

Abstract
Aurora kinase family of serine/threonine kinases are important regulators of mitosis that are frequently over expressed in human cancers and have been implicated in oncogenic transformation including development of chromosomal instability in cancer cells. In humans, among the three members of the kinase family, Aurora-A, -B and -C, only Aurora-A and -B are expressed in detectable levels in somatic cells undergoing mitotic cell division and have been characterized in greater detail for their involvement in cellular pathways relevant to the development of cancer associated phenotypes. Aurora-A and -B are being investigated as potential targets for anticancer therapy. Development of inhibitors against Aurora kinases as anticancer molecules gained attention because of the facts that aberrant expression of these kinases lead to chromosomal instability and derangement of multiple tumor suppressor and oncoprotein regulated pathways. Pre-clinical studies and early phase I and II clinical trials of multiple Aurora kinase inhibitors as targeted anticancer drugs have provided encouraging results. This article discusses functional involvement of Aurora kinase-A and -B in the regulation of cancer relevant cellular phenotypes together with findings on some of the better characterized Aurora kinase inhibitors in modulating the functional interactions of Aurora kinases. Future possibilities about developing next generation Aurora kinase inhibitors and their clinical utility as anticancer therapeutic drugs are also discussed.
Keywords: Aurora kinase, Mitosis, Chromosomal instability, Tumor suppressor protein, Oncoprotein, Aurora kinase inhibitor, Cancer therapy
1. Introduction
Cancer cells manifest characteristic abnormal growth properties accompanying clonal evolution of cells displaying progressively increasing genomic instability capable of invasion and metastasis to distant organ sites. With the emerging knowledge about the role of known oncogene and tumor suppressor gene mediated pathways in deregulating the growth of cancer cells, novel chemotherapeutic agents targeting these pathways are being developed. Such therapeutic approaches are designed to disrupt the signaling networks involving the respective target genes which are aberrantly expressed to cause uncontrolled growth of the malignant cells. While these strategies have shown promise in the initial treatment outcomes their long term efficacy remain questionable in many instances since prolonged exposure to a specific target inhibiting drug often leads to cancer cells rewiring the aberrantly expressing signaling events to continue proliferation in a deregulated manner. This becomes possible since signaling cascades determining the abnormal growth phenotype are not regulated by linear events but result from complex functional networks constituted of cross talking individual signaling pathways. It is therefore logical to expect that for cancer therapeutics to be maximally effective, multiple signaling pathways converging on the fundamental growth regulatory processes such as DNA replication and/or mitosis need to be targeted in a robust manner. It is noteworthy, in this context, that efficacy of most conventional and novel chemotherapeutic agents rests on the premise that cancer cells can be preferentially eliminated due to their persistent cycling nature by interfering with either their replication/repair of DNA or by disrupting the mitotic division process. Thus, among the commonly used drugs for cancer chemotherapy, agents such as fluoropyramidines, gemcitabine and topoisomerase inhibitors interfere with the DNA replication process while platinum analogs and cyclophosphamide introduce un-resolvable lesions into the replicating DNA of proliferating cells to induce cell death. Another group of effective drugs, which include taxanes, vinca alkaloids and epothilones cause growth inhibition and death of proliferating cells by disrupting the microtubule cytoskeleton essential for the mitotic cell division process (1). However, given the fact that a host of normal cells continuously proliferate in adult tissues, it is not unexpected that cytotoxic drugs indiscriminately targeting proliferating cells inflict varying degree of normal tissue damage and thus cause toxicity to the patients. Functional genomic data from tumors is proving helpful in alleviating the problem by identifying the putative therapeutic target proteins regulating cell cycle that are differentially expressed in tumors compared with the normal cells of the adult tissues. It is plausible that pharmaceutical targeting of such proteins would help the development of a new generation of effective therapeutic drugs that will have minimal host toxicity. These drugs while still interfering with the cell proliferation process would be expected to have a more selective effect on the tumors due to a preferential negative response of signal attenuation in the tumor cells compared with their normal counterparts (2). Based on this rationale, proteins involved in the regulation of cell cycle (3) and cell cycle associated kinases (4,5), expressing at abnormally high levels in tumors, have been proposed as promising novel targets for the development of anti-cancer drugs. In the recent past a number of inhibitors against new mitotic targets have indeed been rapidly moving into clinical trials (6). Among the mitosis regulatory kinases, evolutionarily conserved family of serine/threonine kinases referred to as Aurora kinases has emerged as an exceptionally attractive target for anticancer drug discovery. The interest in designing drugs against Aurora kinase family members stems from the facts that these kinases, expressed at elevated levels in many human cancers, are not only vitally important regulators of mitosis but have also been shown to functionally interact with multiple critical oncoproteins and tumor suppressor proteins. Pre-clinical studies of Aurora kinase inhibitors have shown promising results and the ongoing phase I and II clinical trials for several of these as anticancer molecules have also yielded encouraging results so far.
2. Aurora kinase family
Aurora family of ser/threonine kinases has been recognized as important regulators of mitosis with essential roles in the progressive stages ranging from mitotic entry to cytokinesis (7). These kinases have been highly conserved through evolution playing essential roles in ensuring accurate coordination of chromosomal and cytoskeletal events including centrosome maturation and separation as well as proper spindle assembly leading to faithful partitioning of the chromosomes into daughter cells. The number of Aurora kinase family members varies in different animal phyla. Yeast has one prototypic member Ipl1/Ark1 while in majority of higher eukaryotes the family has two related members Aurora-A and -B represented by conserved orthologs in different species. Aurora-A and -B kinases display different sub-cellular localizations and functions. A third member of the kinase family, Aurora-C, is present only in mammals and is predominantly expressed in testes but has also been reported to rescue in vitro grown human cells depleted of Aurora-B indicating possible functional over lap between the two kinases in somatic cells. The three members of the mammalian Aurora kinase family share similar carboxyl terminus catalytic domains but divergent amino terminal ends of variable lengths displaying little or no similarity. Although all three Aurora kinases have been found to be over expressed in human cancer cells, possible involvement of Aurora-C in the development of tumorigenic phenotypes, if any, remains unknown in view of its minimal expression and function detected in somatic cells. This review, therefore, discusses only Aurora-A and -B as potential anticancer drug targets along with the description of the inhibitors being developed as anticancer molecules targeting the two kinases. A number of comprehensive reviews have been written on the structure and function of Aurora kinases and for the purpose of this article we will be mainly focusing on the cancer relevant functional interactions of Aurora-A and -B kinases with a brief description of structural characteristics and functional involvement in specific cellular pathways.
Aurora-A and -B share about 70% identity in the carboxyl terminus catalytic domain and three conserved Aurora box motifs (A-box I, A-box II and A-box III) in their varying amino terminal domain. The functional significance of A-box motifs is not yet well defined although dephosphorylation of a serine residue in the A-box II is required for degradation of Aurora-A and there is suggestive evidence that the A-box motifs are involved in substrate recognition and subcellular localization of the two kinases. Despite conserved structural characteristics, Aurora-A and -B manifest predominantly different localization and function during mitosis interacting with distinct set of proteins. Aurora-A is localized primarily on spindle poles and transiently along the spindle microtubules as cells progress through mitosis. The kinase functions in mitotic entry, centrosome maturation-separation, bipolar spindle organization and recovery from spindle damage (8). Aurora-B is associated with the Chromsomal Passenger Complex comprising of the scaffolding protein INCENP and the targeting proteins Survivin and Borealin/DasraB. The CPC localizes to the inner centromere during prophase through metaphase and then transfers to the spindle midzone and the midbody during late mitosis and cytokinesis (9). Aurora-B functions in regulating attachment of kintochore to spindle microtubules, sister chromatid cohesion and cytokinesis (7,9). The diverse localization and functions of the two related kinases are determined by their binding partners some of which also regulate their kinase activities. Activation of Aurora-A has been shown to be regulated by multiple protein binding cofactors among which the role of TPX2 is well characterized. While the N-terminus of TPX2 induces conformational change in Aurora-A facilitating activation through auto-phosphorylation of Thr288 in the T-loop, the TPX2 bound kinase is also protected from de-phosphorylation by PP1 on entry into mitosis (10,11). Aurora-B activation involves auto-phosphorylation of Thr232 in the T-loop and requires interaction with the CPC consisting of the INCENP, Survivin and the Borealin/DasraB proteins. The three CPC proteins in a stable core complex target to the centromere (12) interacting with Aurora-B through the C-terminus IN-box of the INCENP protein (13). Intriguingly, most of the interacting proteins with Aurora-A and -B associate with conserved residues in their similar catalytic domains rather than in the variable amino terminus domains and a single amino acid difference in the catalytic domain of the two kinases (G198 in human Aurora-A and N142 in human Aurora-B) was shown to be critical in controlling the intrinsic activity and selective activation of Aurora-A by its binding partner and activator TPX2 (14,15). Site directed mutant of this Aurora-A residue (G198N) revealed classical Aurora-B localization and association with the CPC components INCENP and Survivin partially rescuing Aurora-B loss of function (14,15). Such subtle structural specificity regulating interaction of Aurora-A and -B with binding proteins explain limited functional interchangeability, including some shared substrates such as MCAK, INCENP, Kif2 and RASSF1A, observed for the two kinases. Although functional interactions with the common substrates, most likely, occur at different times during mitosis yet such overlapping activities between the two kinases is expected to have significant implications in terms of affecting proliferation and chromosomal ploidy when the two proteins are aberrantly expressed, as is the case in many human malignant cell types.
Elevated expression of Aurora-A and -B frequently detected in a wide variety of human cancers strongly indicate that high expression of these kinases play roles in the development of cancer associated phenotypes. While Aurora-A has been shown to function as an oncogene when over expressed in mammalian cells in classical in vitro transformation assays and in rodent models (16,17), there is also suggestive evidence of high Aurora-B expression being oncogenic in vivo (18). Since the two proteins are important regulators of mitosis it is not unexpected that malignant cells over expressing the two kinases always manifest chromosomal instability. A list of proteins interacting with Aurora-A and -B in the regulation of different mitotic events is summarized in the Figure 1 below. As mentioned above, for the purpose of this article, we are not discussing the functional details of all these interactions but focusing only on those more directly implicated in the origin of cancer associated phenotypes including chromosomal instability.
Figure 1.

