

Modeling of genome organization in mouse embryonic stem cells (mESCs) shows that deletion of lamin Bs in mESCs causes a reduced interaction between the nuclear lamina and specific lamina-associated regions. The findings reveal the importance of analyzing specific chromatin types when studying the function of NL proteins in chromatin tether and regulation.
Abstract
Lamins, the major structural components of the nuclear lamina (NL) found beneath the nuclear envelope, are known to interact with most of the nuclear peripheral chromatin in metazoan cells. Although NL–chromatin associations correlate with a repressive chromatin state, the role of lamins in tethering chromatin to NL and how such tether influences gene expression have remained challenging to decipher. Studies suggest that NL proteins regulate chromatin in a context-dependent manner. Therefore understanding the context of chromatin states based on genomic features, including chromatin–NL interactions, is important to the study of lamins and other NL proteins. By modeling genome organization based on combinatorial patterns of chromatin association with lamin B1, core histone modification, and core and linker histone occupancy, we report six distinct large chromatin landscapes, referred to as histone lamin landscapes (HiLands)-red (R), -orange (O), -yellow (Y), -green (G), -blue (B), and -purple (P), in mouse embryonic stem cells (mESCs). This HiLands model demarcates the previously mapped lamin-associated chromatin domains (LADs) into two HiLands, HiLands-B and HiLands-P, which are similar to facultative and constitutive heterochromatins, respectively. Deletion of B-type lamins in mESCs caused a reduced interaction between regions of HiLands-B and NL as measured by emerin–chromatin interaction. Our findings reveal the importance of analyzing specific chromatin types when studying the function of NL proteins in chromatin tether and regulation.
INTRODUCTION
Metazoan nucleus contains a network of nuclear lamina (NL) proteins beneath the nuclear envelope. The major structural components of the NL are the type V intermediate filament proteins, lamins, which can be divided into A and B types. Mammals such as mice have three lamin genes, Lmna, Lmnb1, and Lmnb2, which encode lamin A/C (A-type lamins) and lamin B1 and lamin B2/B3 (B-type lamins), respectively. Recent studies of lamin B–null mouse embryonic stem cells (mESCs) and mice show that B-type lamins are not required for mESC self-renewal or lineage specification in vitro, but they are essential for the proper development and function of several organs, including the lung, brain, and diaphragm (Kim et al., 2011). Because lamins interact with chromatin (Dechat et al., 2008), B-type lamins may contribute to organogenesis by regulating gene expression.
The development of DNA adenine methylation (Dam)-based identification (Dam-ID) has allowed the fine mapping of lamin-associated chromatin domains (LADs) in different cell types (Pickersgill et al., 2006; Guelen et al., 2008; Peric-Hupkes et al., 2010). These studies reveal that LADs cover large regions of the mammalian genome that are generally AT rich and gene poor and exhibit repressive chromatin features (Meuleman et al., 2013). This suggests that NL proteins such as B-type lamins repress gene expression within LADs, which, in turn, contributes to organogenesis. Analyses of gene expression in mESCs and differentiated trophectoderm cells, however, show that B-type lamins do not generally repress their bound genes (Kim et al., 2011). Studies in mammalian tissue culture cells also show that tethering of genes to NL leads to the repression of only some genes (Finlan et al., 2008; Kumaran and Spector, 2008; Reddy et al., 2008). Therefore, despite the overall correlation between NL association and low level of gene expression, neither lamin B nor NL association causes consistent gene repression.
Considering the intricate interactions between the NL and chromatin, it is important to take a comprehensive view to study how lamin B (or other NL proteins) regulates chromatin organization and gene expression. For example, lamin B tethering may differentially influence gene expression by forming complexes with different chromatin-binding proteins, epigenetic regulators, and transcription factors in specific chromatin regions (Wilson and Foisner, 2010). Lamin B may also indirectly regulate genes that are near the nuclear periphery but do not physically contact the NL, because different lamin B–associated complexes could create different environments for epigenetic and transcriptional regulation. It is thus too simplistic to assume that NL disruption such as lamin B deletion only leads to derepression of genes in the LADs. In addition, since chromatin could associate with different NL proteins besides lamin B, it is important to not presume that disruption of lamin B would lead to the detachment of all LADs from the remaining NL proteins.
Consistent with the idea that lamins may regulate specific chromatin regions both within and outside of LADs, lamin A was shown to associate with chromatin regions with specific features of chromatin that lead to different transcriptional outcomes both within and outside of LADs (Lund et al., 2013). In addition, recent genome-wide analyses show that human fibroblasts containing a lamin A mutation (called progerin) exhibit large-scale changes of histone H3 lysine 27 trimethylation (H3K27me3) in certain facultative heterochromatin regions (Shumaker et al., 2006; McCord et al., 2013). Lamin B1 loss during cellular senescence also correlates with epigenetic landscape changes in a region-specific manner (Sadaie et al., 2013; Shah et al., 2013). If lamins do not uniformly regulate all LADs, then, by treating different LADs in the same way, one might not appreciate how lamin deletion would affect chromatin–NL interaction based on a simple global analysis. Indeed, based on similar overall patterns of chromatin–NL interactions in wild-type and lamin B1/B2 double-knockout (LBDKO) mESCs, a recent study concluded that B-type lamins do not play a role in regulating LAD–NL interactions (Amendola and van Steensel, 2015).
To better understand how lamins and other NL proteins differentially regulate individual genomic regions, it is important to establish the relationship between the local epigenetic features and their NL association. Although a chromatin model that includes NL association and epigenetic modifications has been reported for Drosophila S2 cells (Filion et al., 2010), such a model is lacking for mammalian cells. All published chromatin models for mammalian cells are based on the open chromatin assay, histone modifications, and chromatin-binding proteins that interact with the open chromatin. These models have failed, however, to provide useful information within most of the heterochromatin regions covered by LADs (Hoffman et al., 2013). A common difficulty in defining heterochromatin characteristics is the lack of signals for many epigenetic markers (Hoffman et al., 2013; Zhu et al., 2013). In addition, LADs have only been mapped in a few mammalian cell types, which limits the development of chromatin models that include NL association.
Among all mammalian cell types, mESCs represent one of the best characterized with regard to core histone modifications and LAD map. Recently the interactions between DNA and linker histones have been mapped in mESCs (Cao et al., 2013). The linker histones, which bind to DNA that enters and exits nucleosomes, have been shown to stabilize the high-order heterochromatin structures (Izzo et al., 2008; Harshman et al., 2013). Therefore the differential association of linker histones with genomic DNA could help to better define different chromatin features, especially in LADs. Using hidden Markov model (HMM) analysis based on the available core histone modifications, core and linker histone occupancies, and LAD maps, we defined six unique chromatin landscapes in mESCs, which we refer to as histone lamina landscapes (HiLands). Using this model, we show that B-type lamins are required for NL–chromatin association of selected LADs mostly covered by HiLands-B.
