

Abstract
Purpose of review
Despite the application of prophylactic antimicrobial therapy and advanced technologies, infection remains one of the most common causes of morbidity and mortality in surgical patients. Understanding the pathogenesis of surgical infection would offer new insights into the development of biomarkers to predict and stratify infection in patients, and to explore specific strategies to minimize this serious postoperative complication.
Recent findings
The acute nonspecific inflammatory response triggered by endogenous danger signals evoked by surgical insult is beneficial, while paradoxically associated with reduced resistance to infection. There is growing evidence indicating that primed inflammation by surgical insult exaggerates the dysregulation of the immune-inflammatory response to the invasion of pathogens postoperatively. Innate immune receptors, such as Toll-like receptors (TLRs), contribute to detecting both pathogen-associated molecular patterns and endogenous damage-associated molecular patterns, and to further amplifying inflammatory responses to infection. Current evidence shows the fascinating role of non-TLRs in the process of infection. Non-TLRs, such as membrane-associated triggering receptor expressed on myeloid cells family, cytosolic nucleotide-binding oligomerization domain-like receptors and nuclear receptor nuclear family 4 subgroup A receptors, are also crucial in triggering the immune responses and mounting an effective defense against surgical insults and the second hit of infection.
Summary
Understanding the pivotal role of non-TLRs in sensing exogenous and endogenous molecules, and the influence of primed systemic inflammation and depressed immune status on the defense against pathogen after surgical insult, would be helpful to fully explore the relevant sophisticated phenomena of surgical infection, and to elucidate the occurrence of heterogeneous constellations of clinical signs and symptoms among this special population.
Keywords: inflammation, non-Toll-like receptor, pathogenesis, surgical infection
INTRODUCTION
It is estimated that approximately 234.2 million major surgical operations are undertaken in the global healthcare annually [1]. The inpatient surgical complications, ranging from 3 to 17.4%, have substantial adverse influence on clinical processes and outcomes, dramatically prolong hospitalization and increase medical care cost [2–4]. The most frequent types of surgical complications include infection, postoperative bleeding, pulmonary embolism, deep vein thrombosis, stroke and cardiovascular events [5]. It was reported that around 11.9% of surgical patients experienced a postoperative infection episode, with an in-hospital mortality of 14.5%, which is hugely different to anesthesia-attributed mortality (34 per million) and total perioperative mortality (0.8%) [6–8]. Medical and surgical patients represent two different populations, and infection has a greater impact on the mortality in the surgical sets [9]. As the leading cause of morbidity and mortality in patients who underwent surgery, infections were shown to increase hospital costs by an average of $1398 per capita compared with those without in US hospitals [10]. The pathophysiology of surgical infection is a complex process, conducted by the primed and pretriggered host immune-inflammatory response to pathogen, predisposed by genetic factors and tailored by the location, the load and the virulence of the invading microbes in surgical patients [11–13]. Furthermore, increasing age, underlying illnesses such as diabetes mellitus, type of invasive procedure, prolonged duration of the surgical manipulation, ischemia and reperfusion, and transfusion might make the patients more susceptible to infection [14,15]. Despite numerous strategies oriented to defense against pathogens, surgical infection remains a challenging issue [16]. Better understanding of pathogenesis would be important for the improvement of infection outcomes following surgery.
Box 1.

no caption available
PRIMED INFLAMMATION AND IMMUNE SUPPRESSION
Surgical procedures evoke the innate immune system, and a systemic inflammatory response syndrome is usually initiated within hours after the surgical injury. This acute nonspecific response is a sterile inflammatory response to tissue damage and blood loss, and is triggered by endogenous danger signals massively released from the damaged tissues [11,12,17]. It has been proven that most danger-associated molecular patterns (DAMPs) and alarmins can be mobilized from the injured tissues or cells into circulation by operative insult [18–20]. These DAMPs and alarmins include heat shock proteins, reactive oxygen species, high mobility group box 1, as well as mitochondrial DAMPs which are evolutionarily conserved patterns in pathogens. The DAMPs interact with various cell-surfaces and intracellular receptors in immune effector cells to activate these cells and then lead to overwhelming inflammatory processes, which include inducing neutrophils and macrophages to migrate across damaged endothelial cells into the injured sites to produce proinflammatory mediators [17,21,22].