Major mitotic signaling pathways controlled by Aurora kinases
a. Aurora kinase-A and -B interacting/substrate proteins. Proteins in blue and yellow areas interact with Aurora-A and -B respectively. Proteins in green area interact with both Aurora-A and -B.
b. Schematic model of Aurora kinase regulated mitotic signalling pathways
The underlying mechanisms giving rise to chromosome instability phenotype are, however, different for the two kinases. Aurora-A gain of function primarily deregulates mitotic entry, centrosome maturation and spindle assembly allowing aberrant progression through mitosis due to hyperactive centrosomes and multipolar spindle assembly in addition to facilitating recovery from spindle damage in cells treated with spindle poisoning drugs. Aurora-B over expression, on the other hand, interferes with chromosome bi-orientation and spindle assembly checkpoint due to enhanced disruption of kinetochore-microtubule attachments and sister chromatid cohesion besides causing abnormal cytokinesis resulting in chromosome segregation errors. Whether or not induction of chromosome ploidy alteration plays a role in the malignant transformation process remains a matter of debate although contribution of chromosomal instability, often associated with centrosome amplification, in progression of disease and acquiring resistance to chemotherapeutic drugs is now fairly well accepted. The latter possibility is also implicit from the observations that Aurora-A facilitates checkpoint recovery and mitotic entry after spindle damage by activating Polo like kinase-1 (19,20) as well as by mediating the formation of kinetochore/chromatin associated microtubule assembly (21). Additionally, Aurora-A has been implicated in the induction of centrosome amplification, frequently detected in tumor cells. Although the underlying mechanism of this phenomenon remains unknown, identification of multiple centrosome associated Aurora-A substrates implicated in the maturation and separation of centrosomes as well as stabilization of centrosomal microtubules such as, NDEL1, TACC3 and ASAP indicate that deregulated expression of Aurora-A induces chromosome segregation defects and aneuploidy in tumor cells also by causing anomalies in centrosome biogenesis and function. It is, therefore, reasonable to suggest that Aurora kinase inhibitors offer the promise of being developed into effective components of anticancer therapeutic regimens possibly in combination with drugs targeting additional aberrantly expressing cancer associated signaling pathways.
In addition to their mitosis specific substrates Aurora kinases, primarily Aurora-A, have also been found to functionally interact with proteins involved in critical cancer associated pathways. A list of these proteins and the functional consequence of their interactions with Aurora kinases is mentioned in the Table-1.
Table 1.
Functional regulation of tumor suppressor and oncogenic proteins by Aurora kinases
| Modification/Regulation | Consequence | Ref | |
|---|---|---|---|
| p53 | Phosphorylation at Ser215 and Ser315 by Aur-A | Down-regulation of transactivation activity and protein stability of p53, and resistance to DNA damage induced cell death | 22,23 |
| BRCA1 | Phosphorylation of Ser308 by Aur-A | Loss of G2/M checkpoint arrest | 24 |
| hTERT | Transcriptional control by Aur-A | mRNA up-regulation through increase in c-myc transactivation activity | 26 |
| GSK3β | Phosphorylation at Ser9 by Aur-A | Activation of β-catenin/TCF transcription complex and induction of their down stream target genes such as Cyclin D1 and c-Myc | 27 |
| IκBα | Induction of phosphorylation at Ser32 and Ser 36 by Aur-A | NF-κB activation and chemo-resistance due to protein degradation of phospho-IκBα | 28,47,113 |
| IκB kinase2 (IKK2) | Phosphorylation of Aur-A by IKK2 (site has not been yet determined) | Targeting of Aur-A for b-TRCP-mediated proteasomal degradation | 29 |
| RaIA | Phosphorylation at Ser194 by Aur-A | Canonical oncogenic Ras signaling through activating RaIA activity and sequential activation of RaIBP1 by activated RaIA | 30,31 |
| NORE1A | Phosphorylation at Ser277 by Aur-A | Inhibition of NORE1A induced microtubule nucleation and polymerization | 32 |
| N-Myc | Interaction with Aur-A | Protein stabilization of N-Myc by inhibiting SCFFoxw7-mediated degradation in kinase activity independent manner | 33 |
| BARD1β | Interaction with Aur-B | Promote complex formation of Aur-B and BRCA2 for midbody formation and abscission | 25 |
Aurora-A phosphorylation of the tumor suppressor proteins BRCA1 and p53 have been reported to cause their loss of function making cells resistant to DNA damage and over ride checkpoint response (22–24). The BRCA1 associated ring domain protein 1, BARD1, and its cancer cell specific variant isoform BARD1β form complexes with Aurora-B eliciting anti-proliferation and pro-proliferation responses respectively (25). The findings taken together indicate that Aurora kinase-A and -B antagonize the p53 and BRCA1 functions and thus tumors with elevated expression of the two kinases acquire loss of function phenotypes for these two critical tumor suppressor pathways even in the absence of any inactivating mutation in the respective tumor suppressor proteins.
Role of Aurora kinases in directly activating multiple oncogenic pathways and promoting proliferation as well as transformation has also been demonstrated. Aurora-A has been shown to up-regulate telomerase reverse transcriptase mRNA through c-myc (26). The kinase was also implicated in activating the Akt pathway and directly phosphorylate GSK3β leading to activation of the β-catenin-TCF transcription complex (27) underscoring the involvement of Aurora-A in this oncogenic pathway. Additionally, positive regulation of NF-κB signaling has been demonstrated as a consequence of Aurora-A mediated IκBα phosphorylation leading to activation of the NF-κB complexes (28). Interestingly, IκB Kinase2, a component of the IκK complex responsible for physiologic phosphorylation and degradation of IκB appears to negatively regulate stability of Aurora-A protein and thus have a role in bipolar spindle assembly (29). These findings support the notion that under normal physiological conditions IκB Kinase2 positively regulates NF-κB but antagonizes Aurora-A signaling to ensure proper mitotic assembly and chromosome segregation while over expression of Aurora-A leads to derangement of this regulatory network. Furthermore, oncogenic effects of Aurora-A is also mediated through phosphorylation of RalA in the Ras signaling pathway (30,31) and the tumor suppressor NORE1A that functions as a regulatory node between Ras signaling and microtubule nucleation (32). Gain of function of Aurora-A, therefore, appears to be a seminal defect in cancer cells that can cause dual problems of aberrantly activating the Ras and the NF-κB signaling and also induction of abnormal mitotic spindle assembly. Finally, a kinase activity independent role of Aurora-A in N-Myc amplified neuroblastoma tumors, showing frequent amplification of Aurora-A was revealed that involved stabilization of the oncogenic N-Myc protein (33). Intriguingly, though, an Aurora-A inhibitor has demonstrated broad and unprecedented robust anti-tumor activity in all neuroblastomas in N-Myc independent manner (34). These findings implicating Aurora-A’s pleiotropic role in regulating mitotic cell division cycle and multiple oncogenic signaling networks justify the current interest in developing effective Aurora kinase inhibitors for anti-cancer therapy. Future work will determine if the currently developed ATP competitive kinase activity inhibitors alone or in combination with allosteric inhibitors interfering with Aurora kinase binding proteins would be required to achieve maximal efficacy in the treatment of human cancers.
3. Aurora kinase inhibitors
Given their pivotal roles in mitotic process during cell cycle, over expression in malignancy and cross talks with tumor suppressor as well as oncogenic signaling pathways, as described above, members of the Aurora kinase family have emerged as promising chemotherapeutic targets for cancer. As indicated earlier, the in vivo target selectivity of Aurora kinases for cancer cells is proposed because these proteins express preferentially only in progressively proliferating cells (35). Supporting this idea, early in vitro experiments of Aurora kinase inhibitor ZM447439 demonstrated selective loss of viability only of rapidly dividing cells while the non-dividing cells remained viable (36). A number of Aurora kinase inhibitors displaying differential IC50 values toward the three family members have since then been developed and utilized to dissect the functional role of Aurora kinases in mitotic progression. Anti-tumor activities of these inhibitors have also been evaluated in preclinical studies and a few of them have progressed to early stage clinical trials for treatment of different types of cancers. In this section, we present an overview of the biological effects of Aurora kinase inhibitors on mitotic processes and the signaling pathways based on the available published literature (Table-2). For detailed information on the results of Aurora kinase inhibitors in clinical trials readers may refer to recent reviews written on the subject (37,38).
Table 2.
Aurora kinase inhibitors in clinical trial
| Structure | Source | Specificity | Administration | Clinical trial | |
|---|---|---|---|---|---|
| VX-680/MK-0457 |
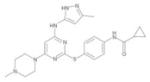
|
Vertex/Merck | Pan-Aurora | I.V. | Phase I–II |
| PHA-739358 |
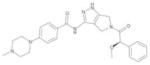
|
Nerviano/Pfizer | Pan-Aurora | I.V. | Phase I–II |
| MLN8054 |
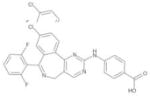
|
Millennium | Aurora-A | Oral | Phase I |
| MLN8237 |
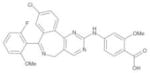
|
Millennium | Aurora-A | Oral | Phase I–II |
| AZD1152 |
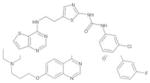
|
AstraZeneca | Aurora-B and -C | I.V. | Phase I–II |
3.1. Hesperadin, ZM447439 and VX-680/MK-0457: First generation inhibitors
ZM447439 (36), Hesperadin (39) and VX-680/MK-0457 (40) were the first generation of Aurora kinase inhibitors identified or designed through different strategies. These three small molecule chemical inhibitors occupy the ATP biding site to inhibit the catalytic activity of the enzyme. Hesperadin was initially identified as a general kinase inhibitor in the screening for novel indolinone derivatives with antiproliferative activity (41) and later shown to have selective inhibitory activity towards Aurora-B (39). ZM447437 was identified by screening of more than 250,000 chemical compounds inhibiting Aurora-A kinase activity (36). Chemical structure of VX-680/MK-0457, on the other hand, was designed to have binding affinity to ATP binding pocket of Aurora kinases which is identical among all three member of Aurora kinase family with no structural similarity to other kinases (40). As theoretically predicted, VX-680/MK-0457 exhibits greater sensitivity toward Aurora kinases compared to Hesperadin and ZM447439 in enzymatic assays in vitro although Hesperadin appears to be specific to Aurora-B and its potency against Aurora-A has not yet been established. Interestingly, the phenotypic defects in mitosis observed in presence of ZM447439 and Hesperadin mimic those seen in case of silencing of Aurora-B and not Aurora-A even though ZM447439 can inhibit Aurora-A with similar or greater potency compared with Aurora-B. The altered phenotypes in presence of these inhibitors include loss of histone H3 phosphorylation, defect in correction of kinetochore-microtubule attachment, spindle checkpoint inactivation and tetraploidization due to error in cytokinesis (36,39). On the other hand, VX-680/MK-0457 treatment phenocopies loss of both Aurora-A and –B functions (40,42). These effects include delayed cell cycle progression and formation of monopolar spindles which are characteristic of loss of Aurora-A kinase activity in addition to mitotic defects resulting from Aurora-B inactivation as mentioned above.
Tetraploid cells induced by these inhibitors usually precede one cell division without cytokinesis (endoreduplication) in most cancer cells types and consequent polyploid cells ultimately arrest in pseudo-G1 state or undergo continued endoreduplication and result in cell death. ZM447439, VX-680/MK-0457 and a structural analogue VE-465 which has comparable potency in Aurora kinase inhibition (see below), were shown to induce DNA damage due to an unknown mechanism accompanied with induction of p53 and p21 proteins in an ATM/ATR dependent manner. It is plausible that inhibition of Aurora-A kinase activity that promotes p53 degradation (22) also facilitates induction of this response. Thus, determination of these different cell fates of whether polyploid cells arrest in pseudo-G1 state or undergo endoreduplication appears to depend on the integrity of the p53-p21 dependent post-mitotic signaling pathway (43,44). Ultimately, accumulated p53 is expected to induce cell death through mitochondria dependent cell death pathway in which both Bak and Bax are required (45,46). On the other hand, Cell death induction in p53 compromised cells probably occurs due to excess accumulation of chromosomes containing un-repaired damaged DNA resulting in the activation of the p73 mediated cell death signaling pathway, possibly also accompanied by the down regulation of the NF-kB mediated cell survival pathway (47–50). As a consequence, prolonged exposure of cells to the inhibitors finally leads to loss of cell viability and cell death.
