

Abstract
MethylationofhistoneH3lysine4(H3K4me)isanintricatelyregulatedposttranslational modification, which is broadly associated with enhancers and promoters of actively transcribed genomic loci. Recent advances in next-generation sequencing have identified a number of H3K4me regulators mutated in neurodevelopmental disorders including intellectual disabilities, autism spectrum disorders, and schizophrenia. Here, we aim to summarize the molecular function of H3K4me-regulating enzymes in brain development and function. We describe four H3K4me methyltransferases (KMT2A, KMT2C, KMT2D, KMT2F), four demethylases (KDM1A, KDM5A, KDM5B, KDM5C), and two reader proteins (PHF21A, PHF8) mutated in neurodevelopmental disorders. Understanding the role of these chromatin regulators in the development and maintenance of neural connections will advance therapeutic opportunities for prevention and treatment of these lifelong neurodevelopmental disorders.
Keywords: autism spectrum disorders, brain development, chromatin, epigenetics, gene regulation, H3K4 methylation, histone methylation, intellectual disability, schizophrenia
Chromatin remodeling is a dynamic process that regulates gene expression via changes in DNA accessibility. Posttranslational modifications on N-terminal tails of histone proteins are an integral element influencing chromatin structure. Advent of next-generation sequencing led to a rapid growth of the list of histone modifiers that are mutated in human neurodevelopmental disorders. These disorders include a plethora of intellectual disability (ID) syndromes [1] and schizophrenia [2]. Additionally, recent large-scale exome sequencing studies highlighted dysregulation of histone methylation as a major contributing factor of autism spectrum disorders (ASD) [3,4]. Therefore, regulation of histone modifications appears to be essential for the development and function of the central nervous system. However, the roles of these mutated histone modifiers in the brain are not well understood.
Methylation of lysine 4 of histone H3 (H3K4me) is one such modification, which is associated with gene activation. Three statuses of lysine methylation, namely mono-, di-, and tri-methyl groups (me1/2/3), confer an additional layer of complexity in chromatin remodeling events. Using technologies such as chromatin immunoprecipitation coupled with next-generation sequencing (ChIP-seq), the genomic signatures of these epigenetic marks can be uncovered for specific cell types and developmental stages. Early genome-wide studies using cultured cell lines revealed that H3K4me1 is a hallmark of enhancers [5,6], while H3K4me2/3 mark promoters of actively transcribed genes [7,8]. Mono- and di-methylated H3K4 appear as broad ChIP-seq peaks, marking stretches of open chromatin throughout the genome [7]. In the human brain, similar to the observations in cell cultures, tri-methylated H3K4 is concentrated in sharp peaks 1–2 kb in length at transcription start sites of regulatory sequences such as proximal gene promoters [9]. These neuron-specific H3K4me3 peaks are enriched at promoters of genes that control synaptic function [9]. Additionally, unmethylated H3K4 (me0) also recruits a distinct set of proteins with transcriptional repressors [10,11], including DNMT3L, which tethers CpG DNA methylation enzymes [11].
Two families of proteins serve as primary regulators of H3K4me: histone lysine methyltransferases (KMTs) are the ‘writers’, which place the methyl marks onto histones; and histone lysine demethylases (KDMs) are the ‘erasers’, which remove them. An additional class of proteins recognizes and ‘reads’ H3K4me status, serving as effectors to recruit chromatin remodeling proteins and regulate transcriptional state. Remarkably, among other methylation sites on histones, H3K4 appears to be the most extensively targeted position by the largest number of writer and eraser enzymes. To date, in higher eukaryotes, at least six KMTs and six KDMs have been shown to regulate H3K4me with differential substrate preference to me0–3 [12,13]. This intricate balancing act at H3K4 by opposing mechanisms might have evolved to sculpt epigenetic landscapes that achieve delicate developmental and cellular processes in complex organs such as brain. Indeed, pioneering work has shown dynamic gain and loss of H3K4me3 throughout the genome in the neurons of developing human prefrontal cortex [9,14]. While H3K4me regulation has been unequivocally shown to be crucial for normal development in metazoans through Hox gene regulation [15], the function and regulation of this methylation mark in the developing nervous system is not fully understood. The dynamics of chromatin regulation in the brain is the focus of the emerging field of neuroepigenetics.
Among the 12 enzymes that target H3K4me in humans, mutations in four KMT and four KDM genes have been associated with neurodevelopmental disorders to date (Figure 1). An excellent review summarized the implications of H3K4 demethylases in neurodevelopmental disorders with a focus on regulatory mechanisms of the demethylases [16]. Shen et al. provided a comprehensive view on writer and eraser enzymes for H3K4me in brain disorders from a clinical perspective, including data from human samples and animal models [17]. More recently, alterations of KMT2F in schizophrenia [2], KDM1A in Kabuki/ KBG syndrome [18], and KDM5A and KDM5B in ID [1,19] have been reported, which were not covered by these earlier reviews. New exome sequencing of a large cohort of individuals with ASD also identified KMT2C and KDM5B [3,4]. It may only be a matter of time before alterations in the remaining four enzymes are identified in neurological disorders. Furthermore, genetic mutations in two reader proteins for H3K4me status, PHF21A and PHF8, have been associated with ID syndromes [20,21] (Figure 1). Here, we aim to summarize the recent progress in understanding the molecular and cellular consequences of mutations in H3K4me regulators. Although very limited reports described the function of H3K4me regulators at a genomic level in the brain, by reviewing the data obtained from non-brain tissues and cell types, we further discuss how intricate balancing and readout of H3K4me can be engaged in brain development genome-wide.
Figure 1. H3K4me writer, eraser and reader genes in neurodevelopmental disorders.
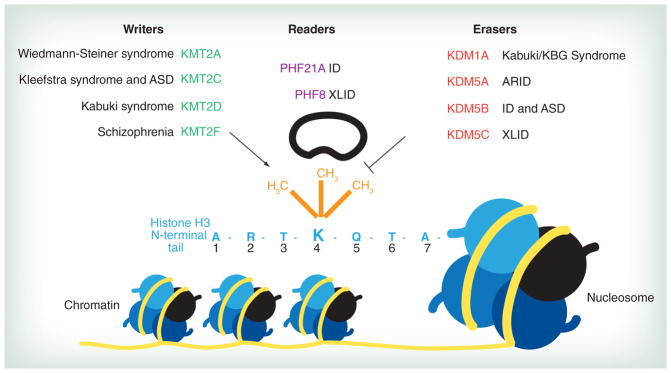
The first seven amino acids of the N-terminal tail of histone H3 (light blue) are extending from a representative nucleosome. Lysine (K) 4 can be differentially methylated (orange) by the writer and eraser proteins shown above. Methyltransferases (green) ‘write’ methyl marks, whereas demethylases (red) ‘erase’ methyl marks. Reader enzymes (purple) recognize and ‘read’ methylation status. Mutations in each of these enzymes lead to neurodevelopmental disorders, indicated in black next to each gene name.
ARID: Autosomal recessive intellectual disability; ASD: Autism spectrum disorder; ID: Intellectual disability; XLID: X-linked ID.
For color images please see online at: www.futuremedicine.com/doi/full/10.2217/EPI.15.1
The six H3K4 methyltransferases
Methylation of H3K4 is generated primarily by the lysine (K)-specific methyltransferase 2 (KMT2) family of enzymes [22,23]. This class of proteins is characterized by the presence of a catalytic Su(var), Enhancer of Zeste, Trithorax (SET) domain, conferring lysine-specific methyltransferase function. While each writer enzyme has been shown to generate all three methylation marks on H3K4 [13], specific substrate preference is often conferred by additional cofactor proteins. For example, while KMT2A itself can generate mono- and di-methyl marks on H3K4, physical contact with cofactors including RbBP5, Ash2L, and WDR5 allows tri-methylation by KMT2A [24].
The six KMT2 family members can be broken into three subclasses of protein pairs that have evolved distinct features and functions. KMT2A and KMT2B are homologues of Trithorax (Trx) gene found in lower organisms such as fly, whereas KMT2C and KMT2D are thought to be duplicated from the common ancestor gene Trithorax-related (Trr) in fly [23]. KMT2F and KMT2G are structurally distinct from the KMT2A-D enzymes. Similar to the KMT2A/B and KMT2C/D pairs, mammalian KMT2F has a cognate paralog, KMT2G, and both share a common ancestor gene SET1 in budding yeast [23]. Four of these six KMTs have been found mutated in neurodevelopmental disorders thus far. Remarkably, evidence suggests that within each protein pair, one enzyme cannot compensate for a mutation in the other, indicating that each KMT in the duo has nonredundant functions.
KMT2A in a developmental disorder with neurological symptoms
The human KMT2A gene (also known as MLL, ALL-1) was first discovered for its involvement in chromosomal translocations in acute leukemia [25,26]. As discussed above, KMT2A is responsible for generating mono-, di-, and tri-methylated H3K4 through its SET domain and by interaction with cofactors [24]. Recently, dominant de novo mutations in KMT2A have been identified in individuals with Wiedemann–Steiner syndrome (Mendelian Inheritance in man [MIM] no. 605130), a developmental disorder with clinical features including ID, microcephaly, and short stature [27,28]. The majority of identified mutations are heterozygous de novo mutations predicting a premature truncation of the protein product (Figure 2), suggesting haploinsufficiency of KMT2A is responsible for these clinical phenotypes. Since the catalytic SET domain is located at the C-terminal end of the protein, the truncations most likely result in a loss of enzymatic activity. Given that H3K4me is a signature of actively transcribed chromatin, the pathogenic mechanism may be insufficient transcription of key genes in the central nervous system. However, several lines of evidence suggest the molecular etiology can be much more complex than this simplistic scenario.
Figure 2. Domain organization of H3K4me regulators mutated in neurodevelopmental disorders.

Mutation types are summarized in inset. Colored blocks correspond to functional protein domains found within each gene. Substrate preference is indicated below each gene name. KMT2D mutations are limited to a subset based on the latest publication [29]. Scale is provided for each category of enzymes.
A: AT-hook; CS: Cleavage site; FC: FY-rich C-terminal domain; FN: FY-rich N-terminal domain; H: High mobility group (HMG) box; LZ: Leucine zipper; P: Plant homeodomain (PHD) finger; RRM: RNA recognition motif; SET: Su(var)3–9 Enhancer-of-zeste Trithorax methyltransferase domain; ZF: Zinc finger.
