodies larger than 450 nm was performed to show the characteristic defect observed after silencing LSM14A (large). Scale bar: 10 μm. The corresponding Western blot analysis is presented in Supplemental Figure 2D.
How arsenite induces P-bodies is unknown. As the drug drastically blocks translation, P-body induction may result from the substantial release of untranslated mRNAs. To investigate whether DDX6, 4E-T, and LSM14A are involved more generally in P-body assembly, we used inducers of different nature. Vinblastine is an antimicrotubule drug reported to induce P-body assembly (Aizer et al., 2008). Its action was controlled by monitoring the appearance of characteristic spiral aggregates of tubulin (Supplemental Figure 2C). In addition, we found that a mild cold shock at 30°C for 2 h is also a P-body inducer, as strong as vinblastine or arsenite treatment (Figure 7A). The three inducers were compared in terms of translation by analyzing polysomes on sucrose gradients (Figure 7B). In contrast to arsenite, vinblastine and 30°C had only moderate effects on polysomes, in agreement with previous reports (Dresios et al., 2005; Carbonaro et al., 2011). The silencing experiments described above were repeated using these new conditions, and the silencing efficiency was controlled by Western blot analysis (Supplemental Figure 2D). In both vinblastine and 30°C conditions, DDX6, 4E-T, and LSM14A were required for P-body induction (Figure 8). On vinblastine treatment, EDC4 facilitated P-body assembly, whereas PAT1B was dispensable. On mild cold shock, both EDC4 and PAT1B facilitated P-body assembly.
FIGURE 7:

P-body induction after treatment with arsenite, vinblastine, and mild cold shock. HeLa cells were treated with arsenite for 30 min, vinblastine for 1 h, or cultured at 30°C for 2 h before lysis or fixation. (A) P-bodies were detected and counted as in Figure 5. Scale bar: 10 μm. (B) Cytoplasmic lysates were separated on sucrose gradients and analyzed by optic densitometry, as in Figure 3C.
FIGURE 8:

Role of P-body proteins in P-body assembly after vinblastine treatment and mild cold shock. HeLa cells were transfected with indicated siRNAs and analyzed, as in Figure 5, except that cells were treated with vinblastine (A) or cultured at 30°C (B) before fixation. P-bodies were counted as in Figure 5 (C). Scale bar: 10 μm. The corresponding Western blot analysis is presented in Supplemental Figure 2D.
In conclusion, in all conditions, the assembly of new P-bodies required DDX6 and 4E-T. LSM14A was also required after vinblastine and 30°C treatments, while after arsenite, very small granules were still induced in its absence. In contrast, EDC3 was dispensable, and EDC4 only facilitated P-body assembly. The requirement for PAT1B depended on the inducer: dispensable after arsenite and vinblastine, it facilitated P-body assembly at 30°C. Overall proteins of the repression complex appeared to be more important for P-body assembly than cofactors of the decapping complex.
DISCUSSION
TAP-tag analysis highlighted the decapping, the CPEB-like, and the ATXN2/2L complexes
Characterization of the DDX6 interactome in human epithelial cells was a starting point to understand DDX6 requirement for P-body assembly. We found a high number of interacting proteins (more than 200) that should be looked at in relation to DDX6 abundance. We showed previously that DDX6 concentration is ∼3.4 μM in HeLa cells and in a ∼30-fold excess over its partner, EDC3 (Ernoult-Lange et al., 2012). According to available quantitative proteomic data (Nagaraj et al., 2011), it is in excess over most of its partners (Supplemental Table 3), indicating the coexistence of a variety of DDX6 complexes. The top 20 partners included at least three groups of proteins belonging to the decapping complex, a CPEB-like repression complex, and an ATXN2/2L complex, which were confirmed in untransfected cells by coimmunoprecipitation with endogenous DDX6. Overall DDX6 interaction with the CPEB-like complex was partly RNA mediated, while interaction with proteins of the decapping complex and the ATXN2/2L complex was independent of RNA. While these RNA-independent interactions might correspond to complexes sitting on mRNAs as well as free in the cytosol, the increased EDC4 and PABPC1 association observed after RNA digestion suggests that these complexes are bound to RNA. Strikingly, a major fraction of EDC3 and DCP1A in the decapping complex and 4E-T in the CPEB-like complex was associated with DDX6.
When DDX6 was isolated from oocytes, it copurified with proteins of the CPEB1 complex, as expected from our previous studies (Minshall et al., 2007, 2009). Interestingly, also predicted was the absence of decapping enzymes, including Dcp2 and its cofactors Dcp1, Edc3, and Edc4, as decapping activity is undetectable in oocytes and only robustly observed in mid-embryogenesis as zygotic transcription is triggered (Gillian-Daniel et al., 1998; Zhang et al., 1999), compatible with the long-term storage of oligoadenylated maternal mRNAs in oocytes. However, DDX6 pulled down the more recently described decapping enzyme Nudt16, raising the possibility that oocytes contain an inactive form of this enzyme bound to DDX6 to be activated following oocyte maturation and fertilization. A second major difference was the absence of the ATXN2/2L complex, though it expressed in the cells, suggesting that these proteins have a different function in oocytes that does not require DDX6.
