

Abstract
Cocaine promotes addictive behavior primarily by blocking the dopamine transporter, thus increasing dopamine transmission in the nucleus accumbens (nAcc); however, additional mechanisms are continually emerging. Sigma-1 receptors (σ1Rs) are known targets for cocaine, yet the mechanisms underlying σ1R-mediated effects of cocaine are incompletely understood. The present study examined direct effects of cocaine on dissociated nAcc neurons expressing phosphatidylinositol-linked D1 receptors. Endoplasmic reticulum-located σ1Rs and inositol 1,4,5-trisphosphate (IP3) receptors (IP3Rs) were targeted using intracellular microinjection. IP3 microinjection robustly elevated intracellular Ca2+ concentration, [Ca2+]i. While cocaine alone was devoid of an effect, the IP3-induced response was σ1R-dependently enhanced by cocaine co-injection. Likewise, cocaine augmented the [Ca2+]i increase elicited by extracellularly applying an IP3-generating molecule (ATP), via σ1Rs. The cocaine-induced enhancement of the P3/ATP-mediated Ca2+ elevation occurred at pharmacologically relevant concentrations and was mediated by transient receptor potential canonical channels (TRPC). IP3 microinjection elicited a slight, transient depolarization, further converted to a greatly enhanced, prolonged response, by cocaine co-injection. The cocaine-triggered augmentation was σ1R-dependent, TRPC-mediated and contingent on [Ca2+]i elevation. ATP-induced depolarization was similarly enhanced by cocaine. Thus, we identify a novel mechanism by which cocaine promotes activation of D1-expressing nAcc neurons: enhancement of IP3R-mediated responses via σ1R activation at the endoplasmic reticulum, resulting in augmented Ca2+ release and amplified depolarization due to subsequent stimulation of TRPC. In vivo, intra-accumbal blockade of σ1R or TRPC significantly diminished cocaine-induced hyperlocomotion and locomotor sensitization, endorsing a physio-pathological significance of the pathway identified in vitro.
Keywords: sigma receptors, transient receptor potential channels, calcium, endoplasmic reticulum, imaging, nucleus accumbens
Graphical Abstract
1. Introduction
Pharmacotherapy of cocaine addiction is particularly ineffective in that addicts invariably relapse to drug use [1–3]. The study of cocaine action in the brain has unraveled newer mechanisms, of continually emerging complexity. The initial rewarding effects of cocaine are attributed to increased dopamine levels in the nucleus accumbens (nAcc)¶ achieved by inhibition of the dopamine transporter [4, 5], however, the transition to cocaine dependence likely involves other cellular processes. Further, a multitude of cocaine-induced responses are strictly contingent on D1 dopamine-receptor activation [6–8], occurring only in D1-expressing neurons [7], supporting the existence of additional mechanisms.
Cocaine is a known ligand of sigma-1 receptors (σ1Rs) [9], which are chaperone proteins residing at the endoplasmic reticulum in a dormant state, but able to relocate to other areas of the cell in response to agonist stimulation, and change their degree of interaction with several other chaperone proteins, favoring activation of various types of receptors and ion channels [10, 11]. Involvement of σ1Rs in modulation of dopaminergic transmission and addictive processes has long been recognized [12, 13]. σ1Rs are expressed in the nAcc, a key node in the circuit that controls reward-directed behavior [14, 15], and have been proposed as a pharmacologic target in the treatment of cocaine abuse [16, 17].
Data from in vivo animal studies point to involvement of σ1Rs in cocaine-induced responses. Cocaine self-administration triggers σ1Rs-mediated reinforcing effects that are absent in subjects without that particular experience with cocaine, and are dopamine-independent [18, 19]. Administration of σ1Rs agonists potentiates the reinforcing effects of cocaine [19]. Conversely,σ1R antagonists attenuate psychomotor and rewarding effects of cocaine [12, 13]. σ1R-blockade inhibits cocaine-induced place conditioning in mice [20, 21].
At the cellular level, cocaine induces an association of σ1Rs and D1 dopamine receptors, which results in cAMP accumulation and ERK1/2 activation in transfected cells and mouse striatal slices [22]. Conversely, cocaine promotes formation of σ1R-D2 dopamine receptor heterooligomers, inhibiting D2-mediated signaling [23]. Thus cocaine putatively destabilizes the balance of D1 and D2 receptor inputs, via σ1Rs, towards the D1 containing, pro-reward and motivating pathway [23]. However, a clear mechanism of cocaine-mediated enhancement of D1-pathway via σ1Rs remains elusive.