KMT2A harbors both transcriptional activator and repressor activities. These two opposite functions appear to be located at the SET-containing C-terminal portion and the PHD-containing N-terminal segment, respectively [30]. A cluster of three plant homeodomain (PHD) fingers, located in the middle of the KMT2A molecule (PHD1–PHD3 in Figure 2), plays an important role in demarcating the two opposite activities. PHD fingers are one of the major modules that can recognize specifically modified histones, and thus can act as ‘readers’ of chromatin modifications [31,32]. Interestingly, KMT2A PHD3 binds to both H3K4me3, a reaction product of KMT2A itself, as well as the transcriptional corepressor protein Cyp33 [33]. While the role of PHD1 remains unknown, PHD2 appears to have ubiquitin ligase activity toward both histones and KMT2A itself, facilitating the degradation of KMT2A [34]. Thus, beyond the canonical function of PHD fingers as reader modules, the KMT2A-PHD cluster represents a novel regulatory hub, which defines the balance between transcriptional activator and repressor functions. Notably, some of the Wiedemann–Steiner syndrome mutations, including a missense mutation, fall within this PHD finger cluster (Figure 2). Therefore, these mutations can potentially lead to both down- and upregulation of KMT2A-target genes in Wiedemann–Steiner syndrome. Future studies can address these possibilities using cells harboring these mutations.
Additionally, alternative splicing events have been reported in the PHD3-encoding area [35]. Yeast two-hybrid experiments revealed that these PHD3 isoforms differ in their ability to interact with Cyp33-containing corepressor complex, suggesting an influence in the transcriptional regulatory activities of KMT2A [35]. Perhaps each transcript type has a unique expression pattern in the developing nervous system. It is plausible that different KMT2A isoforms function as either an activator or a repressor in a cell-type-dependent manner in neurons and glia cells within the central nervous system. Functional studies in developing brains are necessary to determine to what extent this alternative splicing event exists and how it contributes to molecular etiology in vivo.
Expression of KMT2A peaks in the neocortex in human fetal brains, declining slightly through early stages of postnatal development but then gradually increasing into adulthood [36]. This expression signature hints at the importance of KMT2A during neocortical development. Consistently, KMT2A plays an essential role in mouse postnatal neurogenesis [37]. Lim et al. demonstrated that important neurogenesis-promoting genes have bivalent chromatin domains carrying both activating H3K4me3 and repressive H3K27me3 marks, and that presence of KMT2A at these poised loci is necessary to promote an active transcription state [37]. Therefore, impaired neurogenesis by KMT2A mutations might be responsible for the cognitive deficits observed in Wiedemann–Steiner syndrome.
On a genomic scale, KMT2A has been shown to be required for generating 5% of the active H3K4me3 marks on promoters in mouse embryonic fibroblasts [38]. Relatively weak impact on global H3K4me3 landscape upon the loss of KMT2A suggests a redundant, if not completely redundant, function of paralog KMT2B and the four other KMT2 family members. Recent studies in mouse embryonic stem cells (mESCs) reconfirmed the functional redundancy between KMT2A and KMT2B in generating global H3K4me3, whereas removal of KMT2B alone lead to a 2.5-fold reduction in H3K4me levels at bivalent promoters [39]. Reconciling these findings with previous evidence of KMT2A at bivalent promoters in neural precursor cells [37], it is plausible that KMT2A and KMT2B are responsible for generating neural precursor- and mESC-specific bivalent promoters, respectively. Heterozygous de novo mutations in Wiedemann–Steiner syndrome indicate that alteration of H3K4me levels at only limited genomic loci is sufficient to attenuate proper brain development. Further studies to identify direct gene targets of KMT2A during central nervous system development will be imperative in elucidating a functional role for this writer enzyme in the brain.
KMT2C: a new gene for Kleefstra syndrome & ASD
KMT2C (also known as MLL3, HALR) contributes to the implementation of mono- and dimethylation marks on H3K4 in mammalian cells [40,41]. KMT2C was originally discovered as a human homologue of Drosophila gene Trr in the chromosomal region lost in individuals with developmental defects of the fore-brain and myeloid leukemia [42,43]. In recent years, KMT2C variants have been found in two neurodevelopmental disorders. Kleefstra et al. identified a KMT2C mutation in an individual with Kleefstra syndrome (MIM no. 610253), a neurodevelopmental spectrum disorder with hallmarks including severe ID, brachy(micro)cephaly, and epileptic seizures. The female individual carries a heterozygous de novo nonsense mutation, p.Arg1481X [44] (Figure 2), resulting in loss of more than half of the KMT2C protein. This truncation of the C-terminal segment includes loss of the enzymatic SET domain, which likely results in the loss of histone methyltransferase activity. Additionally, five mutations were identified in recent large-scale exome sequencing of individuals with ASD (Figure 2) [3,4]. These new variants will require further studies to examine their consequences on protein function and pathogenic contribution to neurodevelopment. Clearly, disruption of KMT2C and its contribution to a delicate epigenetic regulatory network may underlie broad neurodevelopmental abnormalities including cognitive and social anomalies.
While neuron-specific targets of KMT2C regulation have yet to be discovered, it is known that this writer is highly expressed in the human brain [42,45]. KMT2C expression levels peak during human fetal development and remain steady into adulthood [36]. Notably, expression in the adult brain is stronger in the hippocampus, caudate nucleus, and substantia nigra, regions of the brain associated with learning, memory, and social behaviors [45]. These data implicate roles for KMT2C in the developing brain as well as in mature neuronal circuitries.
Genome-wide functional studies found that KMT2C contributes to installation of mono- and dimethylation of H3K4 at gene enhancers [40,41]. H3K4me1/2 at enhancers is required for full activation of genes. Interestingly, KMT2C can also be found at gene promoters, where promoter-bound KMT2C functions to repress gene transcription [46]. These studies raise a possibility that KMT2C can both activate and repress gene transcription depending on localization along chromatin. Studying the chromatin regulatory network involving KMT2C will lead to a better understanding of the tight gene regulation in brain development, and can pave the way toward targeted therapeutic approaches.
KMT2D mutations are a major cause of Kabuki syndrome
The human histone lysine methyltransferase KMT2D (also known as ALR, MLL2 or MLL4) is partially functionally redundant to KMT2C, and the two enzymes are responsible for a majority of the H3K4me1/2 marks in mammalian cells [40,41]. Exome sequencing revealed KMT2D to be a major cause of Kabuki syndrome (MIM no. 147920) [47], an autosomal-dominant congenital ID disorder characterized by unique facial features. Over 100 unique mutations in KMT2D have been identified in Kabuki syndrome individuals, a majority of which result in premature termination of the protein product [29,47–54] (Figure 2). Though most mutations occur de novo, many are recurrent in unrelated individuals, indicating the importance of these repeatedly mutated amino acids in KMT2D function and/or suggesting the nature of the DNA is particularly mutable.
The large number of mutations appears to cluster into three distinct locations along the protein (Figure 2). One mutation group surrounds a triple PHD finger cluster (PHD4–PHD6 in Figure 2). These three PHD fingers bind unmethylated arginine 3 on histone H4 (H4R3me0), thereby promoting the access of the SET domain to its substrate, namely the histone H3 N-terminus [55]. This trans-tail crosstalk enables KMT2D to efficiently methylate H3K4 in a nucleosomal context [55]. Importantly, two missense mutations in Kabuki syndrome, p.Cys1430Arg and p.Cys1471Tyr in the PHD4–6 cluster, showed decreased binding affinity to H4 and reduced H3K4 methylation activity, when nucleosomes were used for the methyltransferase assays [55]. Thus, Kabuki syndrome mutations in this cluster ultimately result in loss-of-function of KMT2D methyltransferase activity. The second mutation cluster occurs in an area of the protein lacking a characterized functional domain, yet this region may be significant for the 3D structure and function of the enzyme. The third mutation group occurs around the C-terminal PHD finger, whose function remains unknown. Similar to KMT2A and KMT2C mutations, over 30 variants in KMT2D lead to truncation of C-terminal SET catalytic domain, likely resulting in the loss of enzymatic function.
A role for KMT2D in neuronal differentiation has been suggested from recent in vitro studies. Using human pluripotent NT2/D1 carcinoma cells, which commit to a neuronal state upon retinoic acid treatment, Dhar et al. showed that shRNA knockdown of KMT2D results in attenuation of morphological changes and impaired activation of neural differentiation hallmark genes including HOXA1–3 and NES-TIN [55]. The insufficient expression of these KMT2D target genes was associated with decreased H3K4me levels. Thus, loss-of-function mutations in KMT2D in Kabuki syndrome likely result in a decrease in these methyl marks necessary to promote expression of key neuronal differentiation genes, and ultimately hinder commitment of stem cells to a neuronal state. These data suggest a critical role for KMT2D in neuronal differentiation. Whether this writer enzyme also contributes to maintenance of neuronal function later in life remains to be seen.
Several lines of evidence suggest a cooperation of KMT2C and KMT2D in neuronal differentiation. First, knockdown of KMT2C in the NT2/D1 system resulted in downregulation of KMT2D-target genes, although to a lesser extent compared with KMT2D knockdown [55]. Second, KMT2C and KMT2D can be copurified in the same protein complex [56]. Third, both KMT2C and KMT2D are found at gene enhancers and are required for enhancer activation [40,41]. These results raise a possibility that deficiency in common molecular pathways, including enhancer activation of specific genes, impinge on Kabuki and Kleefstra syndromes, though they were originally described as distinct conditions.
KMT2F: a new schizophrenia susceptibility gene
KMT2F (also known as SET1, SETD1A) encodes an H3K4 writer enzyme capable of generating mono-, di-, and tri-methylation marks in vitro [41,57]. Notably, in cells, KMT2F/G and Set1 appear to be responsible for bulk H3K4me3 [58–61].
Recently, Takata et al. have identified KMT2F as a risk gene for schizophrenia (MIM no. 181500), a common and severe psychiatric disorder characterized by delusions, hallucinations, and disorganized thinking [2]. It is significant that two loss-of-function mutations (Figure 2) were found in two phenotypically similar but unrelated schizophrenia individuals with a secondary diagnosis of obsessive-compulsive disorder (OCD) (MIM no. 164230). It is possible that individuals with KMT2F mutations define a new subset of schizophrenia with OCD comorbidity [2]. The first mutation is a de novo frameshift resulting in the introduction of a premature stop codon and likely leads to the loss of enzymatic activity carried in the C-terminal SET domain. The second individual harbors a de novo indel that alters a canonical splice acceptor site. This change is expected to cause a loss of exon 16 and disruption of the N-SET domain, which is critical for proper H3K4 methyltransferase function of the neighboring SET domain in vitro [62].
As previously discussed, KMT2F contributes to deposition of the activating H3K4me3 mark, which shows sharp peaks near transcription start sites of active genes [7]. While H3K4me1 is enriched at gene enhancers, H3K4me3 levels at enhancers appear to be low [5,6]. Hence, KMT2F acts as an H3K4me3 writer enzyme at promoters, in sharp contrast to KMT2C/D function at enhancers. Indeed, KMT2F-interacting protein Cfp1 recruits H3K4me3 activity, likely mediated by KMT2F/G, at CpG islands which are prevalent in mammalian gene promoters [63]. Recent studies found that loss of Cfp1 in mESCs leads to global loss of DNA-damage-induced accumulation of H3K4me3 at the promoters of DNA-damage-responsive genes [64]. However, induction of genes was mildly affected in Cfp1-mutant mESCs except for few dramatically affected genes, suggesting a role for KMT2F in context-dependent transcriptional activity [64]. H3K4me3 plays a crucial part in recruiting the general transcription machinery, TFIID, through an interaction between H3K4me3 and the PHD finger of TAF3, a TFIID component [65,66] Thus, the KMT2F mutations may lead to insufficient gene transcription of important neural circuitry genes via impaired placement of H3K4me3 at their promoters.