For convenience, we described the decapping complex and the repressor complex as distinct. Crystal structures have revealed that some proteins of the two complexes bind DDX6 in a mutually exclusive manner. This is the case for LSM14A and EDC3 (Tritschler et al., 2009) and EDC3 and PAT1B (Sharif et al., 2013). Nevertheless, the capacity of DDX6 to oligomerize (Ernoult-Lange et al., 2012) raises the possibility that oligomers accommodate these partners simultaneously. Moreover, some proteins like PAT1B have an uncertain status and might be shared between the two complexes. Finally, multiple DDX6 binding to repressed RNAs (Ernoult-Lange et al., 2012) should also allow loading of distinct complexes to the same mRNAs.
Furthermore, some DDX6 was also found on polysomes. While at first glance this seems to contradict a function in repression or decay, this observation is consistent with reports in yeast showing some polysomial DDX6 under diauxic growth shift conditions (Drummond et al., 2011) or after mild cross-linking (Sweet et al., 2012). The latter study concluded that DDX6 triggers cotranslational decapping. Nevertheless, the interpretation of the experiment may be more complex if DDX6 simultaneously participates in a variety of complexes of different function. For instance, its function in polysomes might be related to the ATXN2/2L complex rather than to the decapping complex, as ATXN2 too is associated with polysomes (Satterfield and Pallanck, 2006). After cycloheximide treatment, we observed a striking accumulation of DDX6 in polysomes that was stronger than the accumulation of the polysomes themselves as monitored by optical density. Recently Presnyak et al. (2015) showed in yeast that the presence of suboptimal codons leads to mRNA destabilization in addition to slow translational elongation. They proposed that Dhh1/DDX6 could be the sensor for these inefficient polysomes. One interesting possibility would be that cycloheximide-arrested polysomes actively recruit DDX6 in the same manner as suboptimal codon-containing polysomes, leading to extra accumulation of DDX6 in the polysomal fractions.
Only some DDX6 complexes are targeted to P-bodies
We estimated that only 6% of DDX6 is localized in P-bodies (Souquere et al., 2009; Ernoult-Lange et al., 2012). This is nevertheless high compared with other P-body proteins such as Ago2 (1%; Leung and Sharp, 2013). As a consequence, the TAP-tag strategy identified complexes that are mostly cytosolic even if, like DDX6 itself, they can be targeted to P-bodies. In fact, while many of the top DDX6 partners were known components of P-bodies, including proteins of the decapping and CPEB-like complexes, some were excluded from P-bodies, like ATXN2, ATXN2L, and PABPs, demonstrating the existence of two types of DDX6 complexes, which differ by their capacity to be recruited in P-bodies.
The biological meaning of DDX6 interaction with ATXN2 and ATXN2L is unknown. Although their interaction was reported previously (Nonhoff et al., 2007; Kaehler et al., 2012), their top ranking in the mass spectrometry analysis was unexpected. ATXN2L is almost as abundant as DDX6 (Supplemental Table 3). In stressed cells, its silencing was sufficient to prevent DDX6 recruitment to stress granules, suggesting that an important fraction of the DDX6 protein distributed in the cytosol was engaged in ATXN2L complexes. Reciprocally, coimmunoprecipitation experiments indicated that an important fraction of ATXN2L was engaged in DDX6 complexes. Despite possible ATXN2-ATXN2L interactions (Kaehler et al., 2012), ATXN2 silencing did not have a strong effect on DDX6 localization, possibly reflecting its lower expression in the cells (sevenfold; Supplemental Table 3). In Drosophila, ATXN2 is required for optimal repression of several miRNA targets (McCann et al., 2011) and is involved in long-term olfactory habituation via a mechanism depending on RISC and DDX6. Paradoxically, it also activates translation of the clock component PERIOD, but via a DDX6-independent mechanism (Lim and Allada, 2013; Zhang et al., 2013). In our analysis, the RISC complex and the CCR4/NOT complex were almost absent. Yet, DDX6 interactions with AGO1, AGO2, and NOT1 and their functional consequence on the miRNA pathway have been demonstrated by others (Chu and Rana, 2006; Chen et al., 2014; Mathys et al., 2014; Rouya et al., 2014). Again, their absence in our experiment is likely to result from their lower expression compared with DDX6 (50-fold for AGO proteins, 500-fold for NOT1; Supplemental Table 3). Any such interactions would be diluted by the most abundant decay and CPEB-like and ATXN2/2L complexes. Finally, if the DDX6/ATXN2/2L complex is involved in miRNA-mediated repression, it clearly dissociates before repressed mRNAs enter P-bodies. Most models schematizing the role of DDX6 in mRNA metabolism represent translational repression and mRNA decay downstream of translation, seemingly in contradiction to the presence of the EJC and the nuclear cap and poly(A)-binding proteins among DDX6 partners reported here. As these interactions were fully RNA mediated, they are probably not involved in the recruitment of DDX6 to the RNA. Nevertheless, these interactions indicated that DDX6 can also bind mRNAs at their exit from the nucleus, before the first round of translation. In germ cells, the extensive storage of maternal mRNAs most likely precedes their translation. Some repressed mRNAs are stored in DDX6-containing germ granules that are associated with nuclear pores, such as human chromatoid bodies (Nagamori and Sassone-Corsi, 2008), Drosophila sponge bodies (Snee and Macdonald, 2009), and C. elegans P-granules (Pitt et al., 2000). In HEK293 cells, however, the EJC is not found in P-bodies, suggesting that these mRNAs stored outside of P-bodies or rapidly enter translation.
