

Abstract
We have utilized in vitro evolution to identify tRNA variants with significantly enhanced activity for the incorporation of unnatural amino acids into proteins in response to a quadruplet codon in both bacterial and mammalian cells. This approach will facilitate the creation of an optimized and standardized system for the genetic incorporation of unnatural amino acids using quadruplet codons, which will allow the biosynthesis of biopolymers that contain multiple unnatural building blocks.
Keywords: Quadruplet codon, four-base codon, unnatural amino acids, tRNA evolution, genetic code
INTRODUCTION
The genetic code describes how information contained in nucleic acid sequence, i.e., triplet nucleotide codons, is translated into polypeptide sequence. Among the 64 DNA codons, 61 encode the 20 common amino acids and 3 are generally used as stop signals with the rare exceptions of opal (encodes selenocysteine) and amber (encodes pyrrolysine in certain archaebacteria) nonsense codons. Recently, the amber codon has been used to encode unnatural amino acids (UAAs) in living cells using orthogonal aminoacyl-tRNA synthetase (aaRS)–amber suppressor tRNA (tRNACUA) pairs.1–6 Engineered archaeal tyrosyl-tRNA synthetase (MjTyrRS)– and pyrrolysyl-tRNA synthetase (PylRS)– pairs have enabled the incorporation of more than 60 UAAs into proteins in bacteria and higher organisms.1–6 To further realize the potential of this expanded genetic code, more “blank” codons (codons that do not encode a natural proteinogenic amino acid) are needed. To this end, quadruplet (frameshift) codons have been utilized to encode UAAs. The incorporation of UAAs in response to quadruplet codons has been achieved in cell-free systems using in vitro protein translation methods,7–11 in Xenopus oocytes using microinjected, chemically aminoacylated tRNAs,12 and in bacteria.13, 14 However, many of these approaches are hampered by low UAA incorporation efficiency. Here we report a new approach to enhance the efficiency of quadruplet codon suppression based on engineering of the tRNA, rather than other components of the translational machinery. Furthermore, we show for the first time that the evolved tRNA can be readily utilized to incorporate UAAs into proteins in mammalian cells.15
RESULTS AND DISCUSSION
tRNA Library Construction and Selection
To determine whether modifications to tRNA structure can lead to improved quadruplet codon suppression, we chose to investigate , because its anticodon loop may not act as an important recognition element for its cognate PylRS16, 17 (Figure 1A). In fact, a recent report showed that PylRS could charge and as well.18 Therefore, mutation of the CUA anticodon to a quadruplet anticodon in may not significantly affect its interaction with PylRS. In addition, previous work has shown that is not recognized by endogenous aaRSs in both E. coli and mammalian cells as a result of its unique structural features.19, 20 This property allows us to carry out the in vitro evolution experiments in E. coli, and subsequently shuttle the evolved tRNAPyl directly into mammalian cells.
Figure 1.
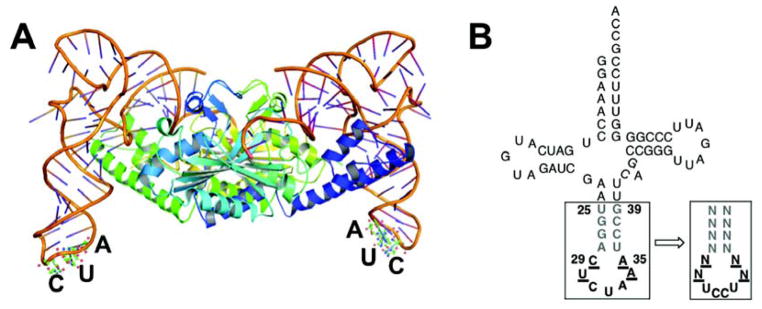
The structure of . (A) Crystal structure of PylRS- from Desulfitobacterium hafniense (PDB, 2ZNI; The co-crystal structure lacks the N-terminal part of the PylRS);16 (B) Clover leaf structures of tRNAPyl from Methanosarcina barkeri. In bold, anticodon; underlined, nucleotides in the anticodon loop that were randomized; grey in bold, nucleotides in the anticodon stem that were randomized.
