

Abstract
A library of sixteen 2nd generation amino- and amido-substituted carboranyl pyrimidine nucleoside analogues, designed as substrates and inhibitors of thymidine kinase 1 (TK1) for potential use in boron neutron capture therapy (BNCT) of cancer, was synthesized and evaluated in enzyme kinetic-, enzyme inhibition-, metabolomic-, and biodistribution studies. One of these 2nd generation carboranyl pyrimidine nucleoside analogues (YB18A [3]), having an amino group directly attached to a meta-carborane cage tethered via ethylene spacer to the 3-position of thymidine, was approximately 3–4 times superior as a substrate and inhibitor of hTK1 than N5-2OH (2), a 1st generation carboranyl pyrimidine nucleoside analogue. Both 2 and 3 appeared to be 5′-monophosphorylated in TK1(+) RG2 cells, both in vitro and in vivo. Biodistribution studies in rats bearing intracerebral RG2 glioma resulted in selective tumor uptake of 3 with an intratumoral concentration that was approximately 4 times higher than that of 2. The obtained results significantly advance the understanding of the binding interactions between TK1 and carboranyl pyrimidine nucleoside analogues and will profoundly impact future design strategies for these agents.
Keywords: Carboranyl Pyrimidine Nucleoside Analogue, Thymidine Kinase 1 (TK1), Carborane, Boron Neutron Capture Therapy (BNCT), DNA Repair Inhibition
1. Introduction
TK1-like enzymes, including human thymidine kinase 1 (hTK1), Bacillus anthracis TK (BaTK), and Staphylococcus aureus TK (SaTK), play essential roles in the biosynthetic salvage pathways of nucleotide synthesis, complementing the de novo pathways particularly in proliferating cells and bacteria [1–6]. In normal human cells, TK1 activity is high only in S-phase. In cancer cells, however, TK1 activity can remain also high in other phases of the cell cycle [7–9].
Clinically relevant agents that rely on TK1 activity include the HIV/AIDS prodrugs zidovudine (AZT) and stavudine (d4T) [10]. Both compounds are analogues of the endogenous TK1 substrate, thymidine (dThd, 1, Fig. 1). They are effectively 5′-monophosphorylated by TK1. Their ultimate mechanism of action, however, is based on the subsequent formation of a cytotoxic triphosphate metabolite by other kinases. Other biomedically relevant analogues of 1 that possibly rely on 5′-monophosphorylation by TK1 are 3-carboranyl thymidine analogues (3CTAs). The agents were developed as boron delivery agents for boron neutron capture therapy (BNCT) of brain tumors [11, 12]. Other types of carboranyl nucleoside derivatives were developed for the same and other purposes [13–17]. BNCT is a binary cancer treatment system that relies on the accumulation of boron-10 (10B) in tumor cells followed by external neutron irradiation. Subsequent capture of neutrons by 10B results predominantly in high linear energy transfer (LET) ionizing radiation, i.e., 4He2+ (a-particle) and 7Li3+ nuclei. These particles can selectively destroy tumor cells because of their limited path lengths of < 10 μm in biological tissue. Prerequisite for the success of BNCT is the selective accumulation of 10B in tumor vs. normal cells [18, 19].
Fig. 1.
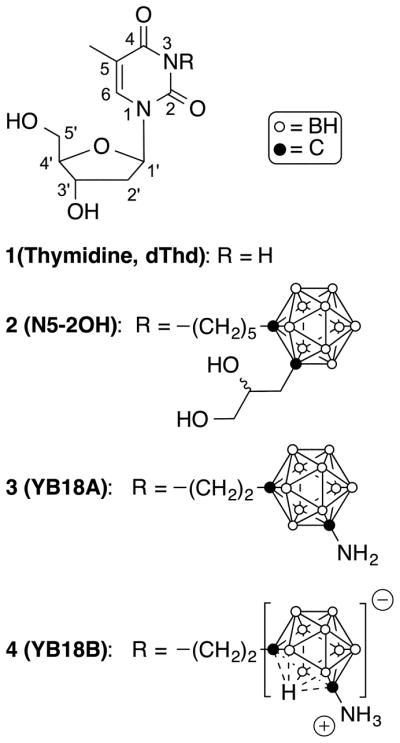
Structures of 1, the first generation 3CTA 2, and the second generation 3CTAs 3 and 4.
First generation 3CTAs, such as N5-2OH (2, Fig. 1), were found to be good substrates of TK1, despite having a bulky and highly lipophilic carboranyl substitutent [20] tethered via alkyl spacer to the 3-position. Preferential uptake of 2 by TK1(+) vs. TK1(−) cells in vitro, presumably involving intracellular trapping of its 5′-monophosphate, was observed [21, 22]. Consequently, a favorable in vivo biodistribution profile of 2 in tumor bearing rodents, possibly encompassing tumor-selective accumulation by intracellular accumulation of the 5′-monophosphate metabolite, led to promising preclinical BNCT of rats with brain tumors [21, 22]. However, the need for improvement of 1st generation 3CTAs became apparent during these studies. They had moderate capacity to inhibit TK1 activity, i.e., to compete with endogenous 1 for phosphorylation at the substrate-binding site [11, 23].
In order to make further progress in achieving the crucial objective of improved TK1 substrate and inhibitory capacities, the design, synthesis, and biological evaluation of carboranyl pyrimidine nucleoside analogues with amino- or amido functionalities in meta (1,7)-carborane cluster-containing side chains either at the 3- or 4-position will be described in this paper (see Fig. 1 for the atom numbering of pyrimidine nucleosides).
2. Results
2.1. Design and Chemistry
All TK1-like enzymes have similar overall 3-D fold including the presence of a structural component that has been designated as the ‘lasso-loop’ [6, 24]. The enzymes undergo significant conformational changes upon binding of 1 and ATP [25, 26]. In the apo state, the lasso loop is folded away from the substrate-binding site whereas in the holo state, the lasso loop covers the substrate-binding site tightly by forming hydrogen bonds with 1, primarily via main chain atoms.
Preliminary computational docking of 1st generation 3CTAs, such as 2, to TK1 crystal structures led to the hypothesis that initial substrate binding results in incomplete closure of the lasso loop, leaving the bulky carborane cage outside of the active site, and that some hydrogen bonds are lost because of the 3-substitution [23, 27, 28]. This model may explain the moderate TK1 inhibitor characteristics of 1st generation 3CTAs because endogenous 1 may still have ample competitive access to the substrate binding site. It is also conceivable that a location of the highly hydrophobic carborane cage outside of the enzyme is disrupting the hydrogen bond network of water molecules and that this is contributing to the lack of binding of 3CTAs to TK1. It needs to be emphasized, however, that the exact molecular interactions between 3CTAs and TK1 amino acid residues remain unknown.
Based on the current hypothesis on 3CTA-TK1 interactions, previous efforts in our laboratories have focused on the introduction of hydrogen donor/acceptors, included hydroxyl-, amidino-, and guanidino groups, in a spacer element between the carborane cluster and the scaffold of 1 in 2nd generation 3CTAs to re-establish hydrogen bonds between substrate and enzyme that were lost due to the 3-substitution [29, 30]. These groups were mostly placed in close proximity to the 3-position. Unfortunately, these 3CTAs did not prove to be superior to 2 as TK1 substrates and inhibitors. In the case of some compounds, lack of stability was another problem [29]. The design concept for carboranyl pyrimidine nucleosides discussed in this paper explores the introduction of hydrogen donor/acceptor amino- or amido groups in spacer elements between meta-carborane cluster and pyrimidine nucleoside scaffold, both at the 3-(Schemes 1–3) and the 4-(Scheme 4) position. In addition, 3 (YB18A) and 4 (YB18B) (Fig. 1), both containing an amino group directly attached to a meta-carborane cluster tethered via ethylene spacer to the 3-position, were prepared for detailed enzyme kinetic and inhibition studies. The syntheses of both compounds and their evaluation in preliminary phosphate transfer assays (PTAs) were described previously by us [31]. Compound 2, synthesized according to a procedure described previously [32], and 1 were used in all biological studies as reference compounds.
Scheme 1.
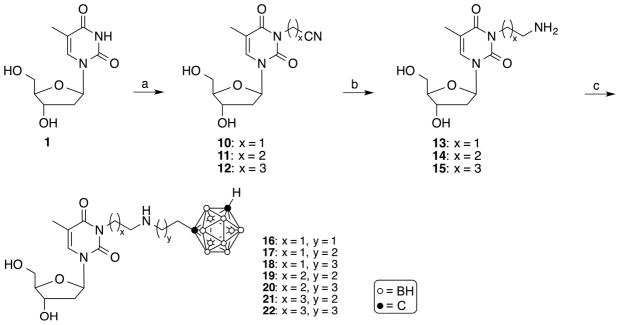
Synthesis of compound 9. Reagents and conditions: (a) NH2NH2 • H2O, 1M KOH in MeOH/H2O (1/2), MeOH, 2 h, rt; (b) tert-butyldimethylsilyl chloride, imidazole, DMF, overnight, rt; (c) 1-(3-iodopropyl)-closo-1,7-carborane [35], NaH, DMF, 90 °C, 2h; (d) TBAF, THF, 2h, rt.
Scheme 3.
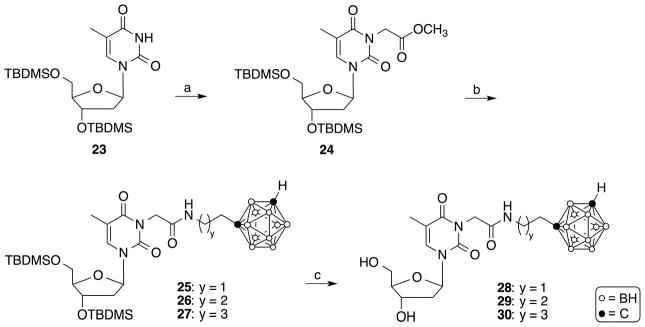
Synthesis of compounds 28–30. Reagents and conditions: (a) Methyl bromoacetate, acetone, NaH, 2 h, rt ; (b) 1-(2-aminoethyl)-closo-1,7-carborane [35], 1-(3-aminopropyl)-closo-1,7-carborane [35], or 1-(4-aminobutyl)-closo-1,7-carborane [35], 1,2,4-triazole, DBU, 72 h, 80 °C; (c) TBAF, THF, rt, 2 h.
Scheme 4.
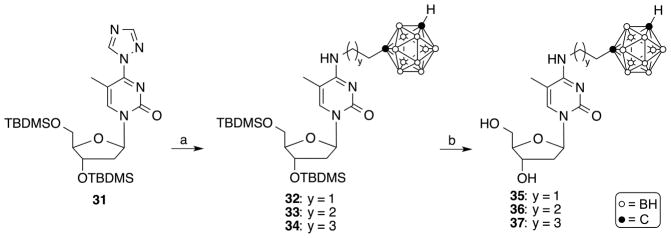
Synthesis of compounds 35–37. Reagents and conditions: (a) 1-(2-Aminoethyl)-closo-1,7-carborane [35], 1-(3-aminopropyl)-closo-1,7-carborane [35], or 1-(4-aminobutyl)-closo-1,7-carborane [35], CH3CN, overnight, 60 °C; (b) TBAF, THF, rt, 2 h.
The synthesis of the target compound 9 is shown in Scheme 1. Modifying a procedure previously described by Ariza et al. for the synthesis of 15N-enriched 5′-O-acetyl-3-amino-2′-3′-isopropylidene uridine [33], 3′,5′-diacetyl-3-nitrothymidine [34] (5) was treated with hydrazine hydrate and potassium hydroxide in a mixture of MeOH and H2O at room temperature for 2 h to yield 3-aminothymidine (6) in 28% yield following purification by reversed phase (RP)-18 HPLC using 0.1% TFA in H2O as the solvent system followed by evaporation. According to 13C-NMR (SM), compound 6 did not retain trifluoroacetate. tert-Butyldimethylsilyl-(TBDMS) protection of the 3′- and 5′-hydroxyl groups of 6 was accomplished by stirring tert-butyldimethylsilyl chloride in the presence of imidazole in DMF at room temperature overnight to produce 7, which was isolated in 90% yield following purification by silica gel column chromatography. Compound 7 was N-alkylated with 1-(3-iodopropyl)-closo-1,7-carborane [35] in the presence of sodium hydride in DMF at 90°C for 2 h to produce compound 8 in 30% yield. The deprotection of the TBDMS-groups was accomplished by treatment with TBAF in THF at room temperature for 2 h to produce target compound 9 in 63% yield. Compounds 8 and 9 were purified by silica gel chromatography. In the synthesis of 9, the N-alkylation at the hydrazine-type primary amine had to be performed using 3′,5′-TBDMS-protected 7 because the reaction did not proceed with its unprotected precursor 6.
The synthesis of target compounds 16–22 is described in Scheme 2. 3-Cyanomethylthymidine (10) [29], 3-(2-cyanoethyl)thymidine (11), and 3-(3-cyanopropyl)thymidine (12) were synthesized by reacting 1 with the appropriate cyanoalkyl halide in the presence of potassium carbonate in DMF/acetone (1/1, v/v) at 60°C for 8 h, as described previously by Agarwal et al.[29]. The compounds were purified by silica gel chromatography in 78–97% yield. Reduction of 10–12 using potassium borohydride and Raney nickel in EtOH at room temperature for 1 h [36] produced compounds 13–15 in 67–82% yield following purification by RP-18 HPLC (MeOH/H2O solvents systems containing 0.1 TFA) followed by evaporation. Compounds 13–15 were 3-alkylated with 1-(2-iodoethyl)-closo-1,7-carborane [35]. 1-(3-iodopropyl)-closo-1,7-carborane [35], and 1-(4-iodobutyl)-closo-1,7-carborane [35], respectively, in the presence of sodium hydride in DMF at 90°C for 2 h to produce target compounds 16–22. Despite the presence of free hydroxyl groups in 13–15, alkylation predominantly resulted in the formation of N-monoalkylated and, to a lesser extent, N,N-dialkylated product. Compounds 16–22 were purified by RP-18 HPLC (MeOH/H2O solvents systems containing 0.1 TFA). Following evaporation, yields ranged from 4–12 % for compound 16–18 and 26–48% for 19–22. The low yields found for 16–18 reflect a significant level of decomposition observed of these compounds especially during reactions and storage. According to 13C-NMR (SM), some of the compounds 13–22 retained traces of trifluoroacetate. The synthesis of amido group containing target compounds 28–30 is shown in Scheme 3. The reaction of 3′,5′-bis-O-(tert-butyldimethylsilyl)thymidine [37] (23) with methyl bromoacetate in the presence of sodium hydride in anhydrous acetone at room temperature for 2 h gave compound 24 in 69% yield following purification by silica gel chromatography. The aminolysis of the ester in 24 was achieved by acyl transfer catalysis [38] using a mixture of 1,2,4-triazole and 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) and either 1-(2-aminoethyl)-closo-1,7-carborane [35], 1-(3-aminopropyl)-closo-1,7-carborane [35], or 1-(4-aminobutyl)-closo-1,7-carborane [35]. The reaction was carried at 80°C for 3 days without solvent to produce compounds 25–27 in 12–48% yields. Aminolysis was carried out with 3′,5′-TBDMS-protected nucleoside in accordance with a previously reported procedure [39].