Psychiatric disorders are often characterized by disorganization of neuronal networks, where alteration of these networks from an early time point leads to lifelong disorders. The inability to activate the correct genes at the correct time during brain development due to a loss of KMT2F function could lead to improper establishment of neuronal networks and, ultimately, impaired brain function manifested by schizophrenia. However, brain-specific roles of KMT2F or specific genomic loci that are targets of KMT2F in brain cells have yet to be defined. The aforementioned genetic studies open an avenue for interrogating chromatin regulatory mechanisms that underlie pathogenesis of psychiatric disorders.
Mutations in H3K4 demethylases
Histone methylation was thought to be an irreversible epigenetic mark, thereby serving as memory of cellular identities. The identification and characterization of histone demethylases revealed that methylation is not a permanent event but rather a dynamic process. Four H3K4me eraser enzymes, KDM1A, KDM5A, KDM5B, and KDM5C, are mutated in neurodevelopmental disorders, evidence that the balance between methylation and demethylation is intricately regulated during brain development and function.
KDM1A in a unique neurodevelopmental syndrome
KDM1A (also known as LSD1, BHC110) was the first histone lysine demethylase gene to be identified [67]. A flavin-containing amine oxidase homologue, KDM1A catalyzes the oxidative demethylation of H3K4me1/2. Recently, a mutation in KDM1A has been identified in a new neurodevelopmental disorder. Tunovic et al. describe two de novo variations in one male with mixed features of Kabuki syndrome and KBG syndrome [18]. As discussed above, Kabuki syndrome is a developmental and cognitive disorder frequently associated with mutations in other histone lysine regulators, KMT2D [47] or KDM6A [29]. KBG syndrome (MIM no. 148050) is characterized by macrodontia, craniofacial dysmorphism, and delay in brain development [68]. One identified variation was p.Tyr785His in the C-terminal amine oxidase domain of KDM1A. The other alteration was a deletion of three nucleotides in ANKRD11, which lead to the loss of an evolutionally conserved lysine residue. Haploinsufficiency of the ANKRD11 gene, which encodes an ankyrin-repeat containing a transcription coactivator, was previously identified as a causative agent for KBG syndrome [68]. Based on the new mixed features of Kabuki and KBG syndromes found in the individual, the authors postulated that the KDM1A mutation might be responsible for the Kabuki-related symptoms such as ptosis and downturned month corners.
How could mutations in counteracting enzymes over H3K4me, namely KMT2D and KDM1A, both result in Kabuki-related syndromes? The majority of KMT2D mutations appear to result in loss of enzymatic activity by truncation of the catalytic SET domain [47]. One possibility is that the identified p.Tyr785His variant may be a gain-of-function mutation. In this scenario, both loss-of-function mutations in a writer and gain-of-function mutations in an eraser would lead to decreased H3K4me levels. Alternatively, developmental change in expression of key genes might require temporal coordination of KMT2D and KDM1A at specific time points. Deletion of Kdm1a in mouse confers embryonic lethality [69], as Kdm1a is required for gastrulation and differentiation of embryonic stem cells [70,71]. Thus, if the KDM1A mutation is a loss-of-function one, it is likely hypomorphic. Further studies including identification of additional mutations in KDM1A are needed to understand this interesting observation from human genetics studies.
While only one KDM1A mutation has been described in a human developmental disorder thus far, studies have demonstrated pivotal roles of this eraser in neurodevelopment. KDM1A was originally discovered as a key subunit of the CoREST complex, which is responsible for the repression of neuronal genes such as Synapsin I (SYN1) in non-neuronal cells [72,73,74]. The CoREST complex is recruited to neuron-specific genes via an interaction with REST, a sequence-specific DNA binding factor [73]. Thus, it is possible that the loss of KDM1A function causes aberrant expression of neuron-specific genes in neural precursor and/or glia cells, thereby eventually perturbing proper brain development. Consistently, leaky expression of neuron-specific genes in HeLa cells was observed upon KDM1A-knockdown [67].
In addition to its involvement in neuronal gene repression, cell-autonomous functions of KDM1A in neurons have also been described. Neuron-specific isoforms of KDM1A have been shown to modulate neurite morphogenesis in zebrafish [75]. The first functional study of KDM1A in the mammalian nervous system demonstrated that the presence of alternatively-spliced exon E8a dynamically regulates neurite maturation. Loss of KDM1A upon shRNA knockdown resulted in decreased dendritic arborizations, secondary branches, and average neurite width, while overexpression lead to an increase in these morphological features in mouse cortical neurons [75]. Inclusion of exon E8a generates a small protruding loop near the catalytic site, which may provide a site for posttranslational modifications and thus confer an additional regulatory element. Taken together, current knowledge supports the idea of a significant role for KDM1A in neurodevelopment.
On a genomic level, ChIP studies on individual genes show that KDM1A localizes to active promoters in HeLa and MCF7 cells, suggesting the enzyme is a locus-specific demethylase [67,76]. Additionally, a ChIP-seq study has found that removal of KDM1A from enhancers of pluripotency genes influences commitment of ES cells to differentiate [71]. Thus, KDM1A may regulate developmental expression of neuronal genes by controlling methylation of promoters and enhancers. What factors recruit KDM1A in specific regulatory regions in neurons remains unanswered.
KDM5 family: a second class of demethylases
The finding that KDM1A is unable to demethylate tri-methylated H3K4 [67] raised the possibility that additional demethylases had yet to be discovered. This lead to the identification and characterization of a second class of H3K4 demethylases, the lysine (K)-specific demethylase 5 (KDM5) family of proteins (formerly JARID1 family), comprising of KDM5A, KDM5B, KDM5C, and KDM5D [77,78,79,80,81,82]. The enzymatic activity of these erasers lies in two Jumonji domains, JmjN and JmjC. This demethylation signature requires Fe(II) and α-ketoglutarate as cofactors to perform the hydroxylation reaction to remove methylation [83]. Notably, to date three of the four KDM5 family members, KDM5A, KDM5B, and KDM5C, are reported mutated in neurodevelopmental disorders, revealing nonredundant functions of this class of enzymes in the brain.
KDM5A: a candidate ID gene
KDM5A (also known as RBP2, JARID1A) was first identified as a binding factor of retinoblastoma RB gene product [84]. Further studies revealed it to be an H3K4me3-specific demethylase from the JmjC-domain-containing family [77,80,81,82]. A mutation in KDM5A was recently identified in an individual with autosomal recessive ID (ARID) [1]. The homozygous de novo missense mutation causes a p.Arg719Gly change, substituting a nonpolar amino acid for a positively charged one. The mutation was predicted to be damaging to KDM5A function [1], as it is located within a zinc-finger motif likely responsible for DNA-binding and possibly contributes to enzymatic activity [85] (Figure 2). It is unknown whether this single amino acid substitution would affect overall stability, DNA-binding property, and/or enzymatic activity of KDM5A. Functional characterization of this mis-sense mutation is necessary to determine its effects on protein function and contribution to ID.
Little work has been done experimentally to investigate the neuron-specific functions of KDM5A; however, a role for this demethylase has been described in early embryonic development [86], cellular differentiation [87], senescence [88], and circadian gene regulation [89]. Genome-wide analysis in mammalian cell lines revealed KDM5A is bound at proximal promoter regions, about half of which are H3K4me3-positive, suggesting an inhibitory role for KDM5A on many genes via direct action on promoters [86]. KDM5A mRNA levels are high during human prenatal brain development and plateau postnatally, with cortical expression levels remaining slightly elevated throughout adulthood [35]. Considering the homozygous mutation in ARID, and the expression data showing KDM5A peaks at a very specific prenatal window, it is plausible that KDM5A serves as an ‘off’ switch for a subset of genes at significant developmental time points.
KDM5B mutations in ID & ASD
KDM5B (also known as PLU1, JARID1B) encodes an H3K4-specific eraser enzyme that directly catalyzes the demethylation of mono-, di-, and tri-methylated H3K4 [77,78,79]. Until recently, all variants in KDM5B had been described in cancer. Next-generation sequencing has since revealed a de novo splicing mutation (c.283A>G) in KDM5B in an individual with nonsyndromic ID [19] (Figure 2). Six additional variants were newly identified in exome sequencing of a large cohort of individuals with ASD [3,4]. These mutations include missense, nonsense, and frameshift variants found all along the protein, many of which lie in key functional domains (Figure 2). Further studies are necessary to assess the functional effects of these mutations and the pathogenicity of mutated KDM5B in ID and ASD.
KDM5B has been shown to regulate cell fate decisions [90] in mESCs to neuronal lineage [91]. Knockdown of KDM5B in mESCs resulted in failure to differentiate to neurons, whereas knockout in mouse neural stem cells (mNSCs) revealed that loss of KDM5B at this later stage does not affect differentiation to mature neurons [91,92]. Knockout of KDM5B in mouse is embryonic lethal [93], and, remarkably, overexpression also leads to impaired neural differentiation [90]. This suggests tight control of this demethylase is critical for cell fate determination. Loss of KDM5B may result in activation of genes that drive proliferation, while over-expression of KDM5B could lead to aberrant demethylation of differentiation genes in uncommitted cells to hinder neural differentiation. Therefore, either loss- or gain-of-function mutations could lead to inefficient neuronal differentiation, which can result in cognitive deficiencies. Identification of additional human mutations in neurological disorders would advance our knowledge about the significance of precise KDM5B regulation in neural differentiation.
How KDM5B regulates neuronal differentiation has become less of a mystery through recent genome-wide studies. ChIP-seq in mESCs shows KDM5B bound to transcription start sites of genes encoding developmental regulators [91]. KDM5B can be recruited to target genomic loci via association with transcription factors such as androgen receptor [79], and act as a transcriptional repressor by stopping H3K4me spreading at gene promoters [78,92,94]. Loss of KDM5B in mESCs results in a global increase in H3K4me3 levels and a failure to silence germ cell genes [91], suggesting that KDM5B is a major H3K4me3 eraser in stem cells. Additionally, KDM5B depletion leads to H3K4me spreading into promoters and gene bodies, revealing a role for KDM5B in restricting H3K4me to specific genomic areas [92,95]. Which specific genes’ misregulation affects neural differentiation will be a focus of future studies.
Additionally, KDM5B has been suggested to regulate genomic stability by dictating the cell’s response to DNA damage [96]. This critical function in DNA repair is likely a fundamental aspect of the pathogenicity of KDM5B mutations in cancer, however if and how this function could contribute to neurological disorders remains open to investigation.