The anticodon of was replaced with UCCU to generate , which recognizes the quadruplet codon AGGA. Based on the notion that mutation of the anticodon loop in might modulate its binding to the ribosome, we created a tRNA library (Library I, theoretical diversity 256, > 99% coverage) in which four bases of the anticodon loop (29, 30, 34, 35) were randomized (Figure 1B). The tRNA library was subjected to a positive selection to identify functional sequences in the presence of BocLysRS, chloramphenicol (34 μg mL−1), and a chloramphenicol acetyltransferase mutant gene containing the AGGA codon at a permissive site (the codon for Gln98 is replaced with AGGA). The survivors were further screened in the presence and absence of Nε-(tert-butyloxycarbonyl)-L-lysine (Boc-Lys),21 and seven individual clones that only survived on chloramphenicol in the presence of Boc-Lys were chosen for DNA sequencing. Five of the sequenced clones converged on a unique sequence (referred to as M1) with the mutations C29A and A35C, while the other two converged to a second related sequence (referred to as M2) with the mutation A35U (Table 1).
Table 1.
Evolved variants with improved AGGA suppression activity. Sequences of each variant at randomized positions are listed.
| Library | tRNA | Positions[a]
|
|||
|---|---|---|---|---|---|
| 25–28 | 29, 30 | 34, 35 | 36–39 | ||
| - | wt | UGGA | CU | AA | UCCG |
|
| |||||
| Lib I | M1 | - | AU | AC | - |
| M2 | - | CU | AU | - | |
|
| |||||
| Lib II | M3 | UCGG | AU | AC | CCUA |
| M4 | UGGG | AU | AC | CCUA | |
|
| |||||
| Lib III | M5 | AGGG | CU | AU | CCGU |
| M6 | UGGG | CU | AU | CCUA | |
| M7 | GGGG | CU | AU | CCGC | |
|
| |||||
| consensus | DSGG | MU | AY | CCKH | |
The positions with mutations are underlined and in bold;
S = G, C; M = A, C; K = G, U; Y = C, U; H = A, U, C; D = U, A, G.
We next sought to further improve the AGGA suppression efficiency by exploring mutation of additional structural elements in . To this end, we constructed two additional tRNA libraries (Library II is based on M1, theoretical diversity 6.6 × 104, > 99% coverage; Library III is based on M2, theoretical diversity 6.6 × 104, > 99% coverage) in which the sequences at positions 25–28 and 36–39 (Figure 1B) of the two hits from Library I were randomized. We hypothesized that sequence changes in the anticodon stem might have an effect on the interaction between the anticodon loop and ribosome, and therefore affect the efficiency of quadruplet codon suppression. Libraries II and III were subjected to the same selection/screening procedure that was used for Library I, but with an increased concentration of chloramphenicol (50 μg mL−1). Out of the more than 103 clones that survived on the selection plate, 96 individual clones were replicated on selection plates with a range of chloramphenicol concentrations (34, 50, 75, 100, 150 μg mL−1). This experiment provided a qualitative comparison of the efficiency of quadruplet codon suppression by mutants. Eight clones that grew with 100 or 150 μg mL−1 chloramphenicol in the presence of Boc-Lys but not with 34 μg mL−1 of chloramphenicol in the absence of Boc-Lys were chosen for sequencing. Three out of eight clones were derived from library II; two of them converged on a unique sequence (referred to as M4; Table 1). The other five clones were derived from library III; two of them converged on a unique sequence (referred to as M6) and another two of them converged on a second unique sequence (referred to as M7; Table 1). All eight clones contain the same A28G and U36C mutations (Table 1), which leads to the replacement of the A28-U36 with a G28-C36 base pair. No mutations were observed at positions 27 and 37, which leaves the G27-C37 base pair intact. The two G-C pairs apparently strengthen the part of the anticodon stem that is directly adjacent to the anticodon loop. Interestingly, all clones contain a mismatched Watson-Crick base paring at position 26–38 (Table 1). Based on molecular simulation with energy minimization using a CHARMM27 forcefield as implemented in Discovery Studio 2.5, the above mutations result in structural changes (Supplementary Figure 1) in the anticodon (especially the last two bases C and U), anticodon loop, and anticodon stem (at base pairs G26-C38, G27-C37, and A28-U36). Such structural changes in may allow a favorable conformational adjustment of tRNA upon its binding to the A site and subsequent translocation to the P and E sites of the ribosome.22, 23
