

Abstract
Genistein has protective effects against prostate cancer (PCa) but whether this protection involves an estrogen receptor (ER) β dependent mechanism has yet to be elucidated. ER-β has a tumor suppressor role in PCa and its levels decline with cancer progression which was linked to ER-β promoter hypermethylation. Genistein has been suggested to have demethylating activities in cancer. However, the ability of genistein to reverse ER-β promoter hypermethylation in PCa has not been studied. In addition, there are great discrepancies among studies that examined the effect of genistein on ER-β gene expression. Therefore, we sought to explore effects of genistein on ER-β promoter methylation as a mechanism of modulating ER-β expression using three PCa cell lines, LNCaP, LAPC-4 and PC-3. We also examined the role of ER-β in mediating the preventive action of genistein. Our data demonstrated that genistein at physiological ranges (0.5–10 µmol/L) reduced ER-β promoter methylation significantly with corresponding dose-dependent increases in ER-β expression in LNCaP and LAPC-4 but not in PC-3 cells, which could be attributed to the low basal levels of ER-β promoter methylation in PC-3 cell line. Genistein induced phosphorylation, nuclear translocation and transcriptional activity of ER-β in all three PCa cell lines. Inhibitory effects of genistein on LAPC-4 and PC-3 cell proliferation were diminished using a specific ER-β antagonist. In conclusion, genistein and ER-β act together to prevent PCa cell proliferation; genistein increases ER-β levels via reducing its promoter methylation and ER-β, in turn, mediates the preventive action of genistein.
Keywords: Genistein, Prostate Cancer, Estrogen Receptor β, promoter methylation
1. Introduction
Prostate cancer (PCa) is the most frequently diagnosed malignancy and the second most common cause of cancer death in men in U.S. [1]. Over 186,000 patients are diagnosed each year and greater than 27,000 will succumb to death by PCa [2]. Despite the high incidence of PCa, little is known about its etiology. Accepted risk factors are age, race, ethnicity, and geographical location. Currently, there is no effective cure for this disease once it has spread beyond the prostate, and more efforts should be devoted for developing preventive strategies to reduce PCa occurrence and impact.
The incidence of PCa is much lower in Asian than in Western populations [1] and Asian migrants to the U.S. have an increased incidence [3]. Therefore, environmental factors including diet have been presumed to play a key role in prostate carcinogenesis. Unlike Western populations, many Asians consume large amounts of soy foods rich in isoflavones. Average concentrations of soy isoflavones in serum and prostatic fluid of Asian men are much higher than those in Western men [4, 5]. Over the past decade, researchers have generated data demonstrating that soy isoflavones and their metabolites have actions that may be useful for the prevention or treatment of PCa [6–8]. Genistein is the predominant and most biologically active isoflavone in soy. It has a structural similarity to 17β-estradiol and it binds to estrogen receptor (ER), with higher affinity to ER-β than to ER-α [9, 10]. Accordingly, it has been suggested that genistein exerts some of its anti-cancer effects through binding to ER-β [11]. ER-β is expressed in prostate epithelial cells, and has a role in the cellular homeostasis that is anti-proliferative [12], pro-differentiative [13], and pro-apoptotic [14]. ER-β expression declines in localized PCa with increasing grade from prostatic intraepithelial neoplasia (PIN) through low to high Gleason scores [15, 16]. This expression pattern supports the tumor suppressor role of ER-β. Therefore, ER-β agonists, such as genistein, have potential as chemopreventive and therapeutic agents for PCa.
One of the proposed mechanisms by which ER-β is modulated in PCa is promoter methylation [17–19]. DNA methylation is an essential regulator of gene transcription and aberrant DNA methylation can result in an unscheduled silencing of genes, such as tumor suppressor genes, which have been associated with a large number of human malignancies. In PCa, the extent of ER-β promoter methylation has been reported to correlate with the degree of aggressiveness of the disease; the more aggressive the tumor is the greater the methylation of the ER-β promoter and consequently the lower the ER-β gene expression [20]. In addition, PCa cells that have been treated with demethylating agents showed increased expression of ER-β relative to untreated cells [21].
Although a number of studies support the notion that genistein can reverse promoter hypermethylation of tumor suppressor genes in PCa cells [22–25], there is no data for the effect of genistein on ER-β gene promoter methylation. Furthermore, data about the effect of genistein on ER-β expression in PCa are inconsistent. In some in vitro studies, ER-β levels increased in response to genistein [26], whereas in others ER-β did not exhibit any apparent changes [11]. Thus, the main objective of this study was to determine the effect of genistein on ER-β methylation and subsequently ER-β expression levels and transcriptional activity. We hypothesize that: (1) genistein is capable of reversing ER-β promoter hypermethylation resulting in an increase in ER-β expression and transcriptional activity in PCa; (2) ER-β is mediating the protective effects of genistein in PCa. The second part of our hypothesis is built on observations from recent studies showing that dietary soy reduced the incidence of PCa in ER-β wild-type TRAMP transgenic mice, but not in ER-β knockout TRAMP mice [27].
In the current study, we tested our hypothesis using three PCa cell lines: LNCaP, LAPC-4 and PC-3. Cells were treated with a physiological range of genistein (0.5–10µmol/L) and 5-Aza-2′- deoxycytidine (5-Aza-dC), a demethylating agent, was used as a positive control. Relative fractions of methylated and unmethylated ER-β promoter were determined in each PCa cell line in basal states and after treatment. ER-β mRNA and protein expression and transcriptional activity were subsequently determined. PCa cell proliferation was analyzed in response to genistein in the presence or absence of the ER-β specific antagonist, 4-(2-phenyl-5, 7-bis (trifluoromethyl) pyrazolo [1, 5-a] pyrimidin-3-yl) phenol (PHTPP), in order to verify the mediating role of ER-β to the action of genistein in PCa cells.
2. Materials and Methods
2.1. Chemicals and antibodies
Genistein, 5-Aza-2′deoxycytidine, MPP dihydrochloride, 17 β-estradiol (E2) and ERB-041 were purchased from Sigma Aldrich (St. Louis, MO). PHTPP (4-[2-Phenyl-5,7-bis(trifluoromethyl)pyrazolo[1,5-a]pyrimidin-3-yl]phenol) was purchased from Santa Cruz Biotechnology (Santa Cruz, CA). ER-β (N-19) goat polyclonal, p-ER-β (serine 87) rabbit polyclonal, DNMT1 (H-300) rabbit polyclonal, DNMT3a (H-295) rabbit polyclonal and DNMT3b (N-19) goat polyclonal antibodies and a pool of five target-specific siRNAs designed to knockdown ER-β gene were purchased from Santa Cruz Biotechnology. ER-β (ab3576) and p-ER-β (serine 105) rabbit polyclonal antibodies were purchased from abcam (Cambridge, MA). Estrogen response element (ERE) reporter luciferase kit was purchased from Qiagen (Germantown, MD) and the Dual–Luciferase Assay System from Promega (Madison, WI).
2.2. Culture of prostate cancer cell lines
LAPC-4 cells [28] were obtained from Dr. R. Reiter via Dr. Karen Knudsen (UCLA). LNCaP and PC-3 cell lines were obtained from the ATCC (Rockville, MD). Our cell lines were recently authenticated using “ATCC cell authentication testing service”. Cells were maintained in phenol red free RPMI media with L-glutamine supplemented with 10% FBS, 100 IU/mL Penicillin, and 100 µg/mL streptomycin. Media were replaced with RPMI containing 10% charcoal stripped fetal bovine serum 24 hours before cell treatments.
2.3. ER-β Silencing in PCa Cells
In order to validate ER-β antibodies used in this study, PCa cells, 5,000 cells/well, were transiently transfected on 24-well plates with 0.5 µg of ER-β siRNA or scramble vector using Lipofectamine™ LTX (Invitrogen Grand Island, NY). Plasmids were purchased from SantaCruz Biotechnology. Four hours later, the transfection media was replaced with fresh growth media. After 72 hours, cells were harvested and the total protein was isolated and probed for ER-β antibodies.
2.4. Cell proliferation assay
Cell survival was assayed using the Cell Titer 96 AQueous Non-Radioactive Cell Proliferation Assay (MTS) (Promega, Madison, WI). Cells were seeded in 96-well plates (3,000 cells/well) in regular media. The media were replaced by charcoal stripped media 24 hours before treating with genistein or other compounds as indicated later. Twenty µL MTS reagent/100 µL medium was added and then left for 4 hours in the humid incubator at 37°C. The absorbance was recorded at 570 nm using a microplate reader (Synergy 2, Biotek, Highland Park, VT).
2.5. Western blot analysis
For Western blot analysis, total protein was isolated from treated cells using 1X cell lysis buffer (Cell Signaling, Danvers, MA) and for semiquantitative analysis of nuclear ER-β; nuclear and cytoplasmic extracts were isolated using a Nuclear Extraction Kit (Active Motif, Carlsbad, CA). Protein concentration was measured at 595 nm with a microplate reader using the Bio-Rad Protein Assay kit (Bio-Rad Laboratories, Hercules, CA). Twenty five µg protein/lane was resolved by NuPAGE 4-12% Bis-Tris Gel (Invitrogen) and transferred to PVDF membranes. The immunoblot was incubated with primary antibodies overnight at 4°C and then with secondary antibodies conjugated to horseradish peroxidase (Promega). ECL Western blotting detection reagents (Amersham Biosciences, Piscataway, NJ) were used to identify the antibody-bound protein bands. β-Tubulin and Tata Binding Protein (TBP) (Cell Signaling, Danvers, MA) were used as loading controls for total protein and nuclear protein, respectively. The blots were exposed to an X-ray film for 30–60 seconds after which the film was developed using an x-ray film developer. The film was then scanned and calibrated using Image J software to calculate the intensity of the bands of protein of interest relative to the β-tubulin housekeeping gene from three independent experiments.
2.6. Real-time PCR (polymerase chain reaction)
Total RNA was extracted using the RNeasy mini kit (Qiagen, Germantown, MD). RNA quantities and qualities were determined by measuring absorbance at 260 and 280 nm by spectrophotometry. Five µg of total RNA were reversely transcribed into cDNA using SuperScript RT III (Invitrogen). ER-β and FOXO1 mRNA expression were determined by real-time PCR using the SYBR Green Assay standard protocol and the step one plus Real-Time PCR System (Applied Biosystems, Foster City, CA) was used. Specific primers for each gene were designed (see table I). Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used as the internal control for normalization of expression data. Threshold cycle numbers (Ct) generated by the real-time RT-PCR were used to calculate the normalized expression ratio of the target genes using the 2−ΔΔCt (Livak) method. Reactions were carried out in triplicates, and the results show three independent experiments.
Table 1.
Sequences of primers used for real-time PCR
| Gene | Forward Primer | Reverse Primer |
|---|---|---|
| ER β | GCCTCAAGATCATCAGCAATGCCT | GTGGTCATGAGTCCTTCCACGAT |
| FOXO1 | AAGAGCGTGCCCTACTTCAA | CTGTTGTTGTCCATGGATGC |
| GAPDH | GCCTCAAGATCATCAGCAATGCCT | TGTGGTCATGAGTCCTTCCACGAT |
2.7. Nuclear localization studies
Cells were plated in 16-well chamber slides (Lab-Tek chamber slide system, Nalge Nunc, Naperville, IL) in regular media until they reach 50 – 70% confluences. Cells were incubated in charcoal stripped media for 24 hours before treating with vehicle (ethanol or DMSO (dimethyl sulfoxide)) or genistein. Thirty minutes - 4 hours after treatment, cells were rinsed with 1x PBS, fixed with 4% formaldehyde/1xPBS for 20 minutes and permeabilized with 0.1% Triton X-100/1xPBS for 10 minutes. Cells were incubated first with 5% serum free protein block for 60 minutes and then with the primary antibody (at a 1:200 dilution overnight at 4°C in a humidified chamber). The next day, cells were washed and incubated with the CY5 conjugated secondary antibodies (Santa Cruz Biotechnology) for 1 hour then incubated with 1:500 Hoechst counterstaining for 20 minutes in the dark. Slides were examined under inverted florescent microscope (Eclipse TE 2000, Nikon, Japan) equipped with a cooled CCD camera (Photometric Cascade II, Tucson, AZ) under control of MetaMorph imaging software (Molecular Devices, Fully Vale, CA). For clear demonstration of ER-β subcellular distribution, another set of experiments were performed where cells were simultaneously labelled for the pan- and the phosphorylated-ER-β antibodies. Secondary antibodies conjugated to fluorophores that have a nonoverlapping emission spectrum (CY5 and FITC) (Santa Cruz Biotechnology) were used to detect the localization of the pan and the phosphorylated ER-β antibodies, respectively. Fluorescence was detected using confocal laser scanning microscope (Zeiss, Inc.).