Hesparadin and ZM447439 have primarily been used to investigate the functional roles of Aurora kinases in regulating specific mitotic events in phenotypic assays performed with in vitro grown cell lines. On the other hand preclinical investigations of VX-680/MK-0457 treatment have revealed ablated colony formation of primary acute myeloid leukemia (AML) cells in vitro and tumor growth inhibition accompanying significant increase in apoptosis and regression of human AML, pancreatic and colon cancer xenografts in mice and rats with minimal cytotoxicity (40). Similarly, antitumor efficacy of VX-680/MK-0457 was also shown in ovarian caner models (51). In addition to targeting Aurora kinases, VX-680/MK-0457 inhibits growth of AML cells harboring mutant FLT3 tyrosine kinase with internal tandem duplication, associated with poor prognosis of AML, as well as manifests anticancer activity towards chronic myeloid leukemia (CML) cells with recurrent imatinib- and dasatinib-resistant T351I and V299L mutant forms of Bcr-Abl fusion protein (40,52,53). It has been reported that VX-680/MK-0457 preferentially induce apoptosis in the leukemic blasts with high Aurora-A expression, but not in normal bone marrow mononuclear cells or Aurora-A low AML cells obtained from AML patients (54). In agreement with this finding, siRNA mediated silencing of Aurora-A led to induction of apoptosis in multiple myeloma cell lines (55). Crystal structural analysis of VX-680/MK-0457 in complex with the imatinib resistant mutant form of Abl tyrosine kinase indicated accommodated binding of VX-680/MK-0457 to the T351I mutant of Bcr-Abl (56). Taken together, these preclinical results indicate that VX680/MK-0457 may be an effective anticancer therapeutic agent for solid tumors expressing elevated levels of Aurora kinases and also for leukemias refractory to the tyrosine kinase inhibitors, thus possibly offering a wide pharmacologic window of therapeutic opportunity for these malignancies.
Results of a phase I–II trial revealed that MK-0457 was active in three patients with T315I refractory CML and Philadelphia chromosome positive acute lymphocytic leukemia (Ph+ ALL), with no significant extramedullary toxicity (57). An alternative treatment protocol of sequential and concomitant treatment of patients with refractory Ph+ acute phase leukemia with dasatinib and VX-680/MK-0457 has also reported success in eliminating the mutant clones with positive haematological response (58,59).
Treatment of hepatocellular carcinoma (HCC) cells with the inhibitor VE-465 showed monopolar and abnormal bipolar mitotic spindles, consistent with functional deficiency of Aurora-A, in addition to phenotypic defects associated with loss of Aurora-B function as described above. The inhibitor induces apoptosis and prevents tumor growth in HCC xenograft models (60). Since HCC is known to frequently overexpress both Aurora-A and -B kinases (61,62), VE-465 is being considered a more potent drug than VX-680/MK-0457 for the treatment of this cancer. Possible application of VE-465 to treat taxol resistant ovarian cancer has also been suggested based on the findings that treatment of ovarian cancer cells overexpressing Aurora-A with the drug in combination with paclitaxel induced apoptosis more efficiently than treatment with paclitaxel alone (63). Furthermore, VE-465 is reported to induce growth inhibition as well as apoptosis in multiple myeloma cells and CML xenograft models in nude mice expressing wild type or T351I mutant form of Bcr-Abl (55,64).
3.2. PHA-739358: Multipotent promising inhibitor
PHA-739358 was designed and developed as an ATP-competitive inhibitor of Aurora kinases based on the x-ray co-crystal structure of a preclinical candidate PHA-680632 in complex with Aurora-A. The compound shows multipotent inhibitory activities against FGFR1, Abl and TRKA with IC50 values similar to those of Aurora kinases (65–67). This multipotency of PHA-739358 is explained by the co-crystal structure study of PHA-739358 and the kinase domain of Abl in which the pyrrolopyrazole scaffold of PHA-739358 associates with an active conformation of the kinase domain in the ATP-binding pocket and lacks the steric hindrance imposed due to substitution of threonine by isoleucine at amino acid 351. The gatekeeper leucine residue at position 210 in the kinase domain of Aurora-A is similar to isoleucine and plays an equivalent role in the interaction with the inhibitor seen in case of the T351I mutant of Abl. (68). Analysis of inhibitory activity of PHA-739358 against both wild type and several mutants of Abl including T315I revealed significantly higher affinity than VX-680/MK-0457 (68). The cellular assay indicated inhibition of both Aurora-A and –B kinase activities although the observed phenotypes reflected Aurora-B rather than Aurora-A inactivation based on loss of histone H3 phosphorylation and induction of endoreduplication (67). Since the dynamics of mitotic architecture in PHA-739358 treated cells have not yet been well investigated, it will be worth while to examine the effect of this inhibitor on mitotic progression in detail. In vivo administration of PHA-739358 exhibited significant antitumor activity at the tolerated doses in several human tumor xenografts as well as spontaneous and transgenic mouse and rat tumor models of CML, ovarian, colon, mammary and hepatocellular carcinomas. The regressing tumors revealed decrease in phosphorylation of histone H3 and increase in expressions of p53 and p21 (67,69,70).
Results of two Phase I clinical studies of advanced or metastatic solid tumors are currently available. In one set of study, 56 patients with advanced solid tumor were treated with PHA-739358 in various dosages with or without G-CSF and the maximum tolerated doze were found as 500 mg/m2 without G-CSF and 750 mg/m2 with G-CSF. At these doze, febrile neutropenia with grade 4 mucositis was observed as the dose-limiting toxicity without G-CSF, and the apparent toxicity was not seen in case with G-CSF. PHA-739358 administration led to disease stabilization in 43% of patients (39% of patients without G-CSF and 50% of patient with G-CSF) and an objective response in one patient with refractory small cell lung cancer was observed lasting for 23 weeks. Tumor regression and CA125 decline was also observed in a patient with refractory ovarian cancer (71). The other set of study enrolled 50 patients for administration of PHA-739358 intravenously on days 1, 8, and 15 every 28 days in 6-hour or 3-hour infusion schedules (IVS). The maximum tolerated dose was 330mg/m2 for 6 hour for IVS. Dose-limiting toxicity was mainly neutropenia in these schedules but resulted in no complete or partial responses. However modest overall disease control rate (23.7%) and long-lasting disease stabilization for 6 or more months in 5 patients were observed. Therefore this study demonstrated that PHA-739358 has limited nonhematologic toxicity and has moderate antitumor activity (72). In both studies, inhibition of histone H3 phosphorylation was detected in skin biopsies, indicating PHA-739358 exhibits active pharmacokinetics against, at least, Aurora-B under such tolerated dozes. These preliminary evidences from preclinical and clinical studies indicate that PHA-739358 is a potentially promising anti cancer compound that may prove to be an effective therapeutic drug for achieving disease stabilization and regression either on its own or in combination depending on the results of additional clinical trial outcomes in the future.
3.3. Aurora-A specific inhibitor, MLN8054 and MLN8237
Unlike pan-Aurora kinase inhibitors, MLN8054, MLN8237 and a second generation of MLN8054 are ATP-competitive and reversible Aurora-A selective inhibitors, which are approximately 40 fold and 200 fold more sensitive towards Aurora-A compared to Aurora-B respectively. The selective inhibition of Aurora-A kinase is also significantly greater than receptor and non-receptor kinases based on in vitro enzymatic assays (34,73). MLN8054 is the first Aurora kinase inhibitor amenable to oral administration which leads to rapid absorbance in the body. The inhibitor, in addition to inducing common phenotypic effects such as G2/M accumulation, spindle defects and chromosome misalignment (73,74) has also allowed identification of previously unknown Aurora-A regulated cellular functions. MLN8054 treatment has uncovered the requirement of Aurora-A kinase activity in the maintenance of spindle assembly checkpoint mediated mitotic delay in response to microtubule perturbing drugs involving a pathway distinct from that mediated by Aurora-B (75). Further, it has been demonstrated that anti-cancer activity of MLN8054 is accompanied with induction of cell death as well as cell senescence in human tumor xenografts (73,76,77). Similar effects of mitotic defects and cell death has been observed in MLN8237 treated T-cell leukemia cells in vitro and human tumor xenografts of pediatric preclinical testing program (PPTP), which included neuroblastomas and ALL models (34,78). The study of PPTP and all 6 ALL models indicated complete response to MLN8237 with respect to reduction in tumor burden and event free survival. Similar results were also obtained from preclinical B-cell non-Hodgkin’s lymphoma models in combination use of MLN8237 and rituximab (79). Based on these initial successes in preclinical models, a number of Phase I clinical trials for anti-cancer therapeutic efficacy of these inhibitors have been initiated in patients with advanced solid tumors and advanced hematological malignancies.
3.4. Aurora-B specific inhibitor, AZD1152
AZD1152 is an Aurora-B selective inhibitor which shows 1000-fold selectivity for Aurora-B over Aurora-A and a panel of 50 additional kinases in enzymatic assays. The compound is rapidly converted to AZD1152-HQPA, the active moiety with advanced pharmacokinetic properties in human plasma (80). A recent study of AZD1152 treatment of primary AML cells and cell lines in vitro observed that mutant FLT3-IDT tyrosine kinase is a secondary target of the inhibitor (81). Treatment of a number of cancer cell types leads to suppression of phosphorylation of histone H3 and chromosome mis-alignment and induction of polyploidy and cell death which are consistent with phenotypes induced by loss of Aurora-B kinase activity (81–84). In view of the reported multipolar spindle formation, a typical phenotype of Aurora-A inhibition (74), seen in HCT116 colorectal cancer cells treated with AZD1152 at 100nM (84), it appears relevant to investigate if AZD1152 also targets Aurora-A at higher concentrations. AZD1152 has recently been shown to reduce protein stability of Aurora-B by enhancing proteasome mediated polyubiquitination and degradation in breast cancer cell line (85). It is currently unknown if this is specific to breast cancer cells or similar effects are also elicited in other types of cancer cells as well. Nonetheless, AZD1152 is the first Aurora kinase inhibitor shown to be negatively regulating both activity and stability of the protein. Administration of AZD1152 to human tumor xenografts of colorectal, breast, HCC, lung and leukemia in mouse and rat, revealed doze- and time- dependent growth inhibition of the tumors in which Aurora-B defective phenotypes were concurrently observed, indicating the ability of AZD1152 in targeting Aurora-B in vivo tumors (80,83,85–87). Further, treatment of cancer cell lines and tumor xenografts with AZD1152 in adjuvant or neoadjuvant setting as well as concomitantly with radio and chemotherapeutic agents demonstrated enhancement of the cytyotoxic effects of ionizing radiation and chemotherapeutic drugs like topoisomerase inhibitors and microtubule polymerizing inhibitors. The findings suggest that combined administration of AZD1152 and conventional therapeutic agents can help improve treatment efficacy for cancer patients (88–92). In a phase I clinical trial in 13 advanced solid tumor patients, AZD1152 was reported to be well tolerated when administrated through IV infusion at doze up to 300mg with significant disease stabilization and no clinically significant toxicities except neutropenia (93). AZD1152 is now undergoing a Phase I clinical trial in patients with AML.