Scheme 2.

Synthesis of compounds 16–22. Reagents and conditions: (a) Iodoacetonitrile, 3-bromopropionitrile, or 4-bromobutanenitrile, K2CO3, DMF/acetone (1/1, v/v), 8 h, 60 °C ; (b) KBH4, Raney nickel, EtOH, rt; (c) 1-(2-iodoethyl)-closo-1,7-carborane [35], 1-(3-iodopropyl)-closo-1,7-carborane [36], or 1-(4-iodobutyl)-closo-1,7-carborane [35], NaH, DMF, 2 h, 90 °C.
Deprotection of the TBDMS-groups of 25–27 was accomplished with TBAF in THF at room temperature for 2 h to produce target compounds 28–30 in 71–93% yields. Compounds 25–30 were purified by silica gel chromatography.
The synthesis of the N4-carboranylalkyl-5-methyl-2′-deoxycytidine analogues 35–37 is shown in Scheme 4. The reaction of compound 31 [39] with 1-(2-aminoethyl)-closo-1,7-carborane [35], 1-(3-aminopropyl)-closo-1,7-carborane [35], or 1-(4-aminobutyl)-closo-1,7-carborane [35] at 60°C in CH3CN overnight gave 32, 33, and 34 in 87%, 40%, and 94% yield, respectively. The TBDMS-groups of 32–34 were removed by treatment with TBAF in THF for 2 h at room temperature to produce N4-carboranylalkyl-5-methyl-2′-deoxycytidine analogues 35, 36, 37 in 86%, 95%, and 97% yield, respectively. Compounds 32–37 were purified by silica gel chromatography.
2.2. Biological Studies
PTAs of 1, 2, 3, 4, 9, 16–22, 28–30 and 35–37 were carried out using [γ-32P]ATP as the phosphate donor and recombinant hTK1 as described previously [29, 31]. 5′-Monophosphorylated products of the test compounds were detected by β-radiography of polyethylenimine (PEI)-cellulose chromatograms that were developed from small quantities of assay mixtures (Figs. 2 and S2/Supplementary Materials [SM]). The phosphorylation rates of the all test compounds are expressed relative to that of 1, the natural substrate of TK1 (Table 1, Fig. 2). We have used such relative phosphorylation rates (rPRs) previously to establish preliminary SARs for larger 3CTA libraries [29, 31]. Relative PRs do not reflect 100% accurately the enzyme kinetic properties of test compounds and they provide, if at all, little information about their enzyme inhibitory capacities [29, 31]. Both properties are important to judge the potential of carboranyl pyrimidine nucleoside analogues as boron delivery agents for BNCT. Therefore, compounds 2, 3, 4, and 21 and 22, which were both stable and had high rPRs, were further evaluated in detailed enzyme kinetic- and inhibitory studies to obtain Km, kcat, rkcat/Km (kcat/Km relative to that of 1) and IC50 of inhibition values (Table 1) [29].
Fig. 2.
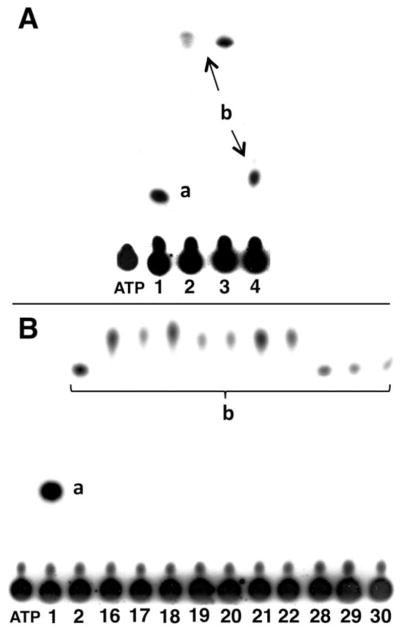
A: β-Autoradiogram of a TLC analysis of the products from phosphate transfer assays using TK1 and [γ-32P]ATP (phosphate donor) as well as 1, 2, 3, and 4 as substrates. B: β-Autoradiogram of a TLC analysis of the products from phosphate transfer assays using TK1 and [γ-32P]ATP (phosphate donor) as well as 1, 2, 16–22, and 28–30 as substrates. a: Monophosphate of 1; b; compound monophosphate.
Table 1.
rPR, rKcat/KM, and IC50 values of 1–4, 16–22, and 28–30
| Compd | rPRa (%) | Km (μM) | kcat (s−1) | rkcat/Km (%) | IC50 (μM) |
|---|---|---|---|---|---|
| 1 | 100 | 2.4 ± 0.7 | 8.6 | 100 | 2.0 ± 0.3 |
| 2 | 40.0 ± 11.2 | 24.3 ± 2.1 | 5.1 | 5.8 | 37.5 ± 6.3 |
| 3 | 78.7 ± 3.4 | 7.5 ± 3.2 | 5.9 | 21.6 | 13.5 ± 2.1 |
| 4 | 79.4 ± 7.8 | 10.7 ± 1.2 | 6.1 | 15.7 | 8.9 ±2.2 |
| 16 | 35.2 ± 5.3 | nd | nd | nd | nd |
| 17 | 24.5 ± 7.8 | nd | nd | nd | nd |
| 18 | 44.4 ± 8.7 | nd | nd | nd | nd |
| 19 | 18.8 ± 4.8 | nd | nd | nd | nd |
| 20 | 19.5 ± 5.3 | nd | nd | nd | nd |
| 21 | 45.2 ± 3.8 | 80.1 ± 37.6 | 5.9 | 2.0 | >250 |
| 22 | 38.0 ± 6.5 | 39.8 ± 10.4 | 5.4 | 3.7 | >250 |
| 28 | 19.2 ± 4.5 | nd | nd | nd | nd |
| 29 | 14.5 ± 4.8 | nd | nd | nd | nd |
| 30 | 12.3 ± 8.7 | nd | nd | nd | nd |
rPr: Phosphorylation rate relative to that of dThd. SDs are based on at least three measurements. nd: not determined. The following compound concentrations were used: rPR values: 100 μM nucleoside analogue and 100 μM ATP; Km and kcat values: 1.95–500 μM nucleoside analogue and 100 μM ATP; IC50 values: 0.5 μM dThd, 5 mM ATP, and 0–250.00 μM nucleoside analogue.
The obtained rPRs for 3 and 4 were about twice as high as that of 2 (78.7 and 79.4% vs. 40.0%) and were in accordance with previously reported rPRs for both compounds [31]. Compounds 16–18, with -(CH2)2-NH-(CH2)2,3,4- spacer had rPRs ranging form 24.5–44.4%. These compounds proved to be unstable during storage in aqueous solutions even at −20°C within 5–7 days. Among the compounds that were found to be stable during storage, compounds 21 and 22, having -(CH2)3-NH-(CH2)3- and -(CH2)3-NH-(CH2)4- spacer, respectively, between carborane and scaffold of 1, had rPRs comparable to that of 2 (45% and 38% vs. 40%), whereas the rPRs of compounds 19, 20 and 28–30 were markedly lower (Table 1). A preliminary PTA for compound 9 resulted in an rPR of ~30 % (SM, S.2). Interestingly, N4-carboranylalkyl-5-methyl-2′-deoxycytidine analogues 35–37 were not substrates of TK1 (data not shown). Overall, there was no clear SAR pattern among compounds 9, 16–22 and 28–30. If at all, the amido-substituted 28–30 had slightly lower rPRs than the amino-substituted 9 and 16–22.
Using rPRs and stability as selection criteria, in-depth enzyme kinetic studies were carried out for compounds 1, 2, 3, 4, 21 and 22 using methods previously described by us (Table 1) [29]. The rkcat/Km values of 3 and 4 were 2.7 – 3.7 times higher than that of 2 (15.7 % and 21.6% vs. 5.8%) whereas those of 21 (2.0 %) and 22 (3.7 %) were markedly lower. Furthermore, competitive inhibition studies were carried for 3, 4, 21 and 22 to determine IC50 values for their ability to compete for phosphorylation with 1 at the substrate-binding site of TK1 (Table 1) [29]. The obtained values were compared with those of 1 and 2. The IC50 values for 21 and 22 (>250 μM) indicated that these compounds did not effectively compete with 1. In contrast, the IC50 values of 3 and 4 were 2.7–4.2 times lower than that of 2 (13.5 μM and 8.9 μM vs. 37.5 μM).
Although 3 and 4 appeared to be equally effective as substrates and inhibitors of TK1, only 3 was initially selected for further in vitro and in vivo these studies because 4 presumably is a mixture of two stable zwitterionic atropoisomers [11, 31], as is evident from, e.g., the split signal for the H1′-proton in the 1H-NMR at 6.31 ppm and 6.39 ppm (SM, S3.3.1). In addition, it is conceivable that the charged character of these structures in this mixture impedes passive diffusion through cell membranes. Compound 3 was evaluated in qualitative in vitro and in vivo metabolomic HRMS studies using RG2 glioma cells and intracerebral RG2 tumors, respectively (Table 2). In addition, biodistribution studies were carried out in rats bearing intracerebral RG2 gliomas (Table 3). In both types of studies, 2 was used as a reference compound. The intracerebral RG2 glioma model was chosen for these studies because 3CTAs were initially designed and developed for BNCT of brain tumors via intracerebral (i.c.) injection [21]. The TK1 protein expression in RG2 cells appeared to be slightly higher than in TK(+) L929 cells, as determined by Western Blot analysis (Fig. 3). Both TK1(+) and TK1(−) L929 cells have been used previously by our group to analyze the intracellular formation of metabolites of 2 by radiodetection techniques [12].
Table 2.
Accurate mass HRMS analysis of 2 and 3 and their monophosphate metabolites in RG2 cells and intracranial RG2 gliomasa
| Comp | Modeb | in vitro/ in vivo | Molecular formula | Foundc (m/z) | Calculatedd (m/z) | Deviation (ppm) |
|---|---|---|---|---|---|---|
| 2 | Positive | in vitro | C20H40N2O7B10 + H+ | 529.3941 | 529.3917 | 4.5 |
| 2 | Negative | in vitro | C20H40N2O7B10 − H+ | 527.3776 | 527.3760 | 3.0 |
| 2 | Negative | in vivo | C20H40N2O7B10 − H+ | 527.3782 | 527.3760 | 4.2 |
| 2-MPe | Negative | in vitro | C20H39N2O10B10P + K+ | 645.3022 | 645.2983 | 6.0 |
| 2-MP | Negative | in vivo | C20H39N2O10B10P + K+ | 645.2966 | 645.2983 | 2.6 |
| 3 | Positive | in vitro | C14H29N3O5B10 + H+ | 428.3195 | 428.3189 | 1.4 |
| 3 | Negative | in vitro | C14H29N3O5B10 + HCOO− | 472.3078 | 472.3087 | 1.9 |
| 3 | Negative | in vivo | C14H29N3O5B10 + HCOO− | 472.3108 | 472.3087 | 4.4 |
| 3-MP | Negative | in vitro | C14H28N3O8B10P + K+ | 544.2322 | 544.2254 | 12.5 |
| 3-MP | Negative | in vivo | C14H28N3O8B10P + K+ | 544.2249 | 544.2254 | 0.9 |
See Supplementary Materials for detailed measured and calculated isotope patterns.
Negative and positive ion modes.
Values corresponding the most intense peak of the theoretical isotopic pattern.
Most intense peak of the theoretical isotopic pattern. Calculated with Isotope Pattern Calculator v4.0 (http://yanjunhua.tripod.com/pattern1.htm).
Monophosphate
Table 3.
Biodistribution of 2 and 3 in rats bearing intracerebral RG2 gliomasa
| Boron concentrations (μg/g) | Ratios | |||||
|---|---|---|---|---|---|---|
|
| ||||||
| Compd | Tumor | R. Brain | L. Brain | Blood | T/B | T/Bl |
| 2 | 8.8 ± 2.9 | 2.1 ± 0.9 | 1.3 ± 0.5 | 1.0 ±0.4 | 4.3 | 9.3 |
| 3 | 32.5 ± 17.9 | 6.6 ± 8.4 | 1.5 ± 1.1 | 0.5 ± 0.03 | 4.9 | 62.5 |
RG2 gliomas (105 cells/10 μL) were implanted stereotactically into the right caudate nucleus of syngeneic Fischer rats. Biodistribution studies were initiated 14 days subsequent to implantation. Quantities of 2 and 3 equivalent to 100 μg of boron, solubilized in 200 μL 35% aqueous DMSO, were administered in a volume of 200 μL were administered i.c. by means of ALZET pumps (model #2001D) over 24 hours at a flow rate of 8.33 μL/h. Rats were euthanized immediately thereafter, and their tumors, brains and blood were taken for boron determinations by ICP-OES. SDs are based on 4 animals per group.
Fig. 3.