Various pathways sharing common factors for P-body assembly
Studies on P-body assembly have been quite confusing so far. First, little attention has been given to partial versus complete requirements. Then, a puzzling observation was that proteins required at first instance for maintaining P-bodies can be fully dispensable upon treatments that induce P-bodies (Serman et al., 2007). As the latter was observed only using an aggressive drug, arsenite, we studied two additional inducers that decreased polysomes only partially, vinblastine, previously used by others (Aizer et al., 2008), and a mild cold shock at 30°C, newly reported in this study. The ATXN2/2L complex did not seem to participate directly in P-body assembly, although overexpressed ATXN2 and ATXN2L was previously shown to indirectly modulate P-bodies by sequestration of DDX6 out of P-bodies (Sweet et al., 2012). The decapping complex also had a minor role compared with DDX6, with no influence of EDC3, as previously found for the decapping enzyme DCP2 itself (Andrei et al., 2005), and a partial requirement for EDC4, which was stronger after arsenite or mild cold shock than after vinblastine. In contrast, three proteins of the CPEB-like complex were necessary to assemble P-bodies in all tested conditions: DDX6, 4E-T, and, to a lesser extent, LSM14A. Indeed, in its absence, tiny P-bodies still formed after arsenite treatment. However, not all components of the CPEB-like complex had such strong effects. PAT1B depletion led to only a partial defect and only in untreated cells or after cold shock, while LSM14B was fully dispensable. Consistently, interfering with the interaction between DDX6 and proteins of the CPEB complex, including LSM14A and PAT1B, impaired P-body assembly. Finally, Lsm14B silencing even induced P-bodies, possibly due to LSM14A up-regulation. While LSM14 has evolved into two paralogues in vertebrates, little is known about their respective function. In Xenopus oocytes, both Lsm14B and Lsm14A are expressed, but only Lsm14B is associated with the CPEB complex (Minshall et al., 2007). In human cells, both were found with similar scores in DDX6 complexes, but only LSM14A is required for P-body assembly.
In summary, we undertook the first comprehensive proteomic study of the important regulatory DDX6 helicase and assessed the role of its interacting partners in P-body assembly. The three identified master genes required for P-body formation in human cells are components of the CPEB-like complex, suggesting that P-body assembly is the result of repressing mRNAs rather than targeting them for decay. Importantly, P-body assembly follows a different pathway in lower eukaryotes. In yeast, it requires limited growth conditions, and DDX6 is dispensable (Teixeira and Parker, 2007), while 4E-T is absent. In human cells, some bona fide P-body proteins, like EDC3 and LSM14B, seem to follow repressed mRNAs passively into P-bodies. Others, like PAT1B and EDC4, participate in P-body assembly only in specific conditions, which most likely reflect the existence of distinct repression pathways. The puzzling differential requirement of some proteins for P-body maintenance and P-body induction, observed here for PAT1B and previously for CPEB1 and GW182 (Serman et al., 2007), may simply reflect such different repression pathways. In contrast, DDX6, 4E-T, and LSM14A, as common factors of all pathways, are excellent candidates to serve as scaffolds during assembly. Indeed, we demonstrated previously that DDX6 has self-oligomerization properties (Ernoult-Lange et al., 2012), and 4E-T and LSM14A have extensive conserved disordered regions that could participate in supramolecular aggregation.
MATERIALS AND METHODS
Cell culture and transfection
Human embryonic kidney HEK293 and epithelioid carcinoma HeLa cells were maintained in DMEM supplemented with 10% fetal calf serum. For P-body induction, cells were cultured with 0.5 mM arsenite (Sigma-Aldrich, Saint-Quentin-Fallavier, France) for 30 min, 10 μM of vinblastine (Sigma-Aldrich) for 1 h, or at 30°C for 2 h.
HEK293 cells were chosen for tandem-affinity purification because of their high transfection efficiency (close to 90% in our conditions). Cells were transfected with 45 μg plasmid DNA per 150-mm-diameter dish using a standard calcium phosphate procedure. Human DDX6 open reading frame (ORF) was subcloned into pcDNA3-FLAG vector (BamHI/NotI restriction sites) so that FLAG is upstream and in-frame with DDX6. The HA tag was then introduced downstream and in-frame with DDX6 ORF, using the In-Fusion Advantage PCR cloning kit (Clontech, Saint-Germain-en-Laye, France).
Because of HEK293's round morphology, HeLa cells were preferred for P-body imaging following silencing. Cells were transfected at the time of their plating (reverse transfection) with 1.5 μg siRNA (Eurofins, Ebersberg, Germany) per 35-mm-diameter dish using Lipofectamine 2000 (Life Technologies, Saint-Aubin, France) and split in two 20 h later. At 48 h after transfection, cells were fixed for immunofluorescence or harvested for protein preparation.