In summary, we have discussed the experimental pre-clinical and clinical trial findings from those Aurora kinase inhibitors, which are relatively well characterized with respect to their target specificity and effects on mitotic phenotypes. While many reports have described evaluation of individual inhibitors in several different tumor models, there is no systematic study yet published on the evaluation of multiple Aurora kinase inhibitors in a single solid tumor model. Such studies, if undertaken in genetically profiled tumors or animal tumor models, may allow critical in vivo evaluation of the effects of the inhibitors on specific cancer associated deranged genetic pathways. Furthermore, the effects of the inhibitors are expected to be influenced by the varying expression levels of the Aurora kinase family members and their substrates in different tumors. The therapeutic efficacy will also depend on whether or not the target substrates in the tumor being treated are shared among the members of the Aurora kinase family besides the expression levels of the Aurora kinase activating proteins such as, TPX2 and INCENP, which have been reported to express at varying levels in different tumors. In addition to the inhibitors described above, a number of structurally unrelated ATP-competitive Aurora kinase inhibitors have also been developed. Functional characterizations and potency towards tumor growth inhibition in vitro and in vivo for these compounds are currently being investigated. These include pan-Aurora inhibitors SNS-314 and Gö6976, (94–97), an Aurora-B and –C inhibitor GSK1070916, (98,99) and AT9283, a broad kinase inhibitor (100,101). Summarized information on these inhibitors can be found in other recent reviews (37,38).
4. The future of cancer therapeutics with Aurora kinase inhibitors
Involvement of Aurora kinases in deregulating multiple tumor suppressor and oncogenic pathways together with the preclinical findings on the efficacy of Aurora kinase inhibitors in attenuating growth of tumor cells suggest that these molecules hold the promise of being developed into effective anticancer drugs in the future. Whether such inhibitors will be effective on their own or in combination with additional drugs targeting other oncogenic deregulated pathways would be decided based on the outcomes of the trials currently underway. The results of these trials will also reveal if tumors initially responding to the drugs acquire clinical resistance on prolonged treatment. The ability of cancer cells to acquire clinical resistance to therapeutic inhibitors is generally associated with mutations in their target protein encoding genes which impair inhibitor binding and this may be encountered in case of Aurora kinase inhibitors as well. Mutation in the oncogenic fusion protein Bcr-Abl tyrosine kinase in leukemia is one such example that makes the disease resistant to therapeutic inhibitors targeting the kinase. Imatinib was the first approved ATP-competitive inhibitor against Bcr-Abl kinase that improved survival of 95% of CML patients when treated during chronic phase of disease whereas treatment initiated during blast phase occasionally led to imatinib-resistance (102,103). Over 50 mutations in the Abl kinase domain have so far been implicated in resistance to imatinib (104). Dasatinib and nilotinib, the second generation inhibitors, targeting imatinib-resistant mutants of Bcr-Abl kinase were developed soon after identification of the mutations and treatment with these compounds have yielded encouraging clinical outcome. However, treatment with these inhibitors results in compound kinase domain mutations that render patients resistant to multiple inhibitors, demonstrating remarkable plasticity of Abl kinase domain and potential hazards of sequential kinase inhibitor treatment (53). As mentioned above, Aurora kinase inhibitors have shown efficacy in the treatment of leukemias expressing clinically resistant mutants of Bcr-Abl. Kinase. Although combinations of different BCR-ABL kinase inhibitors and Aurora kinase inhibitors such as VX-680/MK-0457 and VE-465 might constitute more effective therapeutic regimens for treating Bcr-Abl expressing leukemia, the prospect of long term efficacy of such treatment strategies will be known after additional clinical trials in the future. To circumvent the problems encountered in case of ATP-competitive inhibitors, two alternative approaches are worthy of consideration. One strategy is to find novel inhibitor molecules that inhibit kinase activity by an allosteric non-ATP competitive mechanism and the other is to develop inhibitors that interfere with substrate recognition of the target kinase. GNF2 and GNF-5, analogue of GNF2 were recently identified as allosteric inhibitors of Bcr-Abl that bind to myristate-binding site located near C-terminus of Abl kinase domain resulting in conformational change at the ATP-binding site with loss of kinase activity. These two inhibitors can act cooperatively with imatinib to inhibit both wild type and mutant Bcr-Abl, indicating that treatment with combined allosteric and ATP-competitive inhibitors is possibly an effective way to overcome clinical resistance in mutant kinase harboring refractory disease (105,106). A number of allosteric inhibitors have also been developed for multiple kinases including Akt and IkB kinase (107,108).
What can we learn about the anticancer therapeutic efficacy of Aurora kinase inhibitors from our experience with the Bcr-Abl inhibitors and the long term clinical consequence of their use in treating leukemias? As discussed, all Aurora kinase inhibitors developed to date are ATP-competitive inhibitors although their chemical structures are different in each instance. It is, therefore, possible that clinical resistance due to mutations in Aurora kinase genes may arise in patients after prolonged treatment with the inhibitors. This possibility has gained credence from recent identification of mutations in Aurora-B gene in cells exposed to an Aurora kinase inhibitor (109). In this study, HCT116 colorectal cancer cells developing resistance to ZM447439 after being treated for 3 weeks revealed mutations in the form of amino acid substitution, Y156H, G160E, G160V and H250Y in ATP-binding pocket in the kinase domain of Aurora-B. Indeed, transient expression of Aurora-B allele harboring these mutations resulted in resistance to not only ZM447439 but also VX-680/MK-0457 and AZD1152. Mutation at glycine 216 (G216) in Aurora-A and an equivalent site, G160 in Aurora-B also confer resistance to VX-680/MK-0457 (42). However, prolonged VX-680/MK-0457 treatment of cells expressing G216L mutant Aurora-A or G160L mutant of Aurora-B showed discriminate results in which Aurora-A mutant cells died in contrast to survival of Aurora-B mutant cells. These observations imply that protecting cells from failure to undergo cytokinesis, in other words, minimizing extent of aneuploidy is crucial for cell survival. Interestingly, both Aurora-A and –B mutant cells remained sensitive to Aurora-A specific inhibitor MLN8054 although Aurora-B mutant is insensitive to this inhibitor in the in vitro enzymatic assay. These findings deserve consideration in the development of next generation of Aurora kinase inhibitors and signify the relevance of both Aurora-A and –B as anticancer therapeutic targets, which regulate the cellular phenotypes through complex regulatory interactions. The above results predict that MLN8054 can target VX-680/MK-0457 resistant cancer cells, and Aurora-A may develop mutations at sites different from those of Aurora-B to acquire resistance against the inhibitors. In fact, multiple cancer associated mutations in Aurora-A have been identified, of which, some alter the kinase activity and others create or abolish protein interaction (110–112). As mentioned earlier, I31F polymorphism in Aurora-A can activate NF-κB pathway through preferential UBE2N interaction to down-regulate I B (28,113). Another V57I polymorphism in Aurora-A shows reduced kinase activity and correlates with aneuploidy. The I31F and V57I haplotype is associated with increase in esophageal cancer risk (111). V174M mutant Aurora-A is constitutively active due to stabilization of the activation loop whereas S155R mutant of Aurora-A looses interaction with TPX2 resulting in reduced kinase activity as well as mis-localization on mitotic spindle (112). Moreover, overexpression of the kinase inactivated allele of Aurora-A does not interfere with cell proliferation unless its kinase activity is completely lost (114). To date, the association between Aurora-A mutants and drugability of Aurora kinase inhibitors in enzymatic assays and in vivo studies remains unclear. In contrast to Aurora-A, no cancer associated mutation of Aurora-B has yet been identified. Taken together, the experimental findings demonstrate that contrary to Aurora-B, whose activity is essential for cell viability, both hyper- and reduced activity mutants of Aurora-A are able to induce chromosome instability, suggesting that Aurora-A may be exerting its oncogenic function through both its kinase activity dependent and independent functions. In this regard, it is significant that Aurora-A protein is essential in MYCN gene amplified neuroblastoma cells in which Aurora-A stabilizes N-Myc protein through direct interaction in a kinase activity independent manner (33). In this study, Hesperadin at a concentration sufficient for inhibition of Aurora-A kinase activity failed to destabilize N-Myc. On the other hand, a recent study in pediatric preclinical testing program found that the neuroblastoma cells without MYCN gene amplification remain sensitive to Aurora-A inhibitor MLN8237 thus documenting that N-Myc protein stabilization by Aurora-A is a restricted phenomenon occurring in a minor proportion of neuroblastoma cases (34).
The findings discussed above exemplify the complex mechanisms of action of Aurora kinases in the induction of chromosomal instability and tumorigenesis. Therefore, the most effective way to down regulate the oncogenic functions of Aurora kinases would be to develop inhibitors capable of modulating both the kinase activity and the stability of the proteins. It would also be important to structurally and functionally characterize the cancer associated mutations in Aurora kinases and evaluate the clinical efficacy of different combinations of inhibitor drugs designed to interfere with the diverse functional roles of the kinases relevant to the development of cancer associated phenotypes. Based on our current understanding, improved therapeutic efficacy may be achieved by combining ATP competitive and allosteric inhibitors to down regulate the kinase activity and stability of Aurora kinases. Detailed experimental pre-clinical studies followed by clinical trials of such inhibitors should help design novel targeted therapeutic regimens to achieve effective treatment of cancers and also overcome drug resistance likely to develop during the course of long term therapy.