Western blot analysis of the TK1 expression in RG2, TK1 (+) L929, and TK1 (−) L929 cells.
In general, non-phosphorylated 2 and 3 could be detected both in vitro and in vivo samples by HRMS metabolomic analysis in both positive and negative ion mode (Table 2). In contrast, the corresponding monophosphates, 2-MP and 3-MP, were only measurable in negative ion mode. Di- and triphosphates of 2 and 3 could not be detected in either ion mode. The deviation of 12.5 ppm between calculated and found mass for 3-MP in vitro is somewhat too high for accurate mass HRMS standards. However, the shape of the entire isotope pattern observed in this case is indicative for a 3-MP metabolite (SM, S1.9.).
For a biodistribution study, 2 and 3 were administered intracerebrally (i.c.) by means of ALZET® pumps over a period of 24 h in rats bearing intracerebral RG2 gliomas, as described previously by us [21, 22]. Rats were euthanized immediately thereafter and tumors, normal brain tissue, and blood were taken for boron determination by inductively coupled plasma-optical emission spectroscopy (ICP-OES). The results of this biodistribution study are summarized in Table 3. The boron concentration in brain tumors exposed to 3 (32.5 μg boron (B)/g tumor) was about 4 times higher than that for 2 (8.9 μg B/g tumor). This difference reflects approximately the differences of TK1 substrate and inhibitor capacities between both compounds suggestion that intracellular entrapment of a monophosphate form of 3 is a factor contributing to its increased uptake in the tumor. Boron concentrations in the right and left cerebral hemispheres following administration of 3 were higher than those for 2 (6.6/1.5 μg B/g brain vs. 1.5/1.3 μg B/g brain). However, boron concentrations for both 2 and 3 in surrounding normal brain were significantly lower than in the TK1(+) proliferating tumor cells, as can be expected for non-dividing cells without significant TK1 activity. The boron concentration in the blood was lower than in the tumor and in normal brain (0.5 μg B/g blood vs. 1.0 μg B/g blood), resulting in tumor/blood ratio of 62.4 for 3 compared to 9.3 for 2 The slight differences in uptake between the normal brain of the right and left cerebral hemispheres is explained by the mode of delivery; that is, direct administration to the same site that the tumor cells had been stereotactically implanted, i.e., the right caudate nucleus (see section 4.2.4. for details). The fact that the boron concentrations were low or non-detectable in the left, non-tumor bearing cerebral hemisphere and the blood, indicated that there was little intracerebral transfer of the test agents to the blood or via the corpus collosum from the right to the left cerebral hemisphere.
3. Discussion
Design strategies applied in recent years by our group to improve the capacity of 3CTAs to bind to TK1 focused on the introduction of hydrogen donor/acceptors in a spacer element between the carborane cluster and the 3-position of the scaffold of 1 to re-establish hydrogen bonds between substrate and enzyme [11, 29, 30]. None of these previously evaluated 2nd generation 3CTAs was markedly superior to 2 as a TK1 substrate and inhibitor. Some of the introduced functional groups improved aqueous solubility and/or impeded compound stability [29]. In addition, despite significant differences in structures and also physicochemical properties, no clear-cut TK1 substrate SAR emerged for these 3CTAs [11]. Similar results were obtained for compounds 9, 16–22, and 28–30, having amino- and amido groups in the linker between carborane and 1. Moving the carboranylalkylamino-substituent from the 3- to the 4-position of the pyrimidine nucleoside scaffold, as in compounds 35–37, abolished TK1 substrate characteristics. Lesnikowski et al. recently reported similar observations for a series of triazolo- and ether modified carboranyl pyrimidine nucleoside analogues [13].
A detailed analysis of their kinase interacting properties showed that both 3 and 4 were ~ 3–4 better substrates and inhibitors of TK1 than 2. This is the first SAR study clearly identifying 2nd generation 3CTAs with markedly improved activity compared with 1st generation 2. Biodistribution studies with rats bearing intracerabral RG2 glioma, resulting in a tumor to blood ratio of ~ 62 for 3 compared to ~ 9 for 2, seemed to reflect this superiority.
Metabolomic HRMS studies showed that 2 and 3 were monophosphorylated to 2-MP and 3-MP, respectively, in TK1(+) RG2 cells, both in vitro and in vivo. In addition, non-phosphorylated 2 and 3 were detected in both types of biological samples, whereas the di- and triphosphates of both 3CTAs were not found. These results confirm the recently reported analysis of the intracellular formation of metabolites of 2 in TK1(+)- and TK1(−) L929 cells by radiodetection techniques [12], which showed production of tritiated 2-MP in TK1(+) L929 cells but not in their TK1(−) L929 counterparts. Di- and triphosphorylation did not seem to occur in the TK1(+) L929 cells but a significant quantities of tritiated 2 was found in both cells variants. Therefore, it is conceivable that mechanisms other than phosphorylation contribute to the tumor-selective accumulation of 3CTAs, such 2 and 3.
The most important consequence of the results presented here is that they challenge the current hypothesis for the design of 3CTAs. It seems that depending on an appropriate spacer length between carborane cage and nucleoside scaffold in 3CTAs, the cluster is actually located within the substrate-binding pocket. Furthermore, a single amino substituent at the cluster appeared to interact with amino acid residues within the substrate-binding site leading to markedly enhanced TK1 binding affinity and, thus, improved substrate characteristics and more effective competition with 1. This indicates that the use of the carborane scaffold as a 3D-platform for the introduction of additional/different substituents can lead to 3rd generation 3CTAs with further enhanced TK1 substrate/inhibitor capacities.
Another potential application of 3CTAs in cancer therapy could be that of TK1 inhibitors used in combination with DNA damaging γ-radiation or chemotherapeutics. Recent studies have demonstrated that DNA damage results in increased TK1 expression especially in p53-deficient HCT-116 cancer cells [40]. TK1 expression proved to be nonessential for the growth of p53-deficient HCT-116 cells, but was important in providing sufficient triphosphate pools of 1 for DNA repair triggered by the genotoxic insult of these cells. Consequently, TK1 repression, for example via siRNA-mediated silencing, in p53-deficient HCT-116 cells caused an increase in cell death following DNA damage [40–42]. The apparent importance of TK1 for DNA repair, particularly in the context of p53-gene abnormalities, suggests the possible use of 3CTAs as inhibitors of TK1 in combination therapies, possibly even including BNCT, of cancer via systemic administration.
4. Experimental protocol
4.1. Chemistry
4.1.2. General conditions for chemical experiments
1H- and 13C-NMR spectra were obtained on a Bruker AVIII400HD NMR spectrometer or a Bruker DRX400 NMR spectrometer at The Ohio State University College of Pharmacy. Chemical shifts (δ) are reported in ppm from internal deuterated chloroform or deuterated methanol. Coupling constants are reported in Hz. 13C NMR spectra are fully decoupled. Accurate- and high resolution mass spectra were obtained on a Waters Micromass LCT mass spectrometer or a Waters Micromass Q-TOF II mass spectrometer at The Ohio State University Campus Chemical Instrumentation Center, or a Waters Micromass Q-TOF micro mass spectrometer at The Ohio State University College of Pharmacy. For all carborane-containing compounds, the found mass corresponding to the most intense peak of the theoretical isotopic pattern was reported. Measured patterns agreed with calculated patterns.
Silica gel 60 (0.063 –0.200 mm), used for gravity column chromatography, and silica gel 60 (0.015–0.049 mm), used for flash column chromatograph, were purchased from Dynamic Adsorbents Inc. (Norcross, GA). Reagent-grade solvents were used for silica gel column chromatography. Precoated glass-backed TLC plates with silica gel 60 F254 (0.25-mm layer thickness) from Dynamic Adsorbents (Norcross, GA) were used for TLC. General compound visualization for TLC was achieved by UV light. Carborane-containing compounds were selectively visualized by spraying the plate with a 0.06% PdCl2/1% HCl solution and heating at 120°C, which caused the slow (15–45 s) formation of a gray spot due to the reduction of Pd2+ to Pd0. Preparative HPLC purification was performed with a Gemini 5μ C18 column (21.20 mm x 250 mm, 5 μm particle size) supplied by Phenomenex Inc. CA, USA on a Hitachi HPLC system (L-2130) with a Windows based data acquisition and Hitachi Diode array detector (L-2455). Purification was accomplished using two different solvent systems: 1) 0.1 TFA in H2O and 2) H2O (0.1% TFA)/MeOH (0.1% TFA). The flow rate was 7 mL/min. Detailed gradient conditions for purification of specific compounds are provided in the following. Analytical HPLC was conducted using a Gemini 5μ C18 110A Column (250 x 4.6 mm) supplied by Phenomenex Inc. CA, USA using the Hitachi system used for preparative HPLC. Two different methods were used: 1) 0.1% TFA in H2O, and 2) H2O (0.1% TFA)/MeOH (0.1% TFA). The flow rate was 1 mL/min. For method 2 the following gradient was applied: 100 (H2O): 0 (MeOH) to 0:100 over 20 min and from 0:100 to 100:0 over 10 min. HPLC-grade solvents were used for HPLC. All compounds used for biological studies had a purity >95% at 254 nm detection according to analytical HPLC (SM).
Anhydrous solvents were purchased directly either from Acros Organics (Morris Plains, NJ) or from Sigma Aldrich (Milwaukee, WI). THF was distilled from sodium and benzophenone indicator under argon. closo-1,7-Carborane was purchased from Katchem Ltd. (Prage, Czech Republic). Thymidine (1, Thd) was purchased from Chem-Impex International, Wood Dale, IL. Other chemicals used for synthesis were purchased from standard commercial vendors. 1-(2-Iodoethyl)-closo-1,7-carborane [35], 1-(3-iodopropyl)-closo-1,7-carborane [35], 1-(4-iodobutyl)-closo-1,7-carborane [35], 1-(2-aminoethyl)-closo-1,7-carborane [35], 1-(3-aminopropyl)-closo-1,7-carborane [35], and 1-(4-aminobutyl)-closo-1,7-carborane [35], as well as compounds 2 [32], 3 [31], 4 [31], 5 [34], 10 [29], 23 [37], and 31[39] were synthesized according to previously described procedures. Spectroscopic data for compounds 2–5, 10, and 31 are provided in SM. Procedures for the syntheses of compounds 6 [43, 44], 11 [45], 13 [46], 14 [47], and 15[47] that are different from those described in this paper were reported previously. Unless specified otherwise, all reactions were carried out under argon atmosphere.
4.1.3. 3-Aminothymidine (6)
3′,5′-Diacetyl-3-nitrothymidine [34] (5) (2 g, 5.4 mmol) was dissolved in MeOH (130 mL) and added to the mixture of hydrazine hydrate (400 μL, 8.5 mmol) and 1M KOH in 12 mL of MeOH and 25 mL H2O. The reaction mixture was stirred at room temperature for 2 h. The solvent was removed under reduced pressure and the residue was purified by preparative HPLC (method 1). The solvent was evaporated under reduced pressure to afford a white amorphous solid. Retention time (analytical HPLC method 1): 28.9 min; yield: 400 mg, 28%. 1H NMR (400 MHz, CD3OD) δ 7.83 (q, J = 1.0 Hz 1H), 6.31 (t, J = 6.6 Hz, 1H), 4.57 (br.s, ~3H), 4.41 (dt, J = 3.5, 6.7 Hz, 1H), 3.93 (q, J = 3.5 Hz, 1H), 3.81 (dd, J = 3.2, 12.1 Hz, 1H), 3.74 (dd, J = 3.7 and 12.1 Hz, 1H), 2.17–2.40 (m, 2H), 1.95 (d, J = 1.2 Hz, 3H). 13C NMR (100 MHz, CD3OD) δ 161.92, 149.65, 134.19, 108.99, 87.96, 86.39, 71.02, 61.70, 40.44, 12.07. Accurate mass HRMS (ESI+): m/z calcd for C10H16N3O5 (M+H)+ 258.1090, found 258.1099, 280.0905 (M+Na)+, 296.0677 (M+K)+.
4.1.4. 3-Amino-3′,5′-bis-O-(tert-butyldimethylsilyl)thymidine (7)
3-Aminothymidine (6) (500 mg, 2 mmol), tert-butyldimethylsilyl chloride (1.8 g, 11.5 mmol), and imidazole (0.75 g, 11.5 mmol) were dissolved in DMF (100 mL) and stirred overnight at room temperature. The solvent was evaporated under reduced pressure and the residue purified by silica gel column chromatography using a DCM/MeOH solvent system. The product was obtained as a white amorphous solid at a solvent ratio of DCM/MeOH (24/1, v/v). Rf: 0.47 (DCM/MeOH, 24/1, v/v); yield; 0.8 g, 86%. 1H NMR (400 MHz, CDCl3) δ 7.42–7.46 (m, 1H), 6.32 (dd, J = 5.9, 7.5 Hz, 1H), 4.92 (br.s, ~1.5H), 4.37 (dt, J = 2.7, 5.7 Hz, 1H), 3.92 (q, J = 2.5 Hz, 1H), 3.85 (dd, J = 2.6, 11.4 Hz, 1H), 3.73 (dd, J = 2.6, 11.3 Hz, 1H), 2.27 (ddd, J = 2.8, 5.9, 13.1 Hz, 1H), 1.96–2.03 (m, 1H), 1.94 (s, 3H), 0.86 and 0.89 (two s, 18H), 0.045–0.079 (four s, 12H). 13C NMR (100 MHz, CDCl3) δ 160.55, 148.53, 132.30, 109.30, 88.07, 85.96, 72.22, 62.99, 41.74, 26.03, 25.84, 18.49, 18.10, 13.29, −4.53, −4.74, −5.28, −5.32. Accurate mass HRMS (ESI+): m/z calcd for C22H43N3NaO5Si2 (M+Na)+ 508.2639, found 508.2641.