KDM5C mutations are frequent in X-linked ID
In 2005, the first mutations in KDM5C (also known as SMCX, JARID1C) were identified as a frequent cause of X-linked ID (XLID) (Figure 2) [97]. Currently, mutations in KDM5C are estimated to account for roughly 0.7–2.8% of all XLID [97,98,99]. The function of this gene’s product remained unknown until in 2007 KDM5C was discovered as a specific demethylase for di- and tri-methylated H3K4 [77,100]. To date, 17 mutations have been identified in individuals with XLID, a majority of which are missense or nonsense variants (Figure 2). Of the mutations characterized thus far, functional studies indicate these human variants cause a decrease in KDM5C enzymatic activity, suggesting a loss-of-function pathogenic mechanism [77,100]. In addition to cognitive deficits, aggressive behaviors are frequently observed in affected individuals [97,101] and autistic features have been noted in one case [102]. Along with developmental abnormalities in males, KDM5C variants have also been found in females with short-term memory deficits [103].
KDM5C was originally identified as a gene that escapes X-inactivation [104,105], and both KDM5C and Y-chromosome paralog KDM5D are expressed in a sex-specific manner in mouse brain [106]. These findings suggest a nonredundant function for KDM5C and KDM5D, and that dosage differences between males and females may underlie differential consequence of KDM5C loss between the sexes. Thus, KDM5C appears to be essential for development of a broad spectrum of cognitive and adaptive functions both in males and females.
KDM5C is broadly expressed, with higher levels in human brain and skeletal muscle [97]. Expression levels of KDM5C remain relatively unchanged throughout human brain development, from prenatal to adult stages, suggesting a lifelong critical role of this demethylase [35]. Within the mouse brain, Kdm5c is expressed in areas important for cognitive and emotional behaviors such as the prefrontal cortex, hippocampus, and amygdala [106,107,108]. Additionally, this eraser enzyme has been shown to affect dendritic outgrowth in rat cerebellar granule neurons and neuronal survival in developing zebrafish [77]. Together, these data may help reveal the pathogenic mechanism for mutations in KDM5C leading to neurodevelopmental defects.
Genetic and biochemical interactions of KDM5C with other molecules have provided insights into molecular mechanisms of ID. KDM5C is directly regulated by ARX, a homeobox gene frequently mutated in XLID and epilepsy [109]. A majority of the ARX variants cause a hypomorphic ARX allele, leading to a decrease in KDM5C expression, thus possibly altering the regulation of H3K4me. KDM5C also physically interacts with the transcriptional repressor REST [100]. As discussed above, REST represses neuronal genes in non-neuronal cells. Loss of KDM5C was shown to impair REST-mediated repression of neuronal genes, such as SCN2A and SYN1 [100]. Defective repression of REST-target genes in KDM5C may be a non-cell autonomous mechanism of impaired cognitive development. XLID mutations in KDM5C may cause dysregulation of REST-target genes due to the impaired H3K4me demethylase activity. Further studies will be necessary to understand functions of KDM5C in different cell types in the brain.
Interestingly, KDM5C appears to have dual function depending on its localization along chromatin in mESCs, namely transcription corepressor activity at promoters and transcriptional coactivator activity at enhancers [110]. Thus, it is possible that KDM5C serves as not only a repressor of REST-target genes but also a coactivator for yet unidentified genes in the brain. Jensen et al. recently identified a dozen genes that are commonly dysregulated in multiple lymphocyte lines derived from individuals with KDM5C mutations [111]. However, it is unclear, if gene expression changes in lymphocytes can be translated to the cognitive deficits in individuals with KDM5C mutations. Identifying the KDM5C-target genes and mechanisms of their regulation in a genomic context, especially in relevant cell types in the brain, will greatly advance our understanding of molecular mechanisms underpinning this frequent form of ID and ultimately provide hope for therapeutic interventions.
Recognition of H3K4me by reader molecules
Histone methylation itself does not seem to have an impact on higher-order chromatin structure. In addition to writer and eraser enzymes, reader molecules act at H3K4 by recognizing specific methylation states and recruiting effector proteins to influence transcription. Here, we discuss two H3K4me-specific reader proteins, PHF21A and PHF8, which have been found mutated in intellectual disabilities. Both genes encode for proteins that belong to the PHD finger (PHF) family.
PHF21A: A methyl ‘zero’ reader in ID
PHF21A (also known as BHC80) was the first reader molecule discovered to recognize unmethylated H3K4 (HeK4me0) [10], introducing unmodified H3K4 as an important addition to the histone code in regulating chromatin state. Recognition of unmodified histone H3 tail by PHF21A PHD finger is specifically inhibited by H3K4 methylations [10]. Recently, three unrelated individuals with ID and craniofacial anomalies have been identified harboring de novo balanced translocations disrupting PHF21A [20]. Two of these translocations, t(11;19)(p11.2;p13.2)dn and t(1;11)(p13;p11) dn, are predicted to lead to truncated protein products resulting in loss of the PHD finger (Figure 2) [20]. The translocation breakpoint of the third, t(X;11) (p22.2;p11.2)dn, has not been characterized, yet it is likely that loss of protein function is also the pathogenic mechanism at play [20]. Therefore, H3K4me0 recognition by PHF21A PHD finger appears to be essential for cognitive and craniofacial development. Notably, Potocki–Shaffer syndrome (MIM no. 601224) is a contiguous gene deletion syndrome of the chromosomal region where PHF21A is located, namely chromosome 11p11.2 [112,113]. Potocki–Shaffer syndrome has hallmark clinical features including multiple exostoses, parietal foamina, ID, and craniofacial anomalies. While EXT2 and ALX4 have been identified to be responsible for multiple exostoses and parietal foamina, respectively, it remained unknown which gene(s) deletion leads to ID and craniofacial anomalies. The discovery of heterozygous translocations and resulting truncations of PHF21A strongly suggests that haploinsufficiency of PHF21A is responsible for ID and craniofacial deficits in Potocki–Shaffer syndrome [20].
Transcript levels of PHF21A are high during early stages of human embryonic development, and remain steady throughout development into adulthood [35]. Accordingly, in situ hybridization and immunofluorescence experiments in mouse show Phf21a is expressed highly in both the central nervous system as well as in cranial bones of embryonic and adult mice [20,114], suggesting an important role for this reader protein in both development and function of nervous tissue and craniofacial bones.
To test whether a loss of PHF21A results in defects in neuronal and craniofacial development, Kim et al. studied the effects of phf21a knockdown by morpholino injection in zebrafish [20]. Injection of morpholino resulted in pronounced deficiencies in head development including facial dysmorphism. These abnormalities were rescued upon injection of wild-type human PHF21A mRNA, supporting the specificity of knockdown effect. Interestingly, Phf21a knockout mice show neonatal lethality with an insufficient ability to suckle [115]. Phf21a-null mice were not closely examined for craniofacial defects; therefore, it is also plausible that failure to suckle was a result of craniofacial anomalies, as suggested by the zebrafish studies [20]. In addition, PHF21A appears to be involved in pituitary function through modulation of neurosecretion, based on in vitro studies using mammalian cells [116]. Taken together, these functional studies in model organisms and cells support a critical role for PHF21A in neuro- and craniofacial development and function. It would be informative to generate conditional knockout mice in order to assess the effects of loss of Phf21a specifically in neurons or cranial bones.
PHF21A is a key subunit of the CoREST complex [117], as is demethylase KDM1A, discussed above. This protein complex functions to mediate repression of neuron-specific genes [117], and thus it may not be surprising that loss of PHF21A leads to neurodevelopmental phenotypes. Significantly, RNAi knockdown of PHF21A in HeLa cells results in a derepression of neuron-specific KDM1A-target genes including SCN1A and SYN1, suggesting PHF21A occupancy at unmethylated H3K4 is required for sufficient gene repression [10,118]. A neuron-specific CoREST/KDM1A target gene, SCN3A, showed increased mRNA expression in the lymphoblastoid cell lines from individuals with the translocations mentioned above [20]. Additionally, ChIP results indicated decreased occupancy of KDM1A at the promoter of SCN3A in the PHF21A-translocation cell lines [20]. Together these findings demonstrate that PHF21A is necessary for KDM1A recruitment and the generation of a repressive chromatin state. It remains unknown what aspects of neurodevelopment are perturbed by PHF21A deficiency and whether the role of PHF21A as a neuronal gene repressor contributes to pathogenesis of ID and craniofacial abnormality.
PHF8, another XLID-associated demethylase, reads H3K4me3
Mutations in X-linked gene PHF8 were thought to be responsible for Siderius XLID (MIM no. 300263), a syndromic form of ID often associated with facial anomalies such as cleft palate [21,119,120,121,122]. In 2010, PHF8 was discovered to be a histone demethylase that mainly targets H3K9me1/2 and H4K20me1 [123,124,125,126,127,128]. Further studies identified that PHF8 contains a PHD finger that specifically recognizes H3K4me3 [125,126,127,129]. Thus, PHF8 represents an intriguing example of histone methylation crosstalk engaged in brain development.
All three human PHF8 mutations in XLID are predicted to truncate the entire or large portion of the JmjC domain, which is responsible for demethylase activity (Figure 2). Therefore, loss of histone demethylase function likely underlies pathogenesis of Siderius XLID. Indeed, one missense mutation, p.Phe279Ser, which is located within the JmjC domain (Figure 2), leads to dramatic decrease in the histone demethylase activity [124,125]. Although any mutation in the PHF8 PHD finger has not yet been clinically observed, the H3K4me3 recognition by this PHD finger appears be significant in fulfilling PHF8 function. The presence of H3K4me3 substantially enhances enzymatic activity of PHF8 via increasing affinity between the histone H3 tail and PHF8 [129]. Thus, this PHD finger domain is critical for optimal demethylase activity of PHF8. Future human genetic studies may identify loss-of-function mutations in this PHD finger.
Significantly, neuronal functions of PHF8 have been shown in model systems. Qi et al. demonstrated that PHF8 is essential for brain and craniofacial development, as well as neuronal survival in zebrafish [123]. PHF8 has also been shown to regulate neuronal differentiation in mouse carcinoma P19 cells, which differentiate into neurons after exposure to retinoic acid. Additionally, knockdown of PHF8 in primary mouse cortical neurons results in downregulation of cytoskeleton genes and deficient neurite outgrowth [130]. PHF8 expression levels sharply decline during early stages of human embryonic development, and plateau at this reduced level throughout development into adulthood [35]. Together, these studies support a role for PHF8 in either proliferation of neural progenitor cells or gene expression changes during early differentiation stages in embryonic development.
Genome-wide, PHF8 has been found at the transcription start sites of more than 7000 RefSeq genes, as well as in gene bodies and intergenic regions [123,127], indicating participation of PHF8 in broad epigenome regulation. PHF8 binding sites consist of approximately one-half of total H3K4me3 peaks across the genome. The PHF8 PHD finger, whose reader function permits recognition of H3K4me2/3, likely contributes to recruitment of this reader protein to gene promoters. Additionally, PHF8 interacts with ZNF711, which is also mutated in XLID, and together the two activate reporter gene expression synergistically [127]. Since ZNF711 contains multiple C2H2 zinc-finger domains, ZNF711 may also participate in recruiting PHF8 onto target loci cooperatively with the H3K4me3/PHD interaction. Qi et al. showed PHF8 to be a positive regulator of gene expression, dependent on its H3K4me3-binding PHD and catalytic domains [123]. The function of PHF8 as a transcriptional activator is consistent with its enzymatic activity, which removes repressive histone methylation marks H3K9me1/2 and H4K20me1. Direct PHF8 target genes are involved in various signaling pathways including retinoic acid and Notch, both of which play pivotal roles in neural and craniofacial development [123,131,132].