Acknowledgments
We apologize to those investigators whose work has not been discussed due to space limitations. Work in the author’s lab is supported by grants of the National Institutes of Health (RO1CA089716) and University Cancer Foundation grants from UTMDACC to S.S.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Gautschi O, Heighway J, Mack PC, Purnell PR, Lara PN, Jr, Gandara DR. Aurora kinases as anticancer drug targets. Clin Cancer Res. 2008;14:1639–1648. doi: 10.1158/1078-0432.CCR-07-2179. [DOI] [PubMed] [Google Scholar]
- 2.Sharma SV, Fischbach MA, Haber DA, Settleman J. “Oncogenic shock”: explaining oncogene addiction through differential signal attenuation. Clin Cancer Res. 2006;12:4392–4395. doi: 10.1158/1078-0432.CCR-06-0096. [DOI] [PubMed] [Google Scholar]
- 3.Pérez de Castro I, de Cárcer G, Malumbres M. A census of mitotic cancer genes: new insights into tumor cell biology and cancer therapy. Carcinogenesis. 2007;28:899–912. doi: 10.1093/carcin/bgm019. [DOI] [PubMed] [Google Scholar]
- 4.Keen N, Taylor S. Aurora-kinase inhibitors as anticancer agents. Nat Rev Cancer. 2004;4:927–936. doi: 10.1038/nrc1502. [DOI] [PubMed] [Google Scholar]
- 5.Fu J, Bian M, Jiang Q, Zhang C. Roles of Aurora kinases in mitosis and tumorigenesis. Mol Cancer Res. 2007;5:1–10. doi: 10.1158/1541-7786.MCR-06-0208. [DOI] [PubMed] [Google Scholar]
- 6.Garber K. Divide and conquer: new generation of drugs targets mitosis. J Natl Cancer Inst. 2005;97:874–876. doi: 10.1093/jnci/97.12.874. [DOI] [PubMed] [Google Scholar]
- 7.Carmena M, Ruchaud S, Earnshaw WC. Making the Auroras glow: regulation of Aurora A and B kinase function by interacting proteins. Curr Opin Cell Biol. 2009;21:796–805. doi: 10.1016/j.ceb.2009.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Vader G, Lens SM. The Aurora kinase family in cell division and cancer. Biochim Biophys Acta. 2008;1786:60–72. doi: 10.1016/j.bbcan.2008.07.003. [DOI] [PubMed] [Google Scholar]
- 9.Ruchaud S, Carmena M, Earnshaw WC. Chromosomal passengers: conducting cell division. Nat Rev Mol Cell Biol. 2007;8:798–812. doi: 10.1038/nrm2257. [DOI] [PubMed] [Google Scholar]
- 10.Bayliss R, Sardon T, Vernos I, Conti E. Structural basis of Aurora-A activation by TPX2 at the mitotic spindle. Mol Cell. 2003;12:851–862. doi: 10.1016/s1097-2765(03)00392-7. [DOI] [PubMed] [Google Scholar]
- 11.Eyers PA, Erikson E, Chen LG, Maller JL. A novel mechanism for activation of the protein kinase Aurora A. Curr Biol. 2003;13:691–697. doi: 10.1016/s0960-9822(03)00166-0. [DOI] [PubMed] [Google Scholar]
- 12.Jeyaprakash AA, Klein UR, Lindner D, Ebert J, Nigg EA, Conti E. Structure of a Survivin-Borealin-INCENP core complex reveals how chromosomal passengers travel together. Cell. 2007;131:271–285. doi: 10.1016/j.cell.2007.07.045. [DOI] [PubMed] [Google Scholar]
- 13.Adams RR, Wheatley SP, Gouldsworthy AM, Kandels-Lewis SE, Carmena M, Smythe C, Gerloff DL, Earnshaw WC. INCENP binds the Aurora-related kinase AIRK2 and is required to target it to chromosomes, the central spindle and cleavage furrow. Curr Biol. 2000;10:1075–1078. doi: 10.1016/s0960-9822(00)00673-4. [DOI] [PubMed] [Google Scholar]
- 14.Fu J, Bian M, Liu J, Jiang Q, Zhang C. A single amino acid change converts Aurora-A into Aurora-B-like kinase in terms of partner specificity and cellular function. Proc Natl Acad Sci U S A. 2009;106:939–944. doi: 10.1073/pnas.0900833106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hans F, Skoufias DA, Dimitrov S, Margolis RL. Molecular distinctions between Aurora A and B: a single residue change transforms Aurora A into correctly localized and functional Aurora B. Mol Biol Cell. 2009;20:3491–502. doi: 10.1091/mbc.E09-05-0370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bischoff JR, Anderson L, Zhu Y, Mossie K, Ng L, Souza B, Schryver B, Flanagan P, Clairvoyant F, Ginther C, Chan CS, Novotny M, Slamon DJ, Plowman GD. A homologue of Drosophila aurora kinase is oncogenic and amplified in human colorectal cancers. EMBO J. 1998;17:3052–3065. doi: 10.1093/emboj/17.11.3052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhou H, Kuang J, Zhong L, Kuo WL, Gray JW, Sahin A, Brinkley BR, Sen S. Tumour amplified kinase STK15/BTAK induces centrosome amplification, aneuploidy and transformation. Nat Genet. 1998;20:189–193. doi: 10.1038/2496. [DOI] [PubMed] [Google Scholar]
- 18.Ota T, Suto S, Katayama H, Han ZB, Suzuki F, Maeda M, Tanino M, Terada Y, Tatsuka M. Increased mitotic phosphorylation of histone H3 attributable to AIM-1/Aurora-B overexpression contributes to chromosome number instability. Cancer Res. 2002;62:5168–5177. [PubMed] [Google Scholar]
- 19.Macůrek L, Lindqvist A, Lim D, Lampson MA, Klompmaker R, Freire R, Clouin C, Taylor SS, Yaffe MB, Medema RH. Polo-like kinase-1 is activated by aurora A to promote checkpoint recovery. Nature. 2008;455:119–123. doi: 10.1038/nature07185. [DOI] [PubMed] [Google Scholar]
- 20.Seki A, Coppinger JA, Jang CY, Yates JR, Fang G. Bora and the kinase Aurora a cooperatively activate the kinase Plk1 and control mitotic entry. Science. 2008;320:1655–1658. doi: 10.1126/science.1157425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Katayama H, Sasai K, Kloc M, Brinkley BR, Sen S. Aurora kinase-A regulates kinetochore/chromatin associated microtubule assembly in human cells. Cell Cycle. 2008;7:2691–2704. doi: 10.4161/cc.7.17.6460. [DOI] [PubMed] [Google Scholar]
- 22.Katayama H, Sasai K, Kawai H, Yuan ZM, Bondaruk J, Suzuki F, Fujii S, Arlinghaus RB, Czerniak BA, Sen S. Phosphorylation by aurora kinase A induces Mdm2-mediated destabilization and inhibition of p53. Nat Genet. 2004;36:55–62. doi: 10.1038/ng1279. [DOI] [PubMed] [Google Scholar]
- 23.Liu Q, Kaneko S, Yang L, Feldman RL, Nicosia SV, Chen J, Cheng JQ. Aurora-A abrogation of p53 DNA binding and transactivation activity by phosphorylation of serine 215. J Biol Chem. 2004;279:52175–52182. doi: 10.1074/jbc.M406802200. [DOI] [PubMed] [Google Scholar]
- 24.Ouchi M, Fujiuchi N, Sasai K, Katayama H, Minamishima YA, Ongusaha PP, Deng C, Sen S, Lee SW, Ouchi T. BRCA1 phosphorylation by Aurora-A in the regulation of G2 to M transition. J Biol Chem. 2004;279:19643–19648. doi: 10.1074/jbc.M311780200. [DOI] [PubMed] [Google Scholar]
- 25.Ryser S, Dizin E, Jefford CE, Delaval B, Gagos S, Christodoulidou A, Krause KH, Birnbaum D, Irminger-Finger I. Distinct roles of BARD1 isoforms in mitosis: full-length BARD1 mediates Aurora B degradation, cancer-associated BARD1beta scaffolds Aurora B and BRCA2. Cancer Res. 2009;69:1125–1134. doi: 10.1158/0008-5472.CAN-08-2134. [DOI] [PubMed] [Google Scholar]
- 26.Yang H, Ou CC, Feldman RL, Nicosia SV, Kruk PA, Cheng JQ. Aurora-A kinase regulates telomerase activity through c-Myc in human ovarian and breast epithelial cells. Cancer Res. 2004;64:463–467. doi: 10.1158/0008-5472.can-03-2907. [DOI] [PubMed] [Google Scholar]
- 27.Dar AA, Belkhiri A, El-Rifai W. The Aurora kinase A regulates GSK-3beta in gastric cancer cells. Oncogene. 2009;28:866–875. doi: 10.1038/onc.2008.434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Briassouli P, Chan F, Savage K, Reis-Filho JS, Linardopoulos S. Aurora-A regulation of nuclear factor-kappaB signaling by phosphorylation of IkappaBalpha. Cancer Res. 2007;67:1689–1695. doi: 10.1158/0008-5472.CAN-06-2272. [DOI] [PubMed] [Google Scholar]
- 29.Irelan JT, Murphy TJ, DeJesus PD, Teo H, Xu D, Gomez-Ferreria MA, Zhou Y, Miraglia LJ, Rines DR, Verma IM, Sharp DJ, Tergaonkar V, Chanda SK. A role for IkappaB kinase 2 in bipolar spindle assembly. Proc Natl Acad Sci U S A. 2007;104:16940–16945. doi: 10.1073/pnas.0706493104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wu JC, Chen TY, Yu CT, Tsai SJ, Hsu JM, Tang MJ, Chou CK, Lin WJ, Yuan CJ, Huang CY. Identification of V23RalA-Ser194 as a critical mediator for Aurora-A-induced cellular motility and transformation by small pool expression screening. J Biol Chem. 2005;280:9013–9022. doi: 10.1074/jbc.M411068200. [DOI] [PubMed] [Google Scholar]
- 31.Lim KH, Brady DC, Kashatus DF, Ancrile BB, Der CJ, Cox AD, Counter CM. Aurora-A phosphorylates, activates, and relocalizes the small GTPase RalA. Mol Cell Biol. 2010;30:508–523. doi: 10.1128/MCB.00916-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bee C, Moshnikova A, Mellor CD, Molloy J, Koryakina Y, Stieglitz B, Khokhlatchev A, Herrmann C. The growth and tumor suppressor novel ras effector 1A (NORE1A) is a regulatory node between ras signaling and microtubule nucleation. J Biol Chem. 2010 doi: 10.1074/jbc.M109.081562. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Otto T, Horn S, Brockmann M, Eilers U, Schüttrumpf L, Popov N, Kenney AM, Schulte JH, Beijersbergen R, Christiansen H, Berwanger B, Eilers M. Stabilization of N-Myc is a critical function of Aurora A in human neuroblastoma. Cancer Cell. 2009;15:67–78. doi: 10.1016/j.ccr.2008.12.005. [DOI] [PubMed] [Google Scholar]
- 34.Maris JM, Morton CL, Gorlick R, Kolb EA, Lock R, Carol H, Keir ST, Reynolds CP, Kang MH, Wu J, Smith MA, Houghton PJ. Initial testing of the aurora kinase a inhibitor MLN8237 by the Pediatric Preclinical Testing Program (PPTP) Pediatr Blood Cancer. 2010 doi: 10.1002/pbc.22430. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lapenna S, Giordano A. Cell cycle kinases as therapeutic targets for cancer. Nat Rev Drug Discov. 2009;8:547–566. doi: 10.1038/nrd2907. [DOI] [PubMed] [Google Scholar]
- 36.Ditchfield C, Johnson VL, Tighe A, Ellston R, Haworth C, Johnson T, Mortlock A, Keen N, Taylor SS. Aurora B couples chromosome alignment with anaphase by targeting BubR1, Mad2, and Cenp-E to kinetochores. J Cell Biol. 2003;161:267–280. doi: 10.1083/jcb.200208091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cheung CH, Coumar MS, Hsieh HP, Chang JY. Aurora kinase inhibitors in preclinical and clinical testing. Expert Opin Investig Drugs. 2009;18:379–398. doi: 10.1517/13543780902806392. [DOI] [PubMed] [Google Scholar]
- 38.Pérez Fidalgo JA, Roda D, Roselló S, Rodríguez-Braun E, Cervantes A. Aurora kinase inhibitors: a new class of drugs targeting the regulatory mitotic system. Clin Transl Oncol. 2009;11:787–798. doi: 10.1007/s12094-009-0447-2. [DOI] [PubMed] [Google Scholar]
- 39.Hauf S, Cole RW, LaTerra S, Zimmer C, Schnapp G, Walter R, Heckel A, van Meel J, Rieder CL, Peters JM. The small molecule Hesperadin reveals a role for Aurora B in correcting kinetochore-microtubule attachment and in maintaining the spindle assembly checkpoint. J Cell Biol. 2003;161:281–294. doi: 10.1083/jcb.200208092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Harrington EA, Bebbington D, Moore J, Rasmussen RK, Ajose-Adeogun AO, Nakayama T, Graham JA, Demur C, Hercend T, Diu-Hercend A, Su M, Golec JM, Miller KM. VX-680, a potent and selective small-molecule inhibitor of the Aurora kinases, suppresses tumor growth in vivo. Nat Med. 2004;10:262–267. doi: 10.1038/nm1003. [DOI] [PubMed] [Google Scholar]
- 41.Walter RA, Heckel GJ, Roth J, Kley G, Schnapp M, Lenter JCA, van Meel Spevak W, Weyer-Czernilofsky U. Sulfonylamino substituted 3-(aminomethylide)-2-indolinones as cell proliferation inhibitors. International Publication Number WO 02/36564 Patent Cooperation Treaty. 2002 inventors and assignees.