Activation of σ1Rs is associated with prolonged Ca2+ efflux from the endoplasmic reticulum (ER) through inositol 1,4,5-trisphosphate (IP3) receptors (IP3Rs) [10, 24]. The present study uses calcium imaging and intracellular microinjection to explore the mechanisms of cocaine-triggered activation of D1-expressing neurons, focusing on intracellularly-located σ1Rs.
2. Materials and methods
2.1. Ethical approval
Animal protocols were approved by the Institutional Animal Care and Use Committees from Temple University and Thomas Jefferson University.
2.2. Chemicals
All chemicals were from Sigma Aldrich (St. Louis, MO), unless otherwise mentioned. Cocaine hydrochloride was generously supplied by NIDA; NE-100 hydrochloride was from Santa Cruz Biotechnology (Dallas, TX). In experiments using intracellular microinjection, the reported concentration of chemicals is the calculated final concentration inside the cell. In experiments using ATP and extracellular administration of cocaine, the cells were pretreated with cocaine for 10 minutes, a time sufficient to allow intracellular uptake of cocaine. In experiments using IP3 and cocaine, there was no pretreatment phase.
2.3. Western blotting
Whole-cell lysates obtained from rat nucleus accumbens and NG108-15 cells (mouse neuroblastoma x rat glioma) were separated on Mini-PROTEAN TGX 4–20% gels (Bio-Rad, Hercules, CA) by SDS-PAGE followed by immunoblotting. Proteins were transferred to an Odyssey nitrocellulose membrane (Li-Cor Biosciences; Lincoln, NE). After blocking with Odyssey blocking buffer, the membranes were incubated overnight with primary antibody against σ1R (rabbit polyclonal, 1:100, OriGene Technologies, Rockville, MD), or IP3R3 (mouse monoclonal, 1:1,000, BD Biosciences, San Jose, CA). An antibody against β-actin (mouse monoclonal, 1:10,000; Sigma Aldrich) was used to confirm equal protein loading. Membranes were washed with Tris-buffered saline-Tween 20 (TBST) and incubated with the secondary antibodies: IRDye 800CW conjugated goat anti-rabbit IgG, and IRDye 680 conjugated goat anti-mouse IgG (1:10,000, 1 h at room temperature). Membranes were then washed in TBST and scanned using Li-Cor Odyssey Infrared and Odyssey V.3 software.
2.4. Neuronal cell culture
Nucleus accumbens neurons were dissociated from neonatal (1–2 day old) Sprague Dawley rats (Ace Animal Inc., Boyertown, PA) of both sexes as previously described [25]. Newborn rats were decapitated and the brains quickly removed surgically and immersed in ice-cold Hanks balanced salt solution (HBSS) (Mediatech, Herndon, VA). The nucleus accumbens was identified, removed, minced and subjected to enzymatic digestion (papain, 37°C), followed by mechanical trituration in presence of total medium – Neurobasal A (Invitrogen, Carlsbad, CA) containing 1% GlutaMax (Invitrogen), 2% penicillin-streptomycin-amphotericin B solution (Mediatech) and 10% fetal bovine serum. Cells were cultured on round 25 mm glass coverslips coated with poly-L-lysine (Sigma-Aldrich) in six-well plates. Cultures were maintained at 37°C in a humidified atmosphere with 5% CO2. The mitotic inhibitor cytosine β-arabinofuranoside (1μM) (Sigma-Aldrich) was added to the culture the third day to inhibit glial cell proliferation. Cells were used after 5 days in culture.
2.5. Calcium imaging
[Ca2+]i was measured as previously described [25]. Cells were incubated with 5 μM fura-2 AM (Invitrogen, Carlsbad, CA) in HBSS at room temperature for 45 min, in the dark, washed three times with dye-free HBSS, and then incubated for another 45 min to allow for complete de-esterification of the dye. Coverslips (25 mm diameter) were subsequently mounted in an open bath chamber (RP-40LP, Warner Instruments, Hamden, CT) on the stage of an inverted microscope Nikon Eclipse TiE (Nikon Inc., Melville, NY). The microscope is equipped with a Perfect Focus System and a Photometrics CoolSnap HQ2 CCD camera (Photometrics, Tucson, AZ). During the experiments, the Perfect Focus System was activated. Fura-2 AM fluorescence (emission = 510 nm), following alternate excitation at 340 and 380 nm, was acquired at a frequency of 0.25 Hz. Images were acquired and analyzed using NIS-Elements AR 3.1 software (Nikon Inc.). After appropriate calibration with ionomycin and CaCl2, and Ca2+ free and EGTA, respectively, the ratio of the fluorescence signals (340/380 nm) was converted to Ca2+ concentrations. In Ca2+-free experiments, CaCl2 was omitted.