The primed inflammation by surgical insults like trauma is initially beneficial as it helps to eradicate tissue debris. However, if not balanced by homeostastic anti-inflammatory mechanisms, it is detrimental to the integrity and repair of tissue in surgical patients, and might even elicit an overt depressed immune response due to extensive death of immune effector cells [12,23▪]. Usually, the increased nonspecific inflammatory response in the early phase of surgical hit is accompanied by suppression of surgical patients’ ability to mount an effective defense against invading microbes. Previous clinical data indicate that the expression of major histocompatability complex class II antigens on monocytes is markedly decreased immediately after surgical insult [24]. The dysfunction or inability of monocytes increases the host's susceptibility to infection by the invading pathogens further stimulating immune cells via their pathogen-associated molecular patterns (PAMPs). Thus, a vicious cycle might be initiated postoperatively, with surgical hit resulting in inflammation and immunosuppression, which, in turn, leads to infection with further inflammation, tissue injury and organ failure (Fig. 1) [11,25].
FIGURE 1.

Immune-inflammatory response model in patients with surgical infection, medical infection and surgical insult. The Y axis represents the level of immune-inflammatory response. The ebb represents a dynamic change of the immune-inflammatory status. Tissue damage, as well as a stress humoral and neural response, evoked by operative insult can mobilize danger-associated molecular patterns (DAMPs) and alarmins which subsequently spillover into the circulation, activate immune cells and then lead to overwhelming inflammatory processes [18–22]. Accompany with the increased nonspecific inflammatory response, the surgical patients’ ability to mount an effective defense against invading pathogens is suppressed which increases the susceptibility to infection [11,12]. In surgical patients, DAMPs and alarmins can act synergistically with pathogen-associated molecular patterns (PAMPs) to further stimulate immune cells, and further lead to deteriorative proinflammatory response and immunosuppression with an increased risk of multiorgan dysfunction and death [11,17,23▪,24–26].
Following surgery, when a second hit such as an invasion of pathogens occurs, a rapid cascade of the inflammatory mediators, such as C3a and C5a, ROS and cytokines have been monitored in animal model and patients [11,21,22,26,27]. In subgroups with surgical infection, like in elderly people and diabetic patients who already have a persistent low-grade activated inflammatory status at baseline [28,29], the proinflammatory response has been primed and triggered by already elevated baseline levels of cytokines. As a result of their reduced ability to produce anti-inflammatory mediators such as interleukin (IL)-10, immunosuppression can be aggravated and lead to fatal outcomes in these surgical patients [28,30]. Evidence for this hypothesis has been demonstrated in a large clinical study including more than 36 000 cases [31].
In addition, anesthetic management may influence defense mechanisms to surgical infections as well. There is growing evidence that high doses of opioids, like remifentanil, administered during surgery might induce immunosuppression through the activation of opioid receptors expressed on leucocytes, and also increase susceptibility to infection resulting from opioid withdrawal [32,33]. On the contrary, some interventions such as regional nerve blocks have shown benefits in reducing the primed inflammatory response [34].
INFLAMMATION, INFECTION AND INNATE IMMUNE RECEPTORS
As innate immunity has long been known to detect foreign nonself materials, innate immune receptors sensing endogenous DAMPs have been studied recently, most of which were found to be shared with exogenous PAMPs [35,36]. Toll-like receptors (TLRs), the major family of pattern recognition receptors (PRRs), are responsible for identifying both microbial PAMPs and endogenous DAMPs released from cells under stress such as trauma and damaged cells to trigger the intracellular signaling cascade [37]. The primed TLRs signaling is amenable to recruit the immune cells to the sites of infection and inflammation, to mediate the motivation of the adaptive immune response, to kill the invading pathogens, to halt their proliferation and spread and to repair damaged tissue [37,38]. Ten TLRs members have been identified in humans, so far. These type I transmembrane proteins are characterized by three domains: the extracellular domain, which contains leucine-rich repeats that mediate the recognition of ligands; a transmembrane region; and a cytoplasmic Toll-IL-1 receptor (TIR) domain that activates downstream signaling pathways [39]. Individual TLR can recognize distinct molecular patterns and be stimulated by their respective inducers. Both PAMP and DAMP engage in the induction of TLRs conformational changes including homodimer or heterodimer of TLRs. The resulting TLRs dimers then recruit a specific set of adaptor molecules, such as TIR domain-containing adaptor protein (TIRAP) and myeloid differentiation primary response gene 88 (MyD88), activate the downstream pathway and result in the upregulation or downregulation of inflammation-related gene expression [38–41]. Elevated production of inflammatory cytokines, chemokines and antimicrobial peptides would enhance the bacterial eradication and damaged tissue repairing [37]. For example, the extracellular domain of TLR2 detects peptidoglycan from Gram-positive bacteria and forms heterodimer with either TLR1 or TLR6, and the extracellular domain of the TLR4 assisted by myeloid differentiation factor 2 detects lipopolysaccharides from Gram-negative bacteria and forms TLR4 homodimer [42,43]. Both TLR2-TLR1 or TLR2-TLR6 heterodimer and TLR4 homodimer can further recruit the TIRAP and MyD88 adaptor to transmit signals in the nuclear factor-kappa B (NF-κB) -dependent manner. Recently, it was discovered that TLR2 within the endosome might activate type I interferon (IFN) gene in IFN regulatory factor (IRF)3/IRF7-dependent manner, and TLR4 within phagosome could result in the production of type I IFN through the activation of TNF receptor-associated factor 3-TANK-binding kinase 1-IRF3 pathway [37,44].
