

Abstract
To identify risk variants for glioma, we conducted a meta-analysis of two genome-wide association studies by genotyping 550K tagging SNPs in a total of 1,878 cases and 3,670 controls, with validation in three additional independent series totaling 2,545 cases and 2,953 controls. We identified five risk loci for glioma at 5p15.33 (rs2736100, TERT; P = 1.50 × 10−17), 8q24.21 (rs4295627, CCDC26; P = 2.34 × 10−18), 9p21.3 (rs4977756, CDKN2A-CDKN2B; P = 7.24 × 10−15), 20q13.33 (rs6010620, RTEL1; P = 2.52 × 10−12) and 11q23.3 (rs498872, PHLDB1; P = 1.07 × 10−8). These data show that common low-penetrance susceptibility alleles contribute to the risk of developing glioma and provide insight into disease causation of this primary brain tumor.
Gliomas account for ∼80% of malignant primary brain tumors (PBT). In the United States, ∼21,000 individuals are diagnosed with glioma annually1 and for most the prognosis is dismal1. No lifestyle exposure has consistently been linked to glioma risk except ionizing radiation, which accounts for few cases1. Predicated on the hypothesis that part of the twofold increased glioma risk in relatives of individuals with PBT2 is a consequence of the co-inheritance of multiple low-risk variants, we conducted two genome-wide association (GWA) studies. Pooling data from both scans and following up the best-supported associations, we have identified five glioma risk loci.
The two GWA studies were conducted in the UK and US (UK-GWA and US-GWA studies, respectively; Fig. 1). In both, genotyping of cases was conducted using Illumina Infinium HD Human610-Quad BeadChips. The UK-GWA study was based on genotyping 636 glioma cases ascertained through INTERPHONE3. After application of quality control criteria, genotypes were available for 631 cases (Fig. 1). For controls, we used Illumina Hap550K-BeadChip genotypes on 1,438 individuals from the British 1958 Birth Cohort4. The US-GWA study was based on genotyping 1,281 glioma cases ascertained by The University of Texas M.D. Anderson Cancer Center. After applying quality control criteria, genotypes were available for 1,247 cases (Fig. 1). For controls, we used Illumina Hap550K-BeadChip genotypes on 2,243 individuals from the Cancer Genetic Markers of Susceptibility studies5,6.
Figure 1.

Subjects and single-SNP exclusion schema for genome-wide association studies.
Across both case series, 572,571 SNPs were satisfactorily genotyped (99.4%), with mean individual sample call rates of 99.8%. We excluded 32 individuals because of non-Western European ancestry and two because of cryptic relatedness (Fig. 1 and Supplementary Fig. 1a). For the 1,878 cases and 3,670 controls in the combined data, 521,318 SNP genotypes were available. We considered only the 454,576 autosomal SNPs having call rates >95% in all series, showing mimimal departure from Hardy-Weinberg equilibrium (HWE; P > 10−5 in controls) and minor allele frequencies (MAF) > 1% in cases and controls (Fig. 1).
Comparison of the observed and expected distributions showed little evidence for an inflation of the test statistics in the datasets (inflation factor7 λ = 1.02 and 1.05 in UK-GWA and US-GWA, respectively, based on the 90% least significant SNPs; Supplementary Fig. 1b), thereby excluding significant hidden population substructure or differential genotype calling. Using standard methods and data from both studies, we derived joint odds ratios (ORs) and confidence intervals for each SNP and associated P values.
To identify true risk alleles among the 34 SNPs showing evidence of an association at P < 10−5, we conducted replication in three case-control series involving a total of 5,498 individuals that passed quality control (French series: 1,392 cases, 1,602 controls; German series: 504 cases, 573 controls; Swedish series: 649 cases, 778 controls). We satisfactorily genotyped 31 of the 34 SNPs in all series. In the French series, rs9656979 and rs7257116 were not genotyped. However, an association between these SNPs and glioma risk was not supported in either the German or Swedish series (Supplementary Table 1). Although rs7300686 was not typed in the German or Swedish series, an association with risk was not supported in the French series (Supplementary Table 1). Hence, failure to genotype all additional series for the three SNPs has not affected study findings. Across the five data series, there was a significant association between rs7124728 and glioma risk, but this was not supported in the three replication series (P > 0.1). rs4944840 maps 57 kb from rs7124728 and both SNPs are in strong linkage disequilibrium (LD; D′ = 0.89, r2 = 0.71). No association between rs4944840 and glioma risk was seen in either GWA study (P = 0.13 UK-GWA, P = 0.03 US-GWA). There was a significant difference in MAF of rs7124728 between both GWA cases and replication cases but not between controls, suggesting a genotyping error with the Illumina610-Quad BeadChip.
Excluding rs7124728, in the combined analyses, 14 of the 31 SNPs representing five genomic regions satisfied an accepted threshold for genome-wide statistical significance (that is, P < 5 × 10−7; Table 1 and Supplementary Table 1). Under a fixed-effects model, the strongest signal was attained at rs4295627 (OR = 1.36, 95% CI: 1.29–1.43; P = 2.34 × 10−18), which localizes to 8q24.21 (130,754,639 bp; Fig. 2 and Table 1). There was, however, evidence of between-study heterogeneity (Phet = 0.01) ascribable to the association being modest in the Swedish series. Under a random-effects model, the OR was 1.36 (95% CI: 1.19–1.58; P = 2.01 × 10−5) using all data and 1.45 (95% CI: 1.32–1.57; P = 1.02 × 10−8) excluding the Swedish study. rs4295627 maps to intron 3 of CCDC26, encoding a retinoic acid modulator of differentiation and death8. Retinoic acid induces caspase-8 transcription through phosphorylation of CREB and increases apoptosis induced by death stimuli in neuroblastoma cells9 and in glioblastoma cells with downregulation of telomerase activity10. Some evidence suggests a second 8q24.21 locus defined by rs891835, mapping to intron 1 of CCDC26 207 kb telomeric to rs4295627 (130,560,934 bp; OR = 1.24, 95% CI: 1.17–1.30; P = 7.54 × 10−11) and showing low LD with rs4295627 (D′ = 0.59 and r2 = 0.19 and D′ = 0.58, r2 = 0.26 in HapMap and GWA scan, respectively). The OR for rs4295627 with adjustment for rs891835 was 1.30 (95% CI: 1.20–1.41; P = 2.80 × 10−10). Similarly, adjusting rs891835 for rs4295627 provided evidence of an association, albeit at a nominal significance level (OR = 1.08, 95% CI: 1.00–1.17; P = 0.03). There was also an increasing trend in OR with an increasing number of risk alleles (P = 2.67 × 10−13) and the frequency of the two common haplotypes differed between cases and controls (Supplementary Table 2). When we imputed 8q24.21 genotypes using genome-wide data, rs1077236, mapping to 130,709,683 bp, provided a marginally better association signal than rs4295627 (P = 2.75 × 10−9 and 1.60 × 10−8, respectively). Hence, the possibility remains that rs4295627 and rs891835, although only weakly associated, are in LD with one or more causal variants in this region.