The siRNA sequences were as follows: si4E-T AGACUCUUCUCCCACUACAdTdT; siATXN2 GGAACCGAAAGGCCAAAUUdTdT; siATXN2L CUUCAACUAUGCUACUAAAdTdT; siDDX6, GGAACUAUGAAGACUUAAAdTdT; siEDC3 UCAGGCAACUCCCAAGAAAdTdT; siEDC4 UGAGCAAAGUGACCAUGAUdTdT; siLSM14A, UCAUGGUCCUGAACAUUGAdTdT;
siLSM14B CUACUGAAGUGGCGCAUAAdTdT; siPAT1B CUAGAAGAUCCAGCUAUUAdTdT.
The complementation assay was performed as described previously (Minshall et al., 2009).
Immunoprecipitation and Western blot analysis
Cytoplasmic proteins were extracted as described previously (Ernoult-Lange et al., 2008). Proteins were separated on a NuPage 4–12% gel (Invitrogen, Life Technologies) and transferred to a polyvinylidene fluoride (PVDF) membrane (Perkin Elmer, Courtaboeuf, France). After blocking in PBS-T (phosphate-buffered saline [PBS], 0,1% Tween-20) containing 5% (wt/vol) nonfat dry milk for 1 h at room temperature, the membrane was incubated with the primary antibody for 1 h at 37°C, rinsed in PBS-T, and incubated with horseradish peroxidase–conjugated secondary antibody for 1 h at room temperature. After washing in PBS-T, immune complexes were detected using the Supersignal West Pico Chemiluminescent Signal kit (Pierce, Life Technologies) and visualized by exposure to CL-XPosure film (Pierce).
Primary antibodies included mouse ATXN2 and G3BP from BD Biosciences; rabbit ATXN2L from Bethyl; goat TIA1 and mouse EDC4, eIF4E, and XRN1 from Santa Cruz Biotechnology (Heidelberg, Germany); goat 4E-T and rabbit LSM12 from Abcam (Paris, France); rabbit LSM14A and mouse PABPC1 from Merck-Millipore (Molsheim, France); rabbit LSM14B and mouse α-tubulin from Sigma-Aldrich; rabbit DDX6 from Novus Biologicals (Bio Techne, Lille, France); and rabbit ribosomal S6 from Cell Signaling Technology (Ozyme, Saint-Quentin-en-Yvelynes, France). Rabbit DCP1A antibody was a kind gift from B. Seraphin (IGBMC, Strasbourg, France), rabbit EDC3 from J. Lykke-Andersen (University of California, San Diego), rabbit PABPN1 from C. Trollet (Institut de Myologie, Paris, France), rabbit PAT1B from N. Standard (University of Cambridge, UK), rabbit eIF4A3 and Y14 from H. le Hir (ENS, Paris, France; Saulière et al., 2012), rabbit MLN51 from C. Tomassetto (IGBMC, Strasbourg, France), and rabbit NCBP1 from E. Izaurralde (MPI, Tuebingen). The secondary antibodies were purchased from Jackson ImmunoResearch Laboratories (Suffolk, UK).
For immunoprecipitations, 2–5 mg of the cytoplasmic extracts was incubated at 4°C for 1 h with 0.5–1.5 μg anti-DDX6 antibodies in lysis buffer (without NP-40). Twenty-five microliters of a 50% slurry of protein A Sepharose (Sigma-Aldrich) was added. After 2 h at 4°C with constant rotation, beads were washed, and associated proteins were eluted in SDS sample buffer. For the EJC proteins, Dynabeads protein A magnetic beads (Life Technologies) were used in place of protein A Sepharose. Immunoprecipitated proteins were migrated along with 15 or 30 μg cytoplasmic lysate. For estimating the fraction of each protein which was immunoprecipitated, signals were quantified from scanned X-ray films using Quantity One software (Bio-Rad, Marnes-la-Coquette, France), standardized for the amount of input and immunoprecipitated proteins loaded on the gel, and expressed as a percentage of DDX6 immunoprecipitated in the same experiment.
Immunofluorescence
Cells grown on glass coverslips were fixed in methanol for 3 min at −20°C. After rehydration, cells were incubated with the primary antibody for 1 h, rinsed with PBS, incubated with the fluorochrome-conjugated secondary antibody for 45 min, and rinsed with PBS, all steps being performed at room temperature. Slides were mounted in Citifluor (Citifluor, London, UK). Microscopy was performed on a Leica DMR microscope (Leica, Heidelberg, Germany) using a 63 × 1.32 oil-immersion objective. Photographs were taken using a Micromax CCD camera (Princeton Scientific Instruments, Monmouth Junction, NJ) driven by MetaMorph software (Molecular Devices, Sunnyvale, CA). Images were processed with ImageJ. For the calculation of DDX6 enrichment in P-bodies, a line was drawn across individual P-bodies, and intensity along this line was plotted using ImageJ software. To quantitate P-bodies, we used the plug-in Spot Detector of the open bioimage informatics platform Icy (http://icy.bioimageanalysis.org; de Chaumont et al., 2012), with a size filtering of 250 and 450 nm for all P-bodies and large P-bodies, respectively.
For Supplemental Figure 3, confocal microscopy was performed on a Leica TCS SP5 inverted confocal microscope (Leica) using an Apochromat 63 × 1.40 oil-immersion objective. Fluorescence signals were acquired in 0.13-μm optical sections. Maximal projections were obtained using ImageJ.