Although the interaction of TLRs with PAMPs or DAMPs plays the central role in the initiation of immune responses against invading pathogens, recent accumulating evidence also sheds light on the importance of non-Toll-like receptors (non-TLRs) to sense infection and damaged tissue and to trigger immune response. In addition to TLRs, non-TLRs, such as triggering receptors expressed on myeloid cells (TREMs) and nucleotide-binding oligomerization domain-like receptors (NLRs), involve in recognition of DAMPs and PAMPs, and act solely or cooperatively with TLRs to modulate the immune response after surgical hit or infection [40,45]. This hypothesis is strongly supported in the clinical investigation that patients devoid of functional TLR signaling show limited and transient susceptibility to infection during childhood and have increasingly rare incidence of infection with age [46]. Here, we will review the sensing and signaling pathway by non-TLRs to delineate the course of infection and inflammation (Fig. 2).
FIGURE 2.

Both Toll-like receptors (TLRs) and non-TLRs can sense PAMPs from invading pathogens and/or DAMPs released from stressed or damaged cells, trigger various intracellular signaling pathways to upregulate/downregulate the transcription of specific genes, and further modulate immune-inflammatory response. DAMPs, danger-associated molecular patterns; PAMPs, pathogen-associated molecular patterns.
According to the anchored location of receptors, innate immune receptors are classified as membrane-associated receptors such as the TREM family, cytosolic receptors like NLRs and nuclear receptors like nuclear family 4 subgroup A (NR4A) receptors.
Membrane-associated receptors: triggering receptors expressed on myeloid cells
TREMs are cell surface innate immune receptors which belong to the immunoglobulin superfamily. Two members of the TREM protein family, TREM-1 and TREM-2, were most studied. TREMs consist of a single extracellular immunoglobulin-like domain of the V-type, a transmembrane region with a charged lysine residue, and a short cytoplasmic tail. As the intracytoplasmic domain lacks any signaling capacity, TREMs associate with a transmembrane adaptor protein, DNAX activation protein-12, to activate intracellular pathways, which include activation of phosphatidylinositol 3-kinase, the phosphorylation of phospholipase Cγ and extracellular signal-related kinase1/2, and an increase in intracellular calcium. Finally, these pathways modulate cellular activation and effector function (Fig. 3) [47–49].
FIGURE 3.
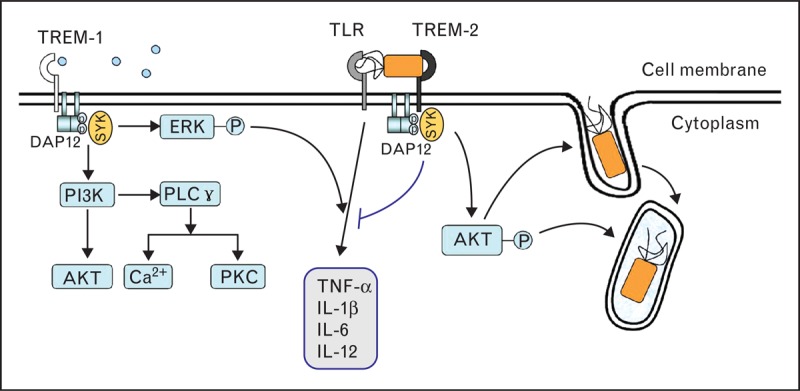
The TREMs signaling pathway during the course of immune-inflammatory response. TREMs associated with the DNAX activation protein-12 (DAP-12) chain subunit can activate phosphatidylinositol 3-kinase (PI3K), the phosphorylation of phospholipase (PL) Cγ and extracellular signal-related kinase (ERK)1/2 [47–49]. TREM-1 can act as an amplifier of the systemic inflammatory response syndrome associated with infection [52–54]. TREM-2 can act as a negative regulator inhibiting TLR-mediated inflammatory response, and enhance the host's ability to eradicate damaged cells and invading pathogens [59–61]. TREMs, triggering receptor expressed on myeloid cells.