Table 1. Summary results for SNPs associated with glioma in GWA and replication samples.
| GWA studies | Replication studies | Combined | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
|
|
|
||||||||||
| SNP | Chr. | Genea | Location (bp) | Ancestral allele frequency | Risk alleleb | OR (95% CI) | P | OR (95% CI) | P | OR (95% CI) | P | P het |
| rs2736100 | 5 | TERT | 1,339,516 | 0.51 | G | 1.20 (1.10–1.33) | 2.21 × 10−6 | 1.33 (1.20–1.49) | 2.87 × 10−13 | 1.27 (1.19–1.37) | 1.50 × 10−17 | 0.18 |
| rs2853676 | 5 | TERT | 1,341,547 | 0.27 | A | 1.22 (1.14–1.31) | 5.30 × 10−6 | 1.30 (1.21–1.38) | 1.06 × 10−9 | 1.26 (1.20–1.32) | 4.21 × 10−14 | 0.67 |
| rs10464870 | 8 | CCDC26 | 130,547,005 | 0.21 | C | 1.24 (1.15–1.34) | 3.90 × 10−6 | 1.22 (1.13–1.31) | 1.77 × 10−5 | 1.23 (1.17–1.30) | 3.04 × 10−10 | 0.05 |
| rs891835 | 8 | CCDC26 | 130,560,934 | 0.22 | G | 1.24 (1.15–1.33) | 3.92 × 10−6 | 1.24 (1.15–1.33) | 4.43 × 10−6 | 1.24 (1.17–1.30) | 7.54 × 10−11 | 0.01 |
| rs6470745 | 8 | CCDC26 | 130,711,103 | 0.20 | G | 1.30 (1.20–1.39) | 5.79 × 10−8 | 1.31 (1.22–1.41) | 9.09 × 10−9 | 1.30 (1.24–1.37) | 2.77 × 10−15 | 0.01 |
| rs16904140 | 8 | CCDC26 | 130,734,825 | 0.21 | A | 1.25 (1.16–1.35) | 1.41 × 10−6 | 1.28 (1.19–1.37) | 1.14 × 10−7 | 1.27 (1.20–1.33) | 7.88 × 10−13 | 0.01 |
| rs4295627 | 8 | CCDC26 | 130,754,639 | 0.17 | G | 1.33 (1.23–1.42) | 1.47 × 10−8 | 1.39 (1.30–1.49) | 2.20 × 10−11 | 1.36 (1.29–1.43) | 2.34 × 10−18 | 0.01 |
| rs1063192 | 9 | CDKN2A/B | 21,993,367 | 0.44 | C | 1.21 (1.13–1.29) | 1.44 × 10−6 | 1.21 (1.14–1.29) | 6.97 × 10−7 | 1.21 (1.16–1.27) | 4.61 × 10−12 | 0.81 |
| rs2157719 | 9 | CDKN2A/B | 22,023,366 | 0.57 | G | 1.22 (1.11–1.35) | 6.80 × 10−7 | 1.22 (1.11–1.33) | 4.42 × 10−7 | 1.22 (1.14–1.30) | 1.41 × 10−12 | 0.68 |
| rs1412829 | 9 | CDKN2A/B | 22,033,926 | 0.42 | C | 1.22 (1.14–1.30) | 7.23 × 10−7 | 1.23 (1.15–1.30) | 1.80 × 10−7 | 1.22 (1.17–1.28) | 6.23 × 10−13 | 0.67 |
| rs4977756 | 9 | CDKN2A/B | 22,058,652 | 0.40 | G | 1.25 (1.17–1.32) | 2.39 × 10−8 | 1.24 (1.16–1.31) | 5.90 × 10−8 | 1.24 (1.19–1.30) | 7.24 × 10−15 | 0.94 |
| rs498872 | 11 | PHLDB1 | 117,982,577 | 0.31 | T | 1.26 (1.17–1.34) | 1.03 × 10−7 | 1.12 (1.04–1.20) | 4.56 × 10−3 | 1.18 (1.13–1.24) | 1.07 × 10−8 | 0.04 |
| rs6010620 | 20 | RTEL1 | 61,780,283 | 0.23 | G | 1.28 (1.18–1.38) | 8.38 × 10−7 | 1.28 (1.18–1.38) | 6.49 × 10−7 | 1.28 (1.21–1.35) | 2.52 × 10−12 | 0.38 |
| rs2297440 | 20 | RTEL1 | 61,782,743 | 0.22 | C | 1.28 (1.18–1.38) | 1.01 × 10−6 | 1.26 (1.16–1.35) | 4.44 × 10−6 | 1.27 (1.20–1.34) | 2.06 × 10−11 | 0.40 |
Gene(s) mapping within 20 kb of each SNP are listed.
Ancestral allele marked in bold.