Tandem-affinity purification and mass spectrometry
HEK293 cells were lysed in a buffer containing 50 mM Tris-HCl (pH 7.4), 150 mM NaCl, 1 mM EDTA, 0.5% NP-40, and 1 mM dithiothreitol (DTT), supplemented with a protease inhibitor cocktail without EDTA (Roche Diagnostics, Meylan, France), for 30 min on ice, in the presence of either 65 U/ml RNaseOut recombinant ribonuclease inhibitor (Promega) or 20 μg/ml RNase A (Euromedex). Nuclei and cytoplasm were separated by centrifugation at 500 × g for 10 min at 4°C. Cytoplasmic proteins were quantified by the Coomassie protein assay (Thermo Scientific). Thirty-five milligrams of proteins was mixed with 120 μl of 50% slurry control agarose resin (Thermo Scientific, Life Technologies) to eliminate resin-binding proteins. Cleared extracts were then incubated with 120 μl of 50% slurry of the anti-FLAG M2 affinity gel (Sigma-Aldrich) for 2 h at 4°C. After beads were washed in the lysis buffer lacking NP-40, bound complexes were eluted in 250 μg/ml of M2 peptide (Sigma-Aldrich) for 1 h at 4°C. Complexes were then incubated overnight with 120 μl of 50% slurry of the monoclonal anti-HA agarose (Sigma-Aldrich). After beads were washed, bound proteins were eluted in Laemmli SDS sample buffer. One-tenth of each sample was separated on a Nu-PAGE 4%–12% Bis-Tris gel and stained with the Pierce Silver stain kit (Thermo Scientific). The remainder was briefly migrated on a Nu-PAGE 4%–12% Bis-Tris gel and stained with SimplyBlue SafeStain (Life Technologies). Each lane was cut into six slices, which were processed for liquid chromatography–tandem mass spectrometry (LC-MS/MS) analysis in the Cambridge Center for Proteomics, at the University of Cambridge (UK). A Mascot search algorithm (Matrix Science, London, UK) was used to search against the UniProt human database using a fixed modification of carbamidomethyl (C), a variable modification of oxidation (M), and a peptide tolerance of 25 ppm.
Mass spectrometry of Xenopus oocyte DDX6 immunoprecipitates
MS2-FLAG–tagged DDX6 mRNA and control MS2-FLAG mRNA was in vitro transcribed as described previously (Minshall et al., 2009; Ernoult-Lange et al., 2012), and 50 nl (at 500 ng/μl) was injected into each stage VI oocyte. Approximately 800 oocytes were injected with control mRNA and 1600 with DDX6 mRNA. After 36 h at 18°C, 10 μg/ml (final) progesterone was added to 800 of DDX6 mRNA-injected oocytes to induce meiotic maturation, and lysates from the three sets were prepared at 48-h postinjection. Extracts were made by addition of 6 ml NET buffer (50 mM Tris-HCl, pH 7.5, 150 mM NaCl, 0.5% NP-40, 1 mM EDTA, pH 8.0, 0.25% gelatin, 0.02% NaN3) and clarification by centrifugation at 10,000 × g, 2 × 10 min, 4°C. S10 was bound to 80 μl protein G Sepharose and 8 μl mouse anti-FLAG antibody (Sigma Aldrich) overnight at 4°C. After washing, bound proteins were eluted with 2 × 400 μl 0.1× TBS plus 100 ng/μl 3×FLAG peptide (Sigma-Aldrich) for 2 h at 4°C. Following concentration, samples were briefly electrophoresed into a 15% SDS–PAGE gel, and several slices from each lane were trypsin digested and analyzed by LC-MS/MS in the Cambridge Centre for Proteomics. Peptides were assessed against merged Xenopus laevis and tropicalis databases.
Polysome gradients
Cells were harvested in ice-cold PBS and lysed in ice-cold lysis buffer (20 mM HEPES, pH 7.5, 250 mM KCl, 10 mM MgCl2, 5 mM DTT, 1 mM EDTA, 0.5% NP-40), supplemented with EDTA-free protease inhibitor cocktail (Roche Diagnostics, Meylan, France) and 65 U/ml of RNaseOut ribonuclease inhibitor (Promega), for 5 min. After centrifugation at 500 × g for 5 min to eliminate nuclei, supernatants were layered onto 10–50% sucrose gradients in polyribosome buffer (20 mM HEPES, pH 7.5, 250 mM KCl, 20 mM MgCl2, 2 mM DTT) and centrifuged at 39,000 rpm for 2.75 h at 4°C in a Beckman SW41-Ti rotor. Optical density at 254 nm was monitored using a density gradient fractionator (Teledyne Isco, Lincoln, NE).
Supplementary Material
Acknowledgments
We are grateful to Sylvie Souquère and Gérard Pierron for their kind gift of an immunoelectron microscopy image of HEK293 cells. J.A. was supported by the Ministère de l'Enseignement Supérieur. This work was supported by the Association pour la Recherche sur le Cancer, a CNRS PICS grant, and Agence Nationale pour la Recherche contract ANR-14-CE09-0013-01. This work was also supported by the Wellcome Trust (084885/Z/08/Z to N.S.).