- 42.Scutt PJ, Chu ML, Sloane DA, Cherry M, Bignell CR, Williams DH, Eyers PA. Discovery and exploitation of inhibitor-resistant aurora and polo kinase mutants for the analysis of mitotic networks. J Biol Chem. 2009;284:15880–15893. doi: 10.1074/jbc.M109.005694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gizatullin F, Yao Y, Kung V, Harding MW, Loda M, Shapiro GI. The Aurora kinase inhibitor VX-680 induces endoreduplication and apoptosis preferentially in cells with compromised p53-dependent postmitotic checkpoint function. Cancer Res. 2006;66:7668–7677. doi: 10.1158/0008-5472.CAN-05-3353. [DOI] [PubMed] [Google Scholar]
- 44.Dreier MR, Grabovich AZ, Katusin JD, Taylor WR. Short and long-term tumor cell responses to Aurora kinase inhibitors. Exp Cell Res. 2009;315:1085–1099. doi: 10.1016/j.yexcr.2009.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kojima K, Konopleva M, Tsao T, Nakakuma H, Andreeff M. Concomitant inhibition of Mdm2-p53 interaction and Aurora kinases activates the p53-dependent postmitotic checkpoints and synergistically induces p53-mediated mitochondrial apoptosis along with reduced endoreduplication in acute myelogenous leukemia. Blood. 2008;1–12:2886–2895. doi: 10.1182/blood-2008-01-128611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Li M, Jung A, Ganswindt U, Marini P, Friedl A, Daniel PT, Lauber K, Jendrossek V, Belka C. Aurora kinase inhibitor ZM447439 induces apoptosis via mitochondrial pathways. Biochem Pharmacol. 2010;79:122–129. doi: 10.1016/j.bcp.2009.08.011. [DOI] [PubMed] [Google Scholar]
- 47.Sun C, Chan F, Briassouli P, Linardopoulos S. Aurora kinase inhibition downregulates NF-kappaB and sensitises tumour cells to chemotherapeutic agents. Biochem Biophys Res Commun. 2007;352:220–225. doi: 10.1016/j.bbrc.2006.11.004. [DOI] [PubMed] [Google Scholar]
- 48.Dar AA, Belkhiri A, Ecsedy J, Zaika A, El-Rifai W. Aurora kinase A inhibition leads to p73-dependent apoptosis in p53-deficient cancer cells. Cancer Res. 2008;68:8998–9004. doi: 10.1158/0008-5472.CAN-08-2658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cheok CF, Kua N, Kaldis P, Lane DP. Combination of nutlin-3 and VX-680 selectively targets p53 mutant cells with reversible effects on cells expressing wild-type p53. Cell Death Differ. 2010 doi: 10.1038/cdd.2010.18. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 50.Ikezoe T, Yang J, Nishioka C, Yokoyama A. p53 is critical for the Aurora B kinase inhibitor-mediated apoptosis in acute myelogenous leukemia cells. Int J Hematol. 2010;91:69–77. doi: 10.1007/s12185-009-0462-7. [DOI] [PubMed] [Google Scholar]
- 51.Lin YG, Immaneni A, Merritt WM, Mangala LS, Kim SW, Shahzad MM, Tsang YT, Armaiz-Pena GN, Lu C, Kamat AA, Han LY, Spannuth WA, Nick AM, Landen CN, Jr, Wong KK, Gray MJ, Coleman RL, Bodurka DC, Brinkley WR, Sood AK. Targeting aurora kinase with MK-0457 inhibits ovarian cancer growth. Clin Cancer Res. 2008;14:5437–5446. doi: 10.1158/1078-0432.CCR-07-4922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Carter TA, Wodicka LM, Shah NP, Velasco AM, Fabian MA, Treiber DK, Milanov ZV, Atteridge CE, Biggs WH, 3rd, Edeen PT, Floyd M, Ford JM, Grotzfeld RM, Herrgard S, Insko DE, Mehta SA, Patel HK, Pao W, Sawyers CL, Varmus H, Zarrinkar PP, Lockhart DJ. Inhibition of drug-resistant mutants of ABL, KIT, and EGF receptor kinases. Proc Natl Acad Sci U S A. 2005;102:11011–11016. doi: 10.1073/pnas.0504952102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Shah NP, Skaggs BJ, Branford S, Hughes TP, Nicoll JM, Paquette RL, Sawyers CL. Sequential ABL kinase inhibitor therapy selects for compound drug-resistant BCR-ABL mutations with altered oncogenic potency. J Clin Invest. 2007;117:2562–2569. doi: 10.1172/JCI30890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Huang XF, Luo SK, Xu J, Li J, Xu DR, Wang LH, Yan M, Wang XR, Wan XB, Zheng FM, Zeng YX, Liu Q. Aurora kinase inhibitory VX-680 increases Bax/Bcl-2 ratio and induces apoptosis in Aurora-A-high acute myeloid leukemia. Blood. 2008;111:2854–2865. doi: 10.1182/blood-2007-07-099325. [DOI] [PubMed] [Google Scholar]
- 55.Evans R, Naber C, Steffler T, Checkland T, Keats J, Maxwell C, Perry T, Chau H, Belch A, Pilarski L, Reiman T. Aurora A kinase RNAi and small molecule inhibition of Aurora kinases with VE-465 induce apoptotic death in multiple myeloma cells. Leuk Lymphoma. 2008;49:559–569. doi: 10.1080/10428190701824544. [DOI] [PubMed] [Google Scholar]
- 56.Young MA, Shah NP, Chao LH, Seeliger M, Milanov ZV, Biggs WH, 3rd, Treiber DK, Patel HK, Zarrinkar PP, Lockhart DJ, Sawyers CL, Kuriyan J. Structure of the kinase domain of an imatinib-resistant Abl mutant in complex with the Aurora kinase inhibitor VX-680. Cancer Res. 2006;66:1007–1014. doi: 10.1158/0008-5472.CAN-05-2788. [DOI] [PubMed] [Google Scholar]
- 57.Giles FJ, Cortes J, Jones D, Bergstrom D, Kantarjian H, Freedman SJ. MK-0457, a novel kinase inhibitor, is active in patients with chronic myeloid leukemia or acute lymphocytic leukemia with the T315I BCR-ABL mutation. Blood. 2007;109:500–502. doi: 10.1182/blood-2006-05-025049. [DOI] [PubMed] [Google Scholar]
- 58.Papayannidis C, Lacobucci L, Soverini S, Paolini S, Santucci S, Cilloni D, Messa F, Pane F, Meneghini V, Giannoulia P, Ottaviani E, Testoni N, Lama B, Pantaleo M, Baccarani M, Martinelli G. Innovative phase I-II study of concomitant and consecutive treatment with Dasatinib and MK-0457 in refractory Ph+ CML and ALL patients. AACR, Abstract meetings. 2009 [Google Scholar]
- 59.Sanchez-Guijo FM, Lopez-Jimenez J, Gonzalez T, Santamaría C, González M, Del Cañizo MC. Multitargeted sequential therapy with MK-0457 and dasatinib followed by stem cell transplantation for T315I mutated chronic myeloid leukemia. Leuk Res. 2009;33:20–22. doi: 10.1016/j.leukres.2008.10.020. [DOI] [PubMed] [Google Scholar]
- 60.Lin ZZ, Hsu HC, Hsu CH, Yeh PY, Huang CY, Huang YF, Chen TJ, Kuo SH, Hsu C, Hu FC, Jeng YM, Chung Y, Cheng AL. The Aurora kinase inhibitor VE-465 has anticancer effects in pre-clinical studies of human hepatocellular carcinoma. J Hepatol. 2009;50:518–527. doi: 10.1016/j.jhep.2008.10.022. [DOI] [PubMed] [Google Scholar]
- 61.Jeng YM, Peng SY, Lin CY, Hsu HC. Overexpression and amplification of Aurora-A in hepatocellular carcinoma. Clin Cancer Res. 2004;10:2065–2071. doi: 10.1158/1078-0432.ccr-1057-03. [DOI] [PubMed] [Google Scholar]
- 62.Sistayanarain A, Tsuneyama K, Zheng H, Takahashi H, Nomoto K, Cheng C, Murai Y, Tanaka A, Takano Y. Expression of Aurora-B kinase and phosphorylated histone H3 in hepatocellular carcinoma. Anticancer Res. 2006;26:3585–3593. [PubMed] [Google Scholar]
- 63.Scharer CD, Laycock N, Osunkoya AO, Logani S, McDonald JF, Benigno BB, Moreno CS. Aurora kinase inhibitors synergize with paclitaxel to induce apoptosis in ovarian cancer cells. J Transl Med. 2008;6:79–91. doi: 10.1186/1479-5876-6-79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Akahane D, Tauchi T, Okabe S, Nunoda K, Ohyashiki K. Activity of a novel Aurora kinase inhibitor against the T315I mutant form of BCR-ABL: in vitro and in vivo studies. Cancer Sci. 2008;99:1251–1257. doi: 10.1111/j.1349-7006.2008.00810.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Fancelli D, Berta D, Bindi S, Cameron A, Cappella P, Carpinelli P, Catana C, Forte B, Giordano P, Giorgini ML, Mantegani S, Marsiglio A, Meroni M, Moll J, Pittalà V, Roletto F, Severino D, Soncini C, Storici P, Tonani R, Varasi M, Vulpetti A, Vianello P. Potent and selective Aurora inhibitors identified by the expansion of a novel scaffold for protein kinase inhibition. J Med Chem. 2005;48:3080–3084. doi: 10.1021/jm049076m. [DOI] [PubMed] [Google Scholar]
- 66.Fancelli D, Moll J, Varasi M, Bravo R, Artico R, Berta D, Bindi S, Cameron A, Candiani I, Cappella P, Carpinelli P, Croci W, Forte B, Giorgini ML, Klapwijk J, Marsiglio A, Pesenti E, Rocchetti M, Roletto F, Severino D, Soncini C, Storici P, Tonani R, Zugnoni P, Vianello P. 1,4,5,6-tetrahydropyrrolo[3,4-c]pyrazoles: identification of a potent Aurora kinase inhibitor with a favorable antitumor kinase inhibition profile. J Med Chem. 2006;49:7247–7251. doi: 10.1021/jm060897w. [DOI] [PubMed] [Google Scholar]