Characterization And Validation of Quadruplet Codon Suppression in E. coli
To further determine the efficiency and fidelity of AGGA suppression by the wild-type and the mutant variants in E. coli, a frameshift mutation (AGGA) was introduced into GFPUV to replace the Asn149 codon (GFPUV-149AGGA). We constructed plasmid pLei-GFPUV-N149AGGA, which contains GFPUV-149AGGA (driven by a T5 promoter) and a copy of a variant (wild-type or a mutant; expressed under the control of a lpp promoter). Plasmid pLei-GFPUV-N149AGGA was cotransformed into E. coli Genehog strain with plasmid pBK-BocLysRS, which carries a PylRS variant (Y384F mutant; from Methanosarcina mazei) that specifically aminoacylates with Boc-Lys.21 Protein expression was carried out in LB medium supplemented with and without 5 mM Boc-Lys. Fluorescence analysis of E. coli cultures showed that full-length GFPUV protein was produced only in the presence of Boc-Lys for all variants that were examined (Figure 2). This result indicates that both the wild-type and evolved variants are not cross-active with any endogenous aaRSs in E. coli. All mutants (M1 and M2 from library I; M4 and M7 from library II and III) exhibited enhanced AGGA suppression in the presence of Boc-Lys relative to that of the wild-type . Two representative mutants (M4 and M7) from library II and III displayed the largest improvement based on the fluorescence assays (Figure 2).
Figure 2.
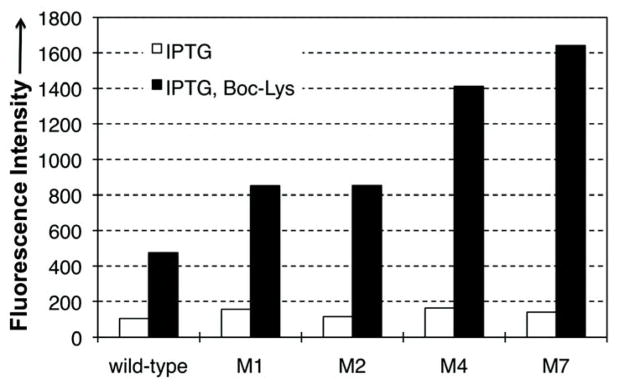
GFPUV fluorescence assays of cells expressing variants. Fluorescence readings of E. coli Genehog cells expressing wt (leftmost bar) or the evolved mutants, each coexpressed with BocLysRS. Fluorescence intensity was normalized to cell growth. Each data point is the average of triplet measurements with standard deviation.
We further confirmed incorporation of Boc-Lys into GFPUV-149AGGA by mass spectrometry (Figure 3). ESI-MS analysis of the partially purified GFPUV mutant showed two major signals (27823.1 Da and 27954.3 Da) corresponding to GFPUV protein containing Boc-Lys with and without the N-terminal Met (Figure 3). The yield of the mutant GFPUV protein was 1 mg/L after partial purification when M7 was used. For comparison, the yield of GFPUV-149TAG mutant in the presence of BocLysRS and under the same conditions was 4 mg/L. It was recently reported that the E. coli could efficiently suppress an AGGA codon at position 134 of a sequence-optimized sfGFP variant.24 However, we did not detect the signal (27752.1 Da) corresponding to the Arg incorporation in GFPUV-149AGGA (Supplementary Figure 3 and 4). Since we also did not detect obvious natural amino acid incorporation in response to AGGA codons in chloramphenicol acetyltransferase (position 98, for positive selection) and in barnase (positions 2 and 44, for negative assays; Supplementary Figure 5) for both wt and mutant in our experiments, we hypothesized that the previous reported suppression of AGGA codon by E. coli might be specific to the codon (position 134) context in sfGFP.24 In addition to Arg, no signals corresponding to the incorporation of other common 20 amino acids at position 149 were observed (Supplementary Figure 3, Supplementary Table 1), either. All the above observations indicate insufficient levels of natural amino acid incorporation into GFPUV-149AGGA and confirmed the good orthogonality of the evolved mutants.
Figure 3.
