

Abstract
Kidney urea transporters are targets for development of small-molecule inhibitors with action as salt-sparing diuretics. A cell-based, functional high-throughput screen identified 2,7-bisacetamido fluorenone 3 as a novel inhibitor of urea transporters UT-A1 and UT-B. Here, we synthesized twenty-two 2,7-disubstituted fluorenone analogs by acylation. Structure-activity relationship analysis revealed: (a) the carbonyl moiety at C9 is required for UT inhibition; (b) steric limitation on C2, 7-substituents; and (c) the importance of a crescent-shape structure. The most potent fluorenones inhibited UT-A1 and UT-B urea transport with IC50 ~ 1 μM. Analysis of in vitro metabolic stability in hepatic microsomes indicated metabolism of 2,7-disubstituted fluorenones by reductase and subsequent elimination. Computational docking to a homology model of UT-A1 suggested UT inhibitor binding to the UT cytoplasmic domain at a site that does not overlap with the putative urea binding site.
The kidney expresses urea transporter (UT) proteins, which facilitate the passive transport of urea across cell plasma membranes in a subset of kidney tubules and microvessels.1 SLc14A1 and SLc14A2 genes encode UT-A and UT-B urea transporters, respectively.2 Studies in mice lacking UTs3–7 and in rodents treated with UT inhibitors8–10 indicate that UT-A1, the UT-A isoform expressed at the apical membrane of epithelial cells in inner medullary collecting duct, is the principal target for diuretic development. Absence or inhibition of UTs impairs urinary concentrating function, producing a diuretic response. UT inhibitors are thus development candidates as first-in-class salt-sparing diuretics for therapy of various edema states and hyponatremias, such as those associated with congestive heart failure and cirrhosis.11–13
Our lab previously developed high-throughput functional assays of UT-A14 and UT-B15 urea transporters. Several classes of small-molecule inhibitors of the target UT-A1 were identified.10, 14 In proof-of-concept studies, two classes of inhibitors with low micromolar IC50 produced a diuretic response in rats;10 however, their inhibition potency and metabolic stability were not optimal for further development. Additional screening reported here identified symmetrical, disubstitutedfluorenones as novel UT inhibitors. Because of the drug-like properties of tricyclicfluorenones and the absence of a commercial source to obtain analogs for structure-activity relationship analysis, here we synthesized 22 symmetrical, disubstitutedfluorenones, measured their UT inhibition activity and selectivity, analyzed their inhibition and metabolism mechanisms, and used homology modeling and computational docking to propose binding sites on UT-A and UT-B.
A UT-A1 inhibition screen of 50,000 compounds identified 2,7-bisacetamidofluorenone 3 as a UT-A1 inhibitor with IC50 ~1 μM that produced complete inhibition at higher concentrations (Fig. 1A). Fluorenone3 also inhibited UT-B with similar potency. The fluorenone scaffold has not been previously reported for the inhibition of urea transporters, though there are prior reports of biological activities of this compound class. Tiloron is an orally bioavailable antiviral agent16, 17 and an immunomodulator.18 The antitumor activity of fluorenone derivatives has been shown to be result from inhibition of telomerase and DNA topoisomerase I.19–21 Most reported fluorenone analogs focused on 2,7-bis-ester or ether moieties, unlike the bis-acetamidofluorenone3 identified from the UT-A1 screen. The 2,7-bis-acetamido fluorenone structure has drug-like properties, including favorable molecular weight (294 Da), topological surface area (75.2 Å2) and cLogP (2.12), which fall within the Lipinski22 and Veber23 criteria for orally bioavailable drugs.
Fig 1.

Discovery of 2,7-disubstituted fluorenone 3 as UT-A1 inhibitor. A. Structure of 3 and concentration-inhibition data for inhibition of rat UT-A1 and UT-B urea transporters. Fitted parameters: IC50 1uM and 1.5uM, Hill coefficient 0.9 and 1.1, for UT-A1 and UT-B respectively. B. Strategy for analogs synthesis for SAR analysis.
Based on the potency and physicochemical properties of 3, a series of 2,7-disubstituted fluorenone analogs were rationally designed to identify more potent urea transport inhibitors and to establish structure-activity relationships. Structurally, fluorenone 3 is a symmetrical, rigid crescent-shaped molecule with a carbonyl group at the 9-position and bisacteamido groups at the 2 and 7 positions.
As diagrammed in Fig. 1B, analogs were designed to include: i) different functional groups on the 2,7-diamino position; ii) different non-carbonyl functional groups at the 9-position; and iii) flexible and ring strain-released scaffolds. In a preliminary study, testing of ~70 commercially available fluorenone analogs did not identify active analogs.
Scheme 1 shows the synthetic approaches for the preparation of 2,7-bis(alkylamido)fluorenones 3–10. Reduction of commercially available 2,7-dinitro-fluoren-9-one 1 using sodium sulfide nonahydrate and sodium hydroxide afforded the key intermediate 2,7-diamino-fluoren-9-one 2.19 The re-synthesis of 3 was accomplished by acetylation of 2 using acetic anhydride. Additional acyl analogs of 3 were similarly prepared by reaction of 2 with the respective acyl reagents under basic condition to yield propionyl, isobutyryl and butyryl analogs 4, 5 and 6 respectively. We next prepared the bio-isostere analogs of 3, trifluoroacetamide 7, carbamate 8 and methanesulfonamide 9. Treatment of 2 with trifluoroacetic acid and heating to reflux in a sealed tube afforded trifluoroacetamide 7.24 The carbamate analog 8 was synthesized by reaction of 2 with potassium isocyanate.25 Sulfonamide analog 9 was prepared by mesylation in pyridine. The γ-sultam 10 was synthesized using chloropropansulfonyl chloride and subsequent cyclization with potassium carbonate. As shown in Scheme 2, analogs with extended chains on the amide bond were synthesized. Bromoacetamido 11 and benzyloxyacetamido 15 were synthesized from 2 using corresponding acyl halides in refluxing xylene. Using maleic anhydride in refluxing chloroform gave the maleamic acid analog 16. Further substitution reaction of 11 using methoxyethanol with sodium hydride afforded 13, and with sodium hydroxide and sodium azide gave the corresponding amide analogs 12 and 14, respectively.
Scheme 1.

Synthesis of 2,7-bisalkylamidofluorenone and 2,7-bissufoneamidofluorenone analogs.
Reaction conditions and reagents: (a) Na2S-9H2O, NaOH, EtOH, 78%; (b) Et3N, THF, Ac2O for 3, propionyl chloride for 4; isobutyryl chloride for 5; butyryl chloride for 6; (c) CF3CO2H in a sealed tube for 7; (d) KNCO, AcOH/H2O for 8; (e) MsCI, pyridine, THF for 9; (f) 3-chloropropansulfonyl chloride, pyridine, CH2CI2; then, K2CO3, CH3CN, reflux, for 10.
Scheme 2.

Synthesis of extended 2,7-bisacetylamidofluorenone analogs. Reaction conditions and reagents: (a) bromoacetyl bromide, xylene, reflux, 40%; (b) benzyloxyacetyl chloride, xylene, reflux, 26% for 15; (c) maleic anhydride, CHCl3, 26% for 16; (d) NaOH, EtOH, 86% for 12; (e) methoxyethanol, NaH, CH3CN, 18% for 13; (f) NaN3, DMF, 24% for 14.
To investigate the importance of the carbonyl at the 9-position for urea transport inhibition, we synthesized oxime 17, hydroxyl 18 and sulfone 20 (Scheme 3). Condensation of 2,7-bisacetamido fluoren-9-one 3 with hydroxylamine hydrochloride in pyridine afforded oxime analog 17. Ketone reduction of 3 with sodium borohydride in methanol gave hydroxyl analog 18. Acylation of commercially available 2,7-diaminodiphenylenesulfone 19 afforded the bisacetamidodiphenylsulfone 20. To prepare flexible ring acyclic analogs, commercially available 3,3′-diaminobenzophenone 21 was acylated and sulfonated to give bisacetamide 22, benzyloxyacetamide 23 and bismethanesulfonamidobenzophenone 24.
Scheme 3.

Synthesis of carbonyl-modified fluorenone analogs. Reaction conditions and reagents: (a) NH2OH-HCI, DMSO/H2O, 22%; (b) NaBH4, CH3CN/MeOH (10/1), 28%; (c) Et3N, THF, Ac2O, 65%; (d) Et3N, THF, Ac2O, 62% for 22; Benzyloxyacetyl chloride, xylene, reflux, 46% for 23; (e) MsCI, pyridine, THF, 23%.
Table 1 summarizes IC50 values for inhibition of UT-A1 and UT-B urea transport by the 2,7-disubstituted fluorenones 1–24. Most analogs showed similar IC50 for inhibition of UT-A1 and UT-B urea transport. Non-substituted fluorenones (1, 2 and 21) were inactive. Elongation of the carbon chain on the amide reduced inhibition, comparing acetamide 3 to propionyl 4, isobutyryl 5, maleamic acid 16 and butyryl 6. Linear ether linkage analogs with elongated side chain also reduced inhibition. Hydroxyacetamide 12 had low activity, and benzyloxyacetamides 15 and 13 were inactive. Replacing acetamide with methanesulfonylamide9 and carbamate8 slightly reduced activity. Addition to the acetamide of one more atom - hydroxyl 12, methyl 4 or bromide 11 - reduced activity. These findings implicate a narrow size limitation for substituents. Interestingly, trifluoroacetamide7, which has the same carbon length as 3, was much less active, indicating that steric and electronic effects also influence inhibition.
Table 1.
Inhibition of UT-A1 and UT-B urea transport by 2,7-disubstituted fluorenoneanalogs.
| Structure | Functional group | UT- A1 IC50 (μM) | UT-B IC50 (μM) |
|---|---|---|---|
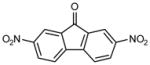
|
1 | >50 | >50 |
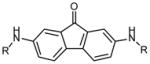
|
R | ||
| 2 H | >50 | >50 | |
| 3 COCH3 | 1 | 1.5 | |
| 4 COCH2CH3 | 15 | 5 | |
| 5 COCH(CH3) 2 | 18 | 50 | |
| 6 CO(CH2) 2CH3 | >50 | >50 | |
| 7 COCF3 | 10 | 7.5 | |
| 8 CONH2 | 2 | 1.2 | |
| 9 SO2CH3 | 1.5 | 2.5 | |
| 10 SO2(CH2)3 | >50 | >50 | |
| 11 COCH2Br | 30 | 10 | |
| 12 COCH2OH | 20 | 25 | |
| 13 COCH2O(CH2)2OMe | >50 | 50 | |
| 14 COCH2N3 | 20 | 30 | |
| 15 COCH2OBn | >50 | >50 | |
| 16 CO(CH) 2CO2H | 30 | >50 | |
|
|
X | ||
| 17 CNHOH | 12 | 25 | |
| 18 CHOH | >50 | 20 | |
| 20 SO2 | 15 | 2 | |

|
R | ||
| 21 H | >50 | >50 | |
| 22 COCH3 | 18 | 20 | |
| 23 COCH2OBn | >50 | >50 | |
| 24 SO2CH3 | >50 | 30 |
The influence of the carbonyl moiety was determined by comparing UT-A1 inhibition by 3, 17, 18 and 20. These analogs contain acetamide on the C2 and C7 positions, but activity was reduced greatly by replacing the carbonyl with oxime 17, sulfone 20 or hydroxyl 18 moieties (IC50 12 to >50 μM), suggesting that a hydrogen bond acceptor is necessary on the carbonyl. Interestingly, sulfone 20 showed UT-B selectivity with IC50 of 2 μM for UT-B compared to 15 μM for UT-A1.
Fig. 2A shows concentration-inhibition data for UT-A1 and UT-B inhibition by 3 and analogs 7, 9 and 22. Fig. 2B shows original concentration-inhibition curves for 3 (left panel). The in vitro characterization of 3 revealed complete reversibility for inhibition of UT-A1 (second panel), rapid inhibition kinetics with complete inhibition by 40 seconds (third panel), and a non-competitive inhibition mechanism in which apparent IC50 was independent of urea concentration (right panel).
Fig. 2.
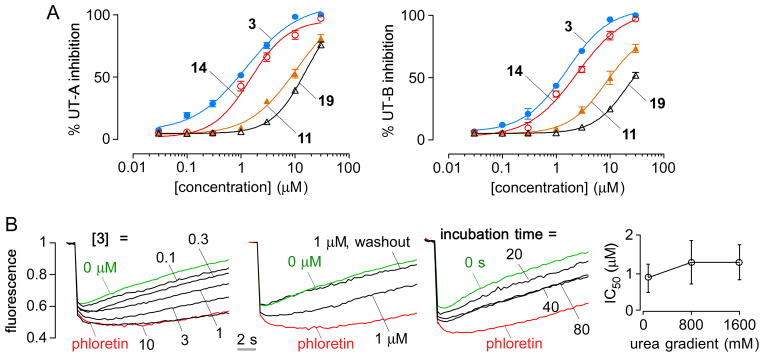
Urea transport inhibition by 2,7-disubstituted fluorenones. A. Concentration-inhibition data for UT-A1 and UT-B for 3, 11, 14 and 19. B. Primary data from UT-A inhibition assay showing reversibility, inhibition kinetics and urea competition for 3. Phloretin concentration was 0.35 mM.
To investigate the potential utility of disubstitutedfluorenones for UT inhibition in animal studies, in vitro microsomal stability measurements were made using an established assay in which compounds were incubated with rat liver microsomes in the presence of NADPH.26 For 3, 60% remained after 15 min (Fig. 3A). Oxidation catalyzed by the cytochrome P450 monooxygenase, a well-known metabolic reaction on aromatic compounds, was not detected from LC/MS analysis. Instead, a peak at [M+H-16] was detected that increased over time (Fig. 3B). Generation of a [M+H-16] metabolite suggests loss of oxygen, suggesting that bisacetamidofluorenone metabolism involves the mechanism proposed in Fig. 4. Carbonyl reduction of 2,7-dinitrofluorenone by rat liver enzymes utilizing NADH/NADPH has been reported, although the specific carbonyl reduction enzyme was not established.27 Following reduction of carbonyl on fluorenone, the amide proton is readily removed by base-catalyzed elimination to give 2-iminofluorene 25. To support this proposed mechanism, fluorenol 18 was subjected to basic condition (1M NaOH). The eliminated product 25 could be isolated after 1 hour in ~60% yield, and matched [M+H-16].28 Also, to confirm this proposed mechanism, metabolic stability was studied for oxime 17, bisacetaminobiphenylacetone 22 and bisacetaminosulfone 20, which do not contain a carbonyl moiety and thus cannot undergo carbonyl reduction/base-catalyzed elimination sequence. We found that 17, 20 and 22 were relatively stable compared to fluorenone 3 (>80% remaining at 15 min), and [M+H-16] was not detected by LC/MS (data not shown).
Fig. 3.

In vitro metabolic stability of disubstituted fluorenones. Compounds incubated with hepatic microsomes in the presence of NADPH. A. LC/MS showing disappearance of 3 and appearance of metabolite over 60 min. B. Kinetics of disappearance of 3, 17, 20, and 22 over 60 min.
Fig. 4.
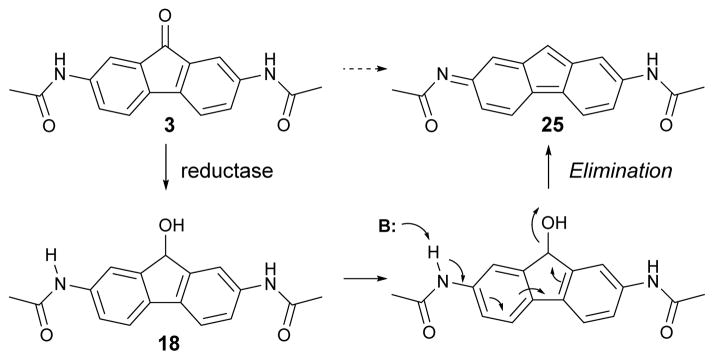
Proposed mechanism of hepatic metabolism of 3.
Interestingly, acetophenone analog 22 had less UT inhibition activity than 3 and sulfonamide 24 was inactive compared to corresponding fluorenone analog 9, which showed strong inhibition. This result suggests that the 3D structure of the fluorenone core is important to UT inhibition. Fluorenones with 2,7-disubstituents have a crescent-like shape, which is lost when substituted with biphenylacetone. Increasing flexiblility of the acetophenone moiety reduced UT inhibition activity.
Computational docking simulations were done to propose binding modes for the most potent UT-A1 inhibitor 3, and the most potent UT-B inhibitor 8. Structures for rat UT-A1 and UT-B were generated by homology modeling as described,10 based on the high resolution X-ray crystal structure of bovine UT-B solved at 2.5Å.29 Our homology models of these two proteins contain sites that appear homologous to narrow constriction (Sm) and low energy urea binding sites (So and Si) observed in the bovine UT-B template structure.
Fig. 5A shows docked conformations of inhibitor 3 bound to UT-A1 and UT-B. The lowest energy conformation of this molecule docked into both proteins in similar orientations. The planar inhibitor scaffold docked into the outer part of the UT-A1 and UT-B pore region, not binding deeply into the channel. In the docked conformation of 3 with UT-A1 the amido NH2 group projects into a pocket surrounded by Glu572, Val603, Asn604, Phe832 and Tyr900. The bis-urea inhibitor 8, which shows a greater potency for UT-B, is shown for comparison (Fig. 5B), and docks in a similar manner as 3. Other active analogs of 3 also docked similarly. The position of the inhibitor scaffolds do not overlap the putative cytoplasmic urea binding site (Si) near Gln599, which is homologous to Gln63 in the selenourea-bound template structure.28 Taken together with urea competition experiments for 3, the proposed binding site in Fig. 5 is consistent with a non-competitive inhibition mechanism.
Fig. 5.
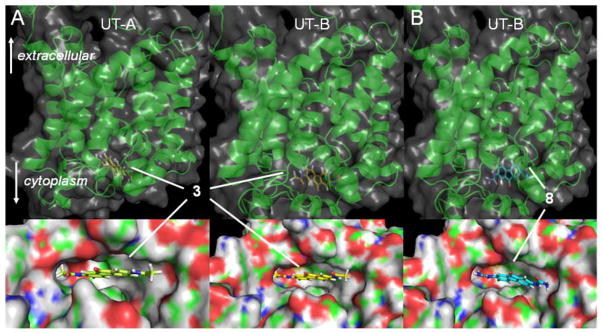
Computational docking of 3 and 8 to homology models of rat UT-A and UT-B. Zoomed -in and zoomed-out representations of (A) inhibitor 3 bound to rat UT-A and UT-B cytoplasmic domains; and (B) the most potent UT-B inhibitor 8 bound to the rat UT-B cytoplasmic domain.
Conclusions
In conclusion, we identified by high-throughput screening symmetrical 2,7-disubstituted fluorenones as novel UT inhibitors, and established structure-activity relationships by synthesis and characterization of 22 analogs. Functional studies indicated reversible inhibition of UT-A1 urea transport by the 2,7-disubstituted fluorenone analogs by a non-competitive inhibition mechanism. Docking computations suggested inhibitor binding at the UT outer pore regions at a site distinct from the putative urea binding site. Finally, analysis of inhibitor metabolism indicated carbonyl reduction by reductase and subsequent base-catalyzed elimination.
Experimentals
Synthesis
All chemical synthetic procedures and characterizations are described in supplementary information.
Cell culture
Triply transfected MDCK cells expressing rat UT-A1, yellow fluorescent protein (YFP)-H148Q/V163S and human aquaporin-1 (AQP1) were grown in Dulbecco’s modified Eagle medium, 10% FBS and three selection antibiotics (zeocin, geneticin and hygromycin B), as described.14
UT-A1 inhibition assay
MDCK-UT-A1-AQP1-YFP cells were used for UT-A1 inhibition assay as described.14 Briefly, after incubation for 15 min with compounds the cells were subjected to a 800 mM urea gradient and cellular YFP fluorescence was continuously measured with a plate reader (model Infinite M1000, Tecan Trading AG, Switzerland). UT-A1 inhibition alters the profile of the curve, increasing the initial shrinkage (decreased fluorescence) and slowing reswelling (increased fluorescence). Percentage UT-A1 inhibition was computed as 100% (Fneg − Ftest)/(Fneg − Fpos), where F is fluorescence measured 7 s after urea injection for the negative control (Fneg), test compound (Ftest) and positive control (Fpos).
UT-B inhibition assay
As described,15 whole rat blood was diluted to a hematocrit of ~1.5% in PBS containing 1.25 M acetamide. Erythrocyte suspensions were incubated for 15 min with test compounds and then rapidly mixed with PBS. Percentage lysis was quantified from absorbance at 710 nm as: 100% (Aneg − Atest)/(Aneg − Apos), where A is absorbance for the negative control (Aneg), test compound (Atest) and positive control (Apos) at 710 nm.
Functional studies
Reversibility was tested by incubation of inhibitors at a concentration near their IC50 and then washing with PBS before UT-A1 inhibition assay. Competition with urea was studied using different concentrations of urea(80 to 1,600 mM) in the UT-A1 inhibition assay.
Supplementary Material
Acknowledgments
Supported by grants DK101373, DK35124, DK72517, EB00415 and EY13574 from the National Institutes of Health. The authors acknowledge OpenEye Scientific (Santa Fe, NM, USA) for its Academic Site License program.
Footnotes
Electronic Supplementary Information (ESI) available: [details of any supplementary information available should be included here]. See DOI: 10.1039/x0xx00000x
Notes and references
- 1.Klein JD, Blount MA, Sands JM. Urea transport in the kidney. Compr Physiol. 2011;2:699–729. doi: 10.1002/cphy.c100030. [DOI] [PubMed] [Google Scholar]
- 2.Smith CP. Mammalian urea transporters. Exp Physiol. 2009;94:180–185. doi: 10.1113/expphysiol.2008.043042. [DOI] [PubMed] [Google Scholar]
- 3.Fenton RA, Chou CL, Stewart GS, Smith CP, Knepper MA. Urinary concentrating defect in mice with selective deletion of phloretin-sensitive urea transporters in the renal collecting duct. Proc Natl Acad Sci. 2004;101:7469–7474. doi: 10.1073/pnas.0401704101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fenton RA, Flynn A, Shodeinde A, Smith CP, Schnermann J, Knepper MA. Renal phenotype of UT-A urea transporter knockout mice. J Am Soc Nephrol. 2005;16:1583–1592. doi: 10.1681/ASN.2005010031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Uchida S, Sohara E, Rai T, Ikawa M, Okabe M, Sasaki S. Impaired urea accumulation in the inner medulla of mice lacking the urea transporter UT-A2. Mol Cell Biol. 2005;25:7357–7363. doi: 10.1128/MCB.25.16.7357-7363.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Klein JD, Frohlich O, Mistry AC, Kent KJ, Martin CF, Sands JM. Transgenic mice expressing UT-A1, but lacking UT-A3, have intact urine concentration ability. FASEB J. 2013;27:1111–1117. [Google Scholar]
- 7.Yang B, Bankir L, Gillespie A, Epstein CJ, Verkman AS. Urea-selective concentrating defect in transgenic mice lacking urea transporter UT-B. J Biol Chem. 2002;277:10633–10637. doi: 10.1074/jbc.M200207200. [DOI] [PubMed] [Google Scholar]
- 8.Yao C, Anderson MO, Zhang J, Yang B, Phuan PW, Verkman AS. Triazolothieno-pyrimidine inhibitors of urea transporter UT-B reduce urine concentration. J Am Soc Nephrol. 2012;23:1210–1220. doi: 10.1681/ASN.2011070751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li F, Lei T, Zhu J, Wang W, Sun Y, Chen J, Dong Z, Zhou H, Yang B. A novel small-molecule thienoquinolin urea transporter inhibitor acts as a potential diuretic. Kidney Int. 2013;83:1076–1086. doi: 10.1038/ki.2013.62. [DOI] [PubMed] [Google Scholar]
- 10.Esteva-Font C, Cil O, Phuan PW, Su T, Lee S, Anderson MO, Verkman AS. Diuresis and reduced urinary osmolality in rats produced by small-molecule UT-A-selective urea transport inhibitors. FASEB J. 2014;28:3878–3890. doi: 10.1096/fj.14-253872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Knepper MA, Miranda CA. Urea channel inhibitors: a new functional class of aquaretics. Kidney Int. 2013;83:991–993. doi: 10.1038/ki.2013.94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sands JM. Renal urea transporters. Curr Opin Nephrol Hypertens. 2004;13:525–532. doi: 10.1097/00041552-200409000-00008. [DOI] [PubMed] [Google Scholar]
- 13.Esteva-Font C, Anderson MO, Verkman AS. Urea transport proteins as drug targets. Nat Rev Nephrol. 2015;11:113–123. doi: 10.1038/nrneph.2014.219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Esteva-Font C, Phuan PW, Anderson MO, Verkman AS. A small molecule screen identifies selective inhibitors of urea transporter UT-A. Chem Biol. 2013;20:1235–1244. doi: 10.1016/j.chembiol.2013.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Levin MH, De la Fuente R, Verkman AS. Urearetics: a small molecule screen yields nanomolar potency inhibitors of urea transporter UT-B. FASEB J. 2007;21:551–563. doi: 10.1096/fj.06-6979com. [DOI] [PubMed] [Google Scholar]
- 16.Mayer GD, Krueger RF. Tilorone hydrochloride: mode of action. Science. 1970;169:1214–1215. doi: 10.1126/science.169.3951.1214. [DOI] [PubMed] [Google Scholar]
- 17.Krueger RF, Mayer GD. Tilorone hydrochloride: an orally active antiviral agent. Science. 1970;169:1213–1214. doi: 10.1126/science.169.3951.1213. [DOI] [PubMed] [Google Scholar]
- 18.Svensson CK, Knowlton PW. Effect of the immunomodulatortilorone on the in vivo acetylation of procainamide in the rat. Pharm Res. 1989;6:477–480. doi: 10.1023/a:1015964306318. [DOI] [PubMed] [Google Scholar]
- 19.Perry PJ, Read MA, Davies RT, Gowan SM, Reszka AP, Wood AA, Kelland LR, Neidle S. 2,7-Disubstituted Amidofluorenone Derivatives as Inhibitors of Human Telomerase. J Med Chem. 1999;42:2679–2684. doi: 10.1021/jm990084q. [DOI] [PubMed] [Google Scholar]
- 20.Lee CC, Chang DM, Huang KF, Chen CL, Chen TC, Lo Y, Guh JH, Huang HS. Design, synthesis and antiproliferative evaluation of fluorenone analogs with DNA topoisomerase I inhibitory properties. Bioorg Med Chem. 2013;21:7125–7133. doi: 10.1016/j.bmc.2013.09.006. [DOI] [PubMed] [Google Scholar]
- 21.Zhou D, Tuo W, Hu H, Xu J, Chen H, Rao Z, Xiao Y, Hua X, Liu P. Synthesis and activity evaluation of tilorone analogs as potential anticancer agents. Eur J Med Chem. 2013;64:432–441. doi: 10.1016/j.ejmech.2013.03.050. [DOI] [PubMed] [Google Scholar]
- 22.Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Deliv Rev. 2001;46:3–26. doi: 10.1016/s0169-409x(00)00129-0. [DOI] [PubMed] [Google Scholar]
- 23.Veber DF, Johnson SR, Cheng HY, Smith BR, Ward KW, Kopple KD. Molecular properties that influence the oral bioavailability of drug Candidates. J Med Chem. 2002;45:2615–2623. doi: 10.1021/jm020017n. [DOI] [PubMed] [Google Scholar]
- 24.Fletcher TL, Taylor ME, Dahl AW. Derivatives of fluorine. I. N-Substituted 2-aminofluorene and 2-aminofluorenone. J Org Chem. 1955;20:1021–1025. [Google Scholar]
- 25.Perry CJ, Holding K, Tyrrell E. Simple, novel synthesis for 1-carbamoyl-1H-benzotriazole and some of its analogs. Syn Commun. 2008;38:3354–3365. [Google Scholar]
- 26.Snyder DS, Tradtrantip L, Yao C, Kurth MJ, Verkman AS. Potent, metabolically stable benzopyrimido-pyrrolo-oxazine-dione (BPO) CFTR inhibitors for polycystic kidney disease. J Med Chem. 2011;54:5468–5477. doi: 10.1021/jm200505e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ritter CL, Decker RW, Malejka-Giganti D. Reduction of nitro and 9-oxo groups of environmental nitrofluorenes by the rat mammary gland in vitro. Chem Res Toxicol. 2000;13:793–800. doi: 10.1021/tx0000567. [DOI] [PubMed] [Google Scholar]
- 28.Compound 25: 1H NMR (DMSO-d6) δ 10.13 (brs, 1H), 7.78 (brs, 1H), 7.57 (d, 1H, J = 8.0 Hz), 7.37 (d, 1H, J = 7.8 Hz), 7.28 (d, 1H, J = 8.0 Hz), 6.79 (brs, 1H), 6.66 (d, 1H, J = 8.1 Hz), 5.57 (brs, 1H), 2.55 (s, 3H), 2.05 (s, 3H); LRMS (ESI) m/z 279 (M+H)+.
- 29.Levin EJ, Cao Y, Enkavi G, Quick M, Pan Y, Tajkhorshid E, Zhou M. Structure and permeation mechanism of a mammalian urea transporter. Proc Natl Acad Sci. 2012;109:11194–11199. doi: 10.1073/pnas.1207362109. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.