RESULTS
Six histone–lamina landscapes divide LADs into two distinct domains in mESCs
We set out to define large-scale chromatin features by combining chromatin–lamin B1 association with other chromatin characteristics in mESCs. For histone modifications, we focused on three epigenetic modifications, histone H3 lysine 4 monomethylation (H3K4me1), H3K9me3, and H3K27me3, which cover large chromatin domains with open chromatin or heterochromatin states. Because nucleosome occupancy itself (represented by H3 enrichment) has been implicated in epigenetic regulation, we included H3 chromatin immunoprecipitation followed by high-throughput sequencing (ChIP-seq) data. LADs may exhibit unique associations with linker histones, because these histones are known to facilitate chromatin compaction, which aids the formation of heterochromatin (Izzo et al., 2008; Harshman et al., 2013). Indeed, different linker histones have different degrees of chromatin condensation activities, and recent studies have mapped the binding of histones H1d and H1c, which exhibit intermediate and weak condensation activities, respectively (Clausell et al., 2009), to chromatin in mESCs (Cao et al., 2013). Our analysis of H1d and H1c enrichment with respect to LADs showed that each linker histone mapped to different regions of the genome, with H1d enriched in most LADs and H1c favoring a subset of LADs (Supplemental Figure S1, A and B).
We reasoned that, by combining histone binding, histone modification, and lamin B1 association, it should be possible to define unique chromatin features that could shed light on how LADs are organized both within themselves and with respect to the rest of the genome in mESCs. We used an HMM to analyze the combinatorial patterns of the mouse genome based on the published maps of LADs (lamin B1 Dam-ID; Peric-Hupkes et al., 2010), H3K4me1, H3K27me3, H3K9me3, H3, H1c, and H1d (Cao et al., 2013) in mESCs. We normalized ChIP-seq signals of various H3 modifications against the H3 ChIP-seq signal to avoid the bias caused by nucleosome occupancy. The genome was divided into nonoverlapping 2-kb windows. The chromatin regions containing no lamin B1 Dam-ID probe or having fewer than two ChIP-seq reads for the other markers were marked as “unknown.” We obtained 1,074,309 informational windows and 215,007 “unknown” windows. On the basis of the enrichment of lamin B1, H3, H1c, and H1d within the informational windows, we found that the seven markers exhibited linear correlations with one another (Figure 1A). These correlations suggest that we can reduce the redundancy of markers using principal component analysis (PCA), which allows more efficient estimation for the modeling parameters (Supplemental Figure S1, C and D).
FIGURE 1:

Modeling of chromatin states. (A) Heatmap showing correlation coefficients between seven markers used to build the chromatin state model. The seven markers used are labeled on the x- and y-axes. (B) Bar plot showing the proportion of variance of the seven principal components calculated from the seven markers shown in A. (C) Bar plot showing the normalized median value of the seven markers in six HiLands. The six colors (red, orange, yellow, green, blue, and purple) in C represent the six HiLands in all figures in this study. (D) Genome browser view showing the enrichment of the seven markers and distribution of HiLands along a region of chromosome 19 (Chr19). A 600-kb region (outlined in red) is zoomed in and shown on the right.
PCA showed that the first three principal components (PCs) accounted for 70% of all the variances (Figure 1B). Thus we trained the HMM using the three PCs in 1,289,316 windows with a Baum–Welch algorithm (Baum et al., 1970), treating the “unknown” windows as missing data. After training with different numbers of states, we found that mESC chromatin could be best separated into six different states, which we refer to as HiLands. We used six rainbow colors—red (R), orange (O), yellow (Y), green (G), blue (B), and purple (P)—to represent each of the states with increasing enrichment of lamin B1 and decreasing enrichment of H3K4me1 (Figure 1, C and D). HiLands-R, -O, -Y, and -G are all outside of LADs, and they exhibit distinct core histone, H1c, and H1d enrichment. HiLands-R and -Y are enriched for active chromatin marker H3K4me1 and are depleted of repressive markers H3K27me3 and H3K9me3, indicating that these two HiLands are active chromatin. HiLands-O is enriched for both H3K4me1 and H3K27me3. Compared to HiLands-R, -O, and -Y, HiLands-G exhibits more interactions with lamin B1 and is enriched for core histones and linker histone H1c. The strongest lamin B1 interaction is found in HiLands-P, followed by HiLands-B. Thus our modeling distinguishes LADs as two regions, HiLands-B and HiLands-P, with distinct chromatin features (Figure 1, C and D).
The genome browser view reveals that the six HiLands have different domain sizes and gene coverage (Figure 2A). As expected, HiLands-P and -B, which correspond to LADs, occupy the largest portion of the genome, followed by HiLands-G, -R, -Y, and then -O (Figure 2B). However, the number of domains for HiLands-P is smaller than that for HiLands-B (Figure 2B). Consequently the average domain size of HiLands-P is larger than that of HiLands-B (Figure 2C; p < 0.001, Student's t test).
FIGURE 2:

Analysis of gene distribution in HiLands. (A) Genome browser view showing the gene coverage and HiLands along chr19. (B) Pie charts showing the genome coverage, number of each HiLands domains, number of genes, and number of promoters in six HiLands. (C) Violin plots showing the size distribution of different HiLands. (D) Bar plot showing gene coverage in six HiLands. Gene coverage is defined as the percentage of nucleotides covered by the gene body. (E) Bar plot showing the TSS density in six HiLands. The y-axis shows the number of TSSs per million bases in six HiLands.
Further analyses show that HiLands-P has a smaller percentage of gene coverage than HiLands-B (Figure 2D). In fact, the gene coverage of HiLands-B is similar to that of HiLands-G and -O, whereas HiLands-R and -Y have the highest gene coverage (Figure 2D). Analyses of promoters show that 34.8% of all promoters are located in HiLands-R, whereas only 24.3% of promoters are found in HiLands-B and -P. HiLands-O occupies the smallest region of the genome (6.85%) and contains the second largest number of promoters (21.9%; Figure 2B). The relatively low gene coverage in HiLands-O is because these domains are enriched for promoters, but the gene bodies are often found in the neighboring HiLands-R (9.9%), -Y (8.6%), or -G (11.2%). Consistent with this, HiLands-O has the second highest density of transcriptional start sites (TSSs; Figure 2E), despite the lower gene coverage compared with HiLands-R and -Y (Figure 2D). Of interest, HiLands-O also has the highest proportion of bivalent promoters (37.5% of total bivalent promoters) as marked by H3K4me3 and H3K27me3 (Bernstein et al., 2006). Therefore, in addition to the distinct association with histones and NL (Figure 1C), the six HiLands also exhibit distinct domain structures.