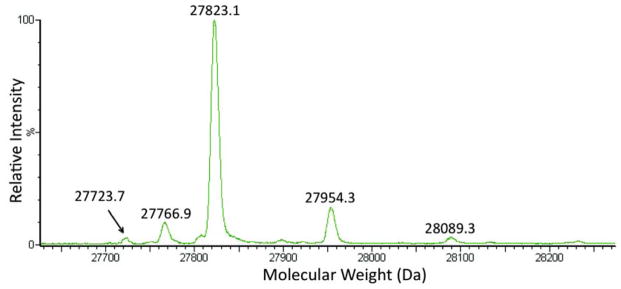
Deconvoluted ESI-MS spectra of the GFPUV-N149-Boc-Lys mutant. Expected masses: 27824.1 Da (without the N-terminal Met) and 27955.2 (with the N-terminal Met); observed masses: 27823.1 Da and 27954.3 Da. The 27723.7 Da signal may correspond to the protein that lost Boc group under mass spectrometry conditions. The 27766.9 Da signal does not correspond to GFPUV mutant that contains any of the 20 proteogenic amino acids at position 149. Supplementary Figure 3 shows the ESI-MS spectra before deconvolution.
An Expanded Genetic Code in Mammalian Cells With A Quadruplet Codon
Next, the activity of the evolved M7 mutant of was examined in mammalian cells. AGGA suppression was monitored using an enhanced GFP (EGFP) with an AGGA mutation at the permissive residue 150 (EGFP-150AGGA). 293T cells were co-transfected with two plasmids, which contained a total of two copies of wild-type or M7 mutant under the control of a human U6 promoter, and BocLysRS and EGFP-150AGGA genes under the control of a nonregulated CMV promoter. Following transfection, cells were cultured in DMEM media (containing 10% FBS and 2 mM L-glutamine) with or without 5 mM Boc-Lys for 12 h before visualization under a fluorescence microscope (Figure 4). Full-length EGFP was detected only in cells supplemented with Boc-Lys, while no EGFP was observed otherwise. Significantly enhanced fluorescence was observed in cell cultures transfected with the M7 mutant (Figure 4, Panel M7) relative to those transfected with wild-type (Figure 4, Panel wt). No obvious detrimental effects on cell viability were observed for either the wild-type or the M7 mutant. These results correlate well with those in E. coli, which indicates that the evolved mutant is also more efficient in decoding AGGA in mammalian cells. Although prokaryotes have 70S ribosomes (30S small subunit, 40S large subunit) whereas eukaryotes have 80S ribosomes (40S small subunit, 50S large subunit), the interaction between ribosome and tRNA may not be that different.
Figure 4.

Suppression of AGGA in EGFP150AGGA protein in 293T cells. The left picture in each panel represents fluorescence image and the right picture in each panel represents overlapped bright field and fluorescence images.
Conclusions
In summary, we have utilized an in vitro evolution approach to identify variants with significantly enhanced activity for the incorporation of unnatural amino acids into proteins in response to a quadruplet codon. We also demonstrated that the evolved variants from bacteria can be readily shuttled into a mammalian system. A similar approach is likely applicable to quadruplet codons other than AGGA, which will facilitate the creation of an optimized and standardized system for the genetic incorporation of unnatural amino acids using quadruplet codons. Generation of additional blank codons will allow the biosynthesis of biopolymers that contain more unnatural elements, which will further facilitate the study and manipulation of biological processes.
METHODS
Materials and General Methods
Boc-Lys was purchased from Bachem. Restriction enzymes, Antarctic phosphatase (AP) and T4 DNA ligase were purchased from New England Biolabs. KOD hot start DNA polymerase was purchased from EMD Millipore. Primers were ordered from Eurofins MWG Operon. Protein mass spectrometric data was collected on a Waters Synapt® G2 mass spectrometry. Data was processed using Masslynx™ software (Waters). Standard molecular biology techniques25 were used throughout. Site-directed mutagenesis was carried out using the QuikChange™ II site-directed mutagenesis kit (Agilent Technologies) by following the manufacturer’s protocol. E. coli DH5α and Genehog were used for routine cloning and DNA propagation. All solutions were prepared in deionized water further treated by Barnstead Nanopure® ultrapure water purification system (Thermo Fisher Scientific Inc). Antibiotics were added where appropriate to following final concentrations: ampicillin, 100 mg L−1; kanamycin, 50 mg L−1; tetracycline, 12.5 mg L−1.