4.1.5. 3′,5′-Bis-O-(tert-butyldimethylsilyl)-3-{[3-(closo-1,7-carboranyl)propyl]amino}thymidine (8)
A mixture of compound 7 (30 mg, 0.06 mmol), 1-(3-iodopropyl)-closo-1,7-carborane [35] (19 mg, 0.06 mmol), and NaH (3 mg, 0.12 mmol, 60% NaH in mineral oil) in DMF (2 mL) was stirred at 80°C for 2 h. The solvent was evaporated and the residue was purified by silica gel column chromatography to give the product as a white amorphous solid. Rf: 0.72 (hexanes/EtOAc, 7/3, v/v); yield: 12.4 mg, 30%; 1H NMR (400 MHz, CDCl3) δ 7.47 (s, 1H), 6.32 (t, J = 7.2 Hz, 1H), 4.33–4.39 (m, 1H), 3.90–3.95 (m, 1H), 3.87 (dd, J = 2.3 and 11.5 Hz, 1H), 3.73 (dd, J = 2.1 and 11.5 Hz, 1H), 3.31-1.55 (br. m, 10H), 2.80–2.90 (m, 3H), 2.22–2.30 (m, 1H), 2.02–2.12 (m, 2H), 1.94–2.12 (m, 1H), 1.92 (s, 3H), 1.54–1.64 (m, 2H), 0.87 and 0.89 (two s, 18H), 0.05 and 0.08 (two s, 12H). 13C NMR (100 MHz, CDCl3) δ 162.1, 149.8, 133.33, 109.68, 88.1, 85.9, 75.9, 72.3, 63.1, 54.9, 49.3, 41.8, 34.4, 28.24, 26.1, 18.3, 13.4, −4.9. Accurate mass HRMS (ESI+): m/z calcd for C27H59B10N3NaO5Si2 (M+Na)+ 692.4910, found 692.4912.
4.1.6. 3-{[3-(closo-1,7-Carboranyl)propyl]amino}thymidine (9)
A mixture of compound 8 (12 mg, 0.018 mmol) and TBAF (38 μL, 0.04 mmol, 1M in THF) was stirred in THF (1 mL) for 2 h. The solvent was evaporated and the residue was purified by silica gel column chromatography to give the product as a white amorphous solid. Rf: 0.49 (DCM/MeOH, 9/1, v/v); yield: 5 mg, 63%. 1H NMR (400 MHz, CD3OD) δ 7.85 (s, 1H), 6.30 (t, J = 6.6 Hz, 1H), 4.34–4.43 (m, 1H), 3.89–3.95 (m, 1H), 3.80 (dd, J = 2.7 and 12.1 Hz, 1H), 3.73 (dd, J = 3.5 and 12.1 Hz), 3.49 (s, 1H), 2.83 (t, J = 6.9 Hz, 2H), 2.08–2.34 (m, 4H), 1.93 (s, 3H), 1.55–3.31 (br. m, 10H), 1.52–1.62 (m, 2H). 13C NMR (100 MHz, CD3OD) δ 164.0, 151.7, 136.0, 110.5, 88.9, 87.4, 77.7, 72.1, 62.7 56.87, 50.0, 41.4, 35.5, 29.1 13.1. Accurate mass HRMS (ESI+): m/z calcd for C15H31B10N3NaO5 (M+Na)+ 464.3173, found 464.3173.
4.1.7. General procedure for the synthesis of compounds 11 and 12
Thymidine (1) (5 g, 20.6 mmol) and potassium carbonate (8.5 g, 61.5 mmol) were dissolved in acetone/DMF (100 mL, 1/1, v/v) and cyanoalkyl halide (45 mmol) was added. The reaction mixture was stirred under reflux for 6 h, cooled to room temperature, added to H2O (750 mL), and extracted with EtOAc (4 × 250 mL). The organic layer was separated, dried with anhydrous magnesium sulfate, evaporated under reduced pressure, and purified by silica gel column chromatography. The product was obtained as a white amorphous solid at a solvent ratio of DCM/MeOH, 24/1, v/v.
4.1.7.1. 3-(2-Cyanoethyl)thymidine (11)
Rf: 0.70 (DCM/MeOH, 9/1, v/v), yield: 5.8 g, 97%. 1H NMR (400 MHz, CD3OD) δ 7.86 (s, 1H), 6.29 (t, J = 6.6 Hz, 1H), 4.36–4.44 (m, 1H), 4.21 (dt, J = 2.7 and 6.7 Hz, 2H), 3.88–3.96 (m, 1H), 3.80 (dd, J = 3.0 and 12.0 Hz, 1H), 3.73 (dd, J = 3.5 and 12.0 Hz, 1H), 2.82 (t, J = 6.7 Hz, 2H), 2.15–2.35 (m, 2H), 1.91 (s, 3H). 13C NMR (100 MHz, CD3OD) δ 165.09, 152.16, 137.02, 119.04, 110.80, 88.99, 87.31, 72.16, 62.86, 41.45, 37.93, 16.64, 13.32. Accurate mass HRMS (ESI+): m/z calcd for C13H17N3NaO5 (M+Na)+ 318.1066, found 318.1033, 334.0800 (M+K)+.
4.1.7.2. 3-(3-Cyanopropyl)thymidine (12)
Rf: 0.70 (DCM/MeOH, 9/1, v/v), yield: 5.0 g, 80%. 1H NMR (400 MHz, CD3OD, δ ppm) δ 7.85 (s, 1H), 6.29 (t, J = 6.6 Hz, 1H), 4.36–4.44 (m, 1H), 4.05 (t, J = 6.7 Hz, 2H), 3.91 (q, J = 3.3 Hz, 1H), 3.80 (dd, J = 3.1 and 12.0 Hz, 1H), 3.73 (dd, J = 3.6 and 12.0 Hz, 1H), 2.50 (t, J = 7.0 Hz, 2H), 2.16–2.34 (m, 2H), 1.92–2.02 (m, 2H), 1.91 (s, 3H). 13C NMR (100 MHz, CD3OD) δ 165.66, 152.57, 136.79, 121.02, 110.78, 88.98, 87.29, 72.13, 62.86, 41.49, 41.34, 24.98, 15.55, 13.41. Accurate mass HRMS (ESI+): m/z calcd for C14H19N3NaO5 (M+Na)+ 332.1222, found 332.1195.
4.1.8. General procedure for the synthesis of compounds 13–15
Potassium borohydride (2 g, 36 mmol) and Raney nickel (~600 mg) were dissolved in anhydrous ethanol (50 mL) [36]. 3-Cyanoalkylthymidine (10–12) (3.6 mmol) was added to the reaction mixture, which was stirred for 2 h at room temperature. The solvent was evaporated under reduced pressure and the residue was purified by preparative HPLC using solvent system 2 with the following gradient: 100 (H2O/TFA):00 (MeOH/TFA) for 15 min, 100:00 to 50:50 over 75 min, and from 50:00 to 100:00 over the following 10 min. The solvents were evaporated under reduced pressure to afford a white amorphous solid.
4.1.8.1. 3-(2-Aminoethyl)thymidine (13)
Rf: 0.10 (DCM/MeOH, 17/3, v/v, retention time (preparative HPLC): 33–42 min, yield: 0.68 g, 67%. 1H NMR (400 MHz, CD3OD) δ 7.86 (s, 1H), 6.28 (t, J = 6.6 Hz, 1H), 4.34–4.40 (m, 1H), 4.20 (t, J = 5.2 Hz, 2H), 3.86–3.92 (m, 1H), 3.77 (dd, J = 2.9 and 12.0 Hz, 1H), 3.70 (dd, J = 3.4 and 12.0 Hz, 1H), 3.18 (t, J = 5.2 Hz, 2H), 2.15–2.30 (m, 2H), 1.89 (s, 3H). 13C NMR (100 MHz, CD3OD) δ 165.76, 152.87, 136.88, 110.99, 88.85, 87.19, 72.10, 62.80, 41.07, 40.03, 39.73, 13.15. Accurate mass HRMS (ESI+): m/z calcd for C12H20N3O5 (M+H)+ 286.1403, found 286.1388, 324.0936 (M+K)+.
4.1.8.2. 3-(3-Aminopropyl)thymidine (14)
Rf: 0.10 (DCM/MeOH, 17/3, v/v), retention time (preparative HPLC): 30–48 min, yield: 0.83 g, 82%. 1H NMR (400 MHz, CD3OD) δ 7.89 (s, 1H), 6.31 (t, J = 6.6 Hz, 1H), 4.36–4.42 (m, 1H), 4.04 (t, J = 6.4 Hz, 2H), 3.86–3.94 (m, 1H), 3.80 (dd, J = 2.9 and 12.0 Hz, 1H), 3.73 (dd, J = 3.5 and 12.0 Hz, 1H), 2.95 (t, J = 7.0 Hz, 2H), 2.15–2.32 (m, 2H), 1.94–2.04 (m, 2H), 1.92 (s, 3H). 13C NMR (100 MHz, CD3OD) δ 165.80, 152.69, 136.99, 110.90, 89.09, 87.34, 72.28, 62.91, 41.39, 39.11, 38.43, 27.07, 13.31. Accurate mass HRMS (ESI+): m/z calcd for C13H22N3O5 (M+H)+ 300.1559, found 300.1549, 322.1449 (M+Na)+.
4.1.8.3. 3-(4-Aminobutyl)thymidine (15)
Rf: 0.10 (DCM/MeOH, 17/3, v/v), retention time (preparative HPLC): 38–60 min, yield: 0.80 g, 79%. 1H NMR (400 MHz, CD3OD: δ 7.84 (s, 1H), 6.29 (t, J = 6.6 Hz, 1H), 4.38–4.44 (m, 1H), 3.90–4.00 (m, 3H), 3.80 (dd, J = 3.0 and 12.0 Hz, 1H), 3.74 (dd, J = 3.7 and 12.0 Hz, 1H), 3.95 (t, J = 6.7 Hz, 2H), 2.16–2.34 (m, 2H), 1.89 (s, 3H), 1.58–1.72 (br s, 4H). 13C NMR (100 MHz, CD3OD) δ 165.65, 152.51, 136.77, 110.03, 88.95, 87.25, 72.30, 62.95, 41.76, 41.33, 40.82, 26.78, 26.81, 13.56. Accurate mass HRMS (ESI+): m/z calcd for C14H24N3O5 (M+H)+ 314.1716, found 314.1698, 336.1479 (M+Na)+.
4.1.9. General procedure for the synthesis of compounds 16–22
Compounds 13–15 (0.18 mmol), iodoalkyl-closo-1,7-carborane [35] (0.18 mmol), and NaH (13 mg, 0.54 mmol, 60% in mineral oil) were dissolved in anhydrous DMF (2 mL) and stirred for 2 h at 80°C. Following the addition of 100 μL H2O the reaction mixture was purified by preparative HPLC using solvent system 2 with the following gradient: 80 (H2O/TFA): 20 (MeOH/TFA) to 00:100 over 90 min and from 00:100 to 100:00 over next 10 min. The solvents were evaporated under reduced pressure to afford a white amorphous solid.
4.1.9.1. 3-({2-[2-(closo-1,7-Carboranyl)ethyl]amino}ethyl)thymidine (16)
Rf: 0.71 (DCM/MeOH, 4/1, v/v), retention time (preparative HPLC): 45–48 min, yield: 3 mg, 4%. 1H NMR (400 MHz, CD3OD) δ 7.93 (s, 1H), 6.33 (t, J = 6.7 Hz, 1H), 4.38–4.44 (m, 1H), 4.20–4.26 (m, 2H), 3.92–3.96 (m, 1H), 3.81 (dd, J = 2.9 and 12.0 Hz, 1H), 3.74 (dd, J = 3.3 and 12.0 Hz, 1H), 3.60–3.70 (m 3H), 3.02–3.10 (m, 2H), 2.30–2.40 (m, 2H), 2.20–2.30 (m, 2H), 1.94 (s, 3H), 1.55–3.31 (br. m, 10H). Accurate mass HRMS (ESI+): m/z calcd for C16H33B10N3O5 (M+H)+ 456.3496, found 456.3539, 478.3349 (M+Na)+.
4.1.9.2. 3-({2-[3-(closo-1,7-Carboranyl)propyl]amino}ethyl)thymidine (17)
Rf: 0.71 (DCM/MeOH, 4/1, v/v), retention time (preparative HPLC): 44–50 min, yield: 6 mg, 7%. 1H NMR (400 MHz, CD3OD) δ 7.92 (s, 1H), 6.33 (t, J = 6.7 Hz, 1H), 4.38–4.45 (m, 1H), 4.25 (t, J = 5.3 Hz, 2H), 3.93 (q, J = 3.1 Hz, 1H), 3.81 (dd, J = 3.1 and 12.0 Hz, 1H), 3.74 (dd, J = 3.5 and 12.0 Hz, 1H), 3.56 (s, 1H), 3.03 (t, J = 8.0 Hz, 2H), 2.95 (t, J = 7.7 Hz, 2H), 2.20–2.30 (m, 2H), 2.07 (t, J = 7.4 Hz, 2H), 1.94 (s, 3H), 1.65–1.85 (m, 2H), 1.55–3.31 (br. m, 10H). 13C NMR (100 MHz, CD3OD) δ 165.78, 152.90, 137.13, 111.01, 89.12, 87.34, 76.34, 72.31, 62.90, 57.93, 57.25, 43.63, 41.39, 39.07, 34.63, 26.34, 13.22. Accurate mass HRMS (ESI+): m/z calcd for C17H36B10N3O5 (M+H)+ 470.3658, found 470.3694.
4.1.9.3. 3-({2-[4-(closo-1,7-Carboranyl)butyl]amino}ethyl)thymidine (18)
Rf: 0.71 (DCM/MeOH, 4/1, v/v), retention time (preparative HPLC): 50–54 min, yield: 10 mg, 12%. 1H NMR (400 MHz, CD3OD) δ 7.92 (s, 1H), 6.33 (t, J = 6.7 Hz, 1H), 4.38–4.45 (m, 1H), 4.26 (t, J = 5.0 Hz, 2H), 3.90–3.96 (m, 1H), 3.81 (dd, J = 3.1 and 12.0 Hz, 1H), 3.74 (dd, J = 3.5 and 12.0 Hz, 1H), 3.52 (s, 1H), 3.31 (m, 2H), 3.00 (t, J = 7.6 Hz, 2H), 2.20–2.35 (m, 2H), 2.00–2.10 (m, 2H), 1.94 (s, 3H), 1.55–1.65 (m, 2H), 1.55–3.31 (br. m, 10H), 1.40–1.50 (m, 2H). 13C NMR (100 MHz, CD3OD) δ 165.70, 152.85, 137.05, 110.93, 89.06, 87.26, 72.24, 71.90, 62.82, 57.02, 47.88, 41.32, 38.99, 37.40, 28.06, 26.76, 13.14. Accurate mass HRMS (ESI+): m/z calcd for C18H38B10N3O5 (M+H)+ 484.3815, found 484.3834.