Nucleosome repeat length and histone enrichment in HiLands correlate with gene expression, histone modification, and NL association
A key feature of HiLands is their differential enrichment of core and linker histones. Each core histone particle is wrapped by 147-nucleotide (nt) DNA. The binding of different linker histones to linker DNA gives rise to nucleosomes, and, depending on the specific H1, the length of DNA between neighboring nucleosomes (called the nucleosome repeat length [NRL]), varies between 162 and 212 nt (Woodcock et al., 2006). We analyzed how the NRL and histone enrichment correlate with other features in different HiLands. Compared to HiLands-R and -O, HiLands-Y, -G, -B, and -P exhibit higher enrichment of H1c and/or H1d (Figure 1C), two linker histones that have similar effect on the NRL. This suggests that the linker histone–enriched HiLands should have long NRL. We analyzed nucleosome phasing on the basis of the available MNase-seq in mESCs (Teif et al., 2012). The phasograms (Figure 3A) show that HiLands-Y, -G, -B, and -P indeed have longer NRLs than HiLands-R and -O (Figure 3B; p < 0.001, Wilcoxon test).
FIGURE 3:

Nucleosome distribution, gene expression, and epigenetic modifications in HiLands. (A) Phasogram showing distribution of nucleosome-–nucleosome distances. The x-axis shows the distance between nucleosome pairs; the y-axis shows the normalized number of nucleosome pairs having that distance between them. Each line is calculated from nucleosome pairs in the type of HiLands corresponding to the line color. (B) Bar plot showing nucleosome repeat lengths in six HiLands estimated from the phasogram. Error bars, SE of estimation. (C) Violin plot showing gene expression levels in six HiLands. The y-axis shows the log10 of mRNA expression based on RNA-seq. (D) Bar plot showing enrichment of four histone modifications in six HiLands.
Next we analyzed gene expression levels of the six HiLands in mESCs. As expected, HiLands-B and -P, which are associated with the NL and have long NRLs, exhibit lower overall gene expression than the other four HiLands (Figure 3C; p < 0.001, Wilcoxon test). Of interest, although HiLands-Y has longer NRLs than HiLands-R and -O, the overall gene expression levels in HiLands-Y are close to those in HiLands-R but are higher than those in HiLands-O (Figure 3C; p < 0.001, Wilcoxon test). Because HiLands-Y is occupied largely by gene bodies (Figure 2, D and E), the relatively long NRL in this domain reflects the feature of nucleosome-associated gene bodies but not the heterochromatin state. HiLands-O has shorter NRLs but exhibits lower gene expression than HiLands-Y, which could be due to the fact that HiLands-O is enriched for both H3K4me3 and H3K27me3, a bivalent state that poises the genes for expression upon induction (Figures 1C and 3D).
To examine whether the NRL and gene expression patterns of the six HiLands also correlate with active or repressive chromatin states, we normalized ChIP-seq signals of various H3 modifications against the H3 ChIP-seq signal to avoid the bias caused by nucleosome occupancy. HiLands-R and -Y, with high levels of gene expression, are enriched for H3 modifications that are characteristic of active euchromatin domains (Figures 1C and 3D). HiLands-O exhibits both active and repressive chromatin features (Figures 1C and 3D), whereas HiLands-G, -B, and -P, with the longest NRLs, are depleted of active chromatin modifications (Figure 3D) but enriched for repressive chromatin marks such as H3K27me3 and H3K9me3 (Figure 1C). These analyses show that different HiLands are associated with different NRLs, which correlates with different gene expression and chromatin features.
The characteristics and relative positions of HiLands in mESCs help to predict LAD alterations upon differentiation
Visual inspection of the genome-wide distributions of all six HiLands suggests that some HiLands frequently neighbor one another, whereas others are far apart (Figure 4A). To quantify the neighborhood relationships among the HiLands, we calculated the frequency of given different HiLands pairs to be next to one another. To visualize the neighborhood relationships along the linear genome, we use a neighbor index (NI) for each HiLands pair (Figure 4B; see Materials and Methods for NI calculation). The higher the NI between a pair of HiLands, the more frequently are they positioned next to one another. We also used the two strongest PCs, which are linear combinations of the seven markers as described earlier, to show the similarity of the six HiLands as displayed on the two-dimensional PCA space (Figure 4B). These analyses show that HiLands-R is most frequently positioned next to HiLands-O, followed by HiLands-Y. HiLands-Y is scattered throughout the genome by connecting with all other HiLands except for HiLands-P. Of interest, whereas the NL-associated HiLands-P is connected to the rest of the genome through the NL-associated HiLands-B, the majority (80%) of HiLands-B border HiLands-G, with the remaining 20% bordering HiLands-Y (Figure 4B).
FIGURE 4:

Neighborhood relationships, GC contents, and differentiation-dependent alterations of HiLands. (A) Genome browser view of six HiLands and the corresponding genes along a section of chromosome 17. (B) A neighborhood plot of six HiLands. The x-axis shows the median PC1 value of six HiLands; the y-axis shows the median PC2 value of six HiLands. Lines connecting the HiLands with the NI numbers show the probability that two HiLands are next to one another along the linear genome. (C) Violin plot showing the distribution of GC content in six HiLands. GC content is calculated in 2-kb windows. (D) Bar plot showing the correlation between the six HiLands and the cLADs/fLADs. The y-axis shows the lamin B1 Dam-ID probes that fall into specific HiLands types (shown by color) and belong to specific LAD/non-LAD types (shown on the x-axis). (E) Comparison between HiLands-B/P and f/c-LADs. Left, the HiLands-B regions overlapping fLADs and cLADs, respectively. Right, HiLands-P regions overlapping fLADs and cLADs, respectively. The distribution of GC content in each region is shown on the y-axis.
Previous studies showed that certain LADs dissociate from NL upon ESC differentiation, whereas some non-LAD regions in ESCs can become LADs upon differentiation (Peric-Hupkes et al., 2010). Because LAD alterations could facilitate proper genome reorganization and gene regulation during differentiation, the ability to accurately predict cell type–specific facultative (f) LADs and constitutive (c) LADs could help us understand the role of lamin–chromatin interaction in development. Previous analyses of fLADs and cLADs using four mouse cell types—mESCs, mouse embryonic fibroblasts (MEFs), neural progenitor cells, and astrocytes—have shown that cLADs have a lower GC content than fLADs (Meuleman et al., 2013). Of interest, of the two subregions of LADs defined by HiLands, HiLands-P has a lower GC content than HiLands-B (Figure 4C; p < 0.001, Wilcoxon test). This suggests that HiLands-P and -B might correspond to cLADs and fLADs, respectively.
To further analyze how each of the HiLands corresponds to the previously defined cLADs, fLADs, and non-LADs regions based on the four cell types, we first divided the fLADs into fLADs-mESC, which refers to fLADs that are LADs in mESCs, and fLADs-new, which are fLADs in differentiated cells but are non-LADs in mESCs. We then mapped all six HiLands to cLADs, fLADs-ESC, fLADs-new, and non-LADs as defined by the four cell types in mESCs. As expected, HiLands-R, -O, and -Y were mostly found in non-LADs regions, whereas HiLands-P and -B were mostly found in cLADs and fLADs, respectively (Figure 4D). Detailed analyses showed that 79.1% of HiLands-P and 61% of HiLands-B corresponded to cLADs and fLADs, respectively, with a significant portion of HiLands-B (27%) found in cLADs.