2.8. ERE Reporter Assay
Cells were seeded into 24 well-plates at a density of 5000 cells/well in standard medium without antibiotics. Cells were transiently co-transfected with ERE-firefly luciferase construct and Renilla luciferase expression vector as a control for transfection efficiency using Lipofectamine 2000 (Invitrogen). A non-inducible reporter construct that does not have the ERE insert was used as a negative control. A constitutively expressing firefly luciferase construct was used as a positive control. All constructs were purchased from Qiagen (Germantown, MD). Eight hours after transfection, cells were treated with vehicle (ethanol and/or DMSO), genistein or the ER-β agonist, ERB-041 in the presence or absence of a concomitant treatment with the selective ER-β antagonist, PHTPP or the selective ER-α antagonist, MPP dihydrochloride. Forty eight hours later, cells were harvested, and reporter and Renilla luciferase activities were determined using the Dual–Luciferase Assay System (Promega).
2.9. Assessment of ER-β promoter methylation by methylation specific PCR (MSP)
Genomic DNA that was extracted from prostate cancer cells after treatment with genistein (0.5 – 10µM) or 5-aza-2′deoxycytidine (5µmol/L) was submitted to bisulfite modification using the EpiTect Bisulfite Kit specific protocol (Qiagen). Modified DNA was then amplified using methylated and unmethylated primers for MSP that were designed using the Methprimer software (http://www.urogene.org/cgibin/methprimer/methprimer.cgi). The human genome sequence for ER-β promoter, exon 0N and exon 1 that has been used to design these primers was retrieved from The National Center for Biotechnology Information (NCBI) website with accession number AF191544. Primers were designed using the following criteria for optimum MSP primer selection: CpG Island size > 100bp, GC Percent > 50.0, and Observed/Expected CpGs > 0.60. The primers are summarized in Table 2. One primer set (U) anneals to unmethylated DNA and a second primer set (M) anneals to methylated DNA. The PCR product was electrophoresed using 2% agarose gel containing nucleic acid gel stain (GelStar) (Cambrex Bio Science, Baltimore, MD). DNA bands were imaged using the ChemiDoc MP System and analyzed using Quantity One 1-D Analysis Software (Bio-Rad Laboratories).
2.10. Statistical Analysis
The data were analyzed using Student's t -test, or one-way ANOVA followed by a post-hoc test as appropriate and p values of <0.05 were considered significant.
3. Results
3.1
Identification of three CpG islands in the promoter and exon 0N region of ER-β
3.2
DNA methylation is a crucial regulator of gene transcription; genes with high levels of 5-methylcytosine in their promoter region are transcriptionally silent. Aberrant DNA methylation represses transcription of tumor suppressor genes in malignancy. Methylation groups are added to the DNA at the CpG sites and the most important CpG islands are those in the promoter region since they are located at the site of initiation of transcription of the gene. Methylation of these CpG islands causes gene silencing due to direct inhibition of transcription factor binding. This silencing could also be mediated by methyl-binding domain (MBD) proteins that recruit other chromatin-modifying proteins to methylated DNA. [29]. Li et al [30] cloned and characterized a 2.1-kilobase 5′-flanking region of the human ER-β gene (AF191544) from PCa cell lines and reported two major transcription start sites. This region consists of ER-β promoter and an untranslated exon referred to as “exon 0N” and contains many CpG sites. Evidence collected from various primary tumors and tumor cell lines suggests that ERβ transcription may be regulated by hypermethylation of CpG islands located within the promoter and exon 0N region of the ERβ gene [20, 31, 32]. In order to predict CpG islands in the promoter and exon 0N region of the ER-β gene and to design specific primers for their detection, we used an online program, Methprimer. Three CpG islands were identified: (island 1, 217 bp (1927–2143); island 2, 162 bp (1674–1835); and island 3, 107 bp (1211–1317) (fig. 1). These three CpG islands contained 52 CpG sites. We used BLAST services from NCBI (www.ncbi.nlm.nih.gov/BLAST/) to compare the sequence we used to predict CpG islands with sequences reported by previous studies that described the structure of this region; Zhu et al [21], Al-Nakhle et al [33] and Rody et al [20]. We were able to deduce the start point of exon 0N which is at 1871 in our sequence. Accordingly, two of the predicted CpG islands are located in the promotor and the third one is in the exon 0N. Using the criteria for MSP primer selection mentioned in the methods section, three primer sets for each CpG island were designed to study the methylation status of the CpG sites (supplementary table 1). When the analyzed sequence was expanded to include 2000bp distal to the original sequence (AF191544) as well as exon 1 (AL161756) that is separated from exon 0N by 10,958-bp intron, there were no CpG islands found using the criteria for optimum MSP primer selection that we described earlier in the method section. Methylation status of the ER-β promoter studies with methylation specific PCR (MSP)
Fig 1. Graphic depiction of the CpG islands in the ER-β gene promoter and exon 0N (AF191544).
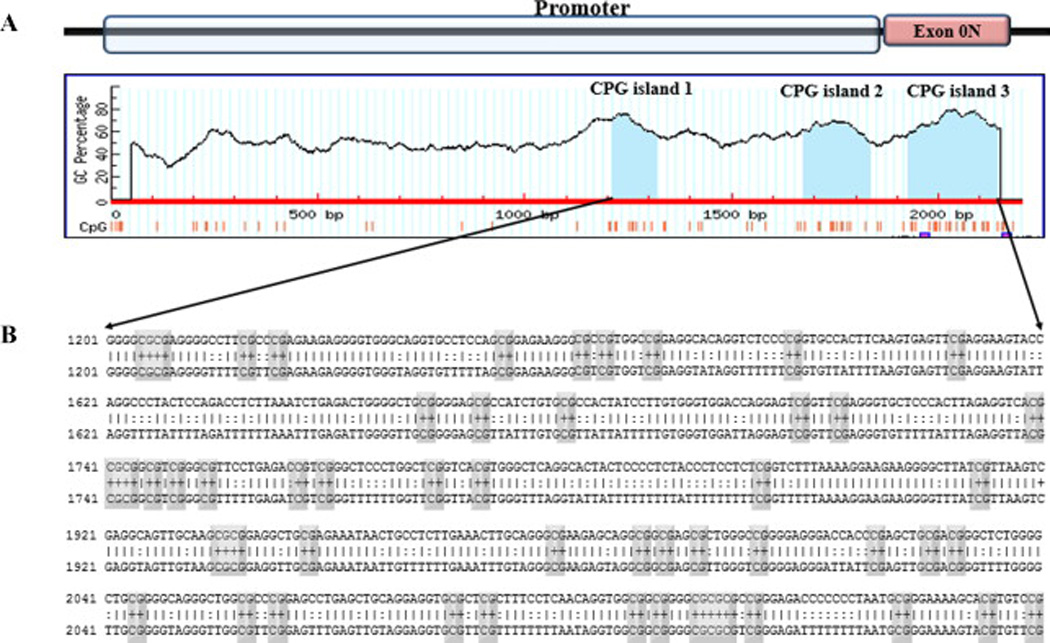
A: Three CpG islands from right to left (island 1, 217 bp (1927–2143); island 2, 162 bp (1674–1835); and island 3, 107 bp (1211–1317)) were identified using the software, Methprimer. Two of the predicted CpG islands are located in the promotor and the third one is in the exon 0N. These three CpG islands contain 52 CpG sites. B: Sequence of CpG island in ER-β promoter and exon 0N region where CpG sites are indicated by grey rectangles.
To determine the ability of genistein to modify ER-β promoter methylation, LAPC-4, LNCaP and PC-3 cells were treated with a physiological range of genistein concentrations (0.5–10 µmol/L) for five days. Cells treated with the demethylating agent, 5-Aza-dC, were used as a positive control. Genomic DNA was extracted from treated cells and submitted to a bisulfite modification. Bisulfite treatment deaminates cytosine in unmethylated DNA and turns it to uracil, while 5-methylcytosine resists this bisulfite action. The modified DNA was then amplified using methylated and unmethylated primers that anneal with the methylated and the unmethylated DNA, respectively. The PCR product was gel electrophoresed and bands were imaged using a ChemiDoc MP System and quantified using image J software. At the basal level (vehicle control), signals detected by methylated primers, corresponding to the methylated promoter, were intense while the unmethylated promoter signals were very faint in LNCaP and LAPC-4 cells (figs. 2A and 2B). In contrast, in PC-3 cells, the methylated promoter signal was much fainter than the unmethylated signal in basal states (fig. 2C). These results indicate that, under basal conditions, the ER-β promoter is hypermethylated in LNCaP and LAPC-4 cells, whereas in PC-3 the ER-β promoter is nearly unmethylated. Treatment with 5-Aza-dC reduced the methylation of ER-β CpG islands in both LNCaP and LAPC-4 cells (figs. 2A and 2B) but had minimal to null effect in PC3 cells (fig. 2C). Partial dose-dependent demethylation of ER-β promoter was observed in LNCaP and LAPC-4 cell lines after genistein treatment as demonstrated by an increase in the unmethylated fraction of the promoter and reduction in the methylated segment (figs. 2A and 2B). Blocking of ER-β or ER-α using PHTPP or MPP, respectively did not abolish this effect. Various doses of genistein did not have any effect on the methylation of ER-β gene promoter in PC-3 cells (fig. 2C). Similar findings were obtained using three designed sets of primers for each CpG island. Figure 2D shows data from one set of primers for islands two and three for the three PCa cell lines.
Fig 2. Changes in ER-β DNA methylation pattern in the promoter and exon 0N region in PCa cells after treatment with genistein.
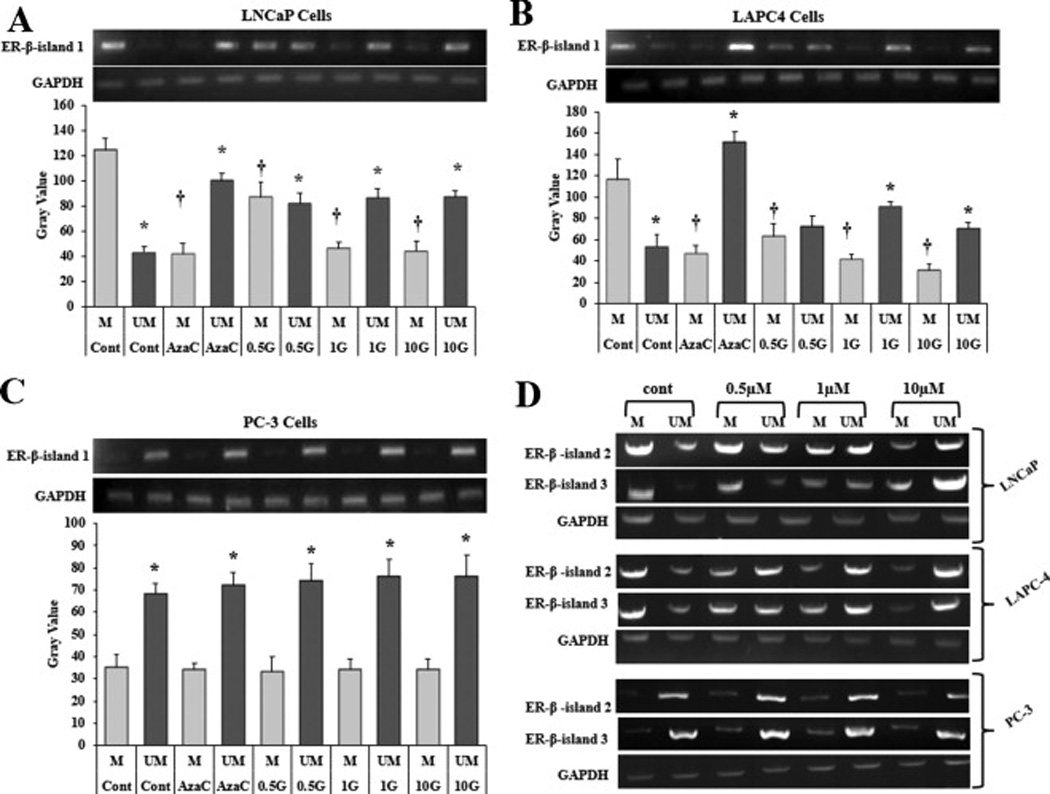
Methylation specific PCR of ER-β gene in LNCaP (A), LAPC-4 (B) and PC-3 (C) cells was performed with corresponding diagrammatic representation of relative intensity of bands quantified by the software, image J. Cells were treated with genistein (0.5–10 µmol/L) or vehicle (DMAO). 5-aza-dC (a demethylating agent) was used as a positive control. DNA from treated cells were bisulfite modified and then PCR was done using methylation specific primers and unmethylation specific primers for the first CpG island (exon 0N). Products from the methylation specific PCR were run on agarose gels. All methylation values were normalized to GAPDH. Results represent the means ± standard deviation (SD) of three independent experiments * (P < 0.05) for comparing the UM bands after genistein and 5-aza-dC treatments with the UM band in the vehicle control. † (P < 0.05) for comparing the M bands after genistein and 5-aza-dC treatments with the M band in the vehicle control. D: Methylation specific PCR of ER-β promoter CpG islands (island 2 and 3) after treatment with genistein in LNCaP, LAPC-4 and PC-3 cells. G, genistein, M, methylated, UM, unmethylated.