4.1.9.4. 3-({3-[3-(closo-1,7-Carboranyl)propyl]amino}propyl)thymidine (19)
Rf: 0.66 (DCM/MeOH, 17/3, v/v), retention time (preparative HPLC): 62–64 min, yield: 22 mg, 26%. 1H NMR (400 MHz, CD3OD) δ 7.92 (s, 1H, H-6), 6.32 (t, J = 6.7 Hz, 1H), 4.38–4.45 (m, 1H), 4.04 (t, J = 6.4 Hz, 2H), 3.88–3.94 (m, 1H), 3.81 (dd, J = 3.0 and 12.0 Hz, 1H), 3.74 (dd, J = 3.5 and 12.0 Hz, 1H), 3.51 (s, 1H), 2.90–3.04 (m, 4H), 2.18–2.32 (m, 2H), 1.98–2.10 (m, 2H), 1.92 (s, 3H), 1.55–3.31 (br. m, 10H), 1.40–1.60 (m, 4H). 13C NMR (100 MHz, CD3OD) δ 165.86, 152.68, 137.06, 110.90, 89.11, 87.31, 77.41, 72.26, 62.87, 57.08, 46.53, 41.43, 39.13, 37.55, 28.21, 26.97, 25.92, 13.38. Accurate mass HRMS (ESI+): m/z calcd for C18H38B10N3O5 (M+H)+ 484.3815, found 484.3837.
4.1.9.5. 3-({3-[4-(closo-1,7-Carboranyl)butyl]amino}propyl)thymidine (20)
Rf: 0.71 (DCM/MeOH, 17/3, v/v), retention time (preparative HPLC): 62–70 min, yield: 26 mg, 30%. 1H NMR (400 MHz, CD3OD) δ 7.91 (s, 1H), 6.31 (t, J = 6.6 Hz, 1H), 4.38–4.45 (m, 1H), 4.04 (t, J = 6.4 Hz, 2H), 3.89–3.95 (m, 1H), 3.80 (dd, J = 3.0 and 12.0 Hz, 1H), 3.74 (dd, J = 3.5 and 12.0 Hz, 1H), 3.50 (s, 1H), 2.93–3.03 (m, 4H), 2.14–2.30 (m, 2H), 1.95–2.05 (m, 4H), 1.92 (s, 3H), 1.55–3.31 (br. m, 10H), 1.40–1.65 (m, 4H). 13C NMR (100 MHz, CD3OD) δ 165.84, 152.70, 137.06, 110.92, 89.13, 87.35, 77.41, 72.25, 62.88, 58.50, 53.17, 48.53, 41.43, 39.13, 37.55, 28.19, 26.95, 25.91, 13.33. Accurate mass HRMS (ESI+): m/z calcd for C19H40B10N3O5 (M+H)+ 498.3966, found 498.4001.
4.1.9.6. 3-({4-[3-(closo-1,7-Carboranyl)propyl]amino}butyl)thymidine (21)
Rf: 0.63 (DCM/MeOH, 17/3, v/v), retention time (preparative HPLC): 58–68 min, yield: 42 mg, 48%. 1H NMR (400 MHz, CD3OD) δ 7.86 (s, 1H), 6.30 (t, J = 6.6 Hz, 1H), 4.38–4.45 (m, 1H), 3.90–4.00 (m, 3H), 3.80 (dd, J = 3.0 and 12.0 Hz, 1H), 3.73 (dd, J = 3.6 and 12.0 Hz, 1H), 3.52 (s, 1H), 3.00–3.10 (m, 2H), 2.92 (t, J = 7.6 Hz, 2H), 2.14–2.30 (m, 2H), 2.09 (t, J = 8.6 Hz, 2H), 1.92 (s, 3H), 1.65–1.85 (m, 6H), 1.55–3.31 (br. m, 10H). 13C NMR (100 MHz, CD3OD) δ 165.34, 152.28, 136.55, 110.66, 88.85, 87.04, 76.28, 72.09, 62.72, 56.98, 47.83, 43.41, 41.19, 41.16, 34.43, 27.67, 25.56, 24.39, 13.16. Accurate mass HRMS (ESI+): m/z calcd for C19H40B10N3O5 (M+H)+ 498.3966, found 498.3962.
4.1.9.7. 3-({4-[4-(closo-1,7-Carboranyl)butyl]amino}butyl)thymidine (22)
Rf: 0.63 (DCM/MeOH, 17/3, v/v), retention time (preparative HPLC): 64–69 min, yield: 32 mg, 35%. 1H NMR (400 MHz, CD3OD) δ 7.87 (s, 1H), 6.31 (t, J = 6.6 Hz, 1H), 4.38–4.45 (m, 1H), 3.90–4.00 (m, 3H), 3.80 (dd, J = 3.0 and 12.0 Hz, 1H), 3.73 (dd, J = 3.6 and 12.0 Hz, 1H), 3.50 (s, 1H), 3.03 (t, J = 6.6 Hz, 2H), 2.97 (t, J = 7.6 Hz, 2H), 2.14–2.30 (m, 2H), 2.03 (t, J = 8.4 Hz, 2H), 1.91 (s, 3H), 1.65–1.75 (m, 4H), 1.55–1.65 (m, 2H), 1.55–3.31 (br. m, 10H), 1.42–1.52 (m, 2H). 13C NMR (100 MHz, CD3OD) δ 165.44, 152.37, 136.65, 110.75, 88.95, 87.14, 72.19, 62.81, 56.96, 49.90, 48.47 and 48.40, 41.29, 37.42, 28.06, 26.79, 25.69, 24.52, 13.23. Accurate mass HRMS (ESI+): m/z calcd for C20H42B10N3O5 (M+H)+ 512.4128, found 512.4112.
4.1.10. 3′,5′-Bis-O-(tert-butyldimethylsilyl)-3-(methoxycarbonylmethyl)thymidine (24)
A mixture of compound 2338 (1 g, 2.12 mmol), methyl bromoacetate (0.36 mL, 4.2 mmol), and sodium hydride (0.16 g, 6.8 mmol, 60% in mineral oil) were stirred in anhydrous acetone (5 mL) for 2 h at room temperature. H2O (100 μL) was added to the reaction mixture, the solvents evaporated under reduced pressure, and the residue purified by silica gel chromatography using hexanes/EtOAc (4/1, v/v) as solvent system to afford a white amorphous solid. Rf: 0.58 (hexanes/EtOAc, 4/1, v/v), yield: 0.8 g, 69%. 1H NMR (400 MHz, CDCl3) δ 7.53 (s, 1H), 6.36 (dd, J = 5.9 and 7.8 Hz, 1H), 4.71 (s, 2H), 4.38–4.45 (m, 1H), 3.92–3.97 (m, 1H), 3.88 (dd, J =2.6 and 11.4 Hz, 1H), 3.77–3.80 (m, 1H), 3.76 (s, 3H), 2.27 (ddd, J = 2.6, 5.7 and 13.1 Hz, 1H), 1.98–2.06 (m, 1H), 1.95 (s, 3H), 0.89 and 0.94 (two s, 18H), 0.08 and 0.12 (two s, 12H). 13C NMR (100 MHz, CDCl3) δ 168.39, 162.91, 150.56, 134.03, 110.93, 87.85, 85.61, 72.24, 62.97, 52.41, 41.92, 41.46, 25.92, 25.70, 18.38, 17.96, 13.19, −4.78, −5.43. Accurate mass HRMS (ESI+): m/z calcd for C25H46N2NaO7Si2 (M+Na)+ 565.2736, found 565.2738.
4.1.11. General procedure for the synthesis of compounds 25–27
A mixture of compound 24 (50 mg, 0.1 mmol), aminoalkyl-closo-1,7-carborane [35] (0.1 mmol), 1,2,4-triazole (2 mg, 0.03 mmol) and 1,8-diazabicyclo[5.4.0]undec-7-ene (5.4 μL, 0.036 mmol) were heated without solvent for 3 days at 80°C. The reaction mixture was purified by silica gel chromatography using DCM/MeOH (19/1, v/v) as the solvent system to afford the desired product as a white amorphous solid.
4.1.11.1. 3′,5′-Bis-O-(tert-butyldimethylsilyl)-3-({2-[2-(closo-1,7-carboranyl)ethyl]amino}-2-oxoethyl)thymidine (25)
Rf: 0.62 (DCM/MeOH, 19/1, v/v), yield: 7.4 mg, 12%. 1H NMR (400 MHz, CDCl3) δ 7.53 (s, 1H), 6.34 (dd, J = 5.9 and 7.7 Hz, 1H), 5.90 (t, J = 5.6 Hz, 1H), 4.57 (s, 2H), 4.38–4.43 (m, 1H), 3.93–3.96 (m, 1H), 3.87 (dd, J = 2.5 and 11.4 Hz, 1H), 3.76 (dd, J = 2.2 and 11.4 Hz), 3.20–3.30 (m, 2H), 2.93 (s, 1H), 2.22 (ddd, J = 2.5, 5.7 and 13.2 Hz, 1H), 2.18 (t, J = 7.4 Hz, 2H), 1.96–2.05 (m, 1H), 1.95 (s, 3H), 1.55–3.31 (br. m, 10H), 0.94 and 0.91 (two s, 18H), 0.08 and 0.12 (two s, 12H). 13C NMR (100 MHz, CDCl3) δ 166.94, 163.22, 150.65, 134.18, 109.92, 87.86, 85.66, 72.22, 72.58, 62.96, 55.11, 43.93, 41.47, 39.13, 35.64, 25.71, 25.91, 17.96, 18.39, 13.20, −5.42, −4.77. Accurate mass HRMS (ESI+): m/z calcd for C28H59B10N3NaO6Si2 (M+Na)+ 720.4843, found 720.4894.
4.1.11.2. 3′,5′-Bis-O-(tert-butyldimethylsilyl)-3-({2-[3-(closo-1,7-carboranyl)propyl]amino}-2-oxoethyl)thymidine (26)
Rf: 0.64 (DCM/MeOH, 19/1, v/v), yield: 13 mg, 20%. 1H NMR (400 MHz, CD3OD) δ 7.63 (s, 1H), 6.28 (dd, J = 6.3 and 7.5 Hz, 1H), 4.54 (s, 2H), 4.45–4.51 (m, 1H), 3.92–3.98 (m, 1H), 3.89 (dd, J = 3.2 and 11.4 Hz, 1H), 3.83 (dd, J = 3.1 and 11.4 Hz, 1H), 3.47 (s, 1H), 3.14 (t, J = 6.6 Hz, 2H), 2.26 (ddd, J = 2.8, 5.9 and 13.3 Hz, 1H), 2.14.–2.22 (m, 1H), 2.02 (t, J = 8.8 Hz, 2H), 1.93 (s, 3H), 1.55–3.31 (br. m, 10H), 1.53–1.66 (m, 2H), 0.94 and 0.96 (two s, 18H), 0.13 and 0.15 (two s, 12H). 13C NMR (100 MHz, CD3OD) δ 169.91, 165.03, 152.22, 136.07, 110.72, 89.36, 87.26, 73.82, 64.26, 56.90, 44.37, 41.96, 39.68, 35.28, 31.11, 26.33, 26.53, 18.93, 19.36, 13.44, −4.49, −5.17. Accurate mass HRMS (ESI+): m/z calcd for C29H61B10N3NaO6Si2 (M+Na)+ 734.5000; found 734.5054.
4.1.11.3. 3′,5′-Bis-O-(tert-butyldimethylsilyl)-3-({2-[4-(closo-1,7-carboranyl)butyl]amino}-2-oxoethyl)thymidine (27)
Rf: 0.63 (DCM/MeOH, 19/1, v/v), yield: 32 mg, 48%. 1H NMR (400 MHz, CDCl3) δ 7.52 (s, 1H), 6.35 (dd, J = 5.9 and 7.7 Hz, 1H), 5.87 (t, J = 5.2 Hz, 1H), 4.57 (s, 2H), 4.34–4.42 (m, 1H), 3.92–3.96 (m, 1H), 3.87 (dd, J = 2.4 and 11.4 Hz, 1H), 3.76 (dd, J = 2.2 and 11.4 Hz, 1H,), 3.22 (q, J = 6.4 Hz, 2H), 2.90 (s, 1H), 2.27 (ddd, J = 2.4, 5.7 and 13.0 Hz, 1H), 1.96–2.06 (m, 3H), 1.94 (s, 3H), 1.55–3.31 (br. m, 10H), 1.24–1.46 (m, 4H), 0.89 and 0.93 (two s, 18H), 0.07 and 0.12 (two s, 12H). 13C NMR (100 MHz, CDCl3) δ 166.83, 163.21, 150.68, 134.06, 109.89, 87.81, 85.61, 72.20, 62.94, 54.77, 43.93, 41.44, 39.10, 36.39, 29.03, 27.11, 25.69, 25.90, 17.95, 18.37, 13.20, −5.44, −4.79. Accurate mass HRMS (ESI+): m/z calcd for C30H63B10N3NaO6Si2 (M+Na)+ 748.5156, found 748.5128.
4.1.12. General procedure for the synthesis of compounds 28–30
THF (1 mL) was added to compounds 25–27 (0.034 mmol) and TBAF (75 μL, 1 M solution in THF, 0.075 mmol). The reaction mixture was stirred at room temperature for 2 h, the solvents were evaporated under reduced pressure, and the residue was purified by silica gel chromatography using DCM/MeOH, 9/1, v/v as solvent system to produce a white amorphous solid.