Abbreviations used:
- DTT
dithiothreitol
- EJC
exon junction complex
- HA
hemagglutinin
- LC-MS/MS
liquid chromatography–tandem mass spectrometry
- miRNA
microRNA
- PBS
phosphate-buffered saline
- RNAi
RNA interference
- siRNA
small interfering RNA
- TAP-tag
tandem-affinity purification
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E15-03-0136) on May 20, 2015.
*Co–first authors.
REFERENCES
- Aizer A, Brody Y, Ler LW, Sonenberg N, Singer RH, Shav-Tal Y. The dynamics of mammalian P body transport, assembly, and disassembly in vivo. Mol Biol Cell. 2008;19:4154–4166. doi: 10.1091/mbc.E08-05-0513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrei MA, Ingelfinger D, Heintzmann R, Achsel T, Rivera-Pomar R, Lührmann R. A role for eIF4E and eIF4E-transporter in targeting mRNPs to mammalian processing bodies. RNA. 2005;11:717–727. doi: 10.1261/rna.2340405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchan JR. mRNP granules: assembly, function, and connections with disease. RNA Biol. 2014:e29034. doi: 10.4161/15476286.2014.972208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carbonaro M, O'Brate A, Giannakakou P. Microtubule disruption targets HIF-1α mRNA to cytoplasmic P-bodies for translational repression. J Cell Biol. 2011;192:83–99. doi: 10.1083/jcb.201004145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Boland A, Kuzuog˘lu-Öztürk D, Bawankar P, Loh B, Chang C-T, Weichenrieder O, Izaurralde E. A DDX6-CNOT1 complex and W-binding pockets in CNOT9 reveal direct links between miRNA target recognition and silencing. Mol Cell. 2014;54:737–750. doi: 10.1016/j.molcel.2014.03.034. [DOI] [PubMed] [Google Scholar]
- Chu C, Rana TM. Translation repression in human cells by microRNA-induced gene silencing requires RCK/p54. PLoS Biol. 2006;4:e210. doi: 10.1371/journal.pbio.0040210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coller J, Parker R. General translational repression by activators of mRNA decapping. Cell. 2005;122:875–886. doi: 10.1016/j.cell.2005.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coller JM, Tucker M, Sheth U, Valencia-Sanchez MA, Parker R. The DEAD box helicase, Dhh1p, functions in mRNA decapping and interacts with both the decapping and deadenylase complexes. RNA. 2001;7:1717–1727. doi: 10.1017/s135583820101994x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Chaumont F, Dallongeville S, Chenouard N, Hervé N, Pop S, Provoost T, Meas-Yedid V, Pankajakshan P, Lecomte T, Le Montagner Y, et al. ICY: an open bioimage informatics platform for extended reproducible research. Nat Methods. 2012;9:690–696. doi: 10.1038/nmeth.2075. [DOI] [PubMed] [Google Scholar]
- Dresios J, Aschrafi A, Owens GC, Vanderklish PW, Edelman GM, Mauro VP. Cold stress-induced protein Rbm3 binds 60S ribosomal subunits, alters microRNA levels, and enhances global protein synthesis. Proc Natl Acad Sci USA. 2005;102:1865–1870. doi: 10.1073/pnas.0409764102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drummond SP, Hildyard J, Firczuk H, Reamtong O, Li N, Kannambath S, Claydon AJ, Beynon RJ, Eyers CE, McCarthy JEG. Diauxic shift-dependent relocalization of decapping activators Dhh1 and Pat1 to polysomal complexes. Nucleic Acids Res. 2011;39:7764–7774. doi: 10.1093/nar/gkr474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ernoult-Lange M, Baconnais S, Harper M, Minshall N, Souquere S, Boudier T, Bénard M, Andrey P, Pierron G, Kress M, et al. Multiple binding of repressed mRNAs by the P-body protein Rck/p54. RNA. 2012;18:1702–1715. doi: 10.1261/rna.034314.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ernoult-Lange M, Wilczynska A, Harper M, Aigueperse C, Dautry F, Kress M, Weil D. Nucleocytoplasmic traffic of CPEB1 and accumulation in Crm1-nucleolar-bodies. Mol Biol Cell. 2008;20:176-187. doi: 10.1091/mbc.E08-09-0904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferraiuolo MA, Basak S, Dostie J, Murray EL, Schoenberg DR, Sonenberg N. A role for the eIF4E-binding protein 4E-T in P-body formation and mRNA decay. J Cell Biol. 2005;170:913–924. doi: 10.1083/jcb.200504039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh T, Peterson B, Tomasevic N, Peculis BA. Xenopus U8 snoRNA binding protein is a conserved nuclear decapping enzyme. Mol Cell. 2004;13:817–828. doi: 10.1016/s1097-2765(04)00127-3. [DOI] [PubMed] [Google Scholar]