- 67.Carpinelli P, Ceruti R, Giorgini ML, Cappella P, Gianellini L, Croci V, Degrassi A, Texido G, Rocchetti M, Vianello P, Rusconi L, Storici P, Zugnoni P, Arrigoni C, Soncini C, Alli C, Patton V, Marsiglio A, Ballinari D, Pesenti E, Fancelli D, Moll J. PHA-739358, a potent inhibitor of Aurora kinases with a selective target inhibition profile relevant to cancer. Mol Cancer Ther. 2007;6:3158–3168. doi: 10.1158/1535-7163.MCT-07-0444. [DOI] [PubMed] [Google Scholar]
- 68.Modugno M, Casale E, Soncini C, Rosettani P, Colombo R, Lupi R, Rusconi L, Fancelli D, Carpinelli P, Cameron AD, Isacchi A, Moll J. Crystal structure of the T315I Abl mutant in complex with the aurora kinases inhibitor PHA-739358. Cancer Res. 2007;67:7987–7990. doi: 10.1158/0008-5472.CAN-07-1825. [DOI] [PubMed] [Google Scholar]
- 69.Gontarewicz A, Balabanov S, Keller G, Colombo R, Graziano A, Pesenti E, Benten D, Bokemeyer C, Fiedler W, Moll J, Brümmendorf TH. Simultaneous targeting of Aurora kinases and Bcr-Abl kinase by the small molecule inhibitor PHA-739358 is effective against imatinib-resistant BCR-ABL mutations including T315I. Blood. 2008;111:4355–4364. doi: 10.1182/blood-2007-09-113175. [DOI] [PubMed] [Google Scholar]
- 70.Benten D, Keller G, Quaas A, Schrader J, Gontarewicz A, Balabanov S, Braig M, Wege H, Moll J, Lohse AW, Brummendorf TH. Aurora kinase inhibitor PHA-739358 suppresses growth of hepatocellular carcinoma in vitro and in a xenograft mouse model. Neoplasia. 2009;11:934–944. doi: 10.1593/neo.09664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Cohen RB, Jones SF, Aggarwal C, von Mehren M, Cheng J, Spigel DR, Greco FA, Mariani M, Rocchetti M, Ceruti R, Comis S, Laffranchi B, Moll J, Burris HA. A phase I dose-escalation study of danusertib (PHA-739358) administered as a 24-hour infusion with and without granulocyte colony-stimulating factor in a 14-day cycle in patients with advanced solid tumors. Clin Cancer Res. 2009;15:6694–6701. doi: 10.1158/1078-0432.CCR-09-1445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Steeghs N, Eskens FA, Gelderblom H, Verweij J, Nortier JW, Ouwerkerk J, van Noort C, Mariani M, Spinelli R, Carpinelli P, Laffranchi B, de Jonge MJ. Phase I pharmacokinetic and pharmacodynamic study of the aurora kinase inhibitor danusertib in patients with advanced or metastatic solid tumors. J Clin Oncol. 2009;27:5094–5101. doi: 10.1200/JCO.2008.21.6655. [DOI] [PubMed] [Google Scholar]
- 73.Manfredi MG, Ecsedy JA, Meetze KA, Balani SK, Burenkova O, Chen W, Galvin KM, Hoar KM, Huck JJ, LeRoy PJ, Ray ET, Sells TB, Stringer B, Stroud SG, Vos TJ, Weatherhead GS, Wysong DR, Zhang M, Bolen JB, Claiborne CF. Antitumor activity of MLN8054, an orally active small-molecule inhibitor of Aurora A kinase. Proc Natl Acad Sci U S A. 2007;104:4106–4111. doi: 10.1073/pnas.0608798104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Hoar K, Chakravarty A, Rabino C, Wysong D, Bowman D, Roy N, Ecsedy JA. MLN8054, a small-molecule inhibitor of Aurora A, causes spindle pole and chromosome congression defects leading to aneuploidy. Mol Cell Biol. 2007;27:4513–4525. doi: 10.1128/MCB.02364-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Wysong DR, Chakravarty A, Hoar K, Ecsedy JA. The inhibition of Aurora A abrogates the mitotic delay induced by microtubule perturbing agents. Cell Cycle. 2009;8:876–88. doi: 10.4161/cc.8.6.7897. [DOI] [PubMed] [Google Scholar]
- 76.Shang X, Burlingame SM, Okcu MF, Ge N, Russell HV, Egler RA, David RD, Vasudevan SA, Yang J, Nuchtern JG. Aurora A is a negative prognostic factor and a new therapeutic target in human neuroblastoma. Mol Cancer Ther. 2009;8:2461–2469. doi: 10.1158/1535-7163.MCT-08-0857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Huck JJ, Zhang M, McDonald A, Bowman D, Hoar KM, Stringer B, Ecsedy J, Manfredi MG, Hyer ML. MLN8054, an inhibitor of Aurora A kinase, induces senescence in human tumor cells both in vitro and in vivo. Mol Cancer Res. 2010;8:373–384. doi: 10.1158/1541-7786.MCR-09-0300. [DOI] [PubMed] [Google Scholar]
- 78.Tomita M, Mori N. Aurora A selective inhibitor MLN8237 suppresses the growth and survival of HTLV-1-infected T-cells in vitro. Cancer Sci. 2010 doi: 10.1111/j.1349-7006.2010.01499.x. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Zhang M, Huck J, Hyer M, Ecsedy J, Manfredi M. Effect of Aurora A kinase inhibitor MLN8237 combined with rituximab on antitumor activity in preclinical B-cell non-Hodgkin’s lymphoma models. ASCO meeting. 2009 [Google Scholar]
- 80.Mortlock AA, Foote KM, Heron NM, Jung FH, Pasquet G, Lohmann JJ, Warin N, Renaud F, De Savi C, Roberts NJ, Johnson T, Dousson CB, Hill GB, Perkins D, Hatter G, Wilkinson RW, Wedge SR, Heaton SP, Odedra R, Keen NJ, Crafter C, Brown E, Thompson K, Brightwell S, Khatri L, Brady MC, Kearney S, McKillop D, Rhead S, Parry T, Green S. Discovery, synthesis, and in vivo activity of a new class of pyrazoloquinazolines as selective inhibitors of aurora B kinase. J Med Chem. 2007;50:2213–2224. doi: 10.1021/jm061335f. [DOI] [PubMed] [Google Scholar]
- 81.Grundy M, Seedhouse C, Shang S, Richardson J, Russell N, Pallis M. The FLT3 internal tandem duplication mutation is a secondary target of the aurora B kinase inhibitor AZD1152-HQPA in acute myelogenous leukemia cells. Mol Cancer Ther. 2010;9:661–672. doi: 10.1158/1535-7163.MCT-09-1144. [DOI] [PubMed] [Google Scholar]
- 82.Mortlock AA, Boyle FT, Green S. AZD1152, a selective inhibitor of Aurora B kinase, inhibits human tumor xenograft growth by inducing apoptosis. Clin Cancer Res. 2007;13:3682–3688. doi: 10.1158/1078-0432.CCR-06-2979. [DOI] [PubMed] [Google Scholar]
- 83.Wilkinson RW, Odedra R, Heaton SP, Wedge SR, Keen NJ, Crafter C, Foster JR, Brady MC, Bigley A, Brown E, Byth KF, Barrass NC, Mundt KE, Foote KM, Heron NM, Jung FH, Mortlock AA, Boyle FT, Green S. AZD1152, a selective inhibitor of Aurora B kinase, inhibits human tumor xenograft growth by inducing apoptosis. Clin Cancer Res. 2007;13:3682–3688. doi: 10.1158/1078-0432.CCR-06-2979. [DOI] [PubMed] [Google Scholar]
- 84.Tao Y, Zhang P, Girdler F, Frascogna V, Castedo M, Bourhis J, Kroemer G, Deutsch E. Enhancement of radiation response in p53-deficient cancer cells by the Aurora-B kinase inhibitor AZD1152. Oncogene. 2008;27:3244–3255. doi: 10.1038/sj.onc.1210990. [DOI] [PubMed] [Google Scholar]
- 85.Gully CP, Zhang F, Chen J, Yeung JA, Velazquez-Torres G, Wang E, Yeung SC, Lee MH. Antineoplastic effects of an Aurora B kinase inhibitor in breast cancer. Mol Cancer. 2010;9:42. doi: 10.1186/1476-4598-9-42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Oke A, Pearce D, Wilkinson RW, Crafter C, Odedra R, Cavenagh J, Fitzgibbon J, Lister AT, Joel S, Bonnet D. AZD1152 rapidly and negatively affects the growth and survival of human acute myeloid leukemia cells in vitro and in vivo. Cancer Res. 2009;69:4150–4158. doi: 10.1158/0008-5472.CAN-08-3203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Tanaka S, Arii S. Medical treatments: in association or alone, their role and their future perspectives: Novel molecular-targeted therapy for hepatocellular carcinoma. J Hepatobiliary Pancreat Surg. 2009 doi: 10.1007/s00534-009-0238-8. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 88.Yang J, Ikezoe T, Nishioka C, Tasaka T, Taniguchi A, Kuwayama Y, Komatsu N, Bandobashi K, Togitani K, Koeffler HP, Taguchi H, Yokoyama A. AZD1152, a novel and selective aurora B kinase inhibitor, induces growth arrest, apoptosis, and sensitization for tubulin depolymerizing agent or topoisomerase II inhibitor in human acute leukemia cells in vitro and in vivo. Blood. 2007;110:2034–2040. doi: 10.1182/blood-2007-02-073700. [DOI] [PubMed] [Google Scholar]
- 89.Evans RP, Naber C, Steffler T, Checkland T, Maxwell CA, Keats JJ, Belch AR, Pilarski LM, Lai R, Reiman T. The selective Aurora B kinase inhibitor AZD1152 is a potential new treatment for multiple myeloma. Br J Haematol. 2008;140:295–302. doi: 10.1111/j.1365-2141.2007.06913.x. [DOI] [PubMed] [Google Scholar]
- 90.Ikezoe T, Takeuchi T, Yang J, Adachi Y, Nishioka C, Furihata M, Koeffler HP, Yokoyama A. Analysis of Aurora B kinase in non-Hodgkin lymphoma. Lab Invest. 2009;89:1364–1373. doi: 10.1038/labinvest.2009.106. [DOI] [PubMed] [Google Scholar]