3.3. Genistein downregulates DNA methyl transferases (DNMTs) in PCs cells
The transfer of methyl groups to DNA is mediated by a group of enzymes known as DNMTs. Currently, three DNMTs have been identified in mammalian cells which are DNMT1, DNMT3a, and DNMT3b [34]. Previous studies have demonstrated the overexpression of DNMTs in hormone-sensitive and hormone-resistant PCa [35]. Also, it has been shown that the majority of gene silencing in PCa is attributed to DNA hypermethylation mediated by this group of DNMTs [36, 37]. Based upon these observations, we hypothesized that the ability of genistein to reduce ER-β promotor methylation is very likely to be attributed to genistein-mediated downregulation of DNMTs. To test this hypothesis, PCa cells were treated with genistein (0.5–10 µmol/L) or DMSO (vehicle control) for 5 days after which the total protein extracted from cells was gel electrophoresed and analyzed for the expression of DNMT1, DNMT3a and DNMT3b using specific antibodies. Our data presented in figure 3 revealed that DNMT1 protein levels were significantly decreased in PCa cells in response to the lowest dose of genistein (0.5 µmol/L) (fig. 3A). This decline was dose-dependent, reaching 60%, 74% and 88% in LNCaP, LAPC-4 and PC-3 cells, respectively, at a genistein dose of 10 µmol/L (fig. 3B). Similarly, DNMT3b protein expression was dramatically reduced by genistein in PCa cells (fig. 3A). At 10 µmol/L of genistein, we observed a decline in DNMT3b protein by 90%, 72%, and 77% in LNCaP, LAPC-4, and PC-3 cells, respectively (fig. 3B). Nevertheless, no significant changes were detected in DNMT3a protein levels in PCa cells in response to treatment with genistein (figs. 3A and 3B).
Fig 3. Effects of genistein on DNMT protein expression in LNCaP, LAPC-4 and PC-3 cells.
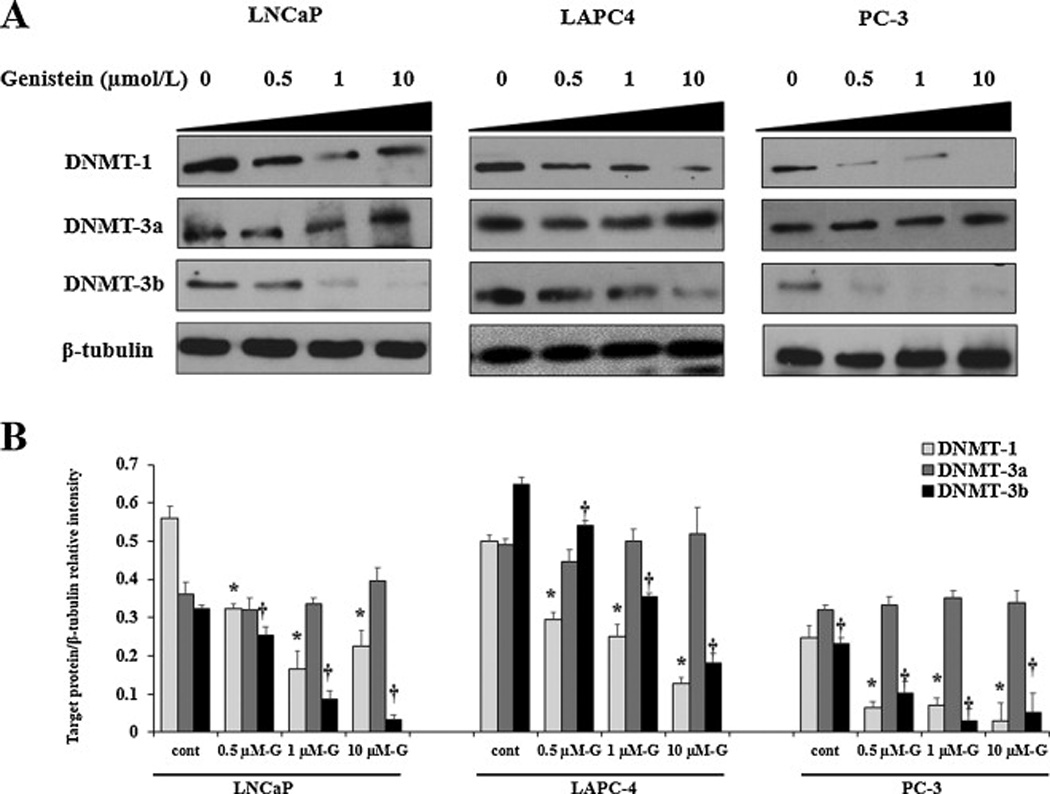
Western blot analysis of the effect of physiological concentrations of genistein (0.5–10 µmol/L) on protein expression of the three DNMT subtypes, DNMT1, DNMT3a and DNMT3b in PCa cells. Signal relative intensity of DNMT bands was normalized to β-tubulin. Results represent the means ± SD of three independent experiments. * (P < 0.05) and † (P < 0.05) for comparing protein levels after genistein stimulation with vehicle control for DNMT1 and DNMT3b, respectively.
3.4. Genistein increases ER-β mRNA and protein expression in LNCaP and LAPC-4 PCa cells
ER-β expression levels have been demonstrated to be lower in PCa than in normal prostate tissue [16]. To further investigate whether the effect of genistein on reducing ER-β promoter methylation, which we demonstrated above, was accompanied by increased ER-β expression in PCa cells, we measured ER-β mRNA and protein in PCa cells after treatment with either increasing doses of genistein (0.5–10 µmol/L) or 5 µmol/L of 5-Aza-dC. ER-β protein was probed using two different antibodies that recognize two different epitopes, ER-β (N-19) (fig. 4A) and ER-β (ab3576) (supplementary figs. 1A and 1B). The antibody specificity was confirmed by the significant down-regulation of the antibody signal after ER-β silencing using a pool of five ER-β-targeting siRNAs (supplementary fig. 1C). 5-Aza-dC, resulted in nearly two-fold increase in ER-β mRNA in both LNCaP and LAPC-4 cells (fig. 4C). It also increased ER-β protein levels by 44% and 73% in LNCaP and LAPC-4 cells (figs. 4A and 4B), respectively. In response to 1µmol/L of genistein, ER-β mRNA and protein levels were increased by 70% and 50%, respectively in LNCaP cells. This increase was dose-dependent, reaching 97% for ER-β mRNA in response to 10 µmol/L. In LAPC-4 cells, ER-β protein expression was increased by 83% in response to 1 µmol/L of genistein, but a dose response was not apparent. Genistein induced dose-dependent increases in ER-β mRNA levels in LAPC-4 cells albeit to a lesser extent than in LNCaP cells. Conversely, in PC-3 cells, neither genistein nor 5-Aza-dC caused any significant changes in ER-β protein or mRNA expression (fig.4).
Fig 4. Effects of increasing doses of genistein on the ER-β protein and mRNA in LNCaP, LAPC-4 and PC-3 cells.
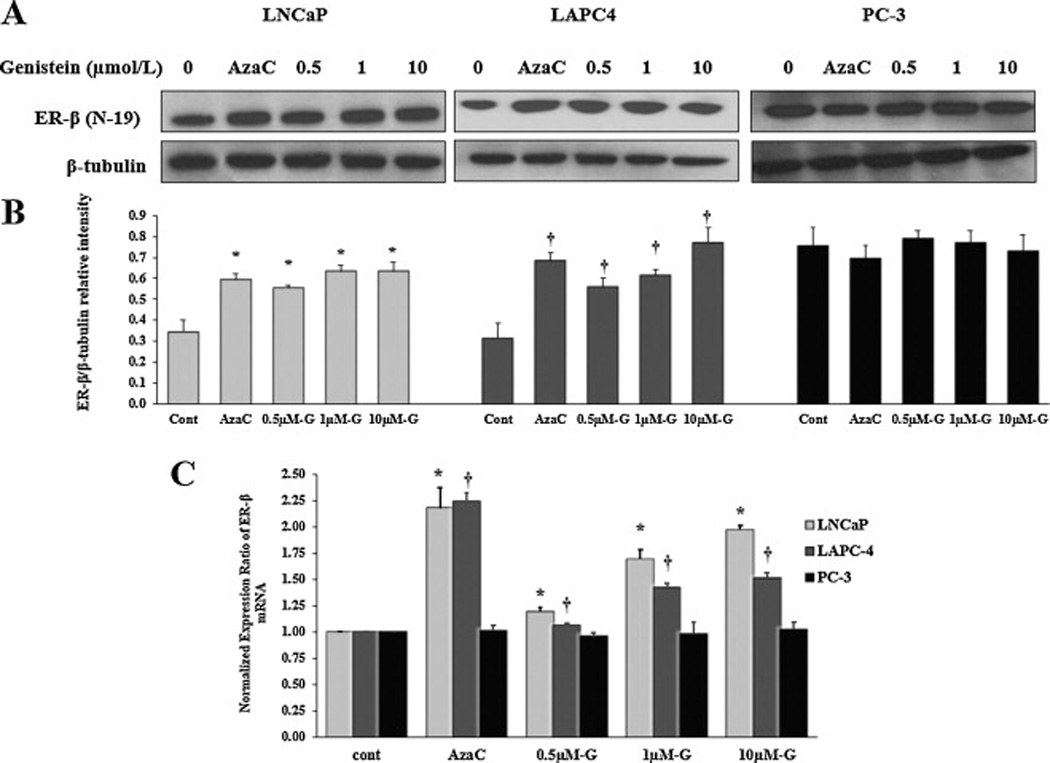
A: Western blot analysis of the effect of different concentrations of genistein on ER-β protein expression in LNCaP, LAPC-4 and PC-3 cells using ER-β (N-19) antibody. B: Signal relative intensity of ER-β bands was normalized to β-tubulin. C: Quantitative assessment by real-time PCR of ER-β mRNA normalized to the house keeping gene, GAPDH. 5-aza-dC is a demethylating agent used as a positive control. Results represent the means ± SD of three independent experiments. * (P < 0.05) and † (P < 0.05) for comparing ER-β expression after genistein or 5-aza-dC treatments with ER-β expression in the vehicle control in LNCaP and LAPC-4 cells, respectively.