Plasmid construction
Plasmid pLei-GFPUV-N149AGGA was obtained by first introducing the AGGA mutation into plasmid pLei-GFPUV-N149TAG26 by site-directed mutagenesis using following primers, 5′-GAGTACAACTATAACTCACACAGGAGTATACATCACGGCAGACAA-3′ and 5′-TTGTCTGCCGTGA-TGTATACTCCTGTGTGAGTTATAGTTGTACTC-3′. The original tRNA on pLei- GFPUV-N149TAG was subsequently replaced with wild-type . Variants of pLei-GFPUV-N149AGGA were constructed by replacing the wild-type with evolved tRNA mutants.
Plasmid pBK-BocLysRS was constructed by inserting BocLysRS-encoding gene behind the constitutive glnS promoter (PglnS) on pBK vector.27
Plasmid pREP-BocLysRS-AGGA (for positive selection) was constructed by modifying plasmid pRepCM12b.26 Specifically, the TAG codon (position 98) in the chloramphenicol acetyltransferase-encoding gene on pRepCM12b was changed to AGGA by site-directed mutagenesis using following primers, 5′-CGCTCTGGAGTGAATACCACAGGAGATTTCCGGCAGTTTCTACA-3′ and 5′-GTAGAAACTGCC-GGAAATCTCCTGTGGTATTCACTCCAGAGCG-3′. The resulting plasmid, pRepCM12b-AGGA, was digested with XbaI and ligated to a DNA fragment containing PglnS-BocLysRS cassette, which was amplified from template pBK-BocLysRS, to yield plasmid pREP-BocLysRS-AGGA.
Plasmid pNEG-AGGA (for negative selection) was constructed by modifying plasmid pNEG(−)tRNA.26 Codons of Gln2 and Asp44 of the barnase-encoding gene were changed to AGGA by overlapping PCR. The PCR product was digested with NdeI and EcoRI, then ligated into pNEG(−)tRNA that was treated with the same restriction enzymes to afford plasmid pNEG-AGGA. The resulting plasmid contains the p15A replication origin, the ampicillin resistance marker, the AraC-encoding gene, and the mutant barnase gene that was under the control of the arabinose promoter (pBAD). The overlapping PCR was carried out using the following primers: 5′-GGAATTCCATATGGCAAGGAGTTATCAACACGTTTGACGGG- 3′, 5′-TGCAAGGTTCCCTTTTGATGCCAC-3′, 5′-GCATCAAAAGGGAACCTTGCAAGGAGTCGCTCCGGGGAAAAGCATC- 3′, and 5′-AGCTTGGCACTGGCCGTCGTT-3′.
Plasmid pCMV-BocLysRS was constructed as follows: The wild-type gene lacking 3′ CCA termini but with a 3′-TTTTTCT sequence was amplified and fused behind a human U6 promoter by overlapping PCR. The resulting gene cassette was inserted into the MfeI site of pcDNA3.1/hygro(+, Life Technologies) to yield plasmid pcDNA3.1- . The BocLysRS-encoding gene was PCR amplified and inserted behind a non-regulated CMV promoter on pcDNA3.1- to afford plasmid pCMV-BocLysRS. Variants of the pCMV-BocLysRS plasmid were constructed by replacing the wild-type with evolved tRNA mutants.
Plasmid pEGFP-150AGGA was constructed similarly as for pCMV-BocLysRS. The EGFP- 150AGGA gene was generated using overlapping PCR to replace the codon of Asn150 with AGGA. The resulting gene was inserted behind the non-regulated CMV promoter on pcDNA3.1- to afford plasmid pEGFP-150AGGA. Variants of the pEGFP-150AGGA plasmid were constructed by replacing the wild-type with evolved tRNA mutants.
tRNA library construction
Procedures for library construction were adapted from Noren and Noren.28 Mutants of tRNA were obtained by overlapping PCR using pBK-mmPylT as the template.29 Resulting PCR products contains DNA cassettes of lpp promoter-tRNA mutant-rrnC terminator. Digestion of PCR products with NcoI and XhoI followed by ligation between NcoI and XhoI sites of pBK vector resulted in the tRNA library. The primers that were used for library construction can be found in supporting information.