4.1.12.1. 3-({2-[2-(closo-1,7-Carboranyl)ethyl]amino}-2-oxoethyl)thymidine (28)
Rf: 0.51 (DCM/MeOH, 9/1, v/v), yield: 15 mg, 93%. 1H NMR (400 MHz, CD3OD) δ 7.91 (s, 1H), 6.28 (t, J = 6.6 Hz, 1H), 4.53 (s, 2H), 4.37–4.44 (m, 1H), 3.89–3.94 (m, 1H), 3.81 (dd, J = 2.9 and 12.0 Hz, 1H), 3.72 (dd, J = 3.6 and 12.0 Hz, 1H), 3.53 (s, 1H), 3.20 (t, J = 7.9 Hz, 2H), 2.21–2.32 (m, 2H), 2.18 (t, J = 7.9 Hz, 2H), 1.92 (s, 3H), 1.55–3.31 (br. m, 10H). 13C NMR (100 MHz, CD3OD) δ 169.90, 165.26, 152.38, 137.03, 110.69, 89.02, 87.38, 72.13, 62.83, 57.28, 44.37, 41.51, 40.24, 36.58, 13.26. Accurate mass HRMS (ESI+): m/z calcd for C16H31B10NaN3O6 (M+Na)+ 492.3114, found 492.3158.
4.1.12.2. 3-({2-[3-(closo-1,7-carboranyl)propyl]amino}-2-oxoethyl)thymidine (29)
Rf: 0.54 (DCM/MeOH, 9/1, v/v), yield: 14 mg, 83%. 1H NMR (400 MHz, CD3OD) δ 7.90 (s, 1H), 6.29 (t, J = 6.5 Hz, 1H), 4.54 (s, 2H), 4.37–4.46 (m, 1H), 3.88–3.96 (m, 1H), 3.82 (dd, J = 2.8 and 12.0 Hz, 1H), 3.74 (dd, J = 3.3 and 12.0 Hz, 1H), 3.49 (s, 1H), 3.13 (t, J = 6.3 Hz, 2H), 2.21–2.34 (m, 2H), 2.02 (t, J = 8.3 Hz, 2H), 1.93 (s, 3H), 1.53–1.66 (m, 2H), 1.55–3.31 (br. m, 10H). 13C NMR (100 MHz, CD3OD) δ 169.93, 165.16, 152.31, 136.88, 110.61, 89.91, 87.25, 72.03, 62.74, 56.95, 44.33, 41.44, 39.70, 35.29, 31.04, 13.21. Accurate mass HRMS (ESI+): m/z calcd for C17H33B10N3NaO6 (M+Na)+ 506.3270, found 506.3274.
4.1.12.3. 3-({2-[4-(closo-1,7-carboranyl)butyl]amino}-2-oxoethyl)thymidine (30)
Rf: 0.47 (DCM/MeOH, 9/1, v/v), yield: 12 mg, 71%. 1H NMR (400 MHz, CD3OD) δ 7.90 (s, 1H), 6.29 (t, J = 6.5 Hz, 1H), 4.54 (s, 2H), 4.32–4.44 (m, 1H), 3.86–3.95 (m, 1H), 3.81 (dd, J = 2.7 and 12.0 Hz, 1H), 3.73 (dd, J = 3.3 and 12.0 Hz, 1H), 3.47 (s, 1H), 3.10–3.21 (m, 2H), 2.18–2.33 (m, 2H), 1.95–2.05 (m, 2H), 1.92 (s, 3H), 1.55–3.31 (br. m, 10H), 1.35–1.49 (m, 4H). 13C NMR (100 MHz, CD3OD) δ 169.88, 165.26, 152.40, 136.94, 110.69, 89.00, 87.32, 78.04, 72.12, 62.83, 56.96, 44.37, 41.51, 39.99, 37.85, 30.09, 28.47, 13.30. Accurate mass HRMS (ESI+): m/z calcd for C18H35B10N3NaO6 (M+Na)+ 520.3427, found 520.3444, 1017.7020 (2M+Na)+.
4.1.13. General procedure for the synthesis of compound 32–35
A mixture of compound 31 [39] (52 mg, 0.1 mmol) and aminoalkyl-closo-1,7-carborane [35] (0.3 mmol) in anhydrous CH3CN (2 mL) was refluxed overnight. The solvent was evaporated and residue was purified by silica gel column chromatography DCM/acetone (9/1, v/v) as the solvent system to produce a white amorphous solid.
4.1.13.1. 3′,5′-Bis-O-(tert-butyldimethylsilyl)-N4-[2-(closo-1,7-carboranyl)ethyl]-5-methyl-2′-deoxycytidine (32)
Rf: 0.52 (DCM/acetone, 9/1, v/v), yield: 53 mg, 87%. 1H NMR (400 MHz, CDCl3) δ 7.59 (s, 1H), 6.32 (t, J = 6.7 Hz, 1H), 5.48 (br s, 1H), 4.37–4.44 (m, 1H), 3.92–3.73 (m, 3H), 3.48–3.57 (m, 2H), 2.97 (s, 1H), 2.32–2.42 (m, 1H), 2.29 (t, J = 7.3, 2H), 1.95–2.04 (m, 1H), 1.93 (s, 3H), 1.55–3.31 (br. m, 10H), 0.92 and 0.89 (two s, 18H), 0.11, 0.10, 0.07 and 0.06 (four s, 12H). 13C NMR (100 MHz, CDCl3) δ 163.87, 157.52, 139.14, 103.98, 90.11, 89.35, 87.71, 87.52, 73.44, 64.34, 56.89, 36.78, 32.50, 27.50, 27.26, 19.96, 19.51, 14.96, 14.66, −3.31, −3.76, −3.8, −3.83. Accurate mass HRMS (ESI+): m/z calcd for C26H58B10N3O4Si2 (M +H)+ 640.4969, found 640.5021.
4.1.13.2. 3′,5′-Bis-O-(tert-butyldimethylsilyl)-N4-[3-(closo-1,7-carboranyl)propyl]-5-methyl-2′-deoxycytidine (33)
Rf: 0.5 (DCM/acetone, 9/1, v/v), yield: 20 mg, 40%. 1H NMR (400 MHz, CDCl3) δ 7.51 (s, 1H), 6.38 (t, J = 6.25 Hz, 1H), 4.85 (br s, 1H), 4.36 (m, 1H), 3.84–3.92 (m, 2H), 3.76 (m, 1H), 3.47 (m, 2H), 2.90 (s, 1H), 2.36 (m, 1H), 1.93–2.02 (m, 3H), 1.88 (s, 3H), 1.66–1.68 (m, 2H), 1.55–3.31 (br. m, 10H), 0.92 and 0.88 (two s, 18H), 0.10, 0.09, 0.06 and 0.05 (four s, 12H). 13C NMR (100 MHz, CDCl3) δ 163.08, 156.22, 137.51, 101.20, 87.61, 85.81, 75.70, 71.83, 62.85, 55.07, 42.17, 40.15, 34.21, 30.21, 25.92, 18.56, 18.17, 13.29, −4.42, −4.74, −5.21, −5.26. Accurate mass HRMS (ESI+): m/z calcd for C27H59B10N3O4Si2Na (M +Na)+ 676.4945, found 676.4980.
4.1.13.3. 3′,5′-Bis-O-(tert-butyldimethylsilyl)-N4-[4-(closo-1,7-carboranyl)butyl]-5-methyl-2′-deoxycytidine (34)
Rf: 0.56 (DCM/acetone, 9/1, v/v), yield: 60 mg, 94%. 1H NMR (400 MHz, CDCl3) δ 7.49 (s, 1H), 6.37 (t, J = 6.51 Hz, 1H), 4.95 (s, 1H), 4.35 (m, 1H), 3.83–3.93 (m, 2H), 3.75 (m, 1H), 3.49 (m, 2H), 2.89 (s, 1H), 2.35 (m, 1H), 1.90–2.00 (m, 3H), 1.87 (s, 3H), 1.48–1.56 (m, 2H), 1.55–3.31 (br. m, 10H), 1.33–1.44 (m, 2H), 0.91 and 0.87 (two s, 18H), 0.10, 0.09, 0.05 and 0.04 (four s, 12H). 13C NMR (100 MHz, CDCl3) δ 163.02, 156.18, 137.30, 101.31, 87.59, 85.78, 76.20, 71.81, 62.84, 54.99, 42.15, 40.70, 36.57, 28.93, 27.37, 25.95, 18.54, 18.16, 13.31, −4.43, −4.75, −5.22, −5.28. Accurate mass HRMS (ESI+): m/z calcd for C28H61B10N3O4Si2Na (M +Na)+ 690.5091, found 691.5049.
4.1.14. General procedure for the synthesis of compounds 35–37
Compounds 32–34 (0.08 mmol) were dissolved in THF (1 mL) and TBAF (0.17 mmol, 1 M solution in THF). The reaction mixture was stirred at room temperature for 2 h, the solvent evaporated under reduced pressure, and the residue was purified by silica gel chromatography using DCM/MeOH (9/1, v/v) as solvent system to produce a white amorphous solid.
4.1.14.1. N4-[2-(closo-1,7-Carboranyl)ethyl]-5-methyl-2′-deoxycytidine (35)
Rf: 0.45 (DCM/MeOH, 9/1, v/v), yield: 27.6 mg, 86%. 1H NMR (400 MHz, CD3OD) δ 8.19 (s, 1H), 6.23 (t, J = 6.3 Hz, 1H), 4.41 (dt, J = 3.8, 7.2 Hz, 1H), 3.91–4.04 (m, 1H), 3.84 (dd, J = 2.9 and 12.1, 1H), 3.75 (dd, J =3.3 and 12.1, 1H), 3.49–3.64 (m, 3H), 2.32–2.50 (m, 2H), 2.24 (m, 2H), 2.02 (s, 3H), 1.50–3.31 (br. m, 10H). 13C NMR (100 MHz, CD3OD) δ 160.39, 160.00, 158.73, 149.08, 140.89, 103.14, 88.53, 86.81, 73.09, 70.76, 61.44, 56.42, 42.22, 40.94, 34.46, 12.08. Accurate mass HRMS (ESI+): m/z calcd for C14H29B10N3O4 (M +H)+ 412.3247, found 412.3212.
4.1.14.2. N4-[3-(closo-1,7-Carboranyl)propyl]-5-methyl-2′-deoxycytidine (36)
Rf: 0.47 (DCM/MeOH, 9/1, v/v), yield: 11.1mg, 95%. 1H NMR (400 MHz, CD3OD) δ 8.18 (s, 1H), 6.23 (t, J = 6.3 Hz, 1H), 4.41 (dt, J = 3.8, 6.2 Hz, 1H), 3.97 (m, 1H), 3.84 (dd, J = 2.9, 12.1 Hz, 1H), 3.75 (dd, J = 3.3, 12.1 Hz, 1H), 3.53 (s, 1H), 3.38 (t, J = 7.4 Hz, 2H), 2.37 (ddd, J = 4.1, 6.2, 13.6 Hz, 1H), 2.24 (m, 1H), 2.09 (m, 2H), 2.03 (s, 3H), 1.71–1.81 (m, 2H), 1.50–3.31 (br. m, 10H). 13C NMR (100 MHz, CD3OD) δ 159.37, 149.56, 141.82, 103.97, 89.56, 87.78, 71.79, 62.48, 57.17, 43.19, 41.93, 34.91, 29.96, 13.11. Accurate mass HRMS (ESI+): m/z calcd for C15H31B10N3O4 (M +H)+ 426.3404, found 426.3378.
4.1.14.3. N4-[4-(closo-1,7-Carboranyl)butyl]-5-methyl-2′-deoxycytidine (37)
Rf: 0.49 (DCM/MeOH, 9/1, v/v), yield: 36.8 mg, 97%. 1H NMR (400 MHz, CD3OD) δ 8.19 (s, 1H), 6.22 (t, J = 6.3 Hz, 1H), 4.41 (m, 1H), 3.97 (m, 1H), 3.84 (dd, J = 2.9, 12.1 Hz, 1H), 3.75 (dd, J = 3.5, 12.3 Hz, 1H), 3.46 (m, 3H), 2.32–2.40 (m, 1H), 2.20–2.28 (m, 1H), 1.97–2.09 (m, 5H), 1.50–3.31 (br.m, 10H), 1.54–1.65 (m, 2H), 1.42–1.52 (m, 2H). 13C NMR (100 MHz, CD3OD) δ 157.93, 148.09, 140.74, 102.90, 88.55, 86.71, 84.55, 76.60, 70.77, 61.45, 56.03, 54.26, 42.61, 41.77, 40.89, 36.59, 27.80, 27.28, 12.09. Accurate mass HRMS (ESI+): m/z calcd for C16H33B10N3O4 (M +H)+ 440.3561, found 440.3516.
4.2. Biology
4.2.1. Phosphate transfer assays (PTAs)
Compound 1, 2, 3, 4, 9, 16–22, 28–30, and 35–37 were dissolved in DMSO (100 mM concentration) and further diluted with H2O to produce stock solutions of concentrations (0.4 mM). The PTAs were carried out according to a previously reported procedure with minor modifications [29, 31]. The reaction mixtures contained 100 μM nucleoside and 100 μM ATP (with a small fraction of 0.13 μM [γ-32P]ATP (10 μCi/μL), 25 mM Tris-HCl (pH 7.6), 5 mM MgCl2, 125 mM KCl, 10 mM DTT, and 0.5 mg/mL BSA in a total volume of 20 μL. [γ-32P]ATP (specific activity: 3000 Ci/mmol) was obtained from Perkin Elmer, Waltham, MA. In all reactions, the final concentration of DMSO was set to < 1%. The reaction mixtures were incubated at 37°C for 20 min in the presence of 0.75 ng of TK1 (Biaffin GmbH & Co. KG, Kassel, Germany [specific activity: 1.7 U/mg – 1 Unit is defined as 1 μmol phosphate transferred to 3′-azido-3′-deoxythymidine per min]). Vials were heated for 2 min at 99°C to deactivate the enzyme. The reaction mixtures were centrifuged and 2 μL supernatants were spotted on PEI–cellulose TLC plates (EMD Chemicals Inc.). The TLC plates were developed in a solvent system containing isobutyric acid:NH4OH:H2O (66:1:33) over a period of 6 h. The radiolabeled spots were visualized by placing the TLC plates on a BioMax XAR film (Kodak) overnight and developing the films using an X-ray film developer (Tiba M6B, Series VI B Rapid processor; Commonwealth X-Ray Inc.). The spot intensities of the phosphorylated compounds were calculated with Adobe Photoshop (Adobe Systems Incorporated, USA).