Genistein has a structural similarity to the natural estrogen in our body, 17 β-estradiol (E2). It has been shown that the affinity of genistein to ER-β is several fold higher than ER-α [38, 39] while E2 has an equal binding affinity for both subtypes of ER [40]. However, it is not clear whether or not E2 induces similar effects on ER-β gene. In order to dissect the effect of genistein from that of E2, PCa cells were treated with E2 (100 nM) and changes in ER-β methylation and expression were assessed. Our data showed that E2 did not induce any significant effects on ER-β methylation (supplementary figs 2A and 2B) or expression (supplementary fig. 2C). In summary, our results indicate that genistein reverses ER-β gene methylation and induces ER-β expression in PCa cells while the ER-β natural ligand, E2, was not capable of inducing similar effects.
1.1. Genistein induces ER-β nuclear localization and transcriptional activity in LNCaP, LAPC-4 and PC-3 PCa cells
ER-β is a nuclear steroid hormone receptor that is activated after ligand binding and then translocated to the nucleus to perform its function as a transcription factor [41]. We examined the ability of genistein to promote ER-β nuclear translocation in PCa cells. LAPC-4, LNCaP, and PC-3 cells were treated with either the specific ER-β agonist, ERB-041 [42], as a positive control for ER-β nuclear translocation, or with a range of physiological genistein concentrations (0.5–10µmol/L) in steroid free media for 30 minutes to four hours. Both ERB-041 and genistein caused a significant increase in ER-β nuclear localization in the all three cell lines (figs. 5A, 6A and 7A). For further confirmation of these observations, PCa cells, treated with genistein (10 µmol/L), were simultaneously immunolabelled for pan-ER-β ((red fluorescence) and phosphorylated ER-β (green fluorescence) antibodies. The subcellular distribution of ER-β was visualized under a high magnification using a confocal microscopy (figs. 5B, 6B, and 7B). Our data showed that ER-β was localized mainly in the cytoplasm in control cells that grow in steroid free media. After treatment with genistein (10 µmol/L), there was a remarkable accumulation of ER-β in the nuclear compartment (yellow, due to an overlap of the red and green fluorescence).
Fig 5. Effects of genistein on ER-β nuclear localization in LAPC-4 cells.
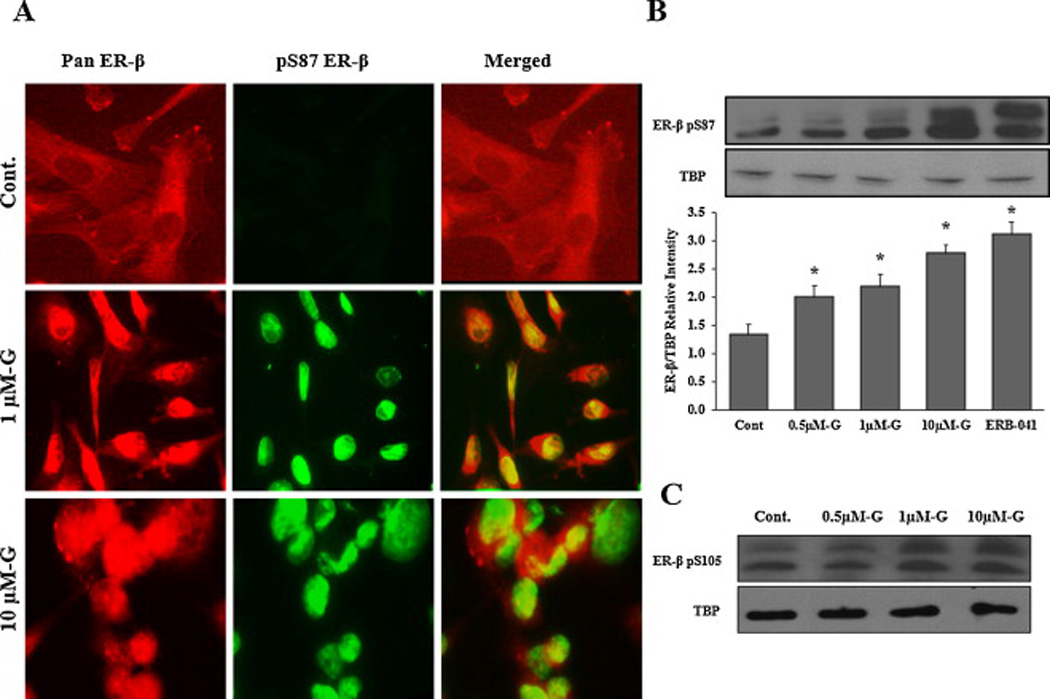
A: Cells were treated with DMSO (vehicle) or genistein 1µmol/L and 10µmol/L for 4hr. Then, cells were fixed, immunostained with pan ER-β and pS87 antibodies simultaneously and visualized by the confocal microscope. The red fluorescent staining in the first column is for pan ER-β, the green fluorescent staining in the second column is for pS87 and the third column is for both images merged together using image J software. B: Western blot analysis of ER-β protein phosphorylation at serine 87 in the nuclear protein fraction of LAPC-4 cells treated with genistein. Signal relative intensity of ER-β bands was normalized to Tata Binding Protein (TBP). Results represent the means ± SD of three independent experiments. * (P < 0.05) for comparisons with the vehicle control. C: Western blot analysis of ER-β protein phosphorylation at serine 105 after treatment with genistein.
Fig 6. Effects of genistein on ER-β nuclear localization in LNCaP cells.
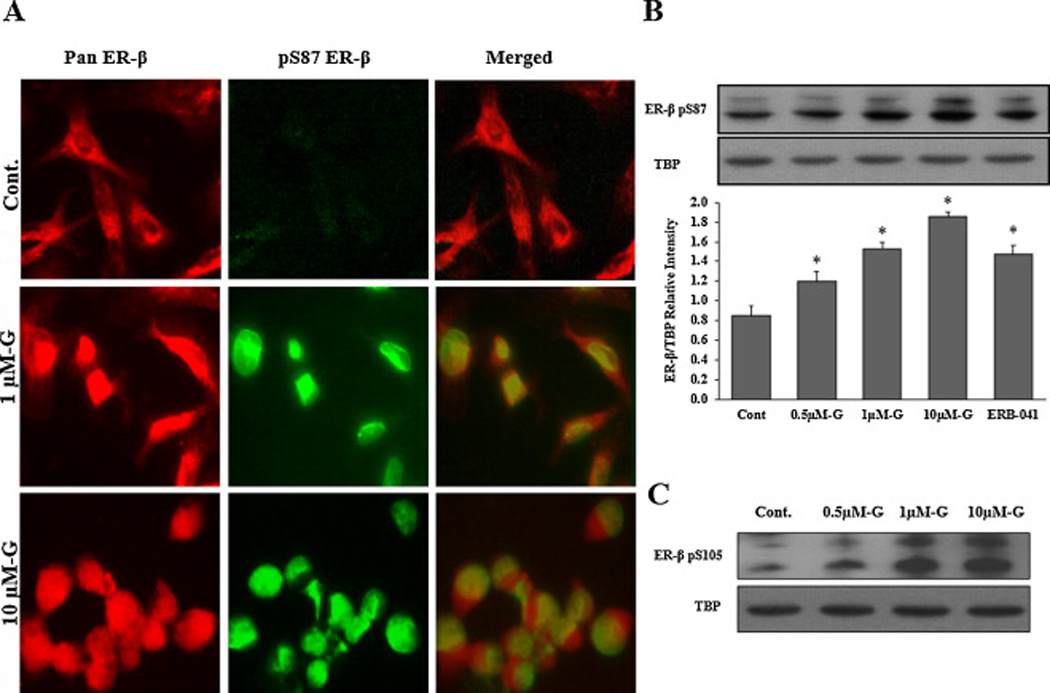
A: Cells were treated with DMSO (vehicle) or genistein 1µmol/L and 10µmol/L for 4hr. Then, cells were fixed and immunostained with pan ER-β and pS87 antibodies simultaneously and visualized by the confocal microscopy. The red fluorescent staining in the first column is for pan ER-β, the green fluorescent staining in the second column is for pS87 and the third column is for both images merged together using image J software. B: Western blot analysis of ER-β protein phosphorylation at serine 87 in the nuclear protein fraction of LAPC-4 cells treated with genistein. Signal relative intensity of ER-β bands was normalized to Tata Binding Protein (TBP). Results represent the means ± SD of three independent experiments. * (P < 0.05) for comparisons with the vehicle control. C: Western blot analysis of ER-β protein phosphorylation at serine 105 after treatment with genistein.
Fig 7. Effects of genistein on ER-β nuclear localization in PC-3 cells.
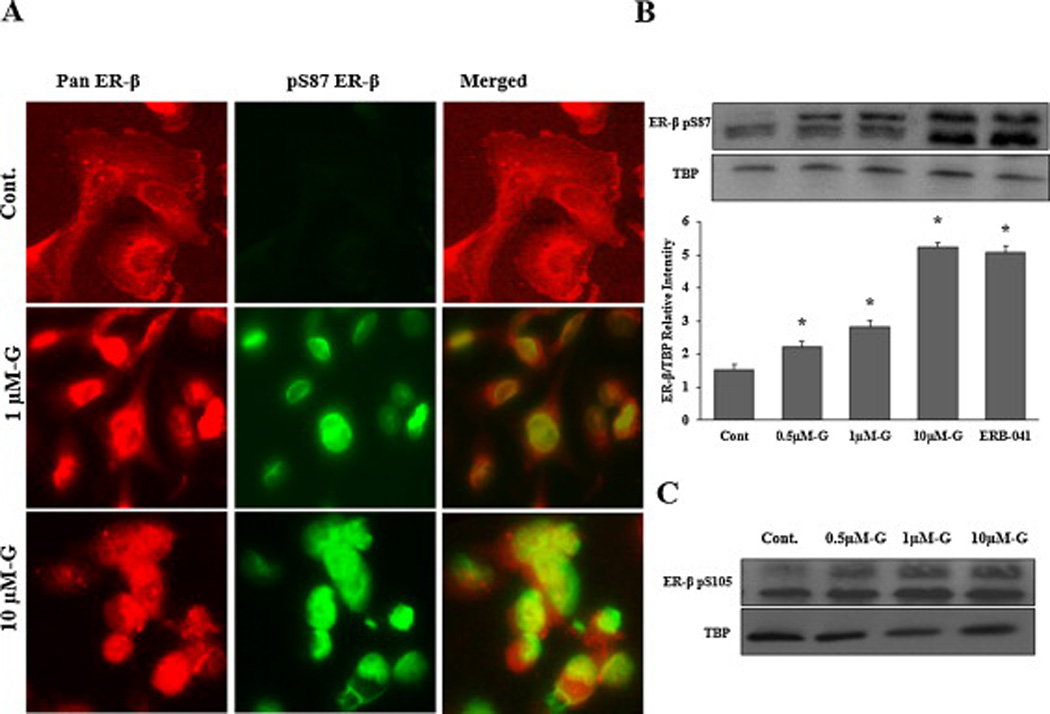
A: Cells were treated with DMSO (vehicle) or genistein 1µmol/L and 10µmol/L for 4hr. Then, cells were fixed and immunostained with pan ER-β and pS87 antibodies simultaneously and visualized by the confocal microscopy. The red fluorescent staining in the first column is for pan ER-β, the green fluorescent staining in the second column is for pS87 and the third column is for both images merged together using image J software. B: Western blot analysis of ER-β protein phosphorylation at serine 87 in the nuclear protein fraction of LAPC-4 cells treated with genistein. Signal relative intensity of ER-β bands was normalized to Tata Binding Protein (TBP). Results represent the means ± SD of three independent experiments. * (P < 0.05) for comparisons with the vehicle control. C: Western blot analysis of ER-β protein phosphorylation at serine 105 after treatment with genistein.