Positive selection
Library DNAs were transformed into DH10B electrocompetent cells containing plasmid pREP-BocLysRS-AGGA. Transformants were cultivated in LB media containing kanamycin and tetracycline. After 12 h of cultivation, cells were harvested. Based on calculation, a certain number of cells (> 4.6 × the size of the library) were plated on LB agar containing kanamycin, tetracycline, Boc-Lys (5 mM), and chloramphenicol (34 mg L−1 for library I, 50 mg L−1 for library II and III). The selection plates were incubated at 37 °C for 48–72 h. Selected numbers of single colonies were further screened by replication onto plates with certain concentrations of chloramphenicol in the presence and absence of Boc-Lys. Only the ones that grew in the presence of Boc-Lys and did not grew in the absence of Boc-Lys were selected for further evaluation.
Negative selection assay
E. coli Genehog was co-transformed with plasmids pNEG-AGGA and pBK-tRNA (a plasmid contains variant of interest, see library construction), and plated on solid LB media containing ampicillin (to maintain the pNEG-AGGA plasmid), kanamycin (to maintain the pBK-tRNA plasmid), and 0% or 0.2% L-arabinose (w/v; to activate the transcription of mutant barnase gene). The cell growth was checked after 12 h of incubation at 37 °C. If the variant of interest is charged by endogenous aaRSs or if the AGGA codon is suppressed by endogenously charged tRNAs, cells should die due to the expression of full-length toxic barnase protein. Normal cell growth should be observed if the variant of interest is not charged by endogenous aaRSs and if the AGGA codon is not suppressed by any endogenously charged tRNAs (See Supplementary Figure 5 for results).
Fluorescence analysis of bacterial culture
E. coli Genehog strain harboring plasmid pBK-BocLysRS and a pLei-GFPUV-N149AGGA variant was cultured in 5 mL LB media containing kanamycin and chloramphenicol at 37 °C. The protein expression was induced at the OD600nm of 0.6 by additions of IPTG (0.5 mM) and Boc-Lys (5 mM). Following cultivation at 37 °C for an additional 16 h, 1 mL of cell culture were collected, washed, then resuspended in 1 mL of potassium phosphate buffer (50 mM, pH 7.4). The processed cells were directly used for fluorescence and cell density measurements using a Synergy™ H1 Hybrid plate reader (BioTek Instruments). The fluorescence of GFPUV was monitored at λEx = 390 nm and λEm = 510 nm. The cell density was estimated by measuring the sample absorbance at 600 nm. Values of fluorescence intensity were normalized to cell growth. Reported data are the average of three measurements with standard deviations.
Protein expression in bacteria
Similar cell cultivation procedure for fluorescence analysis of bacterial culture was applied to E. coli strains that were used for protein purification. The only modification is that 100 mL of culture was grown. Cells were collected by centrifugation at 5 000g and 4 °C for 15 min. Harvested cells were resuspended in lysis buffer containing potassium phosphate (20 mM, pH 7.4), NaCl (150 mM) and imidazole (10 mM). Cells were subsequently disrupted by sonication. Cellular debris was removed by centrifugation (21 000g, 30 min, 4 °C). The cell-free lysate was applied to Ni Sepharose 6 Fast Flow resin (GE Healthcare). Protein purification followed manufacture’s instructions. Protein concentrations were determined by Bradford assay (Bio-Rad). Purified protein was desalted prior to MS analysis.
Protein expression in 293T cells
293T cells were grown in media containing DMEM, 10% FBS (v/v), and 2 mM L-glutamine at 37 °C in a humidified atmosphere of 5% CO2 (v/v). When cells reached 60–70% confluency, cell culture media was exchanged to fresh DMEM media (containing 10% FBS and 2 mM L-glutamine) with or without 5 mM Boc-Lys and the cells were grown for an additional 30 minutes before transfection. Cells were transfected with plasmids pCMV-BocLysRS and pEGFP-150AGGA variants using Lipofectamine™ 2000 (Life Technologies) according to the manufacturer’s protocol (8 μL Lipofectamine + 2 μg of pCMV-BocLysRS + 2 μg of pEGFP-150AGGA for 2 mL cell culture in Costar 6- well cell-culture clusters). Cells were grown for an additional 12 h before being washed with DMEM base medium, fixed with 4% paraformaldehyde (w/v), washed three times with DPBS, and visualized by an Inverted (Olympus IX 81) confocal microscope.
Supplementary Material
Acknowledgments
This work is supported by the New Faculty Startup Fund to J. Guo from the Chemistry Department of University of Nebraska - Lincoln. The authors thank Dr. R. Cerny for help in protein mass spectrometry and Dr. Y. Zhou for help in fluorescence microscope analysis.