4.2.2. Enzyme kinetic studies (Km and kcat)
The enzyme kinetics studies were carried out using the protocol described above for PTAs with varying concentrations of 1, 2, 3, 4, 21, or 22 [29]. The reaction mixtures contained 1.95–500 μM nucleoside and 100 μM ATP (with a small fraction of 0.325 μM [γ-32P]ATP (Perkin Elmer), 25 mM Tris-HCl (pH 7.6), 5 mM MgCl2, 125mM KCl, 10 mM DTT, and 0.5 mg/mL BSA. The kinetic parameters were calculated with the enzyme kinetic module of SigmaPlot 12 (Systat Software Inc, USA) using the Michaelis-Menton equation.
4.2.3. Competitive TK1 inhibition studies (IC50 values)
The IC50 values were determined using a recently published procedure [29]. Compound 1 (0.5 μM) with [methyl-3H]dThd (75 nM, 1 μCi/μL) was reacted with 5 mM ATP in presence of 0.75 ng of TK1, 25 mM Tris-HCl (pH 7.6), 5 mM MgCl2, 10 mM DTT, and 0.5 mg/mL BSA in a total volume of 20 μL. [methyl-3H]dThd (specific activity: 20 Ci/mmol) was obtained from Perkin Elmer, Waltham, MA. The reaction mixtures were incubated at 37°C for 20 min in the presence of 0–250.00 μM of either 1, 2, 3, 4, 21, or 22. The reaction vials were centrifuged and supernatent (10 μL) were spotted on DE81 diethylaminoethanol (DEAE) filter paper (Whatman, USA). Dried filter papers were washed three times with ammonium formate buffer (5 mM) for 5 min and placed in elution buffer (0.5 mL, 0.1 M HCl and 0.2 M KCl). Scintillation cocktail (10 mL) (Fischer Scientific, Columbus, OH) was added and radioactivity was determined by scintillation counting. The IC50 values were determined with SigmaPlot 12 (Systat Software Inc, USA) using the four parameter logistic curve equation [y = min + {max-min}/{1 + (x/EC50)^(−Hillslope)}].
4.2.4. In vivo biodistribution studies
All animal studies were carried out in accordance with the Guide for the Care and Use of Laboratory Animals (National Academy Press, Washington, D.C., 1996) and approved by the Institutional Animal Care and Use Committee (IACUC) of The Ohio State University. The RG2 rat glioma (CRL-2433, American Type Culture Collection (ATCC), Manassus, VA) has been described in detail elsewhere [48]. Cells were propagated in vitro in T-75 flasks (Corning #430641) in Complete Dulbecco’s Modified Eagle’s Medium (DMEM; Life Technology Gibco, Grand Island, NY), high glucose formulation plus 10% Fetal Bovine Serum (FBS; Gemini Bio-Products, West Sacremento, CA) plus 1% penicillin/streptomycin (Gibco) at 100 Units/mL penicillin and 100 μg/mL streptomycin) plus 1% L-glutamine (Gibco). Cells were harvested when they had reached confluency and were disaggregated by treatment with 0.5% trypsin (Sigma Biosciences, St. Louis, MO) and then washed in PBS (pH 7.4). Either 105 or 104 RG2 cells in 10 μL of DMEM were implanted stereotactically into the right caudate nucleus of syngeneic Fischer rats (Charles River, Cambridge, MA). Biodistribution studies were initiated 14 d later for rats receiving 105 cells and 15 d later for rats receiving 104 cells. Quantities of 2 and 3 equivalent to 100 μg of boron, solubilized in 200 μL of 35% aqueous DMSO, were administered intracerebrally (i.c.) by means of ALZET® osmotic pumps (model #2001D, Durect Corporation, Cupertino, CA), which had been implanted subcutaneously (s.c.) into the interscapular dorsal region of Fischer rats. The test agents were administered over 24 hours at a flow rate of 8.33 μL/h via a catheter connected to a 28 gauge needle that had been inserted into the entry port of a plastic screw, which had been inserted into a burr hole in the calvarium prior to stereotactic implantation of the tumor cells. The test agents, therefore, were administered to the very same site as the tumor implantation in all animals. At the end of the administration period the rats were euthanized by exposure to CO2 and immediately thereafter their brains were removed and blood samples were taken. The tumors were carefully dissected out from the tumor bearing right cerebral hemisphere and normal brain and each weighed separately. Boron determinations were carried out, as previously described [49], by means of ICP-OES using an Agilent/Varian spectrometer (Santa Clara, CA). For metabolomic studies, the samples were placed into pre-weighed microcentrifuge tubes, weighed again, snap-frozen in liquid nitrogen, and then stored at −20°C until MS analysis was performed at a later date.
4.2.5. Cell lines for in vitro studies
The cell lines used were the RG2 rat glioma, the L929 TK1(+) wild type cell line (ATCC #CCL1, NCTC clone 929) and its L929 TK1(−) mutant counterpart (ATCC #CCL1.3 L-M [TK-]), which do not express TK1. The TK1(−) phenotype was maintained by continuous culture in complete DMEM in the presence of 10 μM of 5-bromo-2′-deoxyuridine (Sigma Aldrich, St. Louis, MO).
4.2.6. In vitro studies
For in vitro metabolomic studies (see below), RG2 cells were grown to 80–90% confluency in T-75 flasks to an initial density of ~2.5 x 106 cells in 10 mL of complete DMEM as described earlier under ‘in vivo Studies’. In order to propagate the cells in thymidine-free medium, they were washed with PBS, then treated in 10% dialyzed FBS in order to remove thymidine from the media. Complete DMEM was removed and the cells were exposed for 2 hours to either 2 or 3 at concentrations of 17.5 μM in 10 mL of DMEM free of compound 1 containing 10% dialyzed FBS (Gemini Bio-Products, Baltimore, MD). This was done in order to remove 1 from the cell culture medium. At the end of this treatment, the cell monolayers were washed with 5 mL of PBS (pH 7.4). Cells were disaggregated using a mixture of 0.5% trypsin and 0.2% EDTA-4Na (10x trypsin, Sigma’s Biosciences) and this was terminated by adding an equal volume of media containing 10% dialyzed FBS as described above. Cells then were sedimented and resuspended in PBS. A small sample was taken for counting, and then the cells were transferred to pre-weighed microcentrifuge tubes and repetitively sedimented to remove the supernatant. Pellets were weighed, snap-frozen in liquid nitrogen, and stored frozen until analyzed by MS for metabolites of 2 and 3.
4.2.7. Western blot studies
Western blot analysis was carried out on RG2 rat glioma, L929 TK1(+), and L929 TK1(−) cells. Cells were propagated and disaggregated as described above using complete DMEM. Cell lysates were prepared using T-PER tissue protein extraction reagent (Thermo Fisher Scientific, Waltham, MA). Proteins (25 mg) were resolved by electrophoresis on SDS-polyacrylamide gel (4–20%) and transferred on to polyvinylidine difluoride membranes (Bio-Rad Laboratories, Hercules, CA). Western blotting was performed using TK1 primary antibodies (Novus Biologicals, Littleton, CO) and anti-rabbit immunoglobulin G (IgG) horseradish peroxidase (HRP)-linked secondary antibodies (CellSignaling Technology Inc., Danvers, MA). The proteins recognized by the antibodies were visualized with SuperSignal West Dura Chemiluminescent Substrate (Thermo Fisher Scientific). Subsequently, the membranes were incubated with anti-beta actin antibodies for protein loading. The signal intensity of TK1 relative to beta-actin for each cell sample was determined by ImageJ and EXCEL.
4.2.8. Metabolomic studies
Samples included three dissected RG2 gliomas, two from rats that had received 3 (111 mg & 112 mg tumor wet weight) and one from a rat that received 2 (170 mg) as well as four pellets of RG2 cells from in vitro studies, two were exposed to 3 (54 mg & 51 mg cell net weight) and two exposed to 2 (44 mg & 48 mg). Samples were lysed and metabolism quenched by adding 1.0 mL of chilled 1:1 MeOH:H2O mixture, followed by repetitive pipetting. The cell suspensions were vortexed and sonicated 3 x 10 sec/ea. Metabolites were separated from cellular debris by centrifugation at 13K rpm for 5 min at 4 °C. The supernatants were collected, dried in a vacufuge (Eppendorf, Hamburg, Germany) and re-suspended in 100 μL of H2O with 2% CH3CN for LC-MS injection. LC-MS experiments were performed on a Bruker maXis 4G high resolution mass spectrometer (Bremen, Germany) equipped with an electrospray source operated in both positive and negative ion modes. Accurate mass HRMS measurements (with coupled with the LC) were carried out by recalibrating the spectra using well-defined background ions. For comparison, theoretical isotope patterns were calculated with Isotope Pattern Calculator v4.0 (http://yanjunhua.tripod.com/pattern1.htm). ESI was carried out at a capillary voltage 4500 V and a source temperature of 200 °C. The nitrogen drying was set to 8.0 L/min and the nebulizer to 1 bar. The scan range was 100–1100 m/z. The HPLC system was a Dionex Ultimate 3000 (Thermo Fisher Scientific, Waltham, MA). Mobile phase A was H2O containing 0.1% formic acid and mobile phase B was CH3CN with 0.1% formic acid. A 2.1 x 50 mm Accucore Polar Premium column (Thermo Fisher Scientific) with a particle size of 2.6 μ was used for chromatographic separations. The gradient used was 0–80% B over 15 min with a flow rate of 200 μL/min. The total runtime was 26 min.
Supplementary Material
β-Autoradiogram of a TLC analysis of the products from a phosphate transfer assay using TK1 and [γ-32P]ATP (phosphate donor) as well as dThd (1), N5-2OH (2), and compound 9 as substrates. a: dThd (1) monophosphate; b; compound monophosphate.
Highlights.
Fourteen novel carboranyl pyrimidine nucleoside analogues were synthesized.
Thymidine Kinase 1 substrate and inhibitor characteristics were determined.
Metabolomic and biodistribution studies in RG2 cells were carried out.
Understanding of nucleoside analogue-enzyme binding was significantly advanced.
Acknowledgments
The authors thank Jeremy Keirsey and Nanette M. Kleinholz for carrying out the metabolomic experiments and Dr. Arpad Somogyi for helping with the interpretation of the obtained mass spectra. The metabolomic studies were done at the Campus Chemical Instrument Center, Mass Spectrometry and Proteomics Facility, The Ohio State University, Columbus, OH. The present studies were supported by funds from The Ohio State University College of Pharmacy and NIH grant R01 CA127935.
Abbreviations
- 3CTAs
3-carboranyl thymidine analogues
- 10B
boron-10
- ATCC
American type culture collection
- AZT
zidovudine
- B
boron
- BNCT
boron neutron capture therapy
- BaTK
Bacillus anthracis thymidine kinase
- DEAE
diethylaminoethanol
- DMEM
Dulbecco’s modified eagle’s medium
- dThd
thymidine
- d4T
stavudine
- ESI+ or ESI−
electrospray ionization positive or negative ion mode, respectively
- FBS
fetal bovine serum
- HRP
horseradish peroxidase
- hTK1
human thymidine kinase 1
- ICP-OES
inductively coupled plasma-optical emission spectroscopy
- IACUC
institutional animal care and use committee
- LET
linear energy transfer
- MP
monophosphate
- PEI
polyethylenimine
- PTA
phosphate transfer assay
- RP-18
reversed-phase 18
- rPR
relative phosphorylation rate
- SaTK
Staphylococcus aureus thymidine kinase
- siRNA
small interfering ribonucleic acid
- SM
- TBDMS
tert-butyldimethylsilyl
- TK1
thymidine kinase 1
Appendix A. Supplementary material
Supplementary data related to this article can be found online, at ……………...