The phosphorylated part of ER-β is the active part of the protein that is also capable of translocating to the nucleus and exerts it function as a transcription factor. Thus, to verify the effect of genistein on the phosphorylation of ER-β protein, the isolated nuclear fraction of treated cells were immunoblotted with an ER-β antibody specific for the active phosphorylated moiety (serine 87). Genistein increased the phosphorylated nuclear fraction of the ER-β protein (figs. 5C, 6C and 7C), which is consistent with the increased nuclear translocation of the receptor. Quantifying the phosphorylated ER-β protein, using Image J software, showed progressive increments in response to increasing doses of genistein (figs. 5C, 6C and 7C). Despite the lack of any effect of genistein on either ER-β promoter methylation or ER-β expression levels in PC-3 cells, genistein was able to induce ER-β nuclear localization and phosphorylation in this cell line. Since these effects of genistein on ER-β nuclear localization and phosphorylation were comparable to those of the specific ER-β agonist, ERB-041, these results support the conclusion that genistein may act as an agonist for ER-β, promoting its phosphorylation and translocation to the nucleus. Another phosphorylation site that has been shown to influence ER-β activity is serine 105 (pS105) [43]. In order to test the effect of genistein on this phosphorylation spot, the nuclear protein fraction was collected from PCa cells after treatment with genistein (0.5–10 µmol/L) and probed with a specific antibody against phosphorylated S105. Our results indicated that genistein is capable of inducing phosphorylation of ER-β at S105 (figs 5D, 6D, and 7D) which together with phosphorylation of S87 has been reported to potentiate ER-β nuclear localization and transactivation [43].
Once inside the nucleus, ER-β acts as a transcriptional factor that binds to the DNA and regulates the expression of several genes involved in cell proliferation, apoptosis and cell cycle progression. Accordingly, we sought to monitor changes in ER-β transcriptional activity in response to genistein as a next rational step for the enhanced ER-β nuclear localization. To this end, we measured the expression of FOXO-1, a gene known to be regulated by ER-β. It has been shown that activated ER-β induces the expression of FOXO-1 [44]. When LAPC-4 and PC-3 cells were cultured in steroid free media supplemented with genistein (0.5 – 10 µmol/L), they displayed significant dose-dependent increases in FOXO-1 mRNA levels (figs. 8B and 8C), indicative of an increase in ER-β transcriptional activity. Interestingly, this effect was achieved in LNCaP cells only in response to the highest dose that has been used (10 µmol/L) (fig. 8A). In order to verify that genistein-induced changes in FOXO-1 expression were mediated through activated ER-β protein, we performed another set of experiments after blocking ER-β using a specific antagonist, PHTPP [45]. In all PCa cells, the combined treatment with genistein and PHTPP blunted genistein-induced effects on FOXO1 mRNA expression (figs 8A, 8B and 8C).
Fig 8. Effects of genistein on FOXO1 mRNA levels in PCa cells.
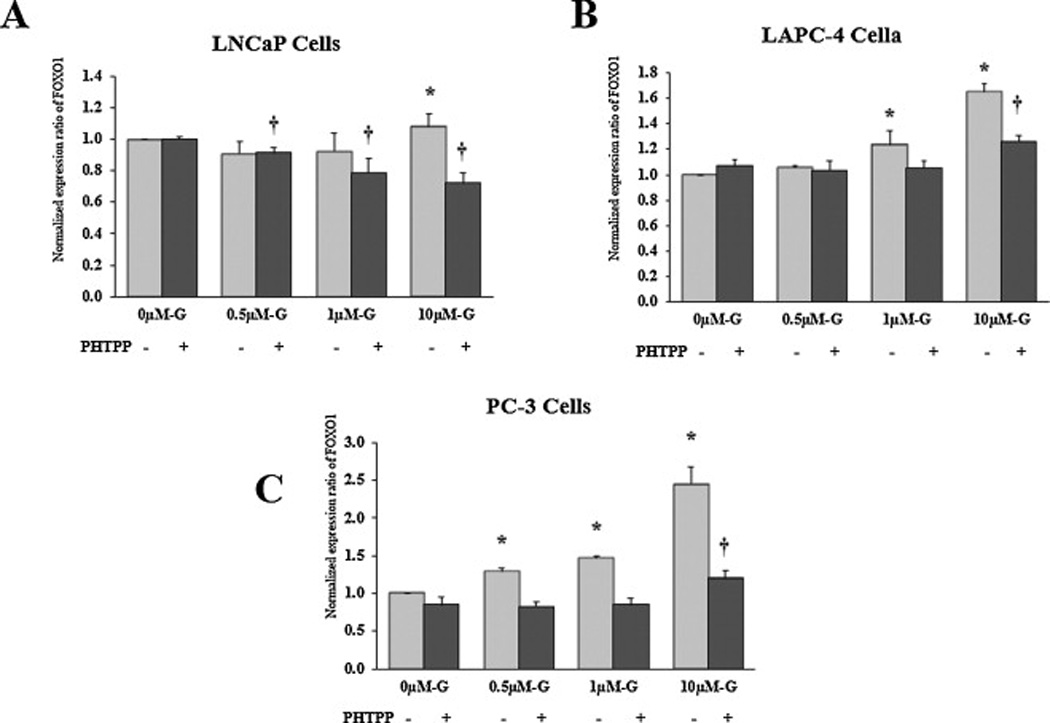
Quantitative assessment by real-time PCR of FOXO1 mRNA in LNCaP (A), LAPC-4 (B) and PC-3 (C) cells treated with genistein with or without the specific ER-β antagonist, PHTPP. FOXO-1 mRNA was normalized to the house keeping gene, GAPDH. Results represent the means ± SD of three independent experiments. * (P < 0.05) and † (P < 0.05) for comparing FOXO1 mRNA levels after genistein treatment with FOXO1 mRNA level in the vehicle control in the absence and presence of PHTPP, respectively.
For further validation of the effect of genistein on ER-β transcriptional activity, PCa cells were transiently transfected with estrogen response element (ERE)-luciferase construct then treated with genistein at increasing doses (0.5–10µmol/L), ERB-041 or DMSO. Since ERE is a common DNA binding site for both ER-β and ER-α, cells were concomitantly treated with genistein and the selective ER-α antagonist, MPP dihydrochloride or genistein plus the selective ER-β antagonist, PHTPP in order to exclude any contamination of the results by ER-α activity and to confirm that the changes obtained are mediated by ER-β protein, respectively. Our data demonstrated a prominent dose-related increase in ERE reporter activity in LAPC-4 and PC-3 cells in response to all genistein concentrations tested. At 10 µmol/L, genistein increased ERE reporter activity by ~ 70% and 85% in LAPC-4 and PC-3 cells, respectively which was comparable with the induction caused by the selective ER-β agonist, ERB-041 (figs. 9A and 9B). In LNCaP cells, only the highest genistein dose (10 µmol/L) resulted in an induction of ERE luciferase activity by 47% which was significantly less than that caused by ERB-041 (85%)(fig. 9C). Genistein-stimulated ERE luciferase activity was dramatically abolished after blocking ER-β in all the three PCa cell lines, however it was not affected by blocking ER-α ((figs. 9A and 9B). Our data show that ER-α protein levels are very low to undetected in PCa cells (fig. 9D) with a very low ER-α to ER-β ratio (fig 9E), which may provide a plausible explanation for the lack of contribution of ER-α to the ERE reporter activity. Collectively, these data indicates that genistein, at physiological concentrations, is capable of activating ER-β through inducing its phosphorylation and nuclear import where it exerts its function as a transcription factor.
Fig 9. Effects of genistein on ER-β transcriptional activity in PCa cells.
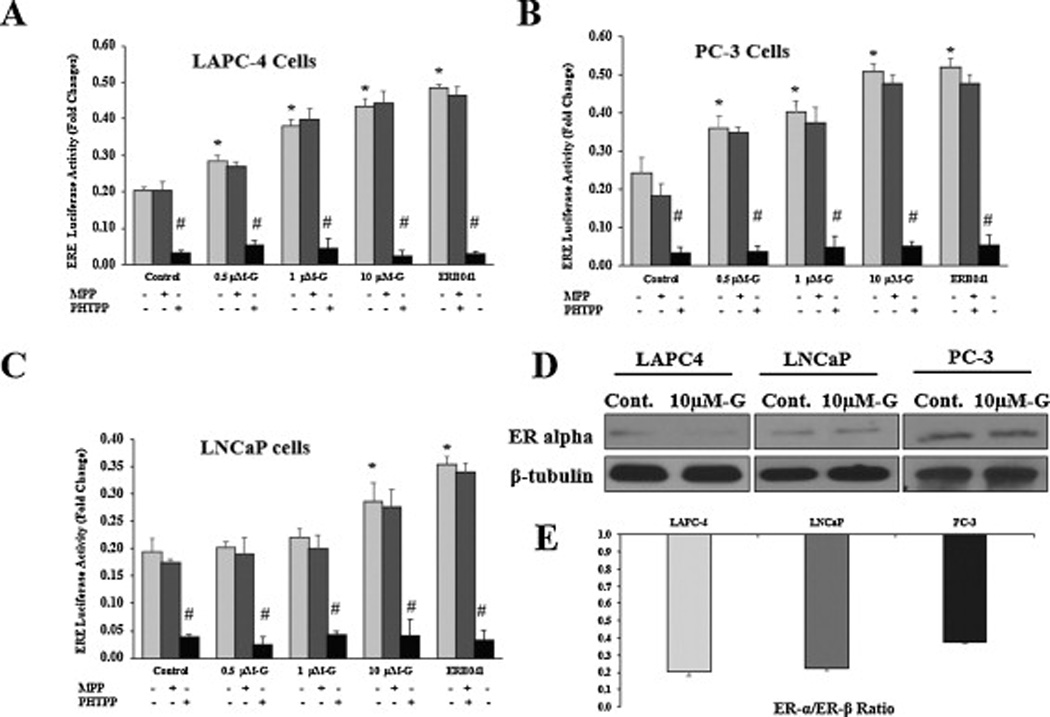
The effect of genistein alone or in combination with either MPP (ER-α antagonist) or PHTPP (ER-β antagonist) on ERE luciferase activity was measured in LAPC-4 (A), PC-3 (B), and LNCaP (C) cells that were transfected with ERE luciferase reporter. Results represent the means ± SD of three independent experiments. * (P < 0.05) for comparing ERE luciferase activity after genistein treatment with that in the vehicle control. # (P < 0.05) for comparing ERE luciferase activity in the presence of PHTPP with that in the absence of PHTPP in each treatment category. D: Western blot analysis of the effect of 10µmol/L of genistein on ER-α protein expression in the three PCa cell lines compared with the vehicle control. E: graphic representation of the ratio between ER-α and ER-β protein quantified from the Western blots in figures 9D and 4A, respectively using image J software.
1.2. Effects of genistein on PCa cell proliferation are mediated by ER-β
In order to verify the involvement of ER-β in genistein’s action, we treated PCa cells with genistein with and without the specific ER-β antagonist, PHTPP, for 24, 48 and 72 hours. Cell proliferation analysis revealed abrogation by PHTPP of the inhibitory effects of low physiological doses of genistein (0.5–10µmol/L) in LAPC-4 and PC-3 cells (figs. 10B and 10C). In LNCaP cells, in contrast, blocking ER-β resulted in augmentation of the stimulatory effects of genistein on cell proliferation (fig. 10A). Based on these results and on our previous data showing that genistein induces a paradoxical activation of AR in LNCaP cells [46], we concluded that ER-β mainly mediates the anti-proliferative effect of genistein in cells with wild-type AR (LAPC-4) and cells lacking AR (PC-3), but not to a much lesser extent in cells with promiscuously mutant AR (LNCaP).
Fig 10. Effects of genistein on PCa cell growth and viability.