Interestingly, KDM5C, an XLID-associated H3K4 demethylase discussed above, appears to be a PHF8 target gene, and consequently is downregulated upon the loss of PHF8. Moreover, PHF8 is localized at nucleoli and required for full activation of rRNA genes [126]. Given the high demand of protein synthesis in rapidly dividing stem cells, it is plausible that insufficient ribosome biogenesis by PHF8 deficiency may underlie impaired maintenance of developing brain cells observed in zebrafish [123]. These findings highlight a role for a novel network of chromatin regulators controlling expression of critical neurodevelopmental genes across the genome.
Conclusion & future perspective
The field of neuroepigenetics is rapidly changing as we continue to define roles for chromatin-regulating proteins in the central nervous system. As the cost of DNA sequencing declines, the number of mutations discovered in neurodevelopmental disorders is dramatically increasing. We believe it is only a matter of time before many more chromatin regulators are added to the growing list of proteins implicated in neurological disorders. Functional studies of the molecular consequences of mutations, alongside thorough investigations of cellular and behavioral abnormalities in animal models of these neurodevelopmental disorders, are essential to uncovering the pathophysiology of brain dysfunction.
We mainly discussed consequences of human mutations in the context of enzymatic activity and protein–protein interactions. It is noteworthy that both writer and eraser enzymes have recently been shown to physically interact with long noncoding RNAs (lncRNAs) [133,134]. lncRNAs appear to play roles in recruitment of associated chromatin modifiers to specific genomic loci [133,134] and stability of protein complexes [135]. Thus, an important direction is to test if mutations affect physical interaction of H3K4me modifiers to lncRNAs. At the epigenomic level, pioneering comparative studies of human and primate brain samples have demonstrated that human-specific H3K4me3 exist in the neurons of the cerebral cortex [136,137]. Given that neurodevelopmental disorders affect higher-order cognitive and adaptive functions, it will be highly informative to test if such human-specific H3K4me landscapes are altered by the mutations.
As summarized in earlier work [17], animal models for neurodevelopmental disorders associated with H3K4me dysregulation will be invaluable tools to dissect molecular and cellular mechanisms, as well as for drug development. Thus far, knockout mouse lines of two H3K4me enzymes, Kmt2a [138] and Kmt2b [139], have been reported to exhibit deficits in learning and memory. Harnessing the contemporary mouse genetics approach, one of the important questions that should be addressed is when the cognitive and adaptive deficits originate. One possibility is that a given chromatin modifier may play roles both in brain development and plasticity of mature circuitry. In such cases, therapeutic intervention would still hold a hope to ameliorate cognitive deficits even after affected individuals reach adulthood.
Great strides are being made with small-molecule inhibitors of writers and erasers in order to modulate H3K4me status [140,141,142]. While these strategies are currently being targeted for cancers, a therapeutic potential for these drugs in brain disorders is evident. Clearly, understanding the intricate nature of H3K4me dynamics, as well as other chromatin modifications, in the developing brain will form the groundwork necessary for targeted therapeutics and prevention for these neurodevelopmental disorders in the future.
Executive summary.
Histone H3 lysine 4 methylation (H3K4me) is one of the most well-characterized chromatin modifications, present at enhancers and promoters of actively transcribed genes throughout the genome.
Mutations in H3K4me regulators have recently been identified in neurodevelopmental disorders, including intellectual disability, autism spectrum disorder, and schizophrenia. These disorders lead to lifelong cognitive, emotional, and social disability, which entail a significant personal and healthcare burden worldwide.
Here, we summarize recent progress in identification of mutations and functional investigations of H3K4me regulators in the context of neurodevelopmental disorders.
Identification of new mutations in neurodevelopmental disorders and determining their consequences at a molecular level will allow us to decipher intricate chromatin signaling that underpins human brain development as well as basic mechanisms of gene regulation.
Insight into the role of these H3K4me regulators in brain development and function will greatly advance therapeutic opportunities for these lifelong neurodevelopmental disorders.
Footnotes
For reprint orders, please contact: reprints@futuremedicine.com
Financial & competing interests disclosure
CN Vallianatos is supported, in part, by an NIH training (T32GM07544), ‘Michigan Predoctoral Training in - Genetics’, and the Farrehi research fund. S Iwase is Cooley’s Anemia Foundation and the University of Medical School. The authors have no other relevant or financial involvement with any organization or financial interest in or financial conflict with - the ter or materials discussed in the manuscript apart from disclosed.
No writing assistance was utilized in the production manuscript.
References
Papers of special note have been highlighted as:
• of interest;
•• of considerable interest
- 1•.Najmabadi H, Hu H, Garshasbi M, et al. Deep sequencing reveals 50 novel genes for recessive cognitive disorders. Nature. 2011;478(7367):57–63. doi: 10.1038/nature10423. First large-scale exome sequencing study identifying numerous chromatin gene mutations in recessive intellectual disabilities. [DOI] [PubMed] [Google Scholar]
- 2.Takata A, Xu B, Ionita-Laza I, Roos JL, Gogos JA, Karayiorgou M. Loss-of-function variants in schizophrenia risk and SETD1A as a candidate susceptibility gene. Neuron. 2014;82(4):773–780. doi: 10.1016/j.neuron.2014.04.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Iossifov I, O’roak BJ, Sanders SJ, et al. The contribution of de novo coding mutations to autism spectrum disorder. Nature. 2014;515(7526):216–221. doi: 10.1038/nature13908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4•.De Rubeis S, He X, Goldberg AP, et al. Synaptic, transcriptional and chromatin genes disrupted in autism. Nature. 2014;515(7526):209–215. doi: 10.1038/nature13772. Recent large-scale exome sequencing study highlighting involvement of histone methylation in pathogenesis of ASD. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Heintzman ND, Hon GC, Hawkins RD, et al. Histone modifications at human enhancers reflect global cell-type-specific gene expression. Nature. 2009;459(7243):108–112. doi: 10.1038/nature07829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Heintzman ND, Stuart RK, Hon G, et al. Distinct and predictive chromatin signatures of transcriptional promoters and enhancers in the human genome. Nat Genet. 2007;39(3):311–318. doi: 10.1038/ng1966. [DOI] [PubMed] [Google Scholar]
- 7.Barski A, Cuddapah S, Cui K, et al. High-resolution profiling of histone methylations in the human genome. Cell. 2007;129(4):823–837. doi: 10.1016/j.cell.2007.05.009. [DOI] [PubMed] [Google Scholar]
- 8.Zhu J, Adli M, Zou JY, et al. Genome-wide chromatin state transitions associated with developmental and environmental cues. Cell. 2013;152(3):642–654. doi: 10.1016/j.cell.2012.12.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cheung I, Shulha HP, Jiang Y, et al. Developmental regulation and individual differences of neuronal H3K4me3 epigenomes in the prefrontal cortex. Proc Natl Acad Sci USA. 2010;107(19):8824–8829. doi: 10.1073/pnas.1001702107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10•.Lan F, Collins RE, et al. Recognition of unmethylated histone H3 lysine 4 links BHC80 to LSD1-mediated gene repression. Nature. 2007;448(7154):718–722. doi: 10.1038/nature06034. First characterization of a reader protein recognizing unmethylated H3K4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ooi SK, Qiu C, Bernstein E, et al. DNMT3L connects unmethylated lysine 4 of histone H3 to de novo methylation of DNA. Nature. 2007;448(7154):714–717. doi: 10.1038/nature05987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ruthenburg AJ, Allis CD, Wysocka J. Methylation of lysine 4 on histone H3: intricacy of writing and reading a single epigenetic mark. Mol Cell. 2007;25(1):15–30. doi: 10.1016/j.molcel.2006.12.014. [DOI] [PubMed] [Google Scholar]
- 13••.Greer EL, Shi Y. Histone methylation: a dynamic mark in health, disease and inheritance. Nat Rev Genet. 2012;13(5):343–357. doi: 10.1038/nrg3173. An overview of histone methylation regulation and consequences on health and disease. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shulha HP, Cheung I, Guo Y, Akbarian S, Weng Z. Coordinated cell type-specific epigenetic remodeling in prefrontal cortex begins before birth and continues into early adulthood. PLoS Genet. 2013;9(4):e1003433. doi: 10.1371/journal.pgen.1003433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Soshnikova N, Duboule D. Epigenetic regulation of Hox gene activation: the waltz of methyls. Bioessays. 2008;30(3):199–202. doi: 10.1002/bies.20724. [DOI] [PubMed] [Google Scholar]
- 16••.Wynder C, Stalker L, Doughty ML. Role of H3K4 demethylases in complex neurodevelopmental diseases. Epigenomics. 2010;2(3):407–418. doi: 10.2217/epi.10.12. Excellent review of H3K4 demethylases in neurodevelopmental disorders. [DOI] [PubMed] [Google Scholar]
- 17••.Shen E, Shulha H, Weng Z, Akbarian S. Regulation of histone H3K4 methylation in brain development and disease. Philos Trans R Soc Lond B Biol Sci. 2014;369(1652) doi: 10.1098/rstb.2013.0514. Excellent recent review of H3K4 writer and eraser enzymes in neurodevelopmental disorders, with an emphasis on human studies and animal models. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tunovic S, Barkovich J, Sherr EH, Slavotinek AM. De novo ANKRD11 and KDM1A gene mutations in a male with features of KBG syndrome and Kabuki syndrome. Am J Med Genet A. 2014;164(7):1744–1749. doi: 10.1002/ajmg.a.36450. [DOI] [PubMed] [Google Scholar]