TREM-1 is an amplifier of inflammatory response to infectious stimuli by synergy with TLR signaling [50]. The ligand for TREM-1 remains unknown. A very recent study found that complex between neutrophil peptidoglycan recognition protein 1 and bacterially derived peptidoglycan constitutes a potent ligand capable of binding TREM-1 and inducing known TREM-1 functions [51], indicating that TREM-1 might detect endogenous DAMPs and/or exogenous PAMPs in the case of surgical hit or infection. The role of TREM-1 in bacterial infection in vivo was evidenced mainly by using agents interfering with TREM-1 signaling. Treating infection models with small peptides or fusion proteins containing the extracellular domain of TREM-1 can fine-tune the inflammatory response and improve survival while preserving the capacity for bacterial clearance [50,52,53]. This protective effect was further confirmed in TREM-1 knockout mice challenged with different pathogens [54]. These studies not only demonstrate that TREM-1 plays a critical role in host immune response, but also suggest that therapeutic targeting of TREM-1 after surgical hit or tissue damage holds considerable promise by dampening excessive inflammation while maintaining effective microbial control. In addition, the soluble triggering receptor expressed on myeloid cells (sTREM-1) was identified as an early marker of infection in the surgical intensive care unit, indicating that sTREM-1 might be a useful tool to diagnose infection in surgical patients [55].
In contrast to TREM-1, TREM-2 was initially identified as a negative regulator inhibiting TLR-mediated inflammatory response [56,57]. TREM-2 can bind anionic carbohydrate molecules from both microorganisms and human cells [58]. Thus, TREM-2 is a key receptor for phagocytosis, which engulfs not only microbes but also apoptotic cells and cell debris [59–61]. This characteristic is especially pivotal in surgical infection to eradicate damaged cells and invading pathogens but keep the inflammatory response balanced.
Cytosolic receptors: nucleotide-binding oligomerization domain-like receptors
Recent studies revealed the emerging roles of NLRs, one of major family of cytosolic PRRs to detect the intracellular pathogens or danger signals in inflammation [62]. NLRs are characterized by three structural domains: a leucine-rich repeat domain at the C-terminus, being the ligand-sensor for recognizing intracellular PAMP and DAMP; the NACHT domain (nucleotide-binding domain or NAIP, CIITA, HET-E and TP1) responsible for NODs oligomerizing and preparing for signal transduction; and the effector domain at N-terminus [63]. The effector domains of human NLRs are structurally variable, which result in the activation of multiple signaling pathways and biological functions [64]. Oligomerization of NODs can activate the inflammatory kinase receptor-interacting protein2, or induce the ubiquitination of NF-κB-essential modulator, which is a key component of the NF-κB signaling complex, further strongly regulating the activity of NF-κB [62,65]. An interesting overlap between the signaling pathways triggered by NLRs and TLRs has been revealed, which suggests cooperation between these pathways and NLRs joining TLRs as crucial innate sensors of pathogens in the process of infection [45,66▪]. Currently, emerging progress has been made in the characterization of a relative novel subfamily of NLRs, such as NACHT-LRR-PYD-containing proteins (NALPs), in inflammation and infection immunity [67]. NALP1 and NALP3 evolve in the sensing of endogenous danger signals independent of the microbial trigger. This is illustrated by the discovery that several stimuli, such as sterile crystals made up of uric acid, asbestos or aluminum hydroxide, can trigger the NALP3 inflammasome, activate caspase-1 and cleave pro-IL-1β to the maturation of IL-1β [68,69]. Recently, it was discovered that activation of NALP3 in intestinal epithelial cells limits pathogen colonization and dampens the ensuing intestinal inflammation during the early course of infection, and the polymorphisms of NALP3 have been demonstrated to functionally link with the susceptibility to inflammatory disease [70▪]. The investigation of the physiological function of those cytosolic NLRs in inflammation and immunity will shed more light on the pathogenesis of infection in surgical patients.