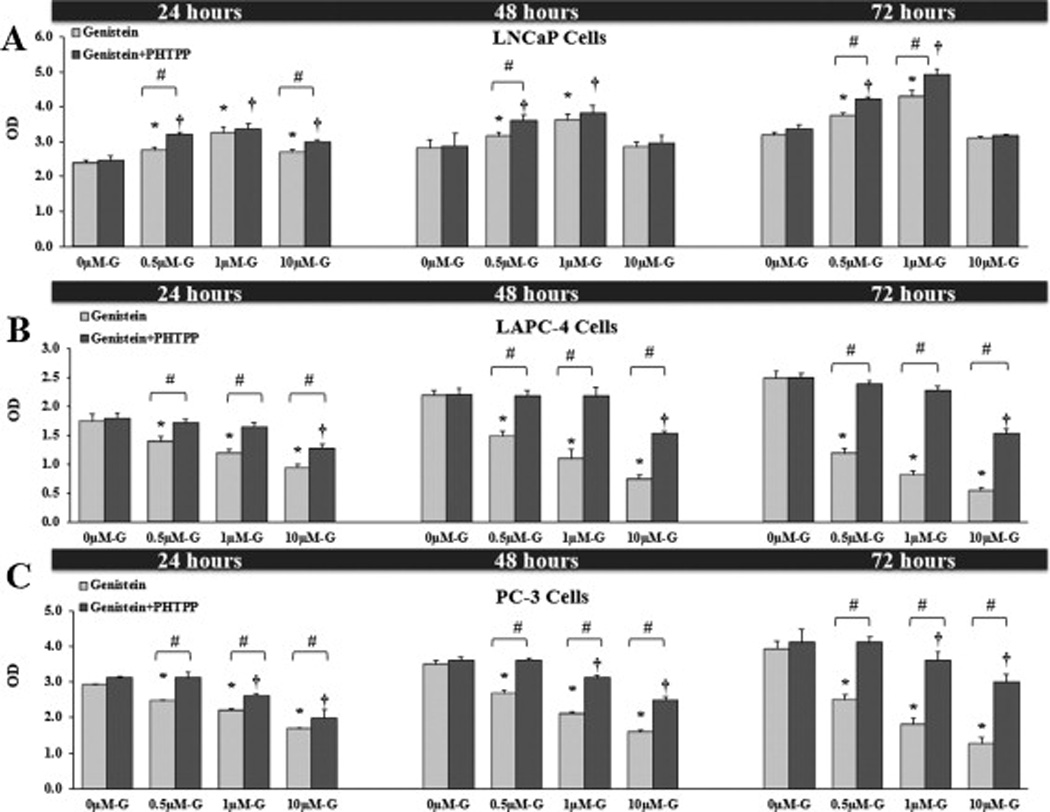
The data represent the mean ± SD of three MTS assay experiments each in triplicate. Graphic presentation of the effects of different concentrations of genistein alone or combined with PHTPP after 24, 48 and 72 hours on LAPC-4 (A), LNCaP (B) and PC-3 (C) cell growth. OD; optical density. * (P < 0.05) and † (P < 0.05) for comparing cell proliferation after genistein treatment with cell proliferation in the vehicle control state in the absence and presence of PHTPP, respectively. # (P < 0.05) for comparing cell proliferation between cells treated with genistein only and cells treated with genistein plus PHTPP within each concentration of genistein.
2. Discussion
This study revealed two important features regarding the effects of genistein on PCa. First, genistein is capable of inducing epigenetic modifications in PCa through reversing ER-β promoter hypermethylation and subsequently increasing ER-β gene expression. Second: ER-β is a major mediator for genistein’s anti-cancer activity in PCa.
Genistein is known to possess several anti-cancer properties in PCa, which have recently been shown to involve epigenetic mechanisms such as DNA methylation. Genistein reversed the promoter hypermethylation in tumor suppressor genes in PCa such as; RASSF1 (Ras association domain family 1), PTEN (phosphatase and tensin homolog), cyclin D, p53 and FOXO3a [22, 23, 25]. Previous studies have shown that genistein inhibits DNA methyltransferase (DNMT) and methyl binding domain (MBD) proteins [24]. Thus, we examined the epigenetic effect of genistein on ER-β gene, known to have antiproliferative and prodifferentiative roles in PCa, suggesting a tumor suppressor function. We also investigated how this epigenetic effect modifies ER-β gene expression and transcriptional activity.
Our study clearly demonstrates that genistein causes demethylation in the ER-β promoter, increases ER-β mRNA and protein levels, and induces ER-β transcriptional activity. To our knowledge, this is the first time that such a role for genistein has been reported in PCa and, importantly, these effects were achieved at physiological doses of genistein. Genistein reversed ER-β promoter hypermethylation and induced ER-β expression only in PCa cells that have higher basal levels of promoter methylation (LNCaP and LAPC-4 cells), but not in PC-3 cells that have low basal levels of promoter methylation. In addition, acting as an agonist, genistein enhanced ER-β nuclear localization and, by inference, its transcriptional activity.
DNA methylation is a critical mechanism by which many tumor suppressor genes get inactivated or silenced in PCa such as E-cadherin [47] and glutathione S-transferase [48]. Our results showed that the ER-β gene promoter is hypermethylated in untreated LNCaP and LAPC-4 PCa cells, but not in PC-3 cells, likely responsible for the low basal levels of ER-β gene expression in these two cell lines relative to PC-3 cells that have high basal levels of ER-β expression. This is consistent with previous studies showing that the extent of methylation in ER-β CpG islands was considerable in LNCaP cells, while this region was unmethylated in PC3 cells [21]. Previous studies have reported transcriptional activity at ER- β promoter and the untranslated region upstream to it, called exon 0N [49]. Our analysis of this region revealed three CpG islands which is partially consistent with a study by Zhu et al [21] where they reported the presence of two CpG islands, one in the ER-β promoter and the other one is in exon 0N. By blasting our sequence with theirs, we found these CpG islands are complementary with our first and second islands which are located in exon 0N and the promoter, respectively. The third island we report is located downstream in the ER-β promoter. These observations highlight DNA methylation at this region as an impending epigenetic mechanism by which ER-β is downregulated in PCa.
The process of methylation is reversible if newly synthesized DNA strands are not methylated. Therefore, demethylating agents such as 5-Aza-dC have been investigated as cancer therapeutic agents in clinical trials [50–54], but their side effects and toxicity are serious concerns. Accordingly, we used genistein, a naturally occurring, nontoxic, dietary isoflavone and compared its effect with 5-Aza-dC. Our results revealed that CpG islands in the promoter and exon 0N region of the ER-β gene are hypermethylated in the LNCaP and LAPC-4 PCa cell lines and that treatment with physiological doses of genistein (5.0–10 µmol/L) caused demethylation of these sites, albeit to a lesser extent than that caused by 5-Aza-dC especially in LAPC-4 cells. Promoter methylation leads to inhibition of gene transcription either directly through blocking the binding of transcription factors to promoters containing methylated CpG sites or indirectly via Methyl-CpG-binding domain (MBD) proteins that cause repression of gene transcription [55]. Thus, genistein significantly induced ER-β gene transcription at the mRNA and protein levels likely by reducing ER-β promoter and exon 0N hypermethylation. These effects were similar to that of 5-Aza-dC in both LNCaP and LAPC-4 cell lines. In PC-3 cells, we found that basal levels of ER-β promoter methylation were much lower than in the other two cell lines. This observation may explains the absence of any tangible increase in ER-β expression by genistein treatment in PC-3 cells and is consistent with the notion that genistein-mediated effects on restoring ER-β levels are linked to a great extent to its ability to reverse promoter hypermethylation of ER-β gene.
To understand how genistein reverse ER-β hypermethylation in PCa cells, we investigated the effect of genistein on the DNMT enzymes, DNMT1, DNMT3a and DNMT3b which are responsible for DNA hypermethylation via transferring the methyl group to cytosine residues. Our data clearly indicate that genistein, at low physiological concentrations, causes a decline in the levels of DNMT1 and DNMT3b without changing the levels of DNMT3a. The effect on DNMT1 and DNMT3a levels was obtained as early as 48 hours after genistein treatment which proceeds changes in DNA methylation by 24 to 36 hours. These observations suggest that DNMT1 and DNMT3a downregulation is a plausible mechanism by which genistein reverses ER-β hypermethylation. Similar effects on DNMT levels have been reported for genistein in breast cancer [56] and for other chemopreventive agents such as Mahanine in PCa [57]. For the first time, our study linked this effect of genistein on DNMT levels to the demethylation of the critical tumor suppressor gene, ER-β in PCa.
Subsequent to the demethylation of ER-β promoter and exon 0N, we found that ER-β expression increased markedly in response to genistein stimulation. This finding is in accordance with the previously reported inverse relationship between ER-β promoter methylation and ER-β expression levels [20]. Nonetheless, our data revealed lack of effect of E2 on ER-β methylation or expression which indicates a distinct effect of genistein on ER-β from that of its primary natural ligand, E2. This phenomena of the distinct effect of ER-β ligands has been proposed by some studies [44, 58, 59], however the mechanism behind these varied effects that different ligands have on ER-β function needs to be thoroughly investigated.
Furthermore, our data showed that genistein induces the phosphorylation of ER-β protein at two different residues, serine 87 and serine 105, which have been previously reported to alter ER-β transcriptional activity [43]. We also found that this induced ER-β phosphorylation by genistein was accompanied by increases in ER-β nuclear translocation. At this point, it is difficult to predict the link between the effect of genistein on ER-β promoter demethylation and its effect on ER-β phosphorylation and nuclear localization. The latter is more likely to be attributed to the ability of genistein to bind to the ER-β as an agonist. However, the two mechanisms converge into one ultimate endpoint which is the increased ER-β transcriptional activity.
ER-β exerts its anti-cancer effects through acting as a transcription factor for genes that limit cell growth or enhance survival. Increasing ER-β expression and/or transcriptional activity has preventive and therapeutic potential in PCa. Thus, we investigated the potential of genistein to induce ER-β transcriptional activity via testing its effects on the expression of FOXO1, known to be a target of ER-β [44, 60], as a proxy for ER-β transcriptional activity. Genistein enhanced the nuclear localization and the phosphorylated fraction of ER-β in all three PCa cell lines, even in PC-3 cells that did not show any ER-β expression response to genistein suggesting that genistein-induced effects on ER-β activation are unrelated to its effects on ER-β expression. The potential of genistein to induce ER-β activation is secondary to its ability to bind to the receptor as a ligand, inducing its phosphorylation and nuclear localization. However, the ability of genistein to increase ER-β expression mostly occurs subsequent to its epigenetic effects on ER-β promoter methylation. Nevertheless, increasing ER-β expression should theoretically increase its transcriptional activity via increasing the quantity of ER-β protein available for activating transcription.
Genistein induced FOXO1 expression in a dose dependent manner in all PCa cell lines. In LNCaP cells, however, a higher dose was needed to achieve the response. This finding might indicate a certain degree of interference by the promiscuously mutant AR in LNCaP cells that has been shown to be paradoxically activated by genistein in our previous studies [61]. The effect of genistein on FOXO1 expression was blunted after blocking ER-β using its specific antagonist PHTPP which indicates a mediatory role of ER-β. Altogether, these results support the notion that ER-β is a major mediator of genistein action and are consistent with previous studies showing that genistein failed to protect against PCa in ER-β knockout mice, albeit reduced the incidence of cancer in ER-β wild-type mice [27]. We demonstrated previously that genistein induces cell proliferation in LNCaP cells. In this study, a co-treatment of LNCaP cells with genistein and the ER-β antagonist augmented cell proliferation which indicates loss of the genistein-mediated inhibitory effect after blocking ER-β.
In order to confirm these findings, we measured ERE luciferase activity in PCa cells after transfecting them with ERE luciferase reporter. Our data showed a robust dose-dependent enhancement of ERE activity in LAPC-4 and PC-3 cells starting from the lowest dose of genistein that has been used (0.5 µmol/L). In LNCaP cells, this effect was achieved only in response to the highest dose of genistein that has been applied (10µmol/L). In our previous work, we demonstrated a potential of genistein to bind to and activate the promiscuously mutant AR in LNCaP cells [61]. This occupation of genistein by the mutant AR might explain the delay in triggering the ERE activity in this cell line. We also found that, in all PCa cells, the induction in ERE luciferase activity was dramatically abolished by ER-β antagonist while not affected by blocking ER-α even in the basal non-stimulated states. These observations indicate that the ERE activity in PCa cells was mainly mediated by ER-β and not by ER-α. Supporting to this notion, we found that ER-α protein levels and their ratio to ER-β protein were very low in PCa cells. These findings was in agreement with Kuiper GG. et al. [38] where they reported very low levels of ER-α mRNA and a very low ratio of ER-α mRNA to ER-β mRNA in prostate tissue. Ito et al. also reported lower levels of ER-α than ER-β in PCa cells [62]. Thus, our study highlights the interaction between genistein and ER-β in absence of any conflicting effects by ER-α. Despite the fact that this pattern of distribution for ER-α and ER-β might be altered in PCa [63, 64], stromal cells are the main source of ER-α production [16] which may explain the preserved ER-α/ER-β ratio in the PCa epithelial cell models that we use. At the same time, this should direct the motivation towards utilizing new models of PCa cells that exhibit different ratios of ER-α and ER-β. The significance of this recommendation is based upon observations form previous studies reporting differential chemopreventive effects of genistein in breast cancer cells depending on the ER-α/ER-β ratio [39].