Footnotes
Supporting Information. Additional figures and data. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Liu CC, Schultz PG. Adding new chemistries to the genetic code. Annu Rev Biochem. 2010;79:413–444. doi: 10.1146/annurev.biochem.052308.105824. [DOI] [PubMed] [Google Scholar]
- 2.Wu X, Schultz PG. Synthesis at the Interface of Chemistry and Biology. J Am Chem Soc. 2009;131:12497–12515. doi: 10.1021/ja9026067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chin JW. Reprogramming the genetic code. Science. 2012;336:428–429. doi: 10.1126/science.1221761. [DOI] [PubMed] [Google Scholar]
- 4.Ai HW. Biochemical analysis with the expanded genetic lexicon. Anal Bioanal Chem. 2012;403:2089–2102. doi: 10.1007/s00216-012-5784-2. [DOI] [PubMed] [Google Scholar]
- 5.Liu WSR, Wang YS, Wan W. Synthesis of proteins with defined posttranslational modifications using the genetic noncanonical amino acid incorporation approach. Mol BioSyst. 2011;7:38–47. doi: 10.1039/c0mb00216j. [DOI] [PubMed] [Google Scholar]
- 6.Mukai T, Kobayashi T, Hino N, Yanagisawa T, Sakamoto K, Yokoyama S. Adding L-lysine derivatives to the genetic code of mammalian cells with engineered pyrrolysyl-tRNA synthetases. Biochem Biophys Res Commun. 2008;371:818–822. doi: 10.1016/j.bbrc.2008.04.164. [DOI] [PubMed] [Google Scholar]
- 7.Hohsaka T, Ashizuka Y, Murakami H, Sisido M. Incorporation of nonnatural amino acids into streptavidin through in vitro frame-shift suppression. J Am Chem Soc. 1996;118:9778–9779. [Google Scholar]
- 8.Hohsaka T, Sisido M. Incorporation of non-natural amino acids into proteins. Curr Opin Chem Biol. 2002;6:809–815. doi: 10.1016/s1367-5931(02)00376-9. [DOI] [PubMed] [Google Scholar]
- 9.Ohtsuki T, Manabe T, Sisido M. Multiple incorporation of non-natural amino acids into a single protein using tRNAs with non-standard structures. FEBS Lett. 2005;579:6769–6774. doi: 10.1016/j.febslet.2005.11.010. [DOI] [PubMed] [Google Scholar]
- 10.Taira H, Fukushima M, Hohsaka T, Sisido M. Four-base codon-mediated incorporation of nonnatural amino acids into proteins in a eukaryotic cell-free translation system. J Biosci Bioeng. 2005;99:473–476. doi: 10.1263/jbb.99.473. [DOI] [PubMed] [Google Scholar]
- 11.Taki M, Tokuda Y, Ohtsuki T, Sisido M. Design of carrier tRNAs and selection of four-base codons for efficient incorporation of various nonnatural amino acids into proteins in Spodoptera frugiperda 21 (Sf21) insect cell-free translation system. J Biosci Bioeng. 2006;102:511–517. doi: 10.1263/jbb.102.511. [DOI] [PubMed] [Google Scholar]
- 12.Rodriguez EA, Lester HA, Dougherty DA. In vivo incorporation of multiple unnatural amino acids through nonsense and frameshift suppression. Proc Natl Acad Sci U S A. 2006;103:8650–8655. doi: 10.1073/pnas.0510817103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Anderson JC, Wu N, Santoro SW, Lakshman V, King DS, Schultz PG. An expanded genetic code with a functional quadruplet codon. Proc Natl Acad Sci U S A. 2004;101:7566–7571. doi: 10.1073/pnas.0401517101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Neumann H, Wang K, Davis L, Garcia-Alai M, Chin JW. Encoding multiple unnatural amino acids via evolution of a quadruplet-decoding ribosome. Nature. 2010;464:441–444. doi: 10.1038/nature08817. [DOI] [PubMed] [Google Scholar]
- 15.Taki M, Matsushita J, Sisido M. Expanding the genetic code in a mammalian cell line by the introduction of four-base codon/anticodon pairs. Chem Bio Chem. 2006;7:425–428. doi: 10.1002/cbic.200500360. [DOI] [PubMed] [Google Scholar]