Nuclear receptors: nuclear receptor nuclear family 4 subgroup A receptors
NR4A1, the member of orphan NR4A subfamily, is a transcription factor which maintains pivotal roles in metabolism, proliferation, apoptosis and inflammation [71]. NR4A1 contains a variable N-terminal region, a conserved DNA binding domain, a hinge region, a ligand binding domain and a C-terminal region [72]. The NR4A1 regulates cytokine production and mediates the growth factor signaling pathway. The expression of NR4A1 in inflamed human synovial tissue, psoriatic skin, atherosclerotic lesions, lung and colorectal cancer cells is aberrant [73]. Recently, it has been demonstrated that NR4A1 participates in hepatitis C virus (HCV) infection via regulating the expression of cellular receptors and apolipoprotein E, to determine HCV replication and facilitate HCV entry and spread [74]. Emerging data and concepts strongly suggest intrinsic links between NR4A1 and inflammatory disease through integrating complex cytokine signals including protein kinases, wingless type and mitogen-activated protein kinase pathways [75]. However, the exact interaction between NR4A1 and infection is poorly understood and needs to be investigated in the future.
CONCLUSION
Infection is one of the most common causes for morbidity and mortality in surgical patients with prolonged hospital length of stay and increased medical care costs, reduced functional independence and impaired long-term outcome. The fundamental mechanisms for the susceptibility to surgical infections remain still to be elucidated. No doubt, a better understanding of the pivotal role of non-TLRs will provide a novel insight for the prevention and the treatment of surgical infections. Surgical infections seem to be different from those in medical populations, with characteristics including the primed systemic inflammatory response by surgical insult, immediate postoperative immune suppression, various invasive interventions and anesthetic techniques, and in addition exposure to specific hetero-pathogens, transfusion and reperfusion. Taking these factors together, the course of surgical infections is much more complex than nonsurgical infections. The strategies to prevent and defend against pathogen invasion and to prevent organ injury should target at reducing inflammation, however, without aggravating immunodepression.
Acknowledgements
None.
Financial support and sponsorship
This study was supported by the National Natural Science Foundation of China (No. 81130036, 81471838), and the National Science & Technology Pillar Program during the Twelfth Five-year Plan Period (No. 2012BAI11B05).
Conflicts of interest
There are no conflicts of interest.
REFERENCES AND RECOMMENDED READING
Papers of particular interest, published within the annual period of review, have been highlighted as:
▪ of special interest
▪▪ of outstanding interest
REFERENCES
- 1.Weiser TG, Regenbogen SE, Thompson KD, et al. An estimation of the global volume of surgery: a modelling strategy based on available data. Lancet 2008; 372:139–144. [DOI] [PubMed] [Google Scholar]
- 2.Khuri SF, Henderson WG, DePalma RG, et al. Determinants of long-term survival after major surgery and the adverse effect of postoperative complications. Ann Surg 2005; 242:326–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ghaferi AA, Birkmeyer JD, Dimick JB. Variation in hospital mortality associated with inpatient surgery. N Engl J Med 2009; 361:1368–1375. [DOI] [PubMed] [Google Scholar]
- 4.Eappen S, Lane BH, Rosenberg B, et al. Relationship between occurrence of surgical complications and hospital finances. JAMA 2013; 309:1599–1606. [DOI] [PubMed] [Google Scholar]
- 5.Bratzler DW, Hunt DR. The surgical infection prevention and surgical care improvement projects: national initiatives to improve outcomes for patients having surgery. Clin Infect Dis 2006; 43:322–330. [DOI] [PubMed] [Google Scholar]
- 6.DiPiro JT, Martindale RG, Bakst A, et al. Infection in surgical patients: effects on mortality, hospitalization, and postdischarge care. Am J Health Syst Pharm 1998; 55:777–781. [DOI] [PubMed] [Google Scholar]
- 7.Bainbridge D, Martin J, Arango M, Cheng D. Evidence-based Peri-operative Clinical Outcomes Research (EPiCOR) Group. Perioperative and anaesthetic-related mortality in developed and developing countries: a systematic review and meta-analysis. Lancet 2012; 380:1075–1081. [DOI] [PubMed] [Google Scholar]