In summary, our study is the first report to show that the ER-β gene, which is epigenetically silenced in PCa, can be reactivated by genistein-induced promoter demethylation. Genistein showed similar effects to that of 5-Aza-dC, which is currently undergoing phase II clinical trials as a treatment for PCa [65]. Furthermore, genistein appeared to cause enhancement of the transcriptional activity of ER-β, an effect that was comparable to that of the specific ERβ agonist ERB-041. Since genistein is a naturally occurring, nontoxic, and dietary soy isoflavone, and we used physiologically attainable genistein concentrations, these results indicate that genistein may be a novel, advantageous therapeutic agent for treating PCa. In addition, this is the first study to demonstrate the role of ER-β in mediating genistein activity on PCa cells in relation to the mutational status of the AR. Further studies are necessary to elucidate the mechanistic role of ER-β in mediating the activity of genistein. Other determinants that could modify this mediation role of ER-β, such as the AR mutations we discuss in this report, should also be investigated. Our findings suggest that the presence of an intact ER-β pathway renders PCa cells more sensitive to the anti-cancer effects of genistein. Thus, maintaining or enhancing the mediatory role of ER-β in the therapeutic effects of genistein on PCa are likely very important.
Furthermore, findings from this study may serve as a platform for future studies that are directed towards exploring the potential variability in the response to genistein among different PCa metastatic sites. We based our assumption on the fact that PCa cells that have been used herein were originally isolated from different metastatic sites and exhibited varied degrees of ER-β promotor methylation. Thus, this study might enlighten our understanding of how the tumor behaves in different beds of metastasis which might affect future therapeutic decisions.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
Highlights.
-
►
Genistein reduces ER-β promoter methylation significantly in prostate cancer cells
-
►
This is accompanied by dose-dependent increases in ER-β protein and mRNA expression
-
►
It also induces nuclear translocation and activity of ER-β in prostate cancer cells
-
►
Genistein inhibits the proliferation of PCa cells with wild type androgen receptor
-
►
Blocking ER-β abolished the inhibitory effects of genistein on cell proliferation
Acknowledgements
We gratefully acknowledge Dr. R. Reiter (UCLA) and Dr. Karen Knudsen (Kimmel Cancer Center, Thomas Jefferson University) for providing us with LAPC-4 cells. This work was supported by NIH Grant No. CA 116195 and by Grant No. GM 0842 from the Egyptian Ministry of Higher Education.
Abbreviations
- 5-Aza-dC
5-Aza-2′-deoxycytidine
- AR
androgen receptor
- DMSO
dimethyl sulfoxide
- DNMT
DNA methyltransferase
- ER-β
estrogen receptor beta
- FOXO-1
forkhead box O-1
- GAPDH
glyceraldehyde-3-phosphate dehydrogenase
- MBD
methyl binding domain
- MSP
methylationspecific PCR
- PCa
prostate cancer
- PCR
polymerase chain reaction
- PHTPP
4-(2-phenyl-5,7-bis(trifluoromethyl)pyrazolo[1,5-a]pyrimidin-3-yl)phenol
- PIN
prostatic intraepithelial neoplasia
- PTEN
phosphatase and tensin homolog
- RASSF1
Ras association domain family 1
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Jemal A, Bray F, Center MM, Ferlay J, Ward E, et al. Global cancer statistics. CA Cancer J Clin. 2011;61:69–90. doi: 10.3322/caac.20107. [DOI] [PubMed] [Google Scholar]
- 2.Siegel R, Desantis C, Virgo K, Stein K, Mariotto A, et al. Cancer treatment and survivorship statistics. CA Cancer J Clin. 2012;2012;62:220–241. doi: 10.3322/caac.21149. [DOI] [PubMed] [Google Scholar]
- 3.Kimura T. East meets West: ethnic differences in prostate cancer epidemiology between East Asians and Caucasians. Chin J Cancer. 2011 doi: 10.5732/cjc.011.10324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Adlercreutz H, Markkanen H, Watanabe S. Plasma concentrations of phyto-oestrogens in Japanese men. Lancet. 1993;342:1209–1210. doi: 10.1016/0140-6736(93)92188-y. [DOI] [PubMed] [Google Scholar]
- 5.Morton MS, Chan PS, Cheng C, Blacklock N, Matos-Ferreira A, et al. Lignans and isoflavonoids in plasma and prostatic fluid in men: samples from Portugal, Hong Kong, and the United Kingdom. Prostate. 1997;32:122–128. doi: 10.1002/(sici)1097-0045(19970701)32:2<122::aid-pros7>3.0.co;2-o. [DOI] [PubMed] [Google Scholar]
- 6.Hwang YW, Kim SY, Jee SH, Kim YN, Nam CM. Soy food consumption and risk of prostate cancer: a meta-analysis of observational studies. Nutr Cancer. 2009;61:598–606. doi: 10.1080/01635580902825639. [DOI] [PubMed] [Google Scholar]
- 7.Yan L, Spitznagel EL. Meta-analysis of soy food and risk of prostate cancer in men. Int J Cancer. 2005;117:667–669. doi: 10.1002/ijc.21266. [DOI] [PubMed] [Google Scholar]
- 8.Jian L. Soy, isoflavones, and prostate cancer. Mol Nutr Food Res. 2009;53:217–226. doi: 10.1002/mnfr.200800167. [DOI] [PubMed] [Google Scholar]
- 9.Kuiper GG, Lemmen JG, Carlsson B, Corton JC, Safe SH, et al. Interaction of estrogenic chemicals and phytoestrogens with estrogen receptor beta. Endocrinology. 1998;139:4252–4263. doi: 10.1210/endo.139.10.6216. [DOI] [PubMed] [Google Scholar]
- 10.Morito K, Hirose T, Kinjo J, Hirakawa T, Okawa M, et al. Interaction of phytoestrogens with estrogen receptors alpha and beta. Biol Pharm Bull. 2001;24:351–356. doi: 10.1248/bpb.24.351. [DOI] [PubMed] [Google Scholar]
- 11.Bektic J, Berger AP, Pfeil K, Dobler G, Bartsch G, et al. Androgen receptor regulation by physiological concentrations of the isoflavonoid genistein in androgen-dependent LNCaP cells is mediated by estrogen receptor beta. Eur Urol. 2004;45:245–251. doi: 10.1016/j.eururo.2003.09.001. discussion 251. [DOI] [PubMed] [Google Scholar]
- 12.Weihua Z, Makela S, Andersson LC, Salmi S, Saji S, et al. A role for estrogen receptor beta in the regulation of growth of the ventral prostate. Proc Natl Acad Sci U S A. 2001;98:6330–6335. doi: 10.1073/pnas.111150898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Imamov O, Morani A, Shim GJ, Omoto Y, Thulin-Andersson C, et al. Estrogen receptor beta regulates epithelial cellular differentiation in the mouse ventral prostate. Proc Natl Acad Sci U S A. 2004;101:9375–9380. doi: 10.1073/pnas.0403041101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.McPherson SJ, Hussain S, Balanathan P, Hedwards SL, Niranjan B, et al. Estrogen receptorbeta activated apoptosis in benign hyperplasia and cancer of the prostate is androgen independent and TNFalpha mediated. Proc Natl Acad Sci U S A. 2010;107:3123–3128. doi: 10.1073/pnas.0905524107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ho SM. Estrogens and anti-estrogens: key mediators of prostate carcinogenesis and new therapeutic candidates. J Cell Biochem. 2004;91:491–503. doi: 10.1002/jcb.10759. [DOI] [PubMed] [Google Scholar]
- 16.Leav I, Lau KM, Adams JY, McNeal JE, Taplin ME, et al. Comparative studies of the estrogen receptors beta and alpha and the androgen receptor in normal human prostate glands, dysplasia, and in primary and metastatic carcinoma. Am J Pathol. 2001;159:79–92. doi: 10.1016/s0002-9440(10)61676-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sasaki M, Tanaka Y, Perinchery G, Dharia A, Kotcherguina I, et al. Methylation and inactivation of estrogen, progesterone, and androgen receptors in prostate cancer. J Natl Cancer Inst. 2002;94:384–390. doi: 10.1093/jnci/94.5.384. [DOI] [PubMed] [Google Scholar]
- 18.Walton TJ, Li G, Seth R, McArdle SE, Bishop MC, et al. DNA demethylation and histone deacetylation inhibition co-operate to re-express estrogen receptor beta and induce apoptosis in prostate cancer cell-lines. Prostate. 2008;68:210–222. doi: 10.1002/pros.20673. [DOI] [PubMed] [Google Scholar]
- 19.Lau KM, LaSpina M, Long J, Ho SM. Expression of estrogen receptor (ER)-alpha and ERbeta in normal and malignant prostatic epithelial cells: regulation by methylation and involvement in growth regulation. Cancer Res. 2000;60:3175–3182. [PubMed] [Google Scholar]
- 20.Rody A, Holtrich U, Solbach C, Kourtis K, von Minckwitz G, et al. Methylation of estrogen receptor beta promoter correlates with loss of ER-beta expression in mammary carcinoma and is an early indication marker in premalignant lesions. Endocr Relat Cancer. 2005;12:903–916. doi: 10.1677/erc.1.01088. [DOI] [PubMed] [Google Scholar]
- 21.Zhu X, Leav I, Leung YK, Wu M, Liu Q, et al. Dynamic regulation of estrogen receptor-beta expression by DNA methylation during prostate cancer development and metastasis. Am J Pathol. 2004;164:2003–2012. doi: 10.1016/s0002-9440(10)63760-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Adjakly M, Bosviel R, Rabiau N, Boiteux JP, Bignon YJ, et al. DNA methylation and soy phytoestrogens: quantitative study in DU-145 and PC-3 human prostate cancer cell lines. Epigenomics. 2011;3:795–803. doi: 10.2217/epi.11.103. [DOI] [PubMed] [Google Scholar]
- 23.Vardi A, Bosviel R, Rabiau N, Adjakly M, Satih S, et al. Soy phytoestrogens modify DNA methylation of GSTP1, RASSF1A, EPH2 and BRCA1 promoter in prostate cancer cells. In Vivo. 2010;24:393–400. [PubMed] [Google Scholar]
- 24.Majid S, Dar AA, Shahryari V, Hirata H, Ahmad A, et al. Genistein reverses hypermethylation and induces active histone modifications in tumor suppressor gene B-Cell translocation gene 3 in prostate cancer. Cancer. 2010;116:66–76. doi: 10.1002/cncr.24662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kikuno N, Shiina H, Urakami S, Kawamoto K, Hirata H, et al. Genistein mediated histone acetylation and demethylation activates tumor suppressor genes in prostate cancer cells. Int J Cancer. 2008;123:552–560. doi: 10.1002/ijc.23590. [DOI] [PubMed] [Google Scholar]
- 26.Kumar R, Verma V, Jain A, Jain RK, Maikhuri JP, et al. Synergistic chemoprotective mechanisms of dietary phytoestrogens in a select combination against prostate cancer. J Nutr Biochem. 2011;22:723–731. doi: 10.1016/j.jnutbio.2010.06.003. [DOI] [PubMed] [Google Scholar]