Figure 2.
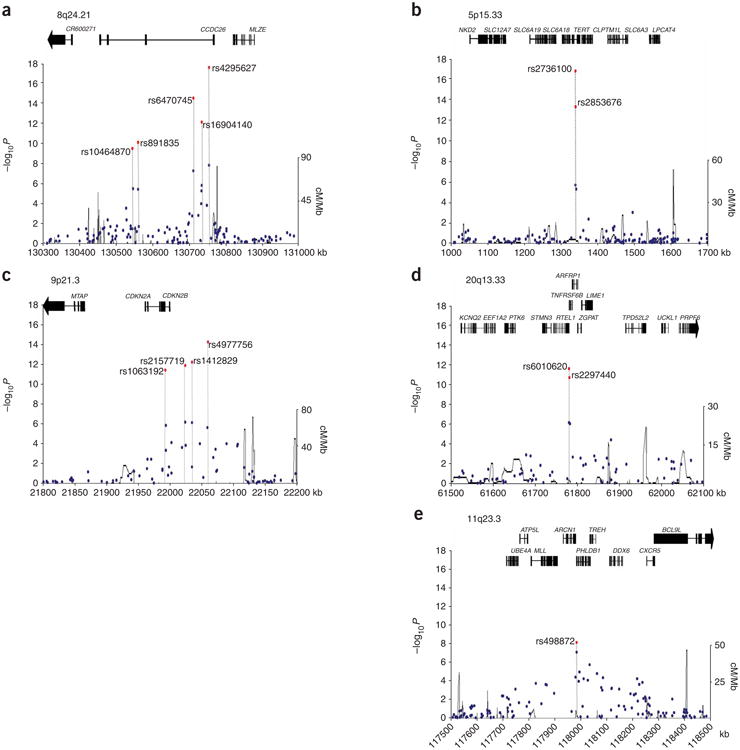
LD structure and association results for the five confirmed glioma-associated regions. (a) 8q24.21. (b) 5p15.33. (c) 9p21.3. (d) 20q13.33. (e) 11q23.3. Chromosomal positions and genes based on NCBI build 36 coordinates. Armitage trend test P values (as −log10 values; left y axis) are shown for SNPs analyzed in the GWA studies in blue and for the combined analysis including the replication series in red. Recombination rates in HapMap CEU across the region are shown in black (right y axis). Also shown are the relative positions of genes mapping to each region of association. Exons of genes have been redrawn to show the relative positions in the gene, and therefore maps are not to physical scale.
Variation at 8q24.21 has been implicated in colorectal11, breast12, prostate6 and bladder cancer risk13. The genomic regions underlying these associations map to the 128- to 129-Mb interval, distinct from the glioma signal. Closer to the 8q24.21 glioma signal is rs987525, a risk factor for cleft lip14. Notably, nonsyndromic cleft lip has been reported to be a risk factor for PBT15. Although rs987525 maps 740 kb centromeric to rs4295627 and LD between rs987525 and rs4295627 is weak (D′ = 0.46, r2 = 0.01), it is possible that these SNPs define a common disease locus.
The second strongest evidence for an association was for rs2736100 (OR = 1.27, 95% CI: 1.19–1.37; P = 1.50 × 10−17) mapping to 5p15.33, which localizes to intron 2 of TERT (1,339,516 bp; Fig. 2 and Table 1). TERT is the reverse transcriptase component of telomerase, essential for telomerase activity in maintaining telomeres and cell immortalization. Although 5p15.33 copy number change is uncommon in glioblastoma multiforme (GBM; Supplementary Fig. 2a), TERT expression correlates with glioma grade and prognosis16. Several lines of evidence raise the possibility of a second locus defined by rs2853676, which also maps to intron 2 of TERT (1,341,547 bp; OR = 1.26, 95% CI: 1.20–1.32; P = 4.21 × 10−14) and is in weak LD with rs2736100 (D′ = 0.67 and r2 = 0.15 and D′ = 0.81 and r2 = 0.24 in HapMap and GWA scan, respectively). The OR for rs2736100 with adjustment for rs2853676 was 1.19 (95% CI: 1.12–1.27; P = 5.33 × 10−8) and adjusting rs2853676 for rs2736100 also provided evidence of an association, with little change in the risk estimate (OR = 1.15, 95% CI: 1.08–1.23; P = 6.90 × 10−5). There was an increasing trend in OR with an increasing number of risk alleles (P = 1.19 × 10−19). Comparison of haplotype frequencies provided evidence of two haplotypes differing between cases and controls (Supplementary Table 2). Recent data has implicated variation at 5p15.33 (TERT-CLPTM1L) defined by rs2736098 with risk of multiple tumor types17. As LD between rs2736098 and both rs2736100 and rs2853676 is poor (D′ = 0.48 and r2 = 0.11 and D′ = 0.28 and r2 = 0.02 in HapMap and GWA scan, respectively), whether the 5p15.33 association with glioma is the same remains to be established.
The third strongest evidence for an association was for rs4977756 (OR = 1.24, 95% CI: 1.19–1.30; P = 7.24 × 10−15), mapping 59 kb telomeric to CDKN2B (22,058,652 bp; Fig. 2 and Table 1) within a 122-kb region of LD at 9p21.3. This region encompasses the CDKN2A-CDKN2B tumor suppressor genes. CDKN2A encodes p16(INK4A) a negative regulator of cyclin-dependent kinases and p14(ARF1), an activator of p53. CDKN2A-CDKN2B has an established role in glioma, with homozygous deletion in CDKN2A detectable in ∼50% of tumors18,19 and loss of expression linked to poor prognosis16. Furthermore, germline mutation of CDKN2A-CDKN2B causes the melanoma-astrocytoma syndrome20,21. Regulation of p16/p14ARF is important for sensitivity to ionizing radiation, the only environmental factor strongly linked to gliomagenesis22. These data provide evidence that common variation in addition to rare mutations in CDKN2A-CDKN2B contributes to glioma predisposition.