- 91.Nair JS, de Stanchina E, Schwartz GK. The topoisomerase I poison CPT-11 enhances the effect of the aurora B kinase inhibitor AZD1152 both in vitro and in vivo. Clin Cancer Res. 2009;15:2022–2030. doi: 10.1158/1078-0432.CCR-08-1826. [DOI] [PubMed] [Google Scholar]
- 92.Tao Y, Leteur C, Calderaro J, Girdler F, Zhang P, Frascogna V, Varna M, Opolon P, Castedo M, Bourhis J, Kroemer G, Deutsch E. The aurora B kinase inhibitor AZD1152 sensitizes cancer cells to fractionated irradiation and induces mitotic catastrophe. Cell Cycle. 2009;8:3172–3181. doi: 10.4161/cc.8.19.9729. [DOI] [PubMed] [Google Scholar]
- 93.Schellens JH, Boss D, Witteveen PO, Zandvliet A, Beijnen JH, Voogel-Fuchs M, Morris C, Wilson D, Voest EE. Phase I and pharmacological study of the novel aurora kinase inhibitor AZD1152. ASCO Annual Meeting. 2006 [Google Scholar]
- 94.Oslob JD, Romanowski MJ, Allen DA, Baskaran S, Bui M, Elling RA, Flanagan WM, Fung AD, Hanan EJ, Harris S, Heumann SA, Hoch U, Jacobs JW, Lam J, Lawrence CE, McDowell RS, Nannini MA, Shen W, Silverman JA, Sopko MM, Tangonan BT, Teague J, Yoburn JC, Yu CH, Zhong M, Zimmerman KM, O’Brien T, Lew W. Discovery of a potent and selective aurora kinase inhibitor. Bioorg Med Chem Lett. 2008;18:4880–4884. doi: 10.1016/j.bmcl.2008.07.073. [DOI] [PubMed] [Google Scholar]
- 95.Stolz A, Vogel C, Schneider V, Ertych N, Kienitz A, Yu H, Bastians H. Pharmacologic abrogation of the mitotic spindle checkpoint by an indolocarbazole discovered by cellular screening efficiently kills cancer cells. Cancer Res. 2009;69:3874–3883. doi: 10.1158/0008-5472.CAN-08-3597. [DOI] [PubMed] [Google Scholar]
- 96.VanderPorten EC, Taverna P, Hogan JN, Ballinger MD, Flanagan WM, Fucini RV. The Aurora kinase inhibitor SNS-314 shows broad therapeutic potential with chemotherapeutics and synergy with microtubule-targeted agents in a colon carcinoma model. Mol Cancer Ther. 2009;8:930–939. doi: 10.1158/1535-7163.MCT-08-0754. [DOI] [PubMed] [Google Scholar]
- 97.Arbitrario JP, Belmont BJ, Evanchik MJ, Flanagan WM, Fucini RV, Hansen SK, Harris SO, Hashash A, Hoch U, Hogan JN, Howlett AR, Jacobs JW, Lam JW, Ritchie SC, Romanowski MJ, Silverman JA, Stockett DE, Teague JN, Zimmerman KM, Taverna P. SNS-314, a pan-Aurora kinase inhibitor, shows potent anti-tumor activity and dosing flexibility in vivo. Cancer Chemother Pharmacol. 2010;65:707–717. doi: 10.1007/s00280-009-1076-8. [DOI] [PubMed] [Google Scholar]
- 98.Anderson K, Lai Z, McDonald OB, Stuart JD, Nartey EN, Hardwicke MA, Newlander K, Dhanak D, Adams J, Patrick D, Copeland RA, Tummino PJ, Yang J. Biochemical characterization of GSK1070916, a potent and selective inhibitor of Aurora B and Aurora C kinases with an extremely long residence time1. Biochem J. 2009;420:259–265. doi: 10.1042/BJ20090121. [DOI] [PubMed] [Google Scholar]
- 99.Hardwicke MA, Oleykowski CA, Plant R, Wang J, Liao Q, Moss K, Newlander K, Adams JL, Dhanak D, Yang J, Lai Z, Sutton D, Patrick D. GSK1070916, a potent Aurora B/C kinase inhibitor with broad antitumor activity in tissue culture cells and human tumor xenograft models. Mol Cancer Ther. 2009;8:1808–1817. doi: 10.1158/1535-7163.MCT-09-0041. [DOI] [PubMed] [Google Scholar]
- 100.Howard S, Berdini V, Boulstridge JA, Carr MG, Cross DM, Curry J, Devine LA, Early TR, Fazal L, Gill AL, Heathcote M, Maman S, Matthews JE, McMenamin RL, Navarro EF, O’Brien MA, O’Reilly M, Rees DC, Reule M, Tisi D, Williams G, Vinković M, Wyatt PG. Fragment-based discovery of the pyrazol-4-yl urea (AT9283), a multitargeted kinase inhibitor with potent aurora kinase activity. J Med Chem. 2009;52:379–388. doi: 10.1021/jm800984v. [DOI] [PubMed] [Google Scholar]
- 101.Curry J, Angove H, Fazal L, Lyons J, Reule M, Thompson N, Wallis N. Aurora B kinase inhibition in mitosis: strategies for optimising the use of aurora kinase inhibitors such as AT9283. Cell Cycle. 2009;8:1921–1929. doi: 10.4161/cc.8.12.8741. [DOI] [PubMed] [Google Scholar]
- 102.Branford S, Rudzki Z, Walsh S, Parkinson I, Grigg A, Szer J, Taylor K, Herrmann R, Seymour JF, Arthur C, Joske D, Lynch K, Hughes T. Detection of BCR-ABL mutations in patients with CML treated with imatinib is virtually always accompanied by clinical resistance, and mutations in the ATP phosphate-binding loop (P-loop) are associated with a poor prognosis. Blood. 2003;102:276–283. doi: 10.1182/blood-2002-09-2896. [DOI] [PubMed] [Google Scholar]
- 103.Goldman JM, Melo JV. Chronic myeloid leukemia--advances in biology and new approaches to treatment. N Engl J Med. 2003;349:1451–1464. doi: 10.1056/NEJMra020777. [DOI] [PubMed] [Google Scholar]
- 104.Weisberg E, Manley PW, Cowan-Jacob SW, Hochhaus A, Griffin JD. Second generation inhibitors of BCR-ABL for the treatment of imatinib-resistant chronic myeloid leukaemia. Nat Rev Cancer. 2007;7:345–356. doi: 10.1038/nrc2126. [DOI] [PubMed] [Google Scholar]
- 105.Adrián FJ, Ding Q, Sim T, Velentza A, Sloan C, Liu Y, Zhang G, Hur W, Ding S, Manley P, Mestan J, Fabbro D, Gray NS. Allosteric inhibitors of Bcr-abl-dependent cell proliferation. Nat Chem Biol. 2006;2:95–102. doi: 10.1038/nchembio760. [DOI] [PubMed] [Google Scholar]
- 106.Zhang J, Adrián FJ, Jahnke W, Cowan-Jacob SW, Li AG, Iacob RE, Sim T, Powers J, Dierks C, Sun F, Guo GR, Ding Q, Okram B, Choi Y, Wojciechowski A, Deng X, Liu G, Fendrich G, Strauss A, Vajpai N, Grzesiek S, Tuntland T, Liu Y, Bursulaya B, Azam M, Manley PW, Engen JR, Daley GQ, Warmuth M, Gray NS. Targeting Bcr-Abl by combining allosteric with ATP-binding-site inhibitors. Nature. 2010;463:501–506. doi: 10.1038/nature08675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Barnett SF, Defeo-Jones D, Fu S, Hancock PJ, Haskell KM, Jones RE, Kahana JA, Kral AM, Leander K, Lee LL, Malinowski J, McAvoy EM, Nahas DD, Robinson RG, Huber HE. Identification and characterization of pleckstrin-homology-domain-dependent and isoenzyme-specific Akt inhibitors. Biochem J. 2005;385:399–408. doi: 10.1042/BJ20041140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Burke JR, Pattoli MA, Gregor KR, Brassil PJ, MacMaster JF, McIntyre KW, Yang X, Iotzova VS, Clarke W, Strnad J, Qiu Y, Zusi FC. BMS-345541 is a highly selective inhibitor of I kappa B kinase that binds at an allosteric site of the enzyme and blocks NF-kappa B-dependent transcription in mice. J Biol Chem. 2003;278:1450–1456. doi: 10.1074/jbc.M209677200. [DOI] [PubMed] [Google Scholar]
- 109.Girdler F, Sessa F, Patercoli S, Villa F, Musacchio A, Taylor S. Molecular basis of drug resistance in aurora kinases. Chem Biol. 2008;15:552–562. doi: 10.1016/j.chembiol.2008.04.013. [DOI] [PubMed] [Google Scholar]
- 110.Ewart-Toland A, Dai Q, Gao YT, Nagase H, Dunlop MG, Farrington SM, Barnetson RA, Anton-Culver H, Peel D, Ziogas A, Lin D, Miao X, Sun T, Ostrander EA, Stanford JL, Langlois M, Chan JM, Yuan J, Harris CC, Bowman ED, Clayman GL, Lippman SM, Lee JJ, Zheng W, Balmain A. Aurora-A/STK15 T+91A is a general low penetrance cancer susceptibility gene: a meta-analysis of multiple cancer types. Carcinogenesis. 2005;26:1368–1373. doi: 10.1093/carcin/bgi085. [DOI] [PubMed] [Google Scholar]
- 111.Kimura MT, Mori T, Conroy J, Nowak NJ, Satomi S, Tamai K, Nagase H. Two functional coding single nucleotide polymorphisms in STK15 (Aurora-A) coordinately increase esophageal cancer risk. Cancer Res. 2005;65:3548–3554. doi: 10.1158/0008-5472.CAN-04-2149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Bibby RA, Tang C, Faisal A, Drosopoulos K, Lubbe S, Houlston R, Bayliss R, Linardopoulos S. A cancer-associated aurora A mutant is mislocalized and misregulated due to loss of interaction with TPX2. J Biol Chem. 2009;284:33177–33184. doi: 10.1074/jbc.M109.032722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Briassouli P, Chan F, Linardopoulos S. The N-terminal domain of the Aurora-A Phe-31 variant encodes an E3 ubiquitin ligase and mediates ubiquitination of IkappaBalpha. Hum Mol Genet. 2006;15:3343–3350. doi: 10.1093/hmg/ddl410. [DOI] [PubMed] [Google Scholar]
- 114.Girdler F, Gascoigne KE, Eyers PA, Hartmuth S, Crafter C, Foote KM, Keen NJ, Taylor SS. Validating Aurora B as an anti-cancer drug target. J Cell Sci. 2006;119:3664–3675. doi: 10.1242/jcs.03145. [DOI] [PubMed] [Google Scholar]