2.6. Intracellular microinjection
Intracellular microinjections were performed using FemtotipsII, InjectManNI2 and FemtoJet systems (Eppendorf) as reported [25]. Pipettes were back-filled with an intracellular solution containing, in mM: 110 KCl, 10 NaCl and 20 HEPES (pH 7.2) or the compounds to be tested. The injection time was 0.4 s at 60 hPa with a compensation pressure of 20 hPa in order to maintain the microinjected volume to less than 1% of cell volume, as measured by microinjection of a fluorescent compound (Fura-2 free acid). The intracellular concentration of chemicals was determined based on the concentration in the pipette and the volume of injection. The cells to be injected were Z-scanned before injection and the cellular volume automatically calculated by the NIS-Elements AR 3.1 software (Nikon, Inc.).
2.7. Measurement of membrane potential
The relative changes in membrane potential of single neurons were evaluated using bis-(1,3-dibutylbarbituric acid) trimethine oxonol, DiBAC4(3), a slow response voltage-sensitive dye, as previously described [25]. Upon membrane hyperpolarization, the dye concentrates in the cell membrane, leading to a decrease in fluorescence intensity, while depolarization induces the sequestration of the dye into the cytosol, resulting in an increase of the fluorescence intensity. Cultured accumbens neurons were incubated for 30 min in HBSS containing 0.5 μM DiBAC4(3) and the fluorescence monitored at 0.17 Hz, excitation/emission: 480 nm/540 nm. Calibration of DiBAC4(3) fluorescence following background subtraction was performed using the Na+-K+ ionophore gramicidin in Na+-free physiological solution and various concentrations of K+ (to alter membrane potential) and N-methylglucamine (to maintain osmolarity). Under these conditions, the membrane potential was approximately equal to the K+ equilibrium potential determined by the Nernst equation. The intracellular K+ and Na+ concentration were assumed to be 130 mM and 10 mM, respectively.
2.8. Data analysis
Data obtained after in vitro experiments are expressed as mean and standard error of mean. One way ANOVA, followed by post-hoc Bonferroni and Tukey tests (Origin 7, OriginLab Corporation, Northampton, MA), were used to assess significant differences between groups; P < 0.05 was considered statistically significant.
2.9. In vivo experiments
Male Sprague–Dawley rats, weighing 250–280 g at the time of surgery, were individually housed under standard conditions. Two 26 gauge stainless steel guide cannulas directed bilaterally at the nucleus accumbens (nAcc) (± 0.9 mm lateral, 1.6 mm anterior, and 5.8 mm ventral to bregma) were stereotaxically implanted under isofluorane anesthesia. Dummy cannulae that extended 1 mm beyond the tip of the guide cannula were inserted immediately after surgery. Rats were handled and habituated to infusion procedures for 2–3 days before testing began. On test Days 1–5, rats were placed in individual automated activity monitors containing 16 infrared light emitters and sensors mounted on a frame within which a standard plastic animal cage was positioned (45 × 20 × 20 cm; AccuScan Instruments, Inc., Columbus, OH, USA). The number of photocell beam breaks was recorded by a computer equipped with Digiscan DMicro software (AccuScan Instruments). Following a 60 min habituation period, bilateral infusions of vehicle (0.5 μl, artificial cerebrospinal fluid (aCSF)) or BD-1063 (80 μg/0.5 μl, ab141323, Abcam, Cambridge, MA) or SKF-96365 (20 μg/0.5 μl, S7809, Sigma-Aldrich, St Louis, MO) were made into the nAcc, at a rate of 0.5 μl/min using a microinfusion pump. The injections cannulae remained in situ for one minute after the infusion. Twenty minutes following infusions, rats were injected with saline (1 ml/kg, intraperitoneal (ip)) or cocaine (15 mg/kg, ip) and behavioral activity monitored for another 60 min. This was repeated once daily for 5 days. Following a 7-day withdrawal period, all rats were then challenged with cocaine (Day 12, 15 mg/kg, ip) in the absence of further intracranial infusions. After testing, brains were removed and fixed in 4% paraformaldehyde for three days. Brains were sliced at 60 μm on a vibratome through the nAcc, and stained with cresyl violet to determine the location of the infusion cannula; injection sites are shown in Fig. 9a. Data from rats with both placements within the nAcc were included in the analysis. Behavioral data were analyzed with two-way repeated measures ANOVA. Significant main effects of treatment, day or interactions between treatment and day were further assessed with a Student-Newman-Keuls (SNK) post hoc test for multiple pair wise comparisons at each time point (Sigmaplot 12.5; Systat Software Inc.).