A 68-kb core region of 9p21.3 incorporating the association signal influences risk of type 2 diabetes (T2D)23 and coronary heart disease (CHD)24. rs564398, a risk factor for T2D and CHD, is in LD with rs4977756 (D′ = 0.77, r2 = 0.50). A possible explanation for a common etiology is that these variants affect determination of birth weight, linked as a risk factor to all three diseases25–27.
The fourth strongest evidence for an association was for rs6010620 (OR = 1.28, 95% CI: 1.21–1.35; P = 2.52 × 10−12), localizing to intron 12 (61,780,283 bp) of the RAD3-like helicase gene RTEL1 (ref 28) and mapping within a 65-kb region of LD at 20q13.33 (Fig. 2 and Table 1). Amplification of 20q13.33 is seen in ∼30% of gliomas with copy number change correlating with RTEL1 expression (Supplementary Fig. 2a). RTEL1 maintains genomic stability directly by suppressing homologous recombination28. These data, coupled with the role of TERT in glioma, make RTEL1 an attractive candidate for the 20q13.33 association but do not preclude the association being through variation in other genes mapping to the region, such as TNFRSF6B, whose expression correlates with glioma grade29.
The fifth-strongest signal was at rs498872 (OR = 1.18, 95% CI: 1.13–1.24; P = 1.07 × 10−8), which maps to the 5′-UTR of PHLDB1 within a 101-kb LD block on 11q23.3 (117982577 bp; Fig. 2 and Table 1). There was, however, evidence of between-study heterogeneity (Phet = 0.04). Under a random-effects model, the combined OR was 1.16 (95% CI: 1.05–1.28; P = 2.24 × 10−3). Most of this heterogeneity could be ascribed to the German series; excluding this, the OR was 1.22 (95% CI: 1.16–1.28; P = 2.56 × 10−10, Phet = 0.40). Although there is no direct evidence for a role of PHLDB1, 11q23.3 is commonly deleted in neuroblastoma30.
We analyzed the association between SNP genotypes and risk of tumors of glial origin, GBM and astrocytic lineage. There was evidence that GBMs have a different risk profile, notably with respect to rs4295627, rs498872 and rs2736100 (Supplementary Table 3). However, because of study heterogeneity, we cannot exclude the possibility that the variants do not influence the risk of developing a specific histological form of glioma without central histopathological review.
We only found evidence of interactive effects between 8q24.21 SNPs (Supplementary Table 4). For all other pairwise combinations, each locus had an independent role in glioma (Pinteraction > 0.10). Glioma risk increases with increasing numbers of variant alleles for the five loci (ORper-allele = 1.31, 95% CI: 1.26–1.36; P = 1.39 × 10−74; Fig. 3 and Supplementary Table 5). Individuals with eight or more risk alleles have an approximately fourfold increase in glioma risk compared to those with a median number of risk alleles. These ORs may be underestimates because the additive model assumes equal weighting across SNPs. We estimate that the five loci we have identified account for ∼7–14% of the excess familial risk of glioma (assuming naïve multiplicative or additive models and an overall familial relative risk of 2.1). However, we acknowledge that the present data provide only crude estimates of the effect on susceptibility attributable to variation at the loci, as the effect of the actual common causal variant responsible for the association will typically be larger.
Figure 3.

Cumulative effects of glioma risk alleles. (a) Distribution of risk alleles in glioma cases (green bars) and controls (blue bars). (b) Plot of the increasing ORs for glioma with increasing number of risk alleles. The ORs are relative to the median number of four risk alleles; vertical bars correspond to 95% confidence intervals. Horizontal line marks the null value (OR = 1). The distribution of risk alleles follows a normal distribution in both case and controls, with a shift toward a higher number of risk alleles in cases.
To investigate the basis of causality for each of the five associations, we interrogated HapMap to identify nonsynonymous SNPs (nsSNPs) correlated with the most strongly associated SNP signals. The strongest LD was between rs6010620 and nsSNP rs3848668 (D′ = 1.0, r2 = 0.05). Although HapMap is not comprehensive, these data suggest the associations identified are probably mediated through LD with sequence changes that influence expression rather than protein sequence or through LD with low-frequency variants not catalogued. We searched for cis-acting regulatory effects of SNPs on nearby genes in lymphoblastoid cell lines and normal brain tissue using publicly available data. Although we did not identify any significant genotype–expression relationships (Supplementary Fig. 2b), this does not exclude subtle effects or the possibility that the variants exert effects through untested transactivation of genes.
Our findings show that common variants influence glioma risk and highlight the importance of variation in genes encoding components of the CDKN2A-CDK4 signaling pathway in glioma. Moreover, this pathway, elucidated through the extended interaction network of CDKN2A, incorporates TERT (through mutual interaction with HSP90) and other genes (including CCDC26) we have identified as risk factors.
Online Methods
Subjects
UK-GWA study
The UK study was based on 636 cases (401 male, 235 female; mean age 46 years; s.d. = 12) ascertained through the INTERPHONE Study3. Briefly, the INTERPHONE Study was an international multicenter case-control study of PBT coordinated by the International Agency for Research on Cancer (IARC), with material collected between September 2000 and February 2004. UK individuals with PBT were collected through neurosurgery, neuropathology, oncology and neurology centers in the Thames regions of Southeast England and the Northern UK including central Scotland, the West Midlands, West Yorkshire and the Trent area. Cases were individuals with glioma (International Classification of Diseases for oncology (ICD-O), 2nd ed., codes 9380-9384, 9390-9411, 9420-9451 and 9505; ICD10 code C71). Cases with previous brain tumors were excluded. To minimize population stratification, cases with self-reported non-western-European ancestry were excluded from the present study. Individuals from the 1958 Birth Cohort served as source of controls4.