- 8.Haynes AB, Weiser TG, Berry WR, et al. A surgical safety checklist to reduce morbidity and mortality in a global population. N Engl J Med 2009; 360:491–499. [DOI] [PubMed] [Google Scholar]
- 9.Toufen CJ, Franca SA, Okamoto VN, et al. Infection as an independent risk factor for mortality in the surgical intensive care unit. Clinics (Sao Paulo) 2013; 68:1103–1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dimick JB, Chen SL, Taheri PA, et al. Hospital costs associated with surgical complications: a report from the private-sector National Surgical Quality Improvement Program. J Am Coll Surg 2004; 199:531–537. [DOI] [PubMed] [Google Scholar]
- 11.Lord JM, Midwinter MJ, Chen YF, et al. The systemic immune response to trauma: an overview of pathophysiology and treatment. Lancet 2014; 384:1455–1465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gentile LF, Cuenca AG, Efron PA, et al. Persistent inflammation and immunosuppression: a common syndrome and new horizon for surgical intensive care. J Trauma Acute Care Surg 2012; 72:1491–1501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cakmakci M. Surgical site infections as a healthcare quality issue. Surg Infect (Larchmt) 2010; 11:1–6. [DOI] [PubMed] [Google Scholar]
- 14.Fakhry SM, Montgomery SC. Peri-operative oxygen and the risk of surgical infection. Surg Infect (Larchmt) 2012; 13:228–233. [DOI] [PubMed] [Google Scholar]
- 15.Leaper DJ. Risk factors for and epidemiology of surgical site infections. Surg Infect (Larchmt) 2010; 11:283–287. [DOI] [PubMed] [Google Scholar]
- 16.Bratzler DW, Houck PM. Antimicrobial prophylaxis for surgery: an advisory statement from the National Surgical Infection Prevention Project. Clin Infect Dis 2004; 38:1706–1715. [DOI] [PubMed] [Google Scholar]
- 17.Manson J, Thiemermann C, Brohi K. Trauma alarmins as activators of damage-induced inflammation. Br J Surg 2012; 99 Suppl 1:12–20. [DOI] [PubMed] [Google Scholar]
- 18.Zhang Q, Raoof M, Chen Y, et al. Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature 2010; 464:104–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Burk AM, Martin M, Flierl MA, et al. Early complementopathy after multiple injuries in humans. Shock 2012; 37:348–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Desborough JP. The stress response to trauma and surgery. Br J Anaesth 2000; 85:109–117. [DOI] [PubMed] [Google Scholar]
- 21.Neher MD, Weckbach S, Flierl MA, et al. Molecular mechanisms of inflammation and tissue injury after major trauma: is complement the ‘bad guy’? J Biomed Sci 2011; 18:90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pugin J. How tissue injury alarms the immune system and causes a systemic inflammatory response syndrome. Ann Intensive Care 2012; 2:27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23▪.Boomer JS, Green JM, Hotchkiss RS. The changing immune system in sepsis: is individualized immuno-modulatory therapy the answer? Virulence 2014; 5:45–56. [DOI] [PMC free article] [PubMed] [Google Scholar]; An excellent update on the immune-inflammatory changes during the course of severe infection such as sepsis which is likely more complex than previously thought.
- 24.Mokart D, Leone M, Sannini A, et al. Predictive perioperative factors for developing severe sepsis after major surgery. Br J Anaesth 2005; 95:776–781. [DOI] [PubMed] [Google Scholar]
- 25.Hietbrink F, Koenderman L, Althuizen M, Leenen LP. Modulation of the innate immune response after trauma visualised by a change in functional PMN phenotype. Injury 2009; 40:851–855. [DOI] [PubMed] [Google Scholar]
- 26.Stoecklein VM, Osuka A, Lederer JA. Trauma equals danger: damage control by the immune system. J Leukoc Biol 2012; 92:539–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li Y, Xiang M, Yuan Y, et al. Hemorrhagic shock augments lung endothelial cell activation: role of temporal alterations of TLR4 and TLR2. Am J Physiol Regul Integr Comp Physiol 2009; 297:R1670–R1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Franceschi C. Inflammaging as a major characteristic of old people: can it be prevented or cured? Nutr Rev 2007; 65:S173–S176. [DOI] [PubMed] [Google Scholar]
- 29.Quante M, Dietrich A, ElKhal A, Tullius SG. Obesity related immune responses and their impact on surgical outcomes. Int J Obes (Lond) 2015; [Epub ahead of print]. [DOI] [PubMed] [Google Scholar]