- 19.Athanasakis E, Licastro D, Faletra F, et al. Next generation sequencing in nonsyndromic intellectual disability: from a negative molecular karyotype to a possible causative mutation detection. Am J Med Genet A. 2014;164(1):170–176. doi: 10.1002/ajmg.a.36274. [DOI] [PubMed] [Google Scholar]
- 20.Kim HG, Kim HT, Leach NT, et al. Translocations disrupting PHF21A in the Potocki-Shaffer syndrome region are associated with intellectual disability and craniofacial anomalies. Am J Hum Genet. 2012;91(1):56–72. doi: 10.1016/j.ajhg.2012.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Laumonnier F, Holbert S, Ronce N, et al. Mutations in PHF8 are associated with X linked mental retardation and cleft lip/ cleft palate. J Med Genet. 2005;42(10):780–786. doi: 10.1136/jmg.2004.029439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Allis CD, Berger SL, Cote J, et al. New nomenclature for chromatin-modifying enzymes. Cell. 2007;131(4):633–636. doi: 10.1016/j.cell.2007.10.039. [DOI] [PubMed] [Google Scholar]
- 23.Shilatifard A. The COMPASS family of histone H3K4 methylases: mechanisms of regulation in development and disease pathogenesis. Annu Rev Biochem. 2012;81:65–95. doi: 10.1146/annurev-biochem-051710-134100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dou YL, Milne TA, Ruthenburg AJ, et al. Regulation of MLL1 H3K4 methyltransferase activity by its core components. Nat Struct Mol Biol. 2006;13(8):713–719. doi: 10.1038/nsmb1128. [DOI] [PubMed] [Google Scholar]
- 25.Zieminvanderpoel S, Mccabe NR, Gill HJ, et al. Identification of a gene, Mll, that spans the breakpoint in 11q23 translocations associated with human leukemias. Proc Natl Acad Sci USA. 1991;88(23):10735–10739. doi: 10.1073/pnas.88.23.10735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cimino G, Moir DT, Canaani O, et al. Cloning of All-1, the locus involved in leukemias with the T(4–11) (Q21-Q23),T(9–11)(P22-Q23), and T(11–19)(Q23-P13) chromosome translocations. Cancer Res. 1991;51(24):6712–6714. [PubMed] [Google Scholar]
- 27.Strom SP, Lozano R, Lee H, et al. De novo variants in the KMT2A (MLL) gene causing atypical Wiedemann-Steiner syndrome in two unrelated individuals identified by clinical exome sequencing. BMC Med Genet. 2014;15:49. doi: 10.1186/1471-2350-15-49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jones WD, Dafou D, Mcentagart M, et al. De novo mutations in MLL cause Wiedemann-Steiner syndrome. Am J Hum Genet. 2012;91(2):358–364. doi: 10.1016/j.ajhg.2012.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Miyake N, Koshimizu E, Okamoto N, et al. MLL2 and KDM6A mutations in patients with Kabuki syndrome. Am J Med Genet A. 2013;161(9):2234–2243. doi: 10.1002/ajmg.a.36072. [DOI] [PubMed] [Google Scholar]
- 30.Zeleznik-Le NJ, Harden AM, Rowley JD. 11q23 translocations split the “AT-hook” cruciform DNA-binding region and the transcriptional repression domain from the activation domain of the mixed-lineage leukemia (MLL) gene. Proc Natl Acad Sci USA. 1994;91(22):10610–10614. doi: 10.1073/pnas.91.22.10610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wysocka J, Swigut T, Milne TA, et al. WDR5 associates with histone H3 methylated at K4 and is essential for H3K4 methylation and vertebrate development. Cell. 2005;121(6):859–872. doi: 10.1016/j.cell.2005.03.036. [DOI] [PubMed] [Google Scholar]
- 32.Musselman CA, Kutateladze TG. Handpicking epigenetic marks with PHD fingers. Nucleic Acids Res. 2011;39(21):9061–9071. doi: 10.1093/nar/gkr613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang Z, Song J, Milne TA, et al. Pro isomerization in MLL1 PHD3-bromo cassette connects H3K4me readout to CyP33 and HDAC-mediated repression. Cell. 2010;141(7):1183–1194. doi: 10.1016/j.cell.2010.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang J, Muntean AG, Wu L, Hess JL. A subset of mixed lineage leukemia proteins has plant homeodomain (PHD)-mediated E3 ligase activity. J Biol Chem. 2012;287(52):43410–43416. doi: 10.1074/jbc.M112.423855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rossler T, Marschalek R. An alternative splice process renders the MLL protein either into a transcriptional activator or repressor. Pharmazie. 2013;68(7):601–607. doi: 10.1055/s-0033-1343653. [DOI] [PubMed] [Google Scholar]
- 36.Johnson MB, Kawasawa YI, Mason CE, et al. Functional and evolutionary insights into human brain development through global transcriptome analysis. Neuron. 2009;62(4):494–509. doi: 10.1016/j.neuron.2009.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lim DA, Huang YC, Swigut T, et al. Chromatin remodelling factor Mll1 is essential for neurogenesis from postnatal neural stem cells. Nature. 2009;458(7237):529–533. doi: 10.1038/nature07726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wang P, Lin C, Smith ER, et al. Global analysis of H3K4 methylation defines MLL family member targets and points to a role for MLL1-mediated H3K4 methylation in the regulation of transcriptional initiation by RNA polymerase II. Mol Cell Biol. 2009;29(22):6074–6085. doi: 10.1128/MCB.00924-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Denissov S, Hofemeister H, Marks H, et al. Mll2 is required for H3K4 trimethylation on bivalent promoters in embryonic stem cells, whereas Mll1 is redundant. Development. 2014;141(3):526–537. doi: 10.1242/dev.102681. [DOI] [PubMed] [Google Scholar]
- 40.Lee JE, Wang CC, Xu SLY, et al. H3K4 mono- and di-methyltransferase MLL4 is required for enhancer activation during cell differentiation. Elife. 2013;2:e01503. doi: 10.7554/eLife.01503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hu DQ, Gao X, Morgan MA, Herz HM, Smith ER, Shilatifard A. The MLL3/MLL4 branches of the COMPASS family function as major histone H3K4 monomethylases at enhancers. Mol Cell Biol. 2013;33(23):4745–4754. doi: 10.1128/MCB.01181-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tan YC, Chow VT. Novel human HALR (MLL3) gene encodes a protein homologous to ALR and to ALL-1 involved in leukemia, and maps to chromosome 7q36 associated with leukemia and developmental defects. Cancer Detect Prev. 2001;25(5):454–469. [PubMed] [Google Scholar]
- 43.Ruault M, Brun ME, Ventura M, Roizes G, De Sario A. MLL3, a new human member of the TRX/MLL gene family, maps to 7q36, a chromosome region frequently deleted in myeloid leukaemia. Gene. 2002;284(1–2):73–81. doi: 10.1016/s0378-1119(02)00392-x. [DOI] [PubMed] [Google Scholar]
- 44.Kleefstra T, Kramer JM, Neveling K, et al. Disruption of an EHMT1-associated chromatin-modification module causes intellectual disability. Am J Hum Genet. 2012;91(1):73–82. doi: 10.1016/j.ajhg.2012.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nagase T, Kikuno R, Ishikawa K, Hirosawa M, Ohara O. Prediction of the coding sequences of unidentified human genes. XVII The complete sequences of 100 new cDNA clones from brain which code for large proteins in vitro. DNA Res. 2000;7(2):143–150. doi: 10.1093/dnares/7.2.143. [DOI] [PubMed] [Google Scholar]
- 46.Cheng J, Blum R, Bowman C, et al. A role for H3K4 monomethylation in gene repression and partitioning of chromatin readers. Mol Cell. 2014;53(6):979–992. doi: 10.1016/j.molcel.2014.02.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ng SB, Bigham AW, Buckingham KJ, et al. Exome sequencing identifies MLL2 mutations as a cause of Kabuki syndrome. Nat Genet. 2010;42(9):790–793. doi: 10.1038/ng.646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Paulussen AD, Stegmann AP, Blok MJ, et al. MLL2 mutation spectrum in 45 patients with Kabuki syndrome. Hum Mutat. 2011;32(2):E2018–E2025. doi: 10.1002/humu.21416. [DOI] [PubMed] [Google Scholar]
- 49.Micale L, Augello B, Fusco C, et al. Mutation spectrum of MLL2 in a cohort of Kabuki syndrome patients. Orphanet J Rare Dis. 2011;6:38. doi: 10.1186/1750-1172-6-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Li Y, Bogershausen N, Alanay Y, et al. A mutation screen in patients with Kabuki syndrome. Hum Genet. 2011;130(6):715–724. doi: 10.1007/s00439-011-1004-y. [DOI] [PubMed] [Google Scholar]
- 51.Hannibal MC, Buckingham KJ, Ng SB, et al. Spectrum of MLL2 (ALR) mutations in 110 cases of Kabuki syndrome. Am J Med Genet A. 2011;155A(7):1511–1516. doi: 10.1002/ajmg.a.34074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Banka S, Veeramachaneni R, Reardon W, et al. How genetically heterogeneous is Kabuki syndrome?: MLL2 testing in 116 patients, review and analyses of mutation and phenotypic spectrum. Eur J Hum Genet. 2012;20(4):381–388. doi: 10.1038/ejhg.2011.220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bogershausen N, Wollnik B. Unmasking Kabuki syndrome. Clin Genet. 2013;83(3):201–211. doi: 10.1111/cge.12051. [DOI] [PubMed] [Google Scholar]
- 54.Banka S, Howard E, Bunstone S, et al. MLL2 mosaic mutations and intragenic deletion-duplications in patients with Kabuki syndrome. Clin Genet. 2013;83(5):467–471. doi: 10.1111/j.1399-0004.2012.01955.x. [DOI] [PubMed] [Google Scholar]
- 55.Dhar SS, Lee SH, Kan PY, et al. Trans-tail regulation of MLL4-catalyzed H3K4 methylation by H4R3 symmetric dimethylation is mediated by a tandem PHD of MLL4. Genes Dev. 2012;26(24):2749–2762. doi: 10.1101/gad.203356.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Cho YW, Hong T, Hong S, et al. PTIP associates with MLL3- and MLL4-containing histone H3 lysine 4 methyltransferase complex. J Biol Chem. 2007;282(28):20395–20406. doi: 10.1074/jbc.M701574200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lee JH, Skalnik DG. CpG-binding protein (CXXC finger protein 1) is a component of the mammalian set1 histone H3-Lys(4) methyltransferase complex, the analogue of the yeast Set1/COMPASS complex. J Biol Chem. 2005;280(50):41725–41731. doi: 10.1074/jbc.M508312200. [DOI] [PubMed] [Google Scholar]
- 58.Wu M, Wang PF, Lee JS, et al. Molecular regulation of H3K4 trimethylation by Wdr82, a component of human Set1/ COMPASS. Mol Cell Biol. 2008;28(24):7337–7344. doi: 10.1128/MCB.00976-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ardehali MB, Mei A, Zobeck KL, Caron M, Lis JT, Kusch T. Drosophila Set1 is the major histone H3 lysine 4 trimethyltransferase with role in transcription. EMBO J. 2011;30(14):2817–2828. doi: 10.1038/emboj.2011.194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hallson G, Hollebakken RE, Li T, et al. dSet1 is the main H3K4 di- and tri-methyltransferase throughout Drosophila development. Genetics. 2012;190(1):91–100. doi: 10.1534/genetics.111.135863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Mohan M, Herz HM, Smith ER, et al. The COMPASS family of H3K4 methylases in Drosophila. Mol Cell Biol. 2011;31(21):4310–4318. doi: 10.1128/MCB.06092-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Mersman DP, Du HN, Fingerman IM, South PF, Briggs SD. Charge-based interaction conserved within histone H3 lysine 4 (H3K4) methyltransferase complexes is needed for protein stability, histone methylation, and gene expression. J Biol Chem. 2012;287(4):2652–2665. doi: 10.1074/jbc.M111.280867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Clouaire T, Webb S, Skene P, et al. Cfp1 integrates both CpG content and gene activity for accurate H3K4me3 deposition in embryonic stem cells. Genes Dev. 2012;26(15):1714–1728. doi: 10.1101/gad.194209.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Clouaire T, Webb S, Bird A. Cfp1 is required for gene expression dependent H3K4me3 and H3K9 acetylation in embryonic stem cells. Genome Biol. 2014;15(9):451. doi: 10.1186/s13059-014-0451-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Lauberth SM, Nakayama T, Wu X, et al. H3K4me3 interactions with TAF3 regulate preinitiation complex assembly and selective gene activation. Cell. 2013;152(5):1021–1036. doi: 10.1016/j.cell.2013.01.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Vermeulen M, Mulder KW, Denissov S, et al. Selective anchoring of TFIID to nucleosomes by trimethylation of histone H3 lysine 4. Cell. 2007;131(1):58–69. doi: 10.1016/j.cell.2007.08.016. [DOI] [PubMed] [Google Scholar]
- 67•.Shi Y, Matson C, et al. Histone demethylation mediated by the nuclear amine oxidase homolog LSD1. Cell. 2004;119(7):941–953. doi: 10.1016/j.cell.2004.12.012. First characterization of a histone demethylase. [DOI] [PubMed] [Google Scholar]
- 68.Sirmaci A, Spiliopoulos M, Brancati F, et al. Mutations in ANKRD11 cause KBG syndrome, characterized by intellectual disability, skeletal malformations, and macrodontia. Am J Hum Genet. 2011;89(2):289–294. doi: 10.1016/j.ajhg.2011.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wang JX, Scully K, Zhu X, et al. Opposing LSD1 complexes function in developmental gene activation and repression programmes. Nature. 2007;446(7138):882–887. doi: 10.1038/nature05671. [DOI] [PubMed] [Google Scholar]
- 70.Wang J, Hevi S, Kurash JK, et al. The lysine demethylase LSD1 (KDM1) is required for maintenance of global DNA methylation. Nat Genet. 2009;41(1):125–129. doi: 10.1038/ng.268. [DOI] [PubMed] [Google Scholar]
- 71.Whyte WA, Bilodeau S, Orlando DA, et al. Enhancer decommissioning by LSD1 during embryonic stem cell differentiation. Nature. 2012;482(7384):221–225. doi: 10.1038/nature10805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Ballas N, Battaglioli E, Atouf F, et al. Regulation of neuronal traits by a novel transcriptional complex. Neuron. 2001;31(3):353–365. doi: 10.1016/s0896-6273(01)00371-3. [DOI] [PubMed] [Google Scholar]
- 73.Andres ME, Burger C, Peral-Rubio MJ, et al. CoREST: a functional corepressor required for regulation of neural-specific gene expression. Proc Natl Acad Sci USA. 1999;96(17):9873–9878. doi: 10.1073/pnas.96.17.9873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Hakimi MA, Dong Y, Lane WS, Speicher DW, Shiekhattar R. A candidate X-linked mental retardation gene is a component of a new family of histone deacetylase-containing complexes. J Biol Chem. 2003;278(9):7234–7239. doi: 10.1074/jbc.M208992200. [DOI] [PubMed] [Google Scholar]
- 75.Zibetti C, Adamo A, Binda C, et al. Alternative splicing of the histone demethylase LSD1/KDM1 contributes to the modulation of neurite morphogenesis in the mammalian nervous system. J Neurosci. 2010;30(7):2521–2532. doi: 10.1523/JNEUROSCI.5500-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Garcia-Bassets I, Kwon YS, Telese F, et al. Histone methylation-dependent mechanisms impose ligand dependency for gene activation by nuclear receptors. Cell. 2007;128(3):505–518. doi: 10.1016/j.cell.2006.12.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77•.Iwase S, Lan F, et al. The X-linked mental retardation gene SMCX/JARID1C defines a family of histone H3 lysine 4 demethylases. Cell. 2007;128(6):1077–1088. doi: 10.1016/j.cell.2007.02.017. Discovery of the first H3K4me3 demethylase and first link between the dynamic nature of histone methylation and human cognitive development. [DOI] [PubMed] [Google Scholar]
- 78.Yamane K, Tateishi K, Klose RJ, et al. PLU-1 is an H3K4 demethylase involved in transcriptional repression and breast cancer cell proliferation. Mol Cell. 2007;25(6):801–812. doi: 10.1016/j.molcel.2007.03.001. [DOI] [PubMed] [Google Scholar]
- 79.Xiang Y, Zhu Z, Han G, et al. JARID1B is a histone H3 lysine 4 demethylase up-regulated in prostate cancer. Proc Natl Acad Sci USA. 2007;104(49):19226–19231. doi: 10.1073/pnas.0700735104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Hayakawa T, Ohtani Y, Hayakawa N, et al. RBP2 is an MRG15 complex component and down-regulates intragenic histone H3 lysine 4 methylation. Genes to Cells. 2007;12(6):811–826. doi: 10.1111/j.1365-2443.2007.01089.x. [DOI] [PubMed] [Google Scholar]
- 81.Klose RJ, Yan Q, Tothova Z, et al. The retinoblastoma binding protein RBP2 is an H3K4 demethylase. Cell. 2007;128(5):889–900. doi: 10.1016/j.cell.2007.02.013. [DOI] [PubMed] [Google Scholar]
- 82.Christensen J, Agger K, Cloos PA, et al. RBP2 belongs to a family of demethylases, specific for tri-and dimethylated lysine 4 on histone 3. Cell. 2007;128(6):1063–1076. doi: 10.1016/j.cell.2007.02.003. [DOI] [PubMed] [Google Scholar]
- 83.Tsukada Y, Fang J, Erdjument-Bromage H, et al. Histone demethylation by a family of JmjC domain-containing proteins. Nature. 2006;439(7078):811–816. doi: 10.1038/nature04433. [DOI] [PubMed] [Google Scholar]
- 84.Defeo-Jones D, Huang PS, Jones RE, et al. Cloning of cDNAs for cellular proteins that bind to the retinoblastoma gene-product. Nature. 1991;352(6332):251–254. doi: 10.1038/352251a0. [DOI] [PubMed] [Google Scholar]
- 85.Chen Z, Zang J, Whetstine J, et al. Structural insights into histone demethylation by JMJD2 family members. Cell. 2006;125(4):691–702. doi: 10.1016/j.cell.2006.04.024. [DOI] [PubMed] [Google Scholar]
- 86.Lopez-Bigas N, Kisiel TA, Dewaal DC, et al. Genome-wide analysis of the H3K4 histone demethylase RBP2 reveals a transcriptional program controlling differentiation. Mol Cell. 2008;31(4):520–530. doi: 10.1016/j.molcel.2008.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Shao GB, Chen JC, Zhang LP, et al. Dynamic patterns of histone H3 lysine 4 methyltransferases and demethylases during mouse preimplantation development. In vitro Cell Dev Biol Anim. 2014;50(7):603–613. doi: 10.1007/s11626-014-9741-6. [DOI] [PubMed] [Google Scholar]
- 88.Chicas A, Kapoor A, Wang X, et al. H3K4 demethylation by Jarid1a and Jarid1b contributes to retinoblastoma-mediated gene silencing during cellular senescence. Proc Natl Acad Sci USA. 2012;109(23):8971–8976. doi: 10.1073/pnas.1119836109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Ditacchio L, Le HD, Vollmers C, et al. Histone lysine demethylase JARID1a activates CLOCK-BMAL1 and influences the circadian clock. Science. 2011;333(6051):1881–1885. doi: 10.1126/science.1206022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Dey BK, Stalker L, Schnerch A, Bhatia M, Taylor-Papidimitriou J, Wynder C. The histone demethylase KDM5b/JARID1b plays a role in cell fate decisions by blocking terminal differentiation. Mol Cell Biol. 2008;28(17):5312–5327. doi: 10.1128/MCB.00128-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Schmitz SU, Albert M, Malatesta M, et al. Jarid1b targets genes regulating development and is involved in neural differentiation. EMBO J. 2011;30(22):4586–4600. doi: 10.1038/emboj.2011.383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Kidder BL, Hu G, Zhao K. KDM5B focuses H3K4 methylation near promoters and enhancers during embryonic stem cell self-renewal and differentiation. Genome Biol. 2014;15(2):R32. doi: 10.1186/gb-2014-15-2-r32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Catchpole S, Spencer-Dene B, Hall D, et al. PLU-1/ JARID1B/KDM5B is required for embryonic survival and contributes to cell proliferation in the mammary gland and in ER+ breast cancer cells. Int J Oncol. 2011;38(5):1267–1277. doi: 10.3892/ijo.2011.956. [DOI] [PubMed] [Google Scholar]
- 94.Scibetta AG, Santangelo S, Coleman J, et al. Functional analysis of the transcription repressor PLU-1/JARID1B. Mol Cell Biol. 2007;27(20):7220–7235. doi: 10.1128/MCB.00274-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Xie L, Pelz C, Wang W, et al. KDM5B regulates embryonic stem cell self-renewal and represses cryptic intragenic transcription. EMBO J. 2011;30(8):1473–1484. doi: 10.1038/emboj.2011.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Li X, Liu L, Yang S, et al. Histone demethylase KDM5B is a key regulator of genome stability. Proc Natl Acad Sci USA. 2014;111(19):7096–7101. doi: 10.1073/pnas.1324036111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Jensen LR, Amende M, Gurok U, et al. Mutations in the JARID1C gene, which is involved in transcriptional regulation and chromatin remodeling, cause X-linked mental retardation. Am J Hum Genet. 2005;76(2):227–236. doi: 10.1086/427563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Goncalves TF, Goncalves AP, Fintelman Rodrigues N, Dos Santos JM, Pimentel MM, Santos-Reboucas CB. KDM5C mutational screening among males with intellectual disability suggestive of X-linked inheritance and review of the literature. Eur J Med Genet. 2014;57(4):138–144. doi: 10.1016/j.ejmg.2014.02.011. [DOI] [PubMed] [Google Scholar]
- 99.Ropers HH, Hamel BC. X-linked mental retardation. Nat Rev Genet. 2005;6(1):46–57. doi: 10.1038/nrg1501. [DOI] [PubMed] [Google Scholar]
- 100.Tahiliani M, Mei PC, Fang R, et al. The histone H3K4 demethylase SMCX links REST target genes to X-linked mental retardation. Nature. 2007;447(7144):601–605. doi: 10.1038/nature05823. [DOI] [PubMed] [Google Scholar]
- 101.Tzschach A, Lenzner S, Moser B, et al. Novel JARID1C/ SMCX mutations in patients with X-linked mental retardation. Hum Mutat. 2006;27(4):389. doi: 10.1002/humu.9420. [DOI] [PubMed] [Google Scholar]
- 102.Adegbola A, Gao H, Sommer S, Browning M. A novel mutation in JARID1C/SMCX in a patient with autism spectrum disorder (ASD) Am J Med Genet A. 2008;146A(4):505–511. doi: 10.1002/ajmg.a.32142. [DOI] [PubMed] [Google Scholar]
- 103.Simensen RJ, Rogers RC, Collins JS, Abidi F, Schwartz CE, Stevenson RE. Short-term memory deficits in carrier females with KDM5C mutations. Genet Couns. 2012;23(1):31–40. [PubMed] [Google Scholar]
- 104.Agulnik AI, Mitchell MJ, Lerner JL, Woods DR, Bishop CE. A mouse Y-chromosome gene encoded by a region essential for spermatogenesis and expression of male-specific minor histocompatibility antigens. Hum Molec Genet. 1994;3(6):873–878. doi: 10.1093/hmg/3.6.873. [DOI] [PubMed] [Google Scholar]
- 105.Kent-First MG, Maffitt M, Muallem A, et al. Gene sequence and evolutionary conservation of human SMCY. Nat Genet. 1996;14(2):128–129. doi: 10.1038/ng1096-128. [DOI] [PubMed] [Google Scholar]
- 106.Xu J, Burgoyne PS, Arnold AP. Sex differences in sex chromosome gene expression in mouse brain. Hum Mol Genet. 2002;11(12):1409–1419. doi: 10.1093/hmg/11.12.1409. [DOI] [PubMed] [Google Scholar]
- 107.Aguilar-Valles A, Vaissiere T, Griggs EM, et al. Methamphetamine-associated memory is regulated by a writer and an eraser of permissive histone methylation. Biol Psychiatry. 2014;76(1):57–65. doi: 10.1016/j.biopsych.2013.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Xu J, Deng X, Disteche CM. Sex-specific expression of the X-linked histone demethylase gene Jarid1c in brain. PLoS ONE. 2008;3(7):e2553. doi: 10.1371/journal.pone.0002553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Poeta L, Fusco F, Drongitis D, et al. A regulatory path associated with X-linked intellectual disability and epilepsy links KDM5C to the polyalanine expansions in ARX. Am J Hum Genet. 2013;92(1):114–125. doi: 10.1016/j.ajhg.2012.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Outchkourov NS, Muino JM, Kaufmann K, et al. Balancing of histone H3K4 methylation states by the Kdm5c/SMCX histone demethylase modulates promoter and enhancer function. Cell Rep. 2013;3(4):1071–1079. doi: 10.1016/j.celrep.2013.02.030. [DOI] [PubMed] [Google Scholar]
- 111.Jensen LR, Bartenschlager H, Rujirabanjerd S, et al. A distinctive gene expression fingerprint in mentally retarded male patients reflects disease-causing defects in the histone demethylase KDM5C. Pathogenetics. 2010;3(1):2. doi: 10.1186/1755-8417-3-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Shaffer LG, Hecht JT, Ledbetter DH, Greenberg F. Familial interstitial deletion 11(p11.12p12) associated with parietal foramina, brachymicrocephaly, and mental retardation. Am J Med Genet. 1993;45(5):581–583. doi: 10.1002/ajmg.1320450512. [DOI] [PubMed] [Google Scholar]
- 113.Potocki L, Shaffer LG. Interstitial deletion of 11(p11.2p12): a newly described contiguous gene deletion syndrome involving the gene for hereditary multiple exostoses (EXT2) Am J Med Genet. 1996;62(3):319–325. doi: 10.1002/(SICI)1096-8628(19960329)62:3<319::AID-AJMG22>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- 114.Iwase S, Januma A, Miyamoto K, et al. Characterization of BHC80 in BRAF-HDAC complex, involved in neuron-specific gene repression. Biochem Biophys Res Commun. 2004;322(2):601–608. doi: 10.1016/j.bbrc.2004.07.163. [DOI] [PubMed] [Google Scholar]
- 115.Iwase S, Shono N, Honda A, et al. A component of BRAF-HDAC complex, BHC80, is required for neonatal survival in mice. FEBS Lett. 2006;580(13):3129–3135. doi: 10.1016/j.febslet.2006.04.065. [DOI] [PubMed] [Google Scholar]
- 116.Klajn A, Ferrai C, Stucchi L, et al. The rest repression of the neurosecretory phenotype is negatively modulated by BHC80, a protein of the BRAF/HDAC complex. J Neurosci. 2009;29(19):6296–6307. doi: 10.1523/JNEUROSCI.5943-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Hakimi MA, Bochar DA, Chenoweth J, Lane WS, Mandel G, Shiekhattar R. A core-BRAF35 complex containing histone deacetylase mediates repression of neuronal-specific genes. Proc Natl Acad Sci USA. 2002;99(11):7420–7425. doi: 10.1073/pnas.112008599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Shi YJ, Matson C, Iwase S, Baba T, Shi Y. Regulation of LSD1 histone demethylase activity by its associated factors. Mol Cell. 2005;19(6):857–864. doi: 10.1016/j.molcel.2005.08.027. [DOI] [PubMed] [Google Scholar]
- 119.Siderius LE, Hamel BCJ, Van Bokhoven H, et al. X-linked mental retardation associated with cleft lip palate maps to Xp11.3-q21.3. Am J Med Genet. 1999;85(3):216–220. [PubMed] [Google Scholar]
- 120.Koivisto AM, Ala-Mello S, Lemmela S, Komu HA, Rautio J, Jarvela I. Screening of mutations in the PHF8 gene and identification of a novel mutation in a Finnish family with XLMR and cleft lip/cleft palate. Clin Genet. 2007;72(2):145–149. doi: 10.1111/j.1399-0004.2007.00836.x. [DOI] [PubMed] [Google Scholar]
- 121.Abidi FE, Miano MG, Murray JC, Schwartz CE. A novel mutation in the PHF8 gene is associated with X-linked mental retardation with cleft lip/cleft palate. Clin Genet. 2007;72(1):19–22. doi: 10.1111/j.1399-0004.2007.00817.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Qiao Y, Liu X, Harvard C, et al. Autism-associated familial microdeletion of Xp11.22. Clin Genet. 2008;74(2):134–144. doi: 10.1111/j.1399-0004.2008.01028.x. [DOI] [PubMed] [Google Scholar]
- 123.Qi HH, Sarkissian M, Hu GQ, et al. Histone H4K20/H3K9 demethylase PHF8 regulates zebrafish brain and craniofacial development. Nature. 2010;466(7305):503–507. doi: 10.1038/nature09261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Loenarz C, Ge W, Coleman ML, et al. PHF8, a gene associated with cleft lip/palate and mental retardation, encodes for an Nepsilon-dimethyl lysine demethylase. Hum Mol Genet. 2010;19(2):217–222. doi: 10.1093/hmg/ddp480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Fortschegger K, De Graaf P, Outchkourov NS, Van Schaik FM, Timmers HT, Shiekhattar R. PHF8 targets histone methylation and RNA polymerase II to activate transcription. Mol Cell Biol. 2010;30(13):3286–3298. doi: 10.1128/MCB.01520-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Feng WJ, Yonezawa M, Ye J, Jenuwein T, Grummt I. PHF8 activates transcription of rRNA genes through H3K4me3 binding and H3K9me1/2 demethylation. Nat Struct Mol Biol. 2010;17(4):445–U483. doi: 10.1038/nsmb.1778. [DOI] [PubMed] [Google Scholar]
- 127.Kleine-Kohlbrecher D, Christensen J, Vandamme J, et al. A functional link between the histone demethylase PHF8 and the transcription factor ZNF711 in X-linked mental retardation. Mol Cell. 2010;38(2):165–178. doi: 10.1016/j.molcel.2010.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Qiu JH, Shi GA, Jia YH, et al. The X-linked mental retardation gene PHF8 is a histone demethylase involved in neuronal differentiation. Cell Res. 2010;20(8):908–918. doi: 10.1038/cr.2010.81. [DOI] [PubMed] [Google Scholar]
- 129.Horton JR, Upadhyay AK, Qi HH, Zhang X, Shi Y, Cheng XD. Enzymatic and structural insights for substrate specificity of a family of jumonji histone lysine demethylases. Nat Struct Mol Biol. 2010;17(1):U38–U52. doi: 10.1038/nsmb.1753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Asensio-Juan E, Gallego C, Martinez-Balbas MA. The histone demethylase PHF8 is essential for cytoskeleton dynamics. Nucleic Acids Res. 2012;40(19):9429–9440. doi: 10.1093/nar/gks716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Lupo G, Harris WA, Lewis KE. Mechanisms of ventral patterning in the vertebrate nervous system. Nat Rev Neurosci. 2006;7(2):103–114. doi: 10.1038/nrn1843. [DOI] [PubMed] [Google Scholar]
- 132.Cau E, Blader P. Notch activity in the nervous system: to switch or not switch? Neural Dev. 2009;4:36. doi: 10.1186/1749-8104-4-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Tsai MC, Manor O, Wan Y, et al. Long noncoding RNA as modular scaffold of histone modification complexes. Science. 2010;329(5992):689–693. doi: 10.1126/science.1192002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Wang KC, Yang YW, Liu B, et al. A long noncoding RNA maintains active chromatin to coordinate homeotic gene expression. Nature. 2011;472(7341):120–124. doi: 10.1038/nature09819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Yang YW, Flynn RA, Chen Y, et al. Essential role of lncRNA binding for WDR5 maintenance of active chromatin and embryonic stem cell pluripotency. Elife. 2014;3:e02046. doi: 10.7554/eLife.02046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Shulha HP, Crisci JL, Reshetov D, et al. Human-specific histone methylation signatures at transcription start sites in prefrontal neurons. PLoS Biol. 2012;10(11):e1001427. doi: 10.1371/journal.pbio.1001427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Bell CG, Wilson GA, Beck S. Human-specific CpG ‘beacons’ identify human-specific prefrontal cortex H3K4me3 chromatin peaks. Epigenomics. 2014;6(1):21–31. doi: 10.2217/epi.13.74. [DOI] [PubMed] [Google Scholar]
- 138.Gupta S, Kim SY, Artis S, et al. Histone methylation regulates memory formation. J Neurosci. 2010;30(10):3589–3599. doi: 10.1523/JNEUROSCI.3732-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Kerimoglu C, Agis-Balboa RC, Kranz A, et al. Histone-methyltransferase MLL2 (KMT2B) is required for memory formation in mice. J Neurosci. 2013;33(8):3452–3464. doi: 10.1523/JNEUROSCI.3356-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Cao F, Townsend EC, Karatas H, et al. Targeting MLL1 H3K4 methyltransferase activity in mixed-lineage leukemia. Mol Cell. 2014;53(2):247–261. doi: 10.1016/j.molcel.2013.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Grembecka J, He S, Shi A, et al. Menin-MLL inhibitors reverse oncogenic activity of MLL fusion proteins in leukemia. Nat Chem Biol. 2012;8(3):277–284. doi: 10.1038/nchembio.773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Helin K, Dhanak D. Chromatin proteins and modifications as drug targets. Nature. 2013;502(7472):480–488. doi: 10.1038/nature12751. [DOI] [PubMed] [Google Scholar]