US-GWA study
The US study was based on 1,247 cases (768 male, 479 female; mean age 47 years; s.d. = 13) ascertained through The University of Texas M.D. Anderson Cancer Center, Texas, between 1990 and 2008. Cases were individuals with glioma (ICD10 code C71; ICD-O codes 9380-9384, 9390-9411, 9420-9451, and 9505). Individuals from CGEMS5,6 served as controls.
Replication series
The French Glioma Collection (FGC) is systematic series of 1,439 individuals with histologically proven glioma (WHO classification AI, AII, AIII, OII, OIII, OAII, OAIII, GBM-IV) ascertained through the Service de Neurologie Mazarin, Groupe Hospitalier Pitié-Salpêtrière Paris. The controls are taken from the SU.VI.MAX (SUpplementation en VItamines et Minraux-AntioXydants) study of 12,735 healthy subjects (aged 35 to 60 years) recruited from across France in 1994 (ref. 31). The Swedish Glioma Collection (SGCCS) comprises 215 glioma cases ascertained as part of the INTERPHONE Study3 conducted between 2000 and 2002 in Sweden, 134 cases from Umeå University Hospital, 122 from the Northern Sweden Health and Disease Study (NSHDS)32 and 203 from neurosurgery university clinics in Sweden. 383 controls for the INTERPHONE study were randomly selected from the population register, frequency-matched to brain tumor cases on age, sex and geographical region. In addition, 400 controls were ascertained from NSHDS that were age, sex and geographically representative of the cohort. The German Glioma Collection (GGC) comprises 564 individuals who underwent surgery for a glioma at the Department of Neurosurgery, University of Bonn Medical Center, between 1996 and 2008. All histological diagnoses were made at the Institute for Neuropathology/German Brain Tumor Reference Center, University of Bonn Medical Center. Control subjects were 576 healthy individuals (288 men, 288 women; mean age 31, range 18–45) of German origin recruited in 2004 by the Institute of Transfusion Medicine and Immunology, Mannheim.
Ethics
Collection of blood samples and clinico-pathological information from case and control subjects was undertaken with informed consent and relevant ethical review board approval in accordance with the tenets of the Declaration of Helsinki.
Genotyping
DNA was extracted from samples using conventional methodologies and quantified using PicoGreen (Invitrogen). Genotyping of both GWA studies was conducted by Illumina Service laboratory using the Illumina Infinium Human610-Quad BeadChips according to Illumina protocols. DNA samples with GenCall scores <0.25 at any locus were considered ‘no calls’. To ensure quality of genotyping, a series of duplicate samples was genotyped in the same batches.
Intensity data from arrays was imported into Illumina's BeadStudio clustering and calling software application. For the small subset of loci that were not clustered properly by the automated algorithm, the data were reviewed to identify loci that needed to be removed, manually edited or left unchanged. Clustered SNPs were evaluated using the metrics listed in the SNP Table of the BeadStudio software. These metrics are based on all samples for each locus and thus provide overall performance information for each locus. To identify loci potentially needing to be edited or removed, each quality metric column in the SNP table was sequentially sorted. Metrics used for identifying poorly or incorrectly clustered data included intensity, cluster separation, position of each cluster (AA, AB, BB), Hardy-Weinberg equilibrium, call frequency and variation of cluster width. The reproducibility of control samples on each plate as well as replicates were also used to identify misclustered loci. Although not all cluster plots were assessed, ∼10% of the lowest performing loci were examined. Of these 10%, ∼20% were edited or annotated (to indicate loci with nearby polymorphisms or hemizygous deletions) and ∼2% were excluded. Review of data was conducted by a second individual to determine if any metrics were missed or if further editing was required. Overall, this process provided substantially increased genotyping accuracy.
Subsequent genotyping of SNPs was conducted using either competitive allele-specific PCR KASPar chemistry (KBiosciences) or single-base primer extension chemistry MALDI-TOF MS detection (Sequenom). All primers and probes used are available on request. Genotyping quality control was further evaluated through inclusion of duplicate DNA samples in SNP assays. For all SNP assays, >99% concordant results were obtained. Samples having SNP call rates <90% were excluded from the analysis.
Statistical analysis
Statistical analyses were undertaken using R (v2.6), STATA (v8; State College, Texas, US) and PLINK (v1.05)33 software. All reported P values are two-sided. Genotype data were used to search for duplicates and closely related individuals among all samples in each of the GWA studies. Identity-by-state (IBS) values were calculated for each pair of individuals, and for any pair with allele sharing of >80%, the sample generating the lowest call rate was removed from further analysis. To identify individuals who might have non-Western European ancestry, we merged our case and control data with the 60 western European (CEU), 60 Nigerian (YRI), 90 Japanese (JPT) and 90 Han Chinese (CHB) individuals from the HapMap Project. For each pair of individuals, we calculated genome-wide IBS distances on markers shared between HapMap and our SNP panel and used these as dissimilarity measures upon which to perform principal component analysis. The first two principal components for each individual were plotted and any individual not present in the main CEU cluster (that is, outside 5% from cluster centroids) was excluded from subsequent analyses.
In the UK-GWA study, genotyped samples were excluded from analyses due to non-CEU ancestry (n = 3). In the US-GWA study, genotyped samples were excluded from analyses for the following reasons: relatedness (n = 2) and non-CEU ancestry (n = 23).
The adequacy of the case-control matching and possibility of differential genotyping of cases and controls were formally evaluated using quantile-quantile plots of test statistics. Deviation of the genotype frequencies in the controls from those expected under Hardy-Weinberg equilibrium (HWE) was assessed by a χ2 test or Fisher's exact test where an expected cell count was < 5.
The association between each SNP and risk of glioma was assessed by the Cochran-Armitage trend test. Odds ratios and associated 95% CIs were calculated by unconditional logistic regression. Relationships between multiple SNPs showing association with glioma risk in the same region were investigated using logistic regression analysis, and the impact of additional SNPs from the same region was assessed by a likelihood-ratio test.