- 30.Duggal NA, Upton J, Phillips AC, et al. An age-related numerical and functional deficit in CD19(+) CD24(hi) CD38(hi) B cells is associated with an increase in systemic autoimmunity. Aging Cell 2013; 12:873–881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Magnotti LJ, Fischer PE, Zarzaur BL, et al. Impact of gender on outcomes after blunt injury: a definitive analysis of more than 36,000 trauma patients. J Am Coll Surg 2008; 206:984–991. [DOI] [PubMed] [Google Scholar]
- 32.Eisenstein TK, Rahim RT, Feng P, et al. Effects of opioid tolerance and withdrawal on the immune system. J Neuroimmune Pharmacol 2006; 1:237–249. [DOI] [PubMed] [Google Scholar]
- 33.Inagi T, Suzuki M, Osumi M, Bito H. Remifentanil-based anesthesia increases the incidence of postoperative surgical site infection. J Hosp Infect 2015; 89:61–68. [DOI] [PubMed] [Google Scholar]
- 34.Wilmore DW. From Cuthbertson to fast-track surgery: 70 years of progress in reducing stress in surgical patients. Ann Surg 2002; 236:643–648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Piccinini AM, Midwood KS. DAMPening inflammation by modulating TLR signalling. Mediators Inflamm 2010; 2010:pii: 672395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Akira S, Uematsu S, Takeuchi O. Pathogen recognition and innate immunity. Cell 2006; 124:783–801. [DOI] [PubMed] [Google Scholar]
- 37.Kawai T, Akira S. The role of pattern-recognition receptors in innate immunity: update on Toll-like receptors. Nat Immunol 2010; 11:373–384. [DOI] [PubMed] [Google Scholar]
- 38.Iwasaki A, Medzhitov R. Toll-like receptor control of the adaptive immune responses. Nat Immunol 2004; 5:987–995. [DOI] [PubMed] [Google Scholar]
- 39.Beutler B. Inferences, questions and possibilities in Toll-like receptor signalling. Nature 2004; 430:257–263. [DOI] [PubMed] [Google Scholar]
- 40.Kawai T, Akira S. Toll-like receptors and their crosstalk with other innate receptors in infection and immunity. Immunity 2011; 34:637–650. [DOI] [PubMed] [Google Scholar]
- 41.Kay E, Scotland RS, Whiteford JR. Toll-like receptors: role in inflammation and therapeutic potential. Biofactors 2014; 40:284–294. [DOI] [PubMed] [Google Scholar]
- 42.Park BS, Song DH, Kim HM, et al. The structural basis of lipopolysaccharide recognition by the TLR4-MD-2 complex. Nature 2009; 458:1191–1195. [DOI] [PubMed] [Google Scholar]
- 43.van Bergenhenegouwen J, Plantinga TS, Joosten LA, et al. TLR2& Co: a critical analysis of the complex interactions between TLR2 and coreceptors. J Leukoc Biol 2013; 94:885–902. [DOI] [PubMed] [Google Scholar]
- 44.Barton GM, Kagan JC. A cell biological view of Toll-like receptor function: regulation through compartmentalization. Nat Rev Immunol 2009; 9:535–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Martinon F, Tschopp J. NLRs join TLRs as innate sensors of pathogens. Trends Immunol 2005; 26:447–454. [DOI] [PubMed] [Google Scholar]
- 46.Ku CL, Yang K, Bustamante J, et al. Inherited disorders of human Toll-like receptor signaling: immunological implications. Immunol Rev 2005; 203:10–20. [DOI] [PubMed] [Google Scholar]
- 47.Sharif O, Knapp S. From expression to signaling: roles of TREM-1 and TREM-2 in innate immunity and bacterial infection. Immunobiology 2008; 213:701–713. [DOI] [PubMed] [Google Scholar]
- 48.Ford JW, McVicar DW. TREM and TREM-like receptors in inflammation and disease. Curr Opin Immunol 2009; 21:38–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Klesney-Tait J, Turnbull IR, Colonna M. The TREM receptor family and signal integration. Nat Immunol 2006; 7:1266–1273. [DOI] [PubMed] [Google Scholar]
- 50.Bouchon A, Facchetti F, Weigand MA, Colonna M. TREM-1 amplifies inflammation and is a crucial mediator of septic shock. Nature 2001; 410:1103–1107. [DOI] [PubMed] [Google Scholar]
- 51.Read CB, Kuijper JL, Hjorth SA, et al. Cutting edge: identification of neutrophil PGLYRP1 as a ligand for TREM-1. J Immunol 2015; 194:1417–1421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gibot S, Alauzet C, Massin F, et al. Modulation of the triggering receptor expressed on myeloid cells-1 pathway during pneumonia in rats. J Infect Dis 2006; 194:975–983. [DOI] [PubMed] [Google Scholar]