- Gillian-Daniel DL, Gray NK, Aström J, Barkoff A, Wickens M. Modifications of the 5′ cap of mRNAs during Xenopus oocyte maturation: independence from changes in poly(A) length and impact on translation. Mol Cell Biol. 1998;18:6152–6163. doi: 10.1128/mcb.18.10.6152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haas G, Braun JE, Igreja C, Tritschler F, Nishihara T, Izaurralde E. HPat provides a link between deadenylation and decapping in metazoa. J Cell Biol. 2010;189:289–302. doi: 10.1083/jcb.200910141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmes LEA, Campbell SG, De Long SK, Sachs AB, Ashe MP. Loss of translational control in yeast compromised for the major mRNA decay pathway. Mol Cell Biol. 2004;24:2998–3010. doi: 10.1128/MCB.24.7.2998-3010.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holt CE, Schuman EM. The central dogma decentralized: new perspectives on RNA function and local translation in neurons. Neuron. 2013;80:648–657. doi: 10.1016/j.neuron.2013.10.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaehler C, Isensee J, Nonhoff U, Terrey M, Hucho T, Lehrach H, Krobitsch S. Ataxin-2-like is a regulator of stress granules and processing bodies. PLoS One. 2012;7:e50134. doi: 10.1371/journal.pone.0050134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamenska A, Lu W-T, Kubacka D, Broomhead H, Minshall N, Bushell M, Standart N. Human 4E-T represses translation of bound mRNAs and enhances microRNA-mediated silencing. Nucleic Acids Res. 2014;42:3298–3313. doi: 10.1093/nar/gkt1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leung AKL, Sharp PA. Quantifying argonaute proteins in and out of GW/P-bodies: implications in microRNA activities. In: Chan EKL, Fritzler MJ, editors. Ten Years of Progress in GW/P Body Research. New York: Springer; 2013. pp. 165–182. [Google Scholar]
- Lim C, Allada R. ATAXIN-2 Activates PERIOD translation to sustain circadian rhythms in Drosophila. Science. 2013;340:875–879. doi: 10.1126/science.1234785. [DOI] [PubMed] [Google Scholar]
- Liu C, Zhang X, Huang F, Yang B, Li J, Liu B, Luo H, Zhang P, Zhang H. APOBEC3G inhibits microRNA-mediated repression of translation by interfering with the interaction between Argonaute-2 and MOV10. J Biol Chem. 2012;287:29373–29383. doi: 10.1074/jbc.M112.354001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marnef A, Maldonado M, Bugaut A, Balasubramanian S, Kress M, Weil D, Standart N. Distinct functions of maternal and somatic Pat1 protein paralogs. RNA. 2010;16:2094–2107. doi: 10.1261/rna.2295410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathys H, Basquin J, Ozgur S, Czarnocki-Cieciura M, Bonneau F, Aartse A, Dziembowski A, Nowotny M, Conti E, Filipowicz W. Structural and biochemical insights to the role of the CCR4-NOT complex and DDX6 ATPase in microRNA repression. Mol Cell. 2014;54:751–765. doi: 10.1016/j.molcel.2014.03.036. [DOI] [PubMed] [Google Scholar]
- McCann C, Holohan EE, Das S, Dervan A, Larkin A, Lee JA, Rodrigues V, Parker R, Ramaswami M. The Ataxin-2 protein is required for microRNA function and synapse-specific long-term olfactory habituation. Proc Natl Acad Sci USA. 2011;108:E655–E662. doi: 10.1073/pnas.1107198108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minshall N, Kress M, Weil D, Standart N. Role of p54 RNA helicase activity and its C-terminal domain in translational repression, P-body localization and assembly. Mol Biol Cell. 2009;20:2464–2472. doi: 10.1091/mbc.E09-01-0035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minshall N, Reiter MH, Weil D, Standart N. CPEB interacts with an ovary-specific eIF4E and 4E-T in early Xenopus oocytes. J Biol Chem. 2007;282:37389–37401. doi: 10.1074/jbc.M704629200. [DOI] [PubMed] [Google Scholar]
- Morita M, Ler LW, Fabian MR, Siddiqui N, Mullin M, Henderson VC, Alain T, Fonseca BD, Karashchuk G, Bennett CF, et al. A novel 4EHP-GIGYF2 translational repressor complex is essential for mammalian development. Mol Cell Biol. 2012;32:3585–3593. doi: 10.1128/MCB.00455-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagamori I, Sassone-Corsi P. The chromatoid body of male germ cells: epigenetic control and miRNA pathway. Cell Cycle. 2008;7:3503–3508. doi: 10.4161/cc.7.22.6977. [DOI] [PubMed] [Google Scholar]
- Nagaraj N, Wisniewski JR, Geiger T, Cox J, Kircher M, Kelso J, Pääbo S, Mann M. Deep proteome and transcriptome mapping of a human cancer cell line. Mol Syst Biol. 2011;7:548. doi: 10.1038/msb.2011.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nonhoff U, Ralser M, Welzel F, Piccini I, Balzereit D, Yaspo M-L, Lehrach H, Krobitsch S. Ataxin-2 interacts with the DEAD/H-box RNA helicase DDX6 and interferes with P-bodies and stress granules. Mol Biol Cell. 2007;18:1385–1396. doi: 10.1091/mbc.E06-12-1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozgur S, Chekulaeva M, Stoecklin G. Human Pat1b connects deadenylation with mRNA decapping and controls the assembly of processing bodies. Mol Cell Biol. 2010;30:4308–4323. doi: 10.1128/MCB.00429-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitt JN, Schisa JA, Priess JR. P granules in the germ cells of Caenorhabditis elegans adults are associated with clusters of nuclear pores and contain RNA. Dev Biol. 2000;219:315–333. doi: 10.1006/dbio.2000.9607. [DOI] [PubMed] [Google Scholar]
- Presnyak V, Alhusaini N, Chen YH, Martin S, Morris N, Kline N, Olson S, Weinberg D, Baker KE, Graveley BR, et al. Codon optimality is a major determinant of mRNA stability. Cell. 2015;160:1111–1124. doi: 10.1016/j.cell.2015.02.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rouya C, Siddiqui N, Morita M, Duchaine TF, Fabian MR, Sonenberg N. Human DDX6 effects miRNA-mediated gene silencing via direct binding to CNOT1. RNA. 2014;20:1398–1409. doi: 10.1261/rna.045302.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satterfield TF, Pallanck LJ. Ataxin-2 and its Drosophila homolog, ATX2, physically assemble with polyribosomes. Hum Mol Genet. 2006;15:2523–2532. doi: 10.1093/hmg/ddl173. [DOI] [PubMed] [Google Scholar]
- Saulière J, Murigneux V, Wang Z, Marquenet E, Barbosa I, Le Tonquèze O, Audic Y, Paillard L, Roest Crollius H, Le Hir H. CLIP-seq of eIF4AIII reveals transcriptome-wide mapping of the human exon junction complex. Nat Struct Mol Biol. 2012;19:1124–1131. doi: 10.1038/nsmb.2420. [DOI] [PubMed] [Google Scholar]
- Schwanhäusser B, Busse D, Li N, Dittmar G, Schuchhardt J, Wolf J, Chen W, Selbach M. Global quantification of mammalian gene expression control. Nature. 2011;473:337–342. doi: 10.1038/nature10098. [DOI] [PubMed] [Google Scholar]
- Serman A, Le Roy F, Aigueperse C, Kress M, Dautry F, Weil D. GW body disassembly triggered by siRNAs independently of their silencing activity. Nucleic Acids Res. 2007;35:4715–4727. doi: 10.1093/nar/gkm491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharif H, Ozgur S, Sharma K, Basquin C, Urlaub H, Conti E. Structural analysis of the yeast Dhh1-Pat1 complex reveals how Dhh1 engages Pat1, Edc3 and RNA in mutually exclusive interactions. Nucleic Acids Res. 2013;41:8377–8390. doi: 10.1093/nar/gkt600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snee MJ, Macdonald PM. Dynamic organization and plasticity of sponge bodies. Dev Dyn. 2009;238:918–930. doi: 10.1002/dvdy.21914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Souquere S, Mollet S, Kress M, Dautry F, Pierron G, Weil D. Unravelling the ultrastructure of stress granules and associated P-bodies in human cells. J Cell Sci. 2009;122:3619–3626. doi: 10.1242/jcs.054437. [DOI] [PubMed] [Google Scholar]
- Sweet T, Kovalak C, Coller J. The DEAD-box protein Dhh1 promotes decapping by slowing ribosome movement. PLoS Biol. 2012;10:e1001342. doi: 10.1371/journal.pbio.1001342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teixeira D, Parker R. Analysis of P-body assembly in Saccharomyces cerevisiae. Mol Biol Cell. 2007;18:2274–2287. doi: 10.1091/mbc.E07-03-0199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tritschler F, Braun JE, Eulalio A, Truffault V, Izaurralde E, Weichenrieder O. Structural basis for the mutually exclusive anchoring of P body components EDC3 and Tral to the DEAD box protein DDX6/Me31B. Mol Cell. 2009;33:661–668. doi: 10.1016/j.molcel.2009.02.014. [DOI] [PubMed] [Google Scholar]
- Wilczynska A, Aigueperse C, Kress M, Dautry F, Weil D. The translational regulator CPEB1 provides a link between dcp1 bodies and stress granules. J Cell Sci. 2005;118:981–992. doi: 10.1242/jcs.01692. [DOI] [PubMed] [Google Scholar]
- Wühr M, Freeman RM, Presler M, Horb ME, Peshkin L, Gygi SP, Kirschner MW. Deep proteomics of the Xenopus laevis egg using an mRNA-derived reference database. Curr Biol. 2014;24:1467–1475. doi: 10.1016/j.cub.2014.05.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang W-H, Yu JH, Gulick T, Bloch KD, Bloch DB. RNA-associated protein 55 (RAP55) localizes to mRNA processing bodies and stress granules. RNA. 2006;12:547–554. doi: 10.1261/rna.2302706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z, Jakymiw A, Wood MR, Eystathioy T, Rubin RL, Fritzler MJ, Chan EK L. GW182 is critical for the stability of GW bodies expressed during the cell cycle and cell proliferation. J Cell Sci. 2004;117:5567–5578. doi: 10.1242/jcs.01477. [DOI] [PubMed] [Google Scholar]
- Yu JH, Yang W-H, Gulick T, Bloch KD, Bloch DB. Ge-1 is a central component of the mammalian cytoplasmic mRNA processing body. RNA. 2005;11:1795–1802. doi: 10.1261/rna.2142405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S, Williams CJ, Wormington M, Stevens A, Peltz SW. Monitoring mRNA decapping activity. Methods. 1999;17:46–51. doi: 10.1006/meth.1998.0706. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Ling J, Yuan C, Dubruille R, Emery P. A role for Drosophila ATX2 in activation of PER translation and circadian behavior. Science. 2013;340:879–882. doi: 10.1126/science.1234746. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.