The combined effect of each pair of risk locus was investigated by logistic regression modeling with evidence for interactive effects between SNPs assessed by a likelihood ratio test. The OR and trend test for increasing numbers of deleterious alleles was estimated by counting two for a homozygote and one for a heterozygote.
Meta-analysis was conducted using standard methods based on weighted average of study-specific estimates of the ORs, using inverse variance weights. Cochran's Q statistic to test for heterogeneity and the I2 statistic to quantify the proportion of the total variation due to heterogeneity were calculated. The sibling relative risk attributable to a given SNP was calculated using the following formula:
| (1) |
where p is the population frequency of the minor allele, q = 1 − p, and r1 and r2 are the relative risks (estimated as OR) for heterozygotes and rare homozygotes relative to common homozygotes. Assuming a multiplicative interaction the proportion of the familial risk attributable to a SNP was calculated as log(λ*)/log(λ0), where λ0 is the overall familial relative risk estimated from epidemiological studies, assumed to be 2.1 (ref. 2). A naïve estimation of the contribution of all of the loci identified to the excess familial risk of glioma under an additive model was calculated using the following formula:
| (2) |
Bioinformatics
LD metrics between SNPs reported in HapMap were based on Data Release 2/phaseIII Feb09 on NCBI B35 assembly, dbSNPb125, except for between rs2736098 and rs2736100 and rs2853676 which were only available in Data Release 23a/phaseII March08.
We used Haploview software (v3.2) to infer the LD structure of the genome in the regions containing loci associated with glioma risk. Prediction of the untyped SNP in the case-control datasets of GWA data was carried out using IMPUTE and SNPTEST on HapMap. In total, 894 HapMap SNPs were successfully imputed in the interval between 130,051,729 bp and 131,225,253 bp using available SNP genotype data from GWA scans (225 SNPs). We verified the integrity of imputed data, where possible, by crosschecking the concordance of imputed genotypes with that of available Illumina SNP genotype data. Directly genotyped SNPs are denoted in blue; those imputed are shown in green.
To examine whether there is a relationship between SNP genotype and mRNA expression in lymphocytes and normal human cortex, we used publicly available data. We analyzed 90 CEU-derived Epstein-Barr virus–transformed lymphoblastoid cell lines using Sentrix Human-6 Expression BeadChips (Illumina). Online recovery of data was carried out using WGAViewer v1.25 Software34,35. We analyzed 193 normal human cortex cells using Illumina HumanRef-seq-8 Expression BeadChip arrays36. Where SNPs genotyped in our study had not been typed, HapMap data were used to identify correlated SNPs in high LD (r2 > 0.8). Differences in the distribution of levels of mRNA expression between SNP genotypes were compared using a Wilcoxon-type test for trend. Relationship between copy number change and mRNA expression at loci in glioma was investigated using data from The Cancer Genome Atlas (TCGA)18. The Bioconductor module CGHcall was used to assign copy number status.
We searched for interactions between proteins encoded by genes mapping to association signals using the program PINA37.
URLs
Detailed information on the tagSNP panel, http://www.illumina.com/; WGAViewer, http://www.genome.duke.edu/centers/pg2/downloads/wgaviewer.php; 1958 Birth Cohort: http://www.cls.ioe.ac.uk/studies.asp?section=0001000200030012; PLINK, http://pngu.mgh.harvard.edu/∼purcell/plink/; Cancer Genetic Markers of Susceptibility (CGEMS), http://cgems.cancer.gov/; The Cancer Genomics Data Portal, http://cbio.mskcc.org/cancergenomics-dataportal; The Cancer Genome Atlas (TCGA) data portal, http://tcga-data.nci.nih.gov/tcga/homepage.htm; A Survey of Genetic Human Cortical Gene Expression, http://labs.med.miami.edu/myers/; IMPUTE, http://mathgen.stats.ox.ac.uk/impute/impute.html; SNPTEST, http://www.stats.ox.ac.uk/∼marchini/software/gwas/snptest.html; PINA (Protein Interaction Network Analysis platform), http://csbi.ltdk.helsinki.fi/pina/.
Supplementary Material
Acknowledgments
The Wellcome Trust provided principal funding for the study. In the UK, additional funding was provided by Cancer Research UK (C1298/A8362 supported by the Bobby Moore Fund) and the European Union (CPRB LSHCCT-2004-503465). The UK and Swedish INTERPHONE studies were supported by the European Union Fifth Framework Program “Quality of Life and Management of Living Resources” (contract number QLK4-CT-1999-01563) and the International Union against Cancer (UICC). The UICC received funds for this purpose from the Mobile Manufacturers' Forum and Groupe Speciale Mobile (GSM) Association. Provision of funds via the UICC was governed by agreements that guaranteed INTERPHONE's complete scientific independence. These agreements are publicly available at http://www.iarc.fr/en/research-groups/RAD/RCAd.html. The Swedish centre was also supported by the Swedish Research Council, the Cancer Foundation of Northern Sweden, the Swedish Cancer Society, and the Nordic Cancer Union and the UK centre by the Mobile Telecommunications and Health Research (MTHR) Programme and the Health and Safety Executive, Department of Health and Safety Executive and the UK Network Operators (O2, Orange, T-Mobile, Vodafone and ‘3’). The views expressed in the publication are those of the authors and not necessarily those of the funding bodies. In the United States, funding was provided by US National Institutes of Health grants 5R01 CA119215 and 5R01 CA070917. Additional support was obtained from the American Brain Tumor Association and the National Brain Tumor Society. The University of Texas M.D. Anderson Cancer Center acknowledges the work of P. Adatto, F. Morice, H. Zhang, V. Levin, A. Yung, M. Gilbert, R. Sawaya, V. Puduvalli, C. Conrad, F. Lang and J. Weinberg from the Brain and Spine Center. In France, funding was provided by the Délégation à la Recherche Clinique (MUL03012), the Association pour la Recherche sur les Tumeurs Cérébrales (ARTC), the Institut National du Cancer (INCa; PL 046) and the French Ministry of Higher Education and Research. In Germany, funding was provided to M. Simon, J.S. and M. Linnebank by the Deutsche Forschungsgemeinschaft (Si 552, Schr 285), the Deutsche Krebshilfe (70-2385-Wi2, 70-3163-Wi3, 10-6262) and BONFOR. In Sweden, the collection of samples from the Northern Sweden Health of disease study were collected with support from Umeå University Hospital and NIH funded part of GLIOGENE collection (R01 CA119215). B.M. was supported by Acta Oncologica Foundation as a research fellow at Royal Swedish Academy of Science. In the UK we acknowledge NHS funding to the NIHR Biomedical Research Centre. The UK-GWA study made use of genotyping data on the 1958 Birth Cohort. Genotyping data on controls was generated and generously supplied to us by P. Deloukas (Wellcome Trust Sanger Institute). A full list of the investigators who contributed to the generation of the data are available from http://www.wtccc.org.uk. The US-GWA study made use of control genotypes from the CGEMS prostate and breast cancer studies. A full list of the investigators who contributed to the generation of the data are available from http://cgems.cancer.gov/. The results published here are in whole or part based upon data generated by The Cancer Genome Atlas pilot project established by the National Cancer Institute and National Human Genome Research Institute. Information about TCGA and the investigators and institutions that constitute the TCGA research network can be found at http://cancergenome.nih.gov. Finally, we are grateful to all the study subjects for their participation. We also thank the clinicians and other hospital staff, cancer registries and study staff who contributed to the blood sample and data collection for this study.