The incomplete correspondence between HiLands-P/-B and c/fLADs prompted us to examine further whether the classification of fLADs and cLADs on the basis of only four cell types can lead to overestimation and underestimation of the respective LADs. We reasoned that if HiLands-B that fell on cLADs were misclassified fLADs, their GC contents should be similar to those of HiLands-B found in fLADs. Indeed, we found that the 27% of HiLands-B mapped to cLADs have similarly high GC contents as the 61% of HiLands-B found in fLADs, and both groups of HiLands-B have higher GC content than HiLands-P found in cLADs (Figure 4E; p < 0.001, Wilcoxon test). Similarly, the HiLands-P that was mapped to fLADs had a similar GC content to the HiLands-P that was mapped to cLADs. This suggests that HiLands-P and -B can better predict cLADs and fLADs, respectively. Of interest, we found that HiLands-G, which often borders HiLands-B in mESCs (Figure 4B), is most likely to become incorporated into fLADs (fLADs-new) upon differentiation, whereas the HiLands that are farther away from LADs in mESCs are less likely to become fLADs-new (Figure 4D). The foregoing analysis suggests that HiLands-B and -P can accurately predict LADs that undergo changes or remain the same, respectively, upon mESC differentiation.
Lamin B regulates the association of HiLands-B with the NL in mESCs
Recently Amendola and van Steensel (2015) reported no change in NL–chromatin interactions upon lamin B1 and B2 double knockout in mESCs. We reason that if B-type lamins only regulate some specific NL-associated chromatin regions, by globally analyzing NL–chromatin interactions one might not be able to detect local changes. On the other hand, by applying the HiLands model described here, we might be able to identify the region-specific changes of NL–chromatin interactions upon lamin B deletion. We first mapped NL–chromatin interactions in wild-type and LBDKO mESCs we generated (Kim et al., 2011). Previous Dam-ID studies showed that emerin and lamin B1 associate with similar chromatin regions in human fibroblasts and mESCs (Guelen et al., 2008; Amendola and van Steensel, 2015). Because emerin expression and localization in wild-type and LBDKO mESCs appear similar (Kim et al., 2011; Guo et al., 2014), we used emerin Dam-ID to determine NL–chromatin interactions in wild-type and LBDKO mESCs.
The emerin Dam-ID data were first normalized by loess normalization and then quantile normalized among the whole-genome tiling arrays used (Ritchie et al., 2015) to avoid bias caused by differences on different arrays. As reported by Amendola and van Steensel (2015), who used the same LBDKO mESCs as in our study here, we found similar overall patterns of emerin-Dam-ID maps between wild-type and LBDKO mESCs (Figure 5A). When the normalized Dam-ID values were compared between the corresponding HiLands-B and -P in LBDKO and wild-type mESCs, however, we found a clear decrease in emerin Dam-ID in HiLands-B in LBDKO mESCs, whereas emerin Dam-ID values in HiLands-P were similar in the two genotypes (Figure 5B). A genome browser view of chromosome 7 shows the reduction of emerin Dam-ID in HiLands-B in LBDKO mESCs as compared with the wild-type control (Figure 5C). We also compared the emerin and lamin B1 Dam-ID maps on HiLands-B and -P by first using quantile normalization of the data sets. The average of lamin B1 and emerin Dam-ID values on each HiLands-B and -P were then calculated and compared (Supplemental Figure S2, A and B). We found that the emerin Dam-ID values are on average slightly lower than lamin B1 dam-ID values on both HiLands-B and -P, which is probably because the HiLands themselves are calculated from lamin B1 data. However, the amount of reduction is the same in both types of HiLands (Supplemental Figure S2C; p > 0.8 for both t test and Wilcoxon rank-sum test). Therefore there is no specific differential binding of emerin and lamin B1 to HiLands-B and -P. Taken together, these analyses show that deletion of B-type lamins causes a reduced interaction of selected HiLands-B chromatin with the NL in mESCs.
FIGURE 5:

Analysis of emerin Dam-ID in wild-type and LBDKO mESCs. (A) Contour plot comparing emerin Dam-ID in WT and LBDKO mESCs. Each dot represents a 2-kb window on the genome. Average Dam-ID values present in each 2-kb window were used. Here r is the Spearman correlation coefficient. (B) Box plot of average changes of normalized emerin Dam-ID values in HiLands-B and HiLands-P. The changes are plotted as the log2 ratio of emerin DamID values of LBDKO over WT. Boxes indicate 25 and 75% quantiles, whiskers indicate 5 and 95% quantiles, and “x”s indicate 1 and 99% quantiles. (C) Browser views showing changes in emerin Dam-ID values (bottom) on chromosome 7 in LBDKO (middle) compared with wild-type (WT, top) mESCs. Top and middle, emerin binding plotted as the log2 ratio of values of Dam-emerin over Dam alone; bottom, changes between LBDKO and WT plotted as the log2 ratio of emerin DamID values of LBDKO over WT.
Confirmation of selected detachment of HiLands-B from the NL upon lamin B deletion by comparative analysis of different emerin Dam-ID data sets
To verify that the lack of change of NL–chromatin interaction in LBDKO mESCs as described by Amendola and van Steensel (2015) was caused by not analyzing subregions of chromatin, we compared our data sets with theirs. This comparison was possible and fair because both Dam-ID data sets were generated from the same mESC lines. They did separately compare emerin Dam-ID values in fLADs, cLADs, and non-LADs regions between wild-type and LBDKO mESCs. They found that the concordance of 79% between fLADs, which largely overlap HiLands-B (Figure 4D), was lower than that between cLADs (98%; see Figure 1F in Amendola and van Steensel, 2015). They also used lamin-A Dam-ID and emerin Dam-ID to analyze wild-type, LBDKO, or lamin-A RNA interference–treated LBDKO (TKO) mESCs. A similarly lower concordance in fLADs than in cLADs was observed in LBDKO (86% fLADs vs. 98.5% cLADs; see Figure 3G in Amendola and van Steensel, 2015) and TKO (82.1% fLADs vs. 98% cLADs; see Figure 4F in Amendola and van Steensel, 2015) mESCs compared with the wild-type control. Although the apparently reduced consistency suggests that lamin deletion could affect NL–chromatin interactions in specific regions within fLADs, Amendola and colleagues argued that the differences were due to random noise.