Figure 9. Cocaine-induced behavioral hyperactivity and sensitization involves σ1R and TRPC.
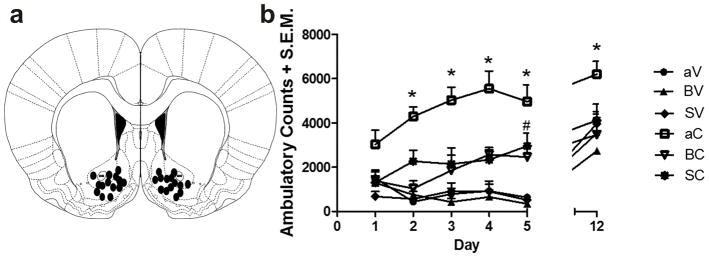
a, Coronal section indicating the distribution of infusion sites in the nAcc for the 35 experimental animals (aCSF-saline, n = 5; aCSF-cocaine, n = 7; BD-1063-saline, n = 5; BD-1063-cocaine, n = 6; SKF96365-saline, n = 6; SKF96365-cocaine, n = 6). Injection sites may appear fewer than the reported number of rats because of overlap of placements. b, Pretreatment with BD-1063 (80 μg/side) or SKF96365 (20 μg/side) administered 20 min prior to each cocaine injection for 5 days significantly inhibited cocaine-induced ambulatory activity on Days 2–5, and the development of repeated cocaine-induced locomotor sensitization on Day 12. Repeated administration of either antagonist alone did not alter levels of locomotion; *P < 0.05, aCSF-cocaine rats were significantly different from all other experimental groups; # SKF96365-cocaine rats were significantly different from aCSF-saline, BD-1063-saline and SKF96365-saline on day 5. Data are presented as mean +/− sem ambulatory counts/60 minutes; abbreviations: aV, intra-nAcc aCSF + ip saline vehicle; BV, intra-nAcc BD-1063 + ip saline vehicle; SVm intra-nAcc SFK96365 + ip saline vehicle; aC, intra-nAcc aCSF + ip cocaine; BC, intra-nAcc BD1063 + ip cocaine; SC, intra-nAcc SKF-96365 + ip cocaine.
3. Results
3.1. Identification of receptors of interest in cultured nucleus accumbens neurons
We used western blotting to confirm the presence of σ1Rs in cultured nAcc neurons (Fig. 1a), which is in agreement with previous reports [14, 15]. σ1Rs have been shown to associate with type 3 of IP3R (IP3R3) to promote increased Ca2+ efflux from the ER [10, 24]. We found that both IP3R3 and σ1Rs are expressed in cultured nAcc neurons (Fig. 1a). NG108 cells were used as a positive control for IP3R3 and σ1Rs [10].
Figure 1. Neurons of interest.

a, Nucleus accumbens neurons express both σ1Rs and IP3R3 proteins; NG108 cells were used as positive control; β-actin was used as an internal control. Results are representative for three independent experiments. b, Functional characterization of phosphatidylinositol-linked D1 dopamine receptor expression in nAcc neurons: left panel -averaged Ca2+ responses induced by D1 agonist SKF83959 (10 μM) upon extracellular administration to D1-expressing nAcc neurons incubated in Ca2+-containing saline (left) or in Ca2+-free saline, in absence (middle) and presence of IP3R blockers xestospongin C (XeC) and 2-APB (right); right panel – comparison of the Ca2+ responses produced by SKF83959 in the mentioned conditions; P < 0.00001 compared to basal Ca2+ levels (*) or to SKF83959 in Ca2+-free HBSS (**). Neurons not responding to SKF83959 with an increase in [Ca2+]i were not used further for experiments. c, Lack of effect of cocaine microinjection alone on [Ca2+]i of D1-positive nAcc neurons: left - averaged tracings of the Ca2+ responses produced by intracellular microinjection of either control buffer or cocaine (C, 100 μM); right - Comparison of the amplitudes and areas under curve (A.U.C.) of the Ca2+ responses; lower concentrations of cocaine were similarly ineffective.
Since several cocaine-mediated