Footnotes
Note: Supplementary information is available on the Nature Genetics website.
Author Contributions: R.S.H. and M.B. designed the study. R.S.H. drafted the manuscript, with extensive contributions from F.J.H. and S.S. F.J.H. and S.S. performed statistical analyses. S.E.D., F.J.H. and S.S. performed bioinformatics analyses. L.B.R. performed laboratory management and oversaw genotyping of UK cases and with A.P. performed genotyping of German and Swedish case-control series. In the UK, A.S., M. Schoemaker, K.M., S.J.H. and R.S.H. developed patient recruitment, sample acquisition and performed sample collection of cases. In the US, M.B. and C.L. developed protocols, M.B. and G.A. developed patient recruitment, G.A., X.G. and R.Y. performed curation and organization of samples and data, and Y.L. performed management of genotyping. In Germany, M. Simon and J.S. developed patient recruitment and blood sample collection, M. Simon oversaw DNA isolation and storage, K.H. and M. Linnebank collected control samples, M. Simon and M. Linnebank performed case ascertainment and supervision of DNA extractions, and K.H. and R.K. procured German control samples. In France, M. Sanson, J.-Y.D., K.H.-X. and A.I. developed patient recruitment, and Y.M., B.B. and S. El-H. developed sample acquisition and performed sample collection of cases. M. Lathrop and D.Z. performed laboratory management and oversaw genotyping of the French samples. In Sweden, for the Swedish INTERPHONE Study, M.F., S.L. and A.A. developed study design and conducted patient recruitment and control selection, M.F., S.L., A.A., B.M. and R.H. organized sample acquisition and performed sample collection of case and controls, U.A. coordinated sample collection and complied information into data files of cases and controls for statistical analyses, and B.M. and R.H. performed laboratory management and oversaw DNA extraction. The NSHDS samples were collected by Umeå University (Principal Investigator Göran Hallmans), and the additional samples were collected at the neurosurgery department in Umeå from 2005 and onwards (A.T.B. and R.H.) and through the national GLIOGENE study (Principal Investigator B.M.). All authors contributed to the final paper.
References
- 1.Bondy ML, et al. Brain tumor epidemiology: consensus from the Brain Tumor Epidemiology Consortium. Cancer. 2008;113:1953–1968. doi: 10.1002/cncr.23741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hemminki K, Li X. Familial risks in nervous system tumors. Cancer Epidemiol Biomarkers Prev. 2003;12:1137–1142. [PubMed] [Google Scholar]
- 3.Cardis E, et al. The INTERPHONE study: design, epidemiological methods, and description of the study population. Eur J Epidemiol. 2007;22:647–664. doi: 10.1007/s10654-007-9152-z. [DOI] [PubMed] [Google Scholar]
- 4.Power C, Elliott J. Cohort profile: 1958 British birth cohort (National Child Development Study) Int J Epidemiol. 2006;35:34–41. doi: 10.1093/ije/dyi183. [DOI] [PubMed] [Google Scholar]
- 5.Hunter DJ, et al. A genome-wide association study identifies alleles in FGFR2 associated with risk of sporadic postmenopausal breast cancer. Nat Genet. 2007;39:870–874. doi: 10.1038/ng2075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yeager M, et al. Genome-wide association study of prostate cancer identifies a second risk locus at 8q24. Nat Genet. 2007;39:645–649. doi: 10.1038/ng2022. [DOI] [PubMed] [Google Scholar]
- 7.Clayton DG, et al. Population structure, differential bias and genomic control in a large-scale, case-control association study. Nat Genet. 2005;37:1243–1246. doi: 10.1038/ng1653. [DOI] [PubMed] [Google Scholar]
- 8.Yin W, Rossin A, Clifford JL, Gronemeyer H. Co-resistance to retinoic acid and TRAIL by insertion mutagenesis into RAM. Oncogene. 2006;25:3735–3744. doi: 10.1038/sj.onc.1209410. [DOI] [PubMed] [Google Scholar]
- 9.Jiang M, Zhu K, Grenet J, Lahti JM. Retinoic acid induces caspase-8 transcription via phospho-CREB and increases apoptotic responses to death stimuli in neuroblastoma cells. Biochim Biophys Acta. 2008;1783:1055–1067. doi: 10.1016/j.bbamcr.2008.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Das A, Banik NL, Ray SK. Differentiation decreased telomerase activity in rat glioblastoma C6 cells and increased sensitivity to IFN-γ and taxol for apoptosis. Neurochem Res. 2007;32:2167–2183. doi: 10.1007/s11064-007-9413-y. [DOI] [PubMed] [Google Scholar]
- 11.Tomlinson I, et al. A genome-wide association scan of tag SNPs identifies a susceptibility variant for colorectal cancer at 8q24.21. Nat Genet. 2007;39:984–988. doi: 10.1038/ng2085. [DOI] [PubMed] [Google Scholar]
- 12.Easton DF, et al. Genome-wide association study identifies novel breast cancer susceptibility loci. Nature. 2007;447:1087–1093. doi: 10.1038/nature05887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kiemeney LA, et al. Sequence variant on 8q24 confers susceptibility to urinary bladder cancer. Nat Genet. 2008;40:1307–1312. doi: 10.1038/ng.229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Birnbaum S, et al. Key susceptibility locus for nonsyndromic cleft lip with or without cleft palate on chromosome 8q24. Nat Genet. 2009;41:473–477. doi: 10.1038/ng.333. [DOI] [PubMed] [Google Scholar]