We therefore first analyzed whether the difference could be due to random noise. All Dam-ID studies using lamins or emerin in wild-type mESCs have shown that fLADs exhibits a weaker Dam-ID signal than that of cLADs. An example is shown in Supplemental Figure S2D. Amendola and van Steensel (2015) suggested that this low signal could reduce the signal/noise ratio, thereby leading to the reduced consistency in fLADs between wild-type and lamin-depleted mESCs. However, if the reduced consistency in fLADs observed was due to random noise, the Dam-ID value changes in lamin-depleted mESCs should also be random in different experiments. Instead, we found that the value changes in fLADs were mostly reductions in their data set (Figure 6A). A comparison of maps for emerin or lamin-A Dam-ID on chromosome 9 between wild-type and LBDKO mESCs based on our and their data sets is shown in Figure 6B. Similarly, when we mapped their data set onto our HiLands model, the reduction of Dam-ID values was strongest in HiLands-B in LBDKO mESCs, which is similar to our data set (Figure 6C). Because Dam-ID was performed in different laboratories and on different genomic tilling arrays, the changes in chromatin–NL interactions as revealed by our analyses are not due to the increased Dam-ID noise in the fLADs.
FIGURE 6:

Comparison of independent data sets using different methods reveals a role of B-type lamins in regulating the interaction between HiLands-B and the NL. (A) A genome browser view showing that emerin Dam-ID changes on fLADs (gray blocks) are more pronounced than those on cLADs (black blocks). Selected regions on chromosome 7 (chr7) are shown. The data set used is indicated in parentheses at the y-axis. (B) A genome browser view of emerin Dam-ID changes on chr9. (C) Box plot showing average changes of normalized emerin Dam-ID values (based on our data and those of Amendola and van Steensel, 2015) in HiLands-B and HiLands-P. Boxes indicate 25 and 75% quantiles, whiskers indicate 5 and 95% quantiles, and “x”s indicate 1 and 99% quantiles. (D) QQ plots for comparison of the Z-scores of HiLands-B to the normal distribution. The Z-scores are calculated based on the statistical method described in Peric-Hupkes et al. (2010).
Using a previously published statistical test (Peric-Hupkes et al., 2010), Amendola and van Steensel (2015) analyzed Dam-ID values on all genes instead of the whole genome. We reason that if lamin B regulates NL–chromatin interactions in specific chromatin regions, by limiting the analysis on genes, one might not have detected changes. Therefore we applied the same statistical test used by Amendola and van Steensel (2015) on their Dam-ID data set based on our HiLands model. We identified 220 HiLands-B regions showing significantly decreased chromatin–NL interactions (p < 0.01). To further demonstrate that the changes of Dam-ID values in LBDKO mESCs are not random in the data set of Amendola and van Steensel (2015), we used a QQ plot to compare the Z-score quantiles of HiLands-B calculated by their statistical method with the theoretical normal quantiles. The Z-score quantiles for HiLands-B are consistently lower than the theoretical normal quantiles (Figure 6D). However, the Z-score quantiles for the other HiLands do not show consistent change (Supplemental Figure S3). This further supports a nonrandom shift of regions of HiLands-B away from the NL in LBDKO mESCs. Therefore deletion of B-type lamins in mESCs resulted in reduced interaction between HiLands-B and the NL.
DISCUSSION
Various studies have suggested that lamins and other NL proteins could influence chromatin modification and gene expression differentially depending on the local chromatin context (Shumaker et al., 2006; Lund et al., 2013; McCord et al., 2013; Sadaie et al., 2013; Shah et al., 2013). Although LADs represents ∼40% of the mammalian genome, the chromatin features of these LADs have been poorly characterized. Therefore it has been difficult to discern the region-specific functions of lamins or other NL proteins. One difficulty in modeling the chromatin landscape in LADs is the lack of strong epigenetic modifications in these regions (Hoffman et al., 2013; Zhu et al., 2013). For example, a 15-state chromatin map based on global ChIP-seq of eight histone modifications and CTCF binding in human cells revealed a very low signal for all of the nine analyzed markers in the heterochromatin regions that include LADs (Ernst et al., 2011). This suggests that other chromatin features besides these markers are needed to characterize properly the differences within LADs. Indeed, by taking into consideration NL–chromatin and DNA–histone interactions, we are able to separate mammalian LADs into two distinct chromatin landscapes, referred to as HiLands-B and -P.
Using our HiLands model, we show that B-type lamins regulate the interaction between regions of HiLands-B chromatin with the NL as measured by emerin Dam-ID. Our finding disagrees with a recent publication by Amendola and van Steensel (2015). By applying our HiLands model and our statistical test to analyze their published data, we found a significant reduction of HiLands-B–NL interaction. Similarly, when applying their statistical test and our HiLands model to analyze their data set, we also found a significant reduction of HiLands-B–NL interaction. Therefore, in order to appreciate the role of lamins and other NL proteins, it is important to analyze their functions in the context of chromatin features. Indeed, the emerin Dam-ID values in fLADs differ between wild-type and LBDKO mESCs in both their study (see Figure 1E in Amendola and van Steensel, 2015) and our analysis and reveal a strong overlap between fLADs and HiLands-B. Therefore, despite opposite conclusions, the different data sets from Amendola and van Steensel (2015) and our study support a role of lamin B in regulating specific LADs as characterized by HiLands-B.
By analyzing the differences of LADs in mESCs and three differentiated cell types, Meuleman et al. (2013) separated LADs into cLADs and fLADs that remain unchanged or undergo changes, respectively, upon mESC differentiation. Although this operational definition of LADs revealed the different GC contents between fLADs and cLADs, it did not take into consideration other chromatin features (Meuleman et al., 2013). Considering that hundreds of cell types exist in mammals, the operational definition of f/cLADs is insufficient to demarcate subregions of LADs. Our studies show that the HiLands model can largely recapitulate the operational definitions of fLADs and cLADs in mESCs. Because the HiLands model includes multiple chromatin features, establishing HiLands models in different cell types should allow a better definition of landscape changes upon differentiation in both LAD and non-LAD regions.
Despite extensive studies, how lamins, other NL proteins, and chromatin modifiers regulate the interaction between chromatin and the NL and how this interaction influences chromatin modification and gene expression have remained unclear. By analyzing specific gene loci in LADs, the GAGA motif is shown to mediate the binding of the loci to a complex containing cKrox, HDAC3, and Lap2β in tissue culture cells. This in turn targets the LADs to NL, at least in part through lamin B and Lap2β interactions (Schirmer and Foisner, 2007), thereby leading to gene silencing (Zullo et al., 2012). However, whether GAGA motifs found in LADs generally mediate NL–chromatin interactions remains unknown. On the other hand, studies have shown that the repressive chromatin state marked by histone H3K9me2 or H3K9me3 facilitates chromatin–NL interactions (Wen et al., 2009; Towbin et al., 2012; Kind et al., 2013). However, whether H3K9me2 and H3K9me3 are essential for chromatin–NL interaction requires further study because gene knockout of G9a, known to methylate H3K9, did not significantly affect chromatin localization in one study (Yokochi et al., 2009), whereas reduction of H3K9me2 or H3K9me3 in Caenorhabditis elegans and tissue culture cells disrupted NL–chromatin binding in another report (Towbin et al., 2012; Kind et al., 2013). Although how NL proteins regulate chromatin requires further investigation, the data show that both DNA sequences and local chromatin modifications can contribute to NL–chromatin binding and gene expression. Our HiLands model provides a starting point that should allow incorporation of unique DNA sequences and new chromatin features to develop improved maps of the chromatin landscape. These maps should in turn facilitate the study of how disrupting each NL protein or chromatin modifier might affect different chromatin regions in the genome. This effort should shed light on how NL proteins such as lamin B regulate tissue building and maintenance in the context of development and aging (Kim et al., 2011, 2013; Chen et al., 2013, 2014).