- 27.Slusarz A, Jackson GA, Day JK, Shenouda NS, Bogener JL, et al. Aggressive prostate cancer is prevented in ERalphaKO mice and stimulated in ERbetaKO TRAMP mice. Endocrinology. 2012;153:4160–4170. doi: 10.1210/en.2012-1030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Klein KA, Reiter RE, Redula J, Moradi H, Zhu XL, et al. Progression of metastatic human prostate cancer to androgen independence in immunodeficient SCID mice. Nat Med. 1997;3:402–408. doi: 10.1038/nm0497-402. [DOI] [PubMed] [Google Scholar]
- 29.Patra SK, Patra A, Rizzi F, Ghosh TC, Bettuzzi S. Demethylation of (Cytosine-5-C-methyl) DNA and regulation of transcription in the epigenetic pathways of cancer development. Cancer Metastasis Rev. 2008;27:315–334. doi: 10.1007/s10555-008-9118-y. [DOI] [PubMed] [Google Scholar]
- 30.Li LC, Yeh CC, Nojima D, Dahiya R. Cloning and characterization of human estrogen receptor beta promoter. Biochem Biophys Res Commun. 2000;275:682–689. doi: 10.1006/bbrc.2000.3363. [DOI] [PubMed] [Google Scholar]
- 31.Skliris GP, Munot K, Bell SM, Carder PJ, Lane S, et al. Reduced expression of oestrogen receptor beta in invasive breast cancer and its re-expression using DNA methyl transferase inhibitors in a cell line model. J Pathol. 2003;201:213–220. doi: 10.1002/path.1436. [DOI] [PubMed] [Google Scholar]
- 32.Zhao C, Lam EW, Sunters A, Enmark E, De Bella MT, et al. Expression of estrogen receptor beta isoforms in normal breast epithelial cells and breast cancer: regulation by methylation. Oncogene. 2003;22:7600–7606. doi: 10.1038/sj.onc.1207100. [DOI] [PubMed] [Google Scholar]
- 33.Al-Nakhle H, Smith L, Bell SM, Burns PA, Cummings M, et al. Regulation of estrogen receptor beta1 expression in breast cancer by epigenetic modification of the 5' regulatory region. Int J Oncol. 2013;43:2039–2045. doi: 10.3892/ijo.2013.2112. [DOI] [PubMed] [Google Scholar]
- 34.Turek-Plewa J, Jagodzinski PP. The role of mammalian DNA methyltransferases in the regulation of gene expression. Cell Mol Biol Lett. 2005;10:631–647. [PubMed] [Google Scholar]
- 35.Patra SK, Patra A, Zhao H, Dahiya R. DNA methyltransferase and demethylase in human prostate cancer. Mol Carcinog. 2002;33:163–171. doi: 10.1002/mc.10033. [DOI] [PubMed] [Google Scholar]
- 36.Gravina GL, Marampon F, Piccolella M, Motta M, Ventura L, et al. Hormonal therapy promotes hormone-resistant phenotype by increasing DNMT activity and expression in prostate cancer models. Endocrinology. 2011;152:4550–4561. doi: 10.1210/en.2011-1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kobayashi Y, Absher DM, Gulzar ZG, Young SR, McKenney JK, et al. DNA methylation profiling reveals novel biomarkers and important roles for DNA methyltransferases in prostate cancer. Genome Res. 2011;21:1017–1027. doi: 10.1101/gr.119487.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kuiper GG, Carlsson B, Grandien K, Enmark E, Haggblad J, et al. Comparison of the ligand binding specificity and transcript tissue distribution of estrogen receptors alpha and beta. Endocrinology. 1997;138:863–870. doi: 10.1210/endo.138.3.4979. [DOI] [PubMed] [Google Scholar]
- 39.Nadal-Serrano M, Pons DG, Sastre-Serra J, Blanquer-Rossello Mdel M, Roca P, et al. Genistein modulates oxidative stress in breast cancer cell lines according to ERalpha/ERbeta ratio: effects on mitochondrial functionality, sirtuins, uncoupling protein 2 and antioxidant enzymes. Int J Biochem Cell Biol. 2013;45:2045–2051. doi: 10.1016/j.biocel.2013.07.002. [DOI] [PubMed] [Google Scholar]
- 40.Zhu BT, Han GZ, Shim JY, Wen Y, Jiang XR. Quantitative structure-activity relationship of various endogenous estrogen metabolites for human estrogen receptor alpha and beta subtypes: Insights into the structural determinants favoring a differential subtype binding. Endocrinology. 2006;147:4132–4150. doi: 10.1210/en.2006-0113. [DOI] [PubMed] [Google Scholar]
- 41.Enmark E, Pelto-Huikko M, Grandien K, Lagercrantz S, Lagercrantz J, et al. Human estrogen receptor beta-gene structure, chromosomal localization, and expression pattern. J Clin Endocrinol Metab. 1997;82:4258–4265. doi: 10.1210/jcem.82.12.4470. [DOI] [PubMed] [Google Scholar]
- 42.Cvoro A, Tatomer D, Tee MK, Zogovic T, Harris HA, et al. Selective estrogen receptor-beta agonists repress transcription of proinflammatory genes. J Immunol. 2008;180:630–636. doi: 10.4049/jimmunol.180.1.630. [DOI] [PubMed] [Google Scholar]
- 43.Lam HM, Suresh Babu CV, Wang J, Yuan Y, Lam YW, et al. Phosphorylation of human estrogen receptor-beta at serine 105 inhibits breast cancer cell migration and invasion. Mol Cell Endocrinol. 2012;358:27–35. doi: 10.1016/j.mce.2012.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Leung YK, Ho SM. Estrogen receptor beta: switching to a new partner and escaping from estrogen. Sci Signal. 2011;4:pe19. doi: 10.1126/scisignal.2001991. [DOI] [PubMed] [Google Scholar]
- 45.Compton DR, Sheng S, Carlson KE, Rebacz NA, Lee IY, et al. Pyrazolo[1,5-a]pyrimidines: estrogen receptor ligands possessing estrogen receptor beta antagonist activity. J Med Chem. 2004;47:5872–5893. doi: 10.1021/jm049631k. [DOI] [PubMed] [Google Scholar]
- 46.Mahmoud AM, Yang W, Bosland MC. Soy Isoflavones and Prostate Cancer: A Review of Molecular Mechanisms. J Steroid Biochem Mol Biol. 2013 doi: 10.1016/j.jsbmb.2013.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Li LC, Zhao H, Nakajima K, Oh BR, Ribeiro Filho LA, et al. Methylation of the E-cadherin gene promoter correlates with progression of prostate cancer. J Urol. 2001;166:705–709. [PubMed] [Google Scholar]
- 48.Enokida H, Shiina H, Urakami S, Igawa M, Ogishima T, et al. Ethnic group-related differences in CpG hypermethylation of the GSTP1 gene promoter among African-American, Caucasian and Asian patients with prostate cancer. Int J Cancer. 2005;116:174–181. doi: 10.1002/ijc.21017. [DOI] [PubMed] [Google Scholar]
- 49.Hirata S, Shoda T, Kato J, Hoshi K. The multiple untranslated first exons system of the human estrogen receptor beta (ER beta) gene. J Steroid Biochem Mol Biol. 2001;78:33–40. doi: 10.1016/s0960-0760(01)00071-1. [DOI] [PubMed] [Google Scholar]
- 50.Stewart DJ, Issa JP, Kurzrock R, Nunez MI, Jelinek J, et al. Decitabine effect on tumor global DNA methylation and other parameters in a phase I trial in refractory solid tumors and lymphomas. Clin Cancer Res. 2009;15:3881–3888. doi: 10.1158/1078-0432.CCR-08-2196. [DOI] [PubMed] [Google Scholar]
- 51.George RE, Lahti JM, Adamson PC, Zhu K, Finkelstein D, et al. Phase I study of decitabine with doxorubicin and cyclophosphamide in children with neuroblastoma and other solid tumors: a Children's Oncology Group study. Pediatr Blood Cancer. 2010;55:629–638. doi: 10.1002/pbc.22607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Raffoux E, Cras A, Recher C, Boelle PY, de Labarthe A, et al. Phase 2 clinical trial of 5- azacitidine, valproic acid, and all-trans retinoic acid in patients with high-risk acute myeloid leukemia or myelodysplastic syndrome. Oncotarget. 2010;1:34–42. doi: 10.18632/oncotarget.106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Christman JK. 5-Azacytidine and 5-aza-2'-deoxycytidine as inhibitors of DNA methylation: mechanistic studies and their implications for cancer therapy. Oncogene. 2002;21:5483–5495. doi: 10.1038/sj.onc.1205699. [DOI] [PubMed] [Google Scholar]
- 54.Zhou L, Cheng X, Connolly BA, Dickman MJ, Hurd PJ, et al. Zebularine: a novel DNA methylation inhibitor that forms a covalent complex with DNA methyltransferases. J Mol Biol. 2002;321:591–599. doi: 10.1016/S0022-2836(02)00676-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bird A. DNA methylation patterns and epigenetic memory. Genes Dev. 2002;16:6–21. doi: 10.1101/gad.947102. [DOI] [PubMed] [Google Scholar]
- 56.Xie Q, Bai Q, Zou LY, Zhang QY, Zhou Y, et al. Genistein inhibits DNA methylation and increases expression of tumor suppressor genes in human breast cancer cells. Genes Chromosomes Cancer. 2014;53:422–431. doi: 10.1002/gcc.22154. [DOI] [PubMed] [Google Scholar]
- 57.Agarwal S, Amin KS, Jagadeesh S, Baishay G, Rao PG, et al. Mahanine restores RASSF1A expression by down-regulating DNMT1 and DNMT3B in prostate cancer cells. Mol Cancer. 2013;12:99. doi: 10.1186/1476-4598-12-99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Nakajima Y, Akaogi K, Suzuki T, Osakabe A, Yamaguchi C, et al. Estrogen regulates tumor growth through a nonclassical pathway that includes the transcription factors ERbeta and KLF5. Sci Signal. 2011;4:ra22. doi: 10.1126/scisignal.2001551. [DOI] [PubMed] [Google Scholar]
- 59.Tremblay A, Tremblay GB, Labrie F, Giguere V. Ligand-independent recruitment of SRC-1 to estrogen receptor beta through phosphorylation of activation function AF-1. Mol Cell. 1999;3:513–519. doi: 10.1016/s1097-2765(00)80479-7. [DOI] [PubMed] [Google Scholar]
- 60.Nakamura Y, Felizola SJ, Kurotaki Y, Fujishima F, McNamara KM, et al. Cyclin D1 (CCND1) expression is involved in estrogen receptor beta (ERbeta) in human prostate cancer. Prostate. 2012 doi: 10.1002/pros.22599. [DOI] [PubMed] [Google Scholar]
- 61.Mahmoud AM, Zhu T, Parray A, Siddique HR, Yang W, et al. Differential effects of genistein on prostate cancer cells depend on mutational status of the androgen receptor. PLoS One. 2013;8:e78479. doi: 10.1371/journal.pone.0078479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ito T, Tachibana M, Yamamoto S, Nakashima J, Murai M. Expression of estrogen receptor (ER-alpha and ER-beta) mRNA in human prostate cancer. Eur Urol. 2001;40:557–563. doi: 10.1159/000049836. [DOI] [PubMed] [Google Scholar]
- 63.Latil A, Bieche I, Vidaud D, Lidereau R, Berthon P, et al. Evaluation of androgen, estrogen (ER alpha and ER beta), and progesterone receptor expression in human prostate cancer by realtime quantitative reverse transcription-polymerase chain reaction assays. Cancer Res. 2001;61:1919–1926. [PubMed] [Google Scholar]
- 64.Megas G, Chrisofos M, Anastasiou I, Tsitlidou A, Choreftaki T, et al. Estrogen receptor (alpha and beta) but not androgen receptor expression is correlated with recurrence, progression and survival in post prostatectomy T3N0M0 locally advanced prostate cancer in an urban Greek population. Asian J Androl. 2015;17:98–105. doi: 10.4103/1008-682X.136445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Thibault A, Figg WD, Bergan RC, Lush RM, Myers CE, et al. A phase II study of 5-aza-2'deoxycytidine (decitabine) in hormone independent metastatic (D2) prostate cancer. Tumori. 1998;84:87–89. doi: 10.1177/030089169808400120. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.