- 15.Bille C, et al. Cancer risk in persons with oral cleft—a population-based study of 8,093 cases. Am J Epidemiol. 2005;161:1047–1055. doi: 10.1093/aje/kwi132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wager M, et al. Prognostic molecular markers with no impact on decision-making: the paradox of gliomas based on a prospective study. Br J Cancer. 2008;98:1830–1838. doi: 10.1038/sj.bjc.6604378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rafnar T, et al. Sequence variants at the TERT-CLPTM1L locus associate with many cancer types. Nat Genet. 2009;41:221–227. doi: 10.1038/ng.296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cancer Genome Atlas Research Network. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature. 2008;455:1061–1068. doi: 10.1038/nature07385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Parsons DW, et al. An integrated genomic analysis of human glioblastoma multiforme. Science. 2008;321:1807–1812. doi: 10.1126/science.1164382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Randerson-Moor JA, et al. A germline deletion of p14(ARF) but not CDKN2A in a melanoma-neural system tumor syndrome family. Hum Mol Genet. 2001;10:55–62. doi: 10.1093/hmg/10.1.55. [DOI] [PubMed] [Google Scholar]
- 21.Bahuau M, et al. Germ-line deletion involving the INK4 locus in familial proneness to melanoma and nervous system tumors. Cancer Res. 1998;58:2298–2303. [PubMed] [Google Scholar]
- 22.Simon M, Voss D, Park-Simon TW, Mahlberg R, Koster G. Role of p16 and p14ARF in radio- and chemosensitivity of malignant gliomas. Oncol Rep. 2006;16:127–132. [PubMed] [Google Scholar]
- 23.Scott LJ, et al. A genome-wide association study of type 2 diabetes in Finns detects multiple susceptibility variants. Science. 2007;316:1341–1345. doi: 10.1126/science.1142382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.McPherson R, et al. A common allele on chromosome 9 associated with coronary heart disease. Science. 2007;316:1488–1491. doi: 10.1126/science.1142447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Harder T, Plagemann A, Harder A. Birth weight and subsequent risk of childhood primary brain tumors: a meta-analysis. Am J Epidemiol. 2008;168:366–373. doi: 10.1093/aje/kwn144. [DOI] [PubMed] [Google Scholar]
- 26.Ferrie JE, Langenberg C, Shipley MJ, Marmot MG. Birth weight, components of height and coronary heart disease: evidence from the Whitehall II study. Int J Epidemiol. 2006;35:1532–1542. doi: 10.1093/ije/dyl184. [DOI] [PubMed] [Google Scholar]
- 27.Whincup PH, et al. Birth weight and risk of type 2 diabetes: a systematic review. J Am Med Assoc. 2008;300:2886–2897. doi: 10.1001/jama.2008.886. [DOI] [PubMed] [Google Scholar]
- 28.Barber LJ, et al. RTEL1 maintains genomic stability by suppressing homologous recombination. Cell. 2008;135:261–271. doi: 10.1016/j.cell.2008.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Arakawa Y, et al. Frequent gene amplification and overexpression of decoy receptor 3 in glioblastoma. Acta Neuropathol. 2005;109:294–298. doi: 10.1007/s00401-004-0956-6. [DOI] [PubMed] [Google Scholar]
- 30.Guo C, et al. Allelic deletion at 11q23 is common in MYCN single copy neuroblastomas. Oncogene. 1999;18:4948–4957. doi: 10.1038/sj.onc.1202887. [DOI] [PubMed] [Google Scholar]
- 31.Hercberg S, et al. The SU.VI.MAX Study: a randomized, placebo-controlled trial of the health effects of antioxidant vitamins and minerals. Arch Intern Med. 2004;164:2335–2342. doi: 10.1001/archinte.164.21.2335. [DOI] [PubMed] [Google Scholar]
- 32.Hallmans G, et al. Cardiovascular disease and diabetes in the Northern Sweden Health and Disease Study Cohort—evaluation of risk factors and their interactions. Scand J Public Health Suppl. 2003;61:18–24. doi: 10.1080/14034950310001432. [DOI] [PubMed] [Google Scholar]
- 33.Purcell S, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81:559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Stranger BE, et al. Genome-wide associations of gene expression variation in humans. PLoS Genet. 2005;1:e78. doi: 10.1371/journal.pgen.0010078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Stranger BE, et al. Relative impact of nucleotide and copy number variation on gene expression phenotypes. Science. 2007;315:848–853. doi: 10.1126/science.1136678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Myers AJ, et al. A survey of genetic human cortical gene expression. Nat Genet. 2007;39:1494–1499. doi: 10.1038/ng.2007.16. [DOI] [PubMed] [Google Scholar]
- 37.Wu J, et al. Integrated network analysis platform for protein-protein interactions. Nat Methods. 2009;6:75–77. doi: 10.1038/nmeth.1282. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.