MATERIALS AND METHODS
PCA analysis
We divided the genome into nonoverlapping 2-kb windows. We use 2-kb windows because the lamin B1 Dam-ID microarray had ∼1.2-kb mean distance between neighboring probes and the distance varies between 0.5 and 2 kb. The 2-kb windows would ensure that most of the windows have at least one lamin B1 probe. For each window, we calculated the enrichment values of lamin B1, H3, H1c, H1d, H3K4me1, H3K27me3, and H3K9me3. For lamin B1, we calculated the average Dam-ID value (after normalization) in each 2-kb window, and if there was no probe of tilling array in the window, we regarded the window as “unknown.” For H3, we calculated the value from EnrichH3 = ln(number of H3 reads/number of input reads). Then we center normalized the enrichment value by subtracting the genomic median value. If the read number in a window was <1 for H3 or input, we regarded the window as “unknown.” H1c and H1d were treated in the same way by normalization to the corresponding input control. H3K4me1, H3K27me3, and H3K9me3 were also treated in a similar way by normalization to the corresponding H3 control. The “unknown” windows are generally due to poor mappability of these genome regions. We pooled all the “unknown” windows together and assumed that no information was available in all these windows. We then performed PCA analyses on the seven markers using the princomp command in R. The first three PCs for each 2-kb window were extracted for further analysis.
Hidden Markov model
By trying different numbers of chromatin states for a hidden Markov model, we found that six states were best at characterizing the differences of the seven chromatin markers that we used. We assumed that the six states represent different biological domains in the genome, and the three PCs described earlier are the observations for each 2-kb window. The emission probability distribution used here is the multivariate normal inverse Gaussian distribution, which has been used in speech recognition and other fields in machine learning (Øigård et al., 2005). This distribution allows both skewness and more variable kurtosis compared to a normal or Student's t distribution. The unknown windows are treated as missing data, so that the emission probability is be always 1. The model is trained with a standard expectation-maximization (EM) algorithm (Baum–Welch algorithm for HMM; Baum et al., 1970) with ensured convergence. Because the EM algorithm can converge to local maximum points, we randomized the initial parameters 20 times and selected the best final results as judged by the highest-likelihood value. Then we assigned the states for each window using the Viterbi algorithm (Viterbi, 1967). Neighboring windows that belonged to the same HiLands type were joined together to make one single HiLands.
Nucleosome repeat length analysis
Paired-end MNase-seq data were mapped to the mouse genome (mm9) using Bowtie. The nucleosome center was defined as the middle position of two boundaries. Reads with <100-nt or >250-nt distance between two boundaries were discarded. To derive the phasogram, we considered all pairs of nucleosomes within 1500 nt of each other. Then we obtained a histogram by counting the number of nucleosome pairs at a specific distance. The histogram was smoothed with a binomial kernel, 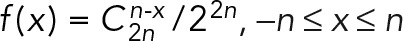 , −n ≤ x ≤ n, where n = 128. Finally, the phasogram was scale normalized to make the highest peak equal to 1. For NRL estimation, we selected the first four (for HiLands-R, -O, and -Y) or seven (for HiLands-G, -B, and -P) peaks on the phasogram (Valouev et al., 2011), and did linear regression against the peak indices (1–4 for R, O, and Y; 1–7 for G, B, and P). Then we used the scopes of the linear regressions as NRL estimations in six HiLands.
, −n ≤ x ≤ n, where n = 128. Finally, the phasogram was scale normalized to make the highest peak equal to 1. For NRL estimation, we selected the first four (for HiLands-R, -O, and -Y) or seven (for HiLands-G, -B, and -P) peaks on the phasogram (Valouev et al., 2011), and did linear regression against the peak indices (1–4 for R, O, and Y; 1–7 for G, B, and P). Then we used the scopes of the linear regressions as NRL estimations in six HiLands.
HiLands neighborhood analysis
To establish a HiLands neighborhood, we went through all HiLands transitions and counted the numbers of neighbors between pairs of specific HiLands types. We defined a neighbor index of two HiLands types as NI(X, Y) = 25,000 × (number of neighbor pairs between HiLands-X and HiLands-Y)/(number of HiLands-X × number of HiLands-Y). For each pair of X and Y, if NI(X, Y) > 0.1, we plotted a line between them in Figure 4B with the indicated NI between two HiLands.
ESC culture and plasmid construction
mESCs were maintained in the absence of feeder cells as described (Kim et al., 2011). To generate the Dam-tagged mouse emerin (Emd) construct, mouse Emd cDNA was amplified from IMAGE clone #40130408 with primers carrying Gateway recombination sequences. The PCR product was cloned into pDONR201 and subsequently into pLgwEcoDamV5 (Vogel et al., 2007). The resulting construct, pLgwEcoDamV5-Ms Emd, was confirmed by DNA sequencing. pLgwV5EcoDam was used as the Dam control construct. All procedures involving recombinant DNA followed the National Institutes of Health guidelines.
Emerin Dam-ID and data analyses
Emerin Dam-ID was performed as described (Kim et al., 2011), with modifications. Briefly, the Dam or Dam-emerin–expressing lentivirus was generated from 293T cells cultured in 10% fetal bovine serum/DMEM. Virus containing supernatant was diluted 2:1 in mESC medium (complete GMEM) containing leukemia inhibitory factor (LIF) (3 × 103 U/ml). Freshly trypsinized mESCs were plated in a six-well plate with the diluted viral supernatant containing either Dam or Dam-emerin–expressing lentivirus, spin infected at 700 × g for 45 min, and incubated at 37°C overnight. Diluted virus supernatant was replaced with fresh mESC medium, followed by an additional 24 h of incubation at 37°C. The genomic DNA was isolated from the transduced mESCs, and the adenine-methylated DNAs were enriched by adaptor-mediated PCR. The enriched DNA was labeled using a dual-color DNA labeling kit (Roche). The labeled DNA was analyzed using 2.1 M mouse whole-genome tiling arrays (NimbleGen #05327911001), which covers the entire genome at ∼250–base pair intervals, according to the manufacturer's recommendations.