Footnotes
The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Arner ES, Eriksson S. Mammalian deoxyribonuclease kinases. Pharmacol Ther. 1995;67:155–186. doi: 10.1016/0163-7258(95)00015-9. [DOI] [PubMed] [Google Scholar]
- 2.Yusa T, Yamaguchi Y, Ohwada H, Hayashi Y, Kuroiwa N, Morita T, Asanagi M, Moriyama Y, Fujimura S. Activity of the cytosolic isozyme of thymidine kinase in human primary lung tumors with reference to malignancy. Cancer Res. 1988;48:5001–5006. [PubMed] [Google Scholar]
- 3.Sandrini MPB, Clausen AR, On SLW, Aarestrup FM, Munch-Petersen B, Piskur J. Nucleoside analogues are activated by bacterial deoxyribonucleoside kinases in a species-specific manner. J Antimicrob Chemother. 2007;60:510–520. doi: 10.1093/jac/dkm240. [DOI] [PubMed] [Google Scholar]
- 4.Saito H, Tomioka H. Thymidine kinase of bacteria: activity of the enzyme in actinomycetes and related organisms. J Gen Microbiol. 1984;130:1863–1870. doi: 10.1099/00221287-130-7-1863. [DOI] [PubMed] [Google Scholar]
- 5.Sandrini MPB, Shannon O, Clausen AR, Bjorck L, Piskur J. Deoxyribonucleoside kinases activate nucleoside antibiotics in severely pathogenic bacteria. Antimicrob Agents Chemother. 2007;51:2726–2732. doi: 10.1128/AAC.00081-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sandrini MPB, Clausen AR, Munch-Petersen B, Piskur J. Thymidine kinase diversity in bacteria. Nucleosides Nucleotides Nucleic Acids. 2006;25:1153–1158. doi: 10.1080/15257770600894469. [DOI] [PubMed] [Google Scholar]
- 7.Hengstschlaeger M, Knoefler M, Muellner EW, Ogris E, Wintersberger E, Wawra E. Different regulation of thymidine kinase during the cell cycle of normal versus DNA tumor virus-transformed cells. J Biol Chem. 1994;269:13836–13842. [PubMed] [Google Scholar]
- 8.Hengstschlaeger M, Muellner EW, Wawra E. Thymidine kinase is expressed differently in transformed versus normal cells: a novel test for malignancy. Int J Oncol. 1994;4:207–210. doi: 10.3892/ijo.4.1.207. [DOI] [PubMed] [Google Scholar]
- 9.Sherley JL, Kelly TJ. Regulation of human thymidine kinase during the cell cycle. J Biol Chem. 1988;263:8350–8358. [PubMed] [Google Scholar]
- 10.De Clercq E. Antiviral drugs in current clinical use. J Clin Virol. 2004;30:115–133. doi: 10.1016/j.jcv.2004.02.009. [DOI] [PubMed] [Google Scholar]
- 11.Khalil A, Ishita K, Ali T, Tjarks W. N3-substituted thymidine bioconjugates for cancer therapy and imaging. Future Med Chem. 2013;5:677–692. doi: 10.4155/fmc.13.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sjuvarsson E, Damaraju VL, Mowles D, Sawyer MB, Tiwari R, Agarwal HK, Khalil A, Hasabelnaby S, Goudah A, Nakkula RJ, Barth RF, Cass CE, Eriksson S, Tjarks W. Cellular influx, efflux, and anabolism of 3-carboranyl thymidine analogs: potential boron delivery agents for neutron capture therapy. J Pharmacol Exp Ther. 2013;347:388–397. doi: 10.1124/jpet.113.207464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wojtczak BA, Olejniczak AB, Wang L, Eriksson S, Lesnikowski ZJ. Phosphorylation of nucleoside-metallacarborane and carborane conjugates by nucleoside kinases. Nucleosides Nucleotides Nucleic Acids. 2013;32:571–588. doi: 10.1080/15257770.2013.838259. [DOI] [PubMed] [Google Scholar]
- 14.Ilinova A, Semioshkin A, Lobanova I, Bregadze VI, Mironov AF, Paradowska E, Studzinska M, Jablonska A, Bialek-Pietras M, Lesnikowski ZJ. Synthesis, cytotoxicity and antiviral activity studies of the conjugates of cobalt bis(1,2-dicarbollide)(−I) with 5-ethynyl-2′-deoxyuridine and its cyclic derivatives. Tetrahedron. 2014;70:5704–5710. [Google Scholar]
- 15.Grimes RN. Carboranes. 2. Elsevier; Amsterdam: 2011. [Google Scholar]
- 16.Bregadze VI, Sivaev IB. Polyhedral boron compounds for BNCT. In: Hosmane NS, editor. Boron Science. New Technologies and Applications. CRC Press; Boca Raton, FL: 2012. pp. 181–207. [Google Scholar]
- 17.Hosmane NS, Maguire JA, Zhu Y, Takagaki M. Boron and Gadolinium Neutron Capture Therapy for Cancer Treatment. World Scientific Publishing Company; Singapore: 2012. [Google Scholar]
- 18.Barth RF, Vicente MGH, Harling OK, Kiger WS, 3rd, Kent JK, Binns JP, Wagner MF, Suzuki M, Aihara T, Kato I, Kawabata S. Current status of boron neutron capture therapy of high grade gliomas and recurrent head and neck cancer. Radiat Oncol. 2012;7:146. doi: 10.1186/1748-717X-7-146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Soloway AH, Tjarks W, Barnum BA, Rong FG, Barth RF, Codogni IM, Wilson JG. The chemistry of neutron capture therapy. Chem Rev. 1998;98:1515–1562. doi: 10.1021/cr941195u. [DOI] [PubMed] [Google Scholar]
- 20.Endo Y, Iijima T, Yaguchi K, Kawachi E, Inoue N, Kagechika H, Kubo A, Itai A. Structure-activity study of retinoid agonists bearing substituted dicarba-closo-dodecaborane. Relation between retinoidal activity and conformation of two aromatic nuclei. Bioorg Med Chem Lett. 2001;11:1307–1311. doi: 10.1016/s0960-894x(01)00204-9. [DOI] [PubMed] [Google Scholar]
- 21.Barth RF, Yang W, Wu G, Swindall M, Byun Y, Narayanasamy S, Tjarks W, Tordoff K, Moeschberger ML, Eriksson S, Binns PJ, Riley KJ. Thymidine kinase 1 as a molecular target for boron neutron capture therapy of brain tumors. Proc Nat Acad Sci USA. 2008;105:17493–17497. doi: 10.1073/pnas.0809569105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Barth RF, Yang W, Al-Madhoun AS, Johnsamuel J, Byun Y, Chandra S, Smith DR, Tjarks W, Eriksson S. Boron-containing nucleosides as potential deliver y agents for neutron capture therapy of brain tumors. Cancer Res. 2004;64:6287–6295. doi: 10.1158/0008-5472.CAN-04-0437. [DOI] [PubMed] [Google Scholar]
- 23.Tjarks W, Tiwari R, Byun Y, Narayanasamy S, Barth RF. Carboranyl thymidine analogues for neutron capture therapy. Chem Commun. 2007:4978–4991. doi: 10.1039/b707257k. [DOI] [PubMed] [Google Scholar]
- 24.Welin M, Kosinska U, Mikkelsen NE, Carnrot C, Zhu C, Wang L, Eriksson S, Munch-Petersen B, Eklund H. Structures of thymidine kinase 1 of human and mycoplasmic origin. Proc Natl Acad Sci USA. 2004;101:17970–17975. doi: 10.1073/pnas.0406332102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Segura-Pena D, Lichter J, Trani M, Konrad M, Lavie A, Lutz S. Quaternary structure change as a mechanism for the regulation of thymidine kinase 1-like enzymes. Structure. 2007;15:1555–1566. doi: 10.1016/j.str.2007.09.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kosinska U, Carnrot C, Sandrini MP, Clausen AR, Wang L, Piskur J, Eriksson S, Eklund H. Structural studies of thymidine kinases from Bacillus anthracis and Bacillus cereus provide insights into quaternary structure and conformational changes upon substrate binding. Febs J. 2007;274:727–737. doi: 10.1111/j.1742-4658.2006.05617.x. [DOI] [PubMed] [Google Scholar]
- 27.Byun Y, Thirumamagal BTS, Yang W, Eriksson S, Barth RF, Tjarks W. Preparation and biological evaluation of 10B-enriched 3-[5-{2-(2,3-dihydroxyprop-1-yl)-o-carboran-1-yl}pentan-1-yl]thymidine (N5–2OH), a new boron delivery agent for boron neutron capture therapy of brain tumors. J Med Chem. 2006;49:5513–5523. doi: 10.1021/jm060413w. [DOI] [PubMed] [Google Scholar]
- 28.Tiwari R, Toppino A, Agarwal HK, Huo T, Byun Y, Gallucci J, Hasabelnaby S, Khalil A, Goudah A, Baiocchi RA, Darby MV, Barth RF, Tjarks W. Synthesis, biological evaluation, and radioiodination of halogenated closo-carboranylthymidine analogues. Inorg Chem. 2012;51:629–639. doi: 10.1021/ic202150b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Agarwal HK, McElroy CA, Sjuvarsson E, Eriksson S, Darby MV, Tjarks W. Synthesis of N3-substituted carboranyl thymidine bioconjugates and their evaluation as substrates of recombinant human thymidine kinase 1. Eur J Med Chem. 2013;60:456–468. doi: 10.1016/j.ejmech.2012.11.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Narayanasamy S, Thirumamagal BTS, Johnsamuel J, Byun Y, Al-Madhoun AS, Usova E, Cosquer GY, Yan J, Bandyopadhyaya AK, Tiwari R, Eriksson S, Tjarks W. Hydrophilically enhanced 3-carboranyl thymidine analogues (3CTAs) for boron neutron capture therapy (BNCT) of cancer. Bioorg Med Chem. 2006;14:6886–6899. doi: 10.1016/j.bmc.2006.06.039. [DOI] [PubMed] [Google Scholar]
- 31.Byun Y, Yan J, Al-Madhoun AS, Johnsamuel J, Yang W, Barth RF, Eriksson S, Tjarks W. Synthesis and biological evaluation of neutral and zwitterionic 3-carboranyl thymidine analogues for boron neutron capture therapy. J Med Chem. 2005;48:1188–1198. doi: 10.1021/jm0491896. [DOI] [PubMed] [Google Scholar]
- 32.Hasabelnaby S, Goudah A, Agarwal HK, Abdalla MS, Tjarks W. Synthesis, chemical and enzymatic hydrolysis, and aqueous solubility of amino acid ester prodrugs of 3-carboranyl thymidine analogs for boron neutron capture therapy of brain tumors. Eur J Med Chem. 2012;55:325–334. doi: 10.1016/j.ejmech.2012.07.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ariza X, Bou V, Vilarrasa J. A new route to 15N-labeled, N-alkyl, and N-amino nucleosides via N-nitration of uridines and inosines. J Am Chem Soc. 1995;117:3665–3673. [Google Scholar]
- 34.Gorchs O, Hernandez M, Garriga L, Pedroso E, Grandas A, Farras J. A new method for the preparation of modified oligonucleotides. Org Lett. 2002;4:1827–1830. doi: 10.1021/ol025643t. [DOI] [PubMed] [Google Scholar]
- 35.Agarwal HK, Buszek B, Ricks KG, Tjarks W. Synthesis of closo-1,7-carboranyl alkyl amines. Tetrahedron Lett. 2011;52:5664–5667. doi: 10.1016/j.tetlet.2011.08.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wu B, Zhang J, Yang M, Yue Y, Ma L-J, Yu X-Q. An efficient and mild system for the reduction of nitriles to amines. ARKIVOC; Gainesville, FL, U. S: 2008. Raney Ni/KBH4; pp. 95–102. [Google Scholar]
- 37.Yamada K, Hattori Y, Inde T, Kanamori T, Ohkubo A, Seio K, Sekine M. Remarkable stabilization of antiparallel DNA triplexes by strong stacking effects of consecutively modified nucleobases containing thiocarbonyl groups. Bioorg Med Chem Lett. 2013;23:776–778. doi: 10.1016/j.bmcl.2012.11.079. [DOI] [PubMed] [Google Scholar]
- 38.Yang X, Birman VB. Acyl transfer catalysis with 1,2,4-triazole anion. Org Lett. 2009;11:1499–1502. doi: 10.1021/ol900098q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jessen HJ, Fendrich W, Meier C. Synthesis and properties of fluorescent cycloSal nucleotides m5K and its. 2′,3′-dideoxy analog dm5K. Eur J Org Chem. 2006:924–931. [Google Scholar]
- 40.Chen YL, Eriksson S, Chang ZF. Regulation and functional contribution of thymidine kinase 1 in repair of DNA damage. J Biol Chem. 2010;285:27327–27335. doi: 10.1074/jbc.M110.137042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Radivoyevitch T, Saunthararajah Y, Pink J, Ferris G, Lent I, Jackson M, Junk D, Kunos CA. dNTP supply gene expression patterns after p53 loss. Cancers. 2012;4:1212–1224. doi: 10.3390/cancers4041212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hu CM, Chang ZF. Synthetic lethality by lentiviral short hairpin RNA silencing of thymidylate kinase and doxorubicin in colon cancer cells regardless of the p53 status. Cancer Res. 2008;68:2831–2840. doi: 10.1158/0008-5472.CAN-07-3069. [DOI] [PubMed] [Google Scholar]
- 43.Kohda K, Kobayashi I, Itano K, Asano S, Kawazoe Y. Syntheses and properties of N-aminopyrimidines. Tetrahedron. 1993;49:3947–3958. [Google Scholar]
- 44.Ashida N, Asano S, Kohda K. 3-Aminothymidine inhibits growth of cultured human T-cell acute lymphoblastoid leukemia cells. Anticancer Res. 1994;14:2061–2062. [PubMed] [Google Scholar]
- 45.Umemoto T, Wada T. Nitromethane as a scavenger of acrylonitrile in the deprotection of synthetic oligonucleotides. Tetrahedron Lett. 2005;46:4251–4253. [Google Scholar]
- 46.Bartholoma MD, Vortherms AR, Hillier S, Ploier B, Joyal J, Babich J, Doyle RP, Zubieta J. Synthesis, cytotoxicity, and insight into the mode of action of Re(CO)3 thymidine complexes. ChemMedChem. 2010;5:1513–1529. doi: 10.1002/cmdc.201000196. [DOI] [PubMed] [Google Scholar]
- 47.Aspland SE, Ballatore C, Castillo R, Desharnais J, Eustaquio T, Goelet P, Guo Z, Li Q, Nelson D, Sun C, Castellino AJ, Newman MJ. Kinase-mediated trapping of bi-functional conjugates of paclitaxel or vinblastine with thymidine in cancer cells. Bioorg Med Chem Lett. 2006;16:5194–5198. doi: 10.1016/j.bmcl.2006.07.003. [DOI] [PubMed] [Google Scholar]
- 48.Barth RF, Kaur B. Rat brain tumor models in experimental neuro-oncology: the C6, 9L, T9, RG2, F98, BT4C, RT-2 and CNS-1 gliomas. J Neurooncol. 2009;94:299–312. doi: 10.1007/s11060-009-9875-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Barth RF, Adams DM, Soloway AH, Mechetner EB, Alam F, Anisuzzaman AKM. Determination of boron in tissues and cells using direct-current plasma atomic emission spectroscopy. Anal Chem. 1991;63:890–893. doi: 10.1021/ac00009a010. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
β-Autoradiogram of a TLC analysis of the products from a phosphate transfer assay using TK1 and [γ-32P]ATP (phosphate donor) as well as dThd (1), N5-2OH (2), and compound 9 as substrates. a: dThd (1) monophosphate; b; compound monophosphate.