- 16.Ambrogelly A, Gundllapalli S, Herring S, Polycarpo C, Frauer C, Söll D. Pyrrolysine is not hardwired for cotranslational insertion at UAG codons. Proc Natl Acad Sci U S A. 2007;104:3141–3146. doi: 10.1073/pnas.0611634104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yanagisawa T, Ishii R, Fukunaga R, Kobayashi T, Sakamoto K, Yokoyama S. Crystallographic studies on multiple conformational states of active-site loops in pyrrolysyl-tRNA synthetase. J Mol Biol. 2008;378:634–652. doi: 10.1016/j.jmb.2008.02.045. [DOI] [PubMed] [Google Scholar]
- 18.Wan W, Huang Y, Wang Z, Russell WK, Pai PJ, Russell DH, Liu WR. A facile system for genetic incorporation of two different noncanonical amino acids into one protein in Escherichia coli. Angew Chem, Int Ed. 2010;49:3211–3214. doi: 10.1002/anie.201000465. [DOI] [PubMed] [Google Scholar]
- 19.Polycarpo C, Ambrogelly A, Berube A, Winbush SM, McCloskey JA, Crain PF, Wood JL, Söll D. An aminoacyl-tRNA synthetase that specifically activates pyrrolysine. Proc Natl Acad Sci U S A. 2004;101:12450–12454. doi: 10.1073/pnas.0405362101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nozawa K, O’Donoghue P, Gundllapalli S, Araiso Y, Ishitani R, Umehara T, Söll D, Nureki O. Pyrrolysyl-tRNA synthetase-tRNAPyl structure reveals the molecular basis of orthogonality. Nature. 2009;457:1163–1167. doi: 10.1038/nature07611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yanagisawa T, Ishii R, Fukunaga R, Kobayashi T, Sakamoto K, Yokoyama S. Multistep engineering of pyrrolysyl-tRNA synthetase to genetically encode Nε-(o-azidobenzyloxycarbonyl) lysine for site-specific protein modification. Chem Biol. 2008;15:1187–1197. doi: 10.1016/j.chembiol.2008.10.004. [DOI] [PubMed] [Google Scholar]
- 22.Phelps SS, Gaudin C, Yoshizawa S, Benitez C, Fourmy D, Joseph S. Translocation of a tRNA with an extended anticodon through the ribosome. J Mol Biol. 2006;360:610–622. doi: 10.1016/j.jmb.2006.05.016. [DOI] [PubMed] [Google Scholar]
- 23.Dunham CM, Selmer M, Phelps SS, Kelley AC, Suzuki T, Joseph S, Ramakrishnan V. Structures of tRNAs with an expanded anticodon loop in the decoding center of the 30S ribosomal subunit. RNA. 2007;13:817–823. doi: 10.1261/rna.367307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.O’Donoghue P, Prat L, Heinemann IU, Ling J, Odoi K, Liu WR, Söll D. Near-cognate suppression of amber, opal and quadruplet codons competes with aminoacyl-tRNAPyl for genetic code expansion. FEBS Lett. 2012;586:3931–3937. doi: 10.1016/j.febslet.2012.09.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sambrook JF, Russell DW. Molecular cloning: A laboratory manual. 3. Cold Spring Harbor Laboratory Press; 2000. [Google Scholar]
- 26.Guo J, Melancon CE, III, Lee HS, Groff D, Schultz PG. Evolution of amber suppressor tRNAs for efficient bacterial production of proteins containing nonnatural amino acids. Angew Chem, Int Ed. 2009;48:9148–9151. doi: 10.1002/anie.200904035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang L, Brock A, Herberich B, Schultz PG. Expanding the genetic code of Escherichia coli. Science. 2001;292:498–500. doi: 10.1126/science.1060077. [DOI] [PubMed] [Google Scholar]
- 28.Noren KA, Noren CJ. Construction of high-complexity combinatorial phage display peptide libraries. Methods. 2001;23:169–178. doi: 10.1006/meth.2000.1118. [DOI] [PubMed] [Google Scholar]
- 29.Chen PR, Groff D, Guo J, Ou W, Cellitti S, Geierstanger BH, Schultz PG. A facile system for encoding unnatural amino acids in mammalian cells. Angew Chem, Int Ed. 2009;48:4052–4055. doi: 10.1002/anie.200900683. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.