The emerin Dam-ID/Dam data were first loess normalized to get the final Dam-ID value using the limma package in R. After quantile normalization of wild-type and LBDKO emerin Dam-ID values, we calculated the differences between quantile-normalized emerin Dam-ID values in wild-type and LBDKO mESCs for each probe. Then, for each HiLands domain, the average emerin Dam-ID change was calculated from all probes within the domain. A box plot was used to plot the distribution of average emerin Dam-ID changes in each HiLands type.
Supplementary Material
Acknowledgments
We thank Minoru Ko, Alexei Sharov, and Haiping Hao for their help with emerin Dam-ID, Ona Martin for technical support, and the members of the Zheng lab for critical comments. This work was supported by National Institutes of Health grants RO1 GM056312 and GM06023 and the Ellison Medical Foundation (to Y.Z.).
Abbreviations used:
- cLAD
constitutive LAD
- Dam-ID
DNA adenine methylation (Dam)-based identification
- fLAD
facultative LAD
- HiLand
histone lamin landscape
- HMM
hidden Markov model
- LAD
lamina-associated domain
- LBDKO
lamin B1/B2 double-knockout
- mESC
mouse embryonic stem cell
- NI
neighbor index
- NL
nuclear lamina
- NRL
nuclear repeat length
- PCA
principal component analysis
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E15-04-0210) on May 20, 2015.
*These authors contributed equally to this work.
†Present address: Soonchunhyang Institute of Medi Bio Science, Soonchunhyang University, Asan-si, Chungcheongnam-do 336-745, Korea.
REFERENCES
- Amendola M, van Steensel B. Nuclear lamins are not required for lamina-associated domain organization in mouse embryonic stem cells. EMBO Rep. 2015;16:610–617. doi: 10.15252/embr.201439789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baum LE, Petrie T, Soules G, Weiss N. A maximization technique occurring in the statistical analysis of probabilistic functions of Markov chains. Ann Math Stat. 1970;41:164–171. [Google Scholar]
- Bernstein BE, Mikkelsen TS, Xie X, Kamal M, Huebert DJ, Cuff J, Fry B, Meissner A, Wernig M, Plath K, et al. A bivalent chromatin structure marks key developmental genes in embryonic stem cells. Cell. 2006;125:315–326. doi: 10.1016/j.cell.2006.02.041. [DOI] [PubMed] [Google Scholar]
- Cao K, Lailler N, Zhang Y, Kumar A, Uppal K, Liu Z, Lee EK, Wu H, Medrzycki M, Pan C, et al. High-resolution mapping of h1 linker histone variants in embryonic stem cells. PLoS Genet. 2013;9:e1003417. doi: 10.1371/journal.pgen.1003417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, Chen X, Zheng Y. The nuclear lamina regulates germline stem cell niche organization via modulation of EGFR signaling. Cell Stem Cell. 2013;13:73–86. doi: 10.1016/j.stem.2013.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, Zheng X, Zheng Y. Age-associated loss of lamin-B leads to systemic inflammation and gut hyperplasia. Cell. 2014;159:829–843. doi: 10.1016/j.cell.2014.10.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clausell J, Happel N, Hale TK, Doenecke D, Beato M. Histone H1 subtypes differentially modulate chromatin condensation without preventing ATP-dependent remodeling by SWI/SNF or NURF. PLoS One. 2009;4:e0007243. doi: 10.1371/journal.pone.0007243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dechat T, Pfleghaar K, Sengupta K, Shimi T, Shumaker DK, Solimando L, Goldman RD. Nuclear lamins: major factors in the structural organization and function of the nucleus and chromatin. Genes Dev. 2008;22:832–853. doi: 10.1101/gad.1652708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ernst J, Kheradpour P, Mikkelsen TS, Shoresh N, Ward LD, Epstein CB, Zhang X, Wang L, Issner R, Coyne M, et al. Mapping and analysis of chromatin state dynamics in nine human cell types. Nature. 2011;473:43–49. doi: 10.1038/nature09906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filion GJ, van Bemmel JG, Braunschweig U, Talhout W, Kind J, Ward LD, Brugman W, de Castro IJ, Kerkhoven RM, Bussemaker HJ, van Steensel B. Systematic protein location mapping reveals five principal chromatin types in Drosophila cells. Cell. 2010;143:212–224. doi: 10.1016/j.cell.2010.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finlan LE, Sproul D, Thomson I, Boyle S, Kerr E, Perry P, Ylstra B, Chubb JR, Bickmore WA. Recruitment to the nuclear periphery can alter expression of genes in human cells. PLoS Genet. 2008;4:e1000039. doi: 10.1371/journal.pgen.1000039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guelen L, Pagie L, Brasset E, Meuleman W, Faza MB, Talhout W, Eussen BH, de Klein A, Wessels L, de Laat W, van Steensel B. Domain organization of human chromosomes revealed by mapping of nuclear lamina interactions. Nature. 2008;453:948–951. doi: 10.1038/nature06947. [DOI] [PubMed] [Google Scholar]
- Guo Y, Kim Y, Shimi T, Goldman RD, Zheng Y. Concentration-dependent lamin assembly and its roles in the localization of other nuclear proteins. Mol Biol Cell. 2014;25:1287–1297. doi: 10.1091/mbc.E13-11-0644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harshman SW, Young NL, Parthun MR, Freitas MA. H1 histones: current perspectives and challenges. Nucleic Acids Res. 2013;41:9593–9609. doi: 10.1093/nar/gkt700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman MM, Ernst J, Wilder SP, Kundaje A, Harris RS, Libbrecht M, Giardine B, Ellenbogen PM, Bilmes JA, Birney E, et al. Integrative annotation of chromatin elements from ENCODE data. Nucleic Acids Res. 2013;41:827–841. doi: 10.1093/nar/gks1284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izzo A, Kamieniarz K, Schneider R. The histone H1 family: specific members, specific functions. Biol Chem. 2008;389:333–343. doi: 10.1515/BC.2008.037. [DOI] [PubMed] [Google Scholar]
- Kim Y, Sharov AA, McDole K, Cheng M, Hao H, Fan CM, Gaiano N, Ko MS, Zheng Y. Mouse B-type lamins are required for proper organogenesis but not by embryonic stem cells. Science. 2011;334:1706–1710. doi: 10.1126/science.1211222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim Y, Zheng X, Zheng Y. Proliferation and differentiation of mouse embryonic stem cells lacking all lamins. Cell Res. 2013;23:1420–1423. doi: 10.1038/cr.2013.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kind J, Pagie L, Ortabozkoyun H, Boyle S, de Vries SS, Janssen H, Amendola M, Nolen LD, Bickmore WA, van Steensel B. Single-cell dynamics of genome-nuclear lamina interactions. Cell. 2013;153:178–192. doi: 10.1016/j.cell.2013.02.028. [DOI] [PubMed] [Google Scholar]