- 53.Gibot S, Buonsanti C, Massin F, et al. Modulation of the triggering receptor expressed on the myeloid cell type 1 pathway in murine septic shock. Infect Immun 2006; 74:2823–2830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Weber B, Schuster S, Zysset D, et al. TREM-1 deficiency can attenuate disease severity without affecting pathogen clearance. PloS Pathog 2014; 10:e1003900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Rivera-Chavez FA, Minei JP. Soluble triggering receptor expressed on myeloid cells-1 is an early marker of infection in the surgical intensive care unit. Surg Infect (Larchmt) 2009; 10:435–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Turnbull IR, Gilfillan S, Cella M, et al. Cutting edge: TREM-2 attenuates macrophage activation. J Immunol 2006; 177:3520–3524. [DOI] [PubMed] [Google Scholar]
- 57.Hamerman JA, Jarjoura JR, Humphrey MB, et al. Cutting edge: inhibition of TLR and FcR responses in macrophages by triggering receptor expressed on myeloid cells (TREM)-2 and DAP12. J Immunol 2006; 177:2051–2055. [DOI] [PubMed] [Google Scholar]
- 58.Daws MR, Sullam PM, Niemi EC, et al. Pattern recognition by TREM-2: binding of anionic ligands. J Immunol 2003; 171:594–599. [DOI] [PubMed] [Google Scholar]
- 59.N’Diaye EN, Branda CS, Branda SS, et al. TREM-2 (triggering receptor expressed on myeloid cells 2) is a phagocytic receptor for bacteria. J Cell Biol 2009; 184:215–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Chen Q, Zhang K, Jin Y, et al. Triggering receptor expressed on myeloid cells-2 protects against polymicrobial sepsis by enhancing bacterial clearance. Am J Respir Crit Care Med 2013; 188:201–212. [DOI] [PubMed] [Google Scholar]
- 61.Takahashi K, Rochford CD, Neumann H. Clearance of apoptotic neurons without inflammation by microglial triggering receptor expressed on myeloid cells-2. J Exp Med 2005; 201:647–657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Di Virqilio F. The therapeutic potential of modifying inflammasomes and NOD-like receptors. Pharmacol Rev 2013; 65:872–905. [DOI] [PubMed] [Google Scholar]
- 63.Schroder K, Tschopp J. The inflammasomes. Cell 2010; 140:821–832. [DOI] [PubMed] [Google Scholar]
- 64.Motta V, Soares F, Sun T, Philpott DJ. NOD-like receptors: versatile cytosolic sentinels. Physiol Rev 2015; 95:149–178. [DOI] [PubMed] [Google Scholar]
- 65.Kanneganti TD, Lamkanfi M, Núñez G. Intracellular NOD-like receptors in host defense and disease. Immunity 2007; 27:549–559. [DOI] [PubMed] [Google Scholar]
- 66▪.Burberry A, Zeng MY, Ding L, et al. Infection mobilizes hematopoietic stem cells through cooperative NOD-like receptor and Toll-like receptor signaling. Cell Host Microbe 2014; 15:779–791. [DOI] [PMC free article] [PubMed] [Google Scholar]; Non-TLRs, such as NOD-like receptor, can act cooperatively with TLRs to modulate the immune-inflammatory response after infection.
- 67.Davis BK, Wen H, Ting JP. The inflammasome NLRs in immunity, inflammation, and associated diseases. Annu Rev Immunol 2011; 29:707–735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.De Nardo D, De Nardo CM, Latz E. New insights into mechanisms controlling the NLRP3 inflammasome and its role in lung disease. Am J Pathol 2014; 184:42–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.De Nardo D, Latz E. NLRP3 inflammasomes link inflammation and metabolic disease. Trends Immunol 2011; 32:373–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70▪.Song-Zhao GX, Srinivasan N, Pott J, et al. NLRP3 activation in the intestinal epithelium protects against a mucosal pathogen. Mucosal Immunol 2014; 7:763–774. [DOI] [PMC free article] [PubMed] [Google Scholar]; Evidence is presented that early activation of NLRP3 could limit pathogen colonization and prevent subsequent pathology associated with the inflammatory response.
- 71.Hanna RN, Shaked I, Hubbeling HG, et al. NR4A1 (Nur77) deletion polarizes macrophages toward an inflammatory phenotype and increases atherosclerosis. Circ Res 2012; 110:416–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Mangelsdorf DJ, Thummel C, Beato M, et al. The nuclear receptor superfamily: the second decade. Cell 1995; 83:835–839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.McMorrow JP, Murphy EP. Inflammation: a role for NR4A orphan nuclear receptors? Biochem Soc Trans 2011; 39:688–693. [DOI] [PubMed] [Google Scholar]
- 74.Zhu W, Pei R, Jin R, et al. Nuclear receptor 4 group A member 1 determines hepatitis C virus entry efficiency through the regulation of cellular receptor and apolipoprotein E expression. J Gen Virol 2014; 95:1510–1521. [DOI] [PubMed] [Google Scholar]
- 75.Ranhotra HS. The NR4A orphan nuclear receptors: mediators in metabolism and diseases. J Recept Signal Transduct Res 2014; 1–5. [DOI] [PubMed] [Google Scholar]