- Kumaran RI, Spector DL. A genetic locus targeted to the nuclear periphery in living cells maintains its transcriptional competence. J Cell Biol. 2008;180:51–65. doi: 10.1083/jcb.200706060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lund E, Oldenburg AR, Delbarre E, Freberg CT, Duband-Goulet I, Eskeland R, Buendia B, Collas P. Lamin A/C-promoter interactions specify chromatin state-dependent transcription outcomes. Genome Res. 2013;23:1580–1589. doi: 10.1101/gr.159400.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCord RP, Nazario-Toole A, Zhang H, Chines PS, Zhan Y, Erdos MR, Collins FS, Dekker J, Cao K. Correlated alterations in genome organization, histone methylation, and DNA-lamin A/C interactions in Hutchinson-Gilford progeria syndrome. Genome Res. 2013;23:260–269. doi: 10.1101/gr.138032.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meuleman W, Peric-Hupkes D, Kind J, Beaudry JB, Pagie L, Kellis M, Reinders M, Wessels L, van Steensel B. Constitutive nuclear lamina-genome interactions are highly conserved and associated with A/T-rich sequence. Genome Res. 2013;23:270–280. doi: 10.1101/gr.141028.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Øigård TA, Hanssen A, Hansen RE, Godtliebsen F. EM-estimation and modeling of heavy-tailed processes with the multivariate normal inverse Gaussian distribution. Signal Processing. 2005;85:1655–1673. [Google Scholar]
- Peric-Hupkes D, Meuleman W, Pagie L, Bruggeman SW, Solovei I, Brugman W, Graf S, Flicek P, Kerkhoven RM, van Lohuizen M, et al. Molecular maps of the reorganization of genome-nuclear lamina interactions during differentiation. Mol Cell. 2010;38:603–613. doi: 10.1016/j.molcel.2010.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pickersgill H, Kalverda B, de Wit E, Talhout W, Fornerod M, van Steensel B. Characterization of the Drosophila melanogaster genome at the nuclear lamina. Nat Genet. 2006;38:1005–1014. doi: 10.1038/ng1852. [DOI] [PubMed] [Google Scholar]
- Reddy KL, Zullo JM, Bertolino E, Singh H. Transcriptional repression mediated by repositioning of genes to the nuclear lamina. Nature. 2008;452:243–247. doi: 10.1038/nature06727. [DOI] [PubMed] [Google Scholar]
- Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, Smyth GK. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015;43:e47. doi: 10.1093/nar/gkv007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadaie M, Salama R, Carroll T, Tomimatsu K, Chandra T, Young AR, Narita M, Perez-Mancera PA, Bennett DC, Chong H, et al. Redistribution of the Lamin B1 genomic binding profile affects rearrangement of heterochromatic domains and SAHF formation during senescence. Genes Dev. 2013;27:1800–1808. doi: 10.1101/gad.217281.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schirmer EC, Foisner R. Proteins that associate with lamins: many faces, many functions. Exp Cell Res. 2007;313:2167–2179. doi: 10.1016/j.yexcr.2007.03.012. [DOI] [PubMed] [Google Scholar]
- Shah PP, Donahue G, Otte GL, Capell BC, Nelson DM, Cao K, Aggarwala V, Cruickshanks HA, Rai TS, McBryan T, et al. Lamin B1 depletion in senescent cells triggers large-scale changes in gene expression and the chromatin landscape. Genes Dev. 2013;27:1787–1799. doi: 10.1101/gad.223834.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shumaker DK, Dechat T, Kohlmaier A, Adam SA, Bozovsky MR, Erdos MR, Eriksson M, Goldman AE, Khuon S, Collins FS, et al. Mutant nuclear lamin A leads to progressive alterations of epigenetic control in premature aging. Proc Natl Acad Sci USA. 2006;103:8703–8708. doi: 10.1073/pnas.0602569103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teif VB, Vainshtein Y, Caudron-Herger M, Mallm JP, Marth C, Hofer T, Rippe K. Genome-wide nucleosome positioning during embryonic stem cell development. Nat Struct Mol Biol. 2012;19:1185–1192. doi: 10.1038/nsmb.2419. [DOI] [PubMed] [Google Scholar]
- Towbin BD, Gonzalez-Aguilera C, Sack R, Gaidatzis D, Kalck V, Meister P, Askjaer P, Gasser SM. Step-wise methylation of histone H3K9 positions heterochromatin at the nuclear periphery. Cell. 2012;150:934–947. doi: 10.1016/j.cell.2012.06.051. [DOI] [PubMed] [Google Scholar]
- Valouev A, Johnson SM, Boyd SD, Smith CL, Fire AZ, Sidow A. Determinants of nucleosome organization in primary human cells. Nature. 2011;474:516–520. doi: 10.1038/nature10002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viterbi AJ. Error bounds for convolutional codes and an asymptotically optimum decoding algorithm. IEEE Trans Inform Theory. 1967;13:260–269. [Google Scholar]
- Vogel MJ, Peric-Hupkes D, van Steensel B. Detection of in vivo protein-DNA interactions using DamID in mammalian cells. Nat Protoc. 2007;2:1467–1478. doi: 10.1038/nprot.2007.148. [DOI] [PubMed] [Google Scholar]
- Wen B, Wu H, Shinkai Y, Irizarry RA, Feinberg AP. Large histone H3 lysine 9 dimethylated chromatin blocks distinguish differentiated from embryonic stem cells. Nat Genet. 2009;41:246–250. doi: 10.1038/ng.297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson KL, Foisner R. Lamin-binding proteins. Cold Spring Harb Perspect Biol. 2010;2:a000554. doi: 10.1101/cshperspect.a000554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodcock CL, Skoultchi AI, Fan Y. Role of linker histone in chromatin structure and function: H1 stoichiometry and nucleosome repeat length. Chromosome Res. 2006;14:17–25. doi: 10.1007/s10577-005-1024-3. [DOI] [PubMed] [Google Scholar]
- Yokochi T, Poduch K, Ryba T, Lu J, Hiratani I, Tachibana M, Shinkai Y, Gilbert DM. G9a selectively represses a class of late-replicating genes at the nuclear periphery. Proc Natl Acad Sci USA. 2009;106:19363–19368. doi: 10.1073/pnas.0906142106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu J, Adli M, Zou JY, Verstappen G, Coyne M, Zhang X, Durham T, Miri M, Deshpande V, De Jager PL, et al. Genome-wide chromatin state transitions associated with developmental and environmental cues. Cell. 2013;152:642–654. doi: 10.1016/j.cell.2012.12.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zullo JM, Demarco IA, Pique-Regi R, Gaffney DJ, Epstein CB, Spooner CJ, Luperchio TR, Bernstein BE, Pritchard JK, Reddy KL, Singh H. DNA sequence-dependent compartmentalization and silencing of chromatin at the nuclear lamina. Cell. 2012;149:1474–1487. doi: 10.1016/j.cell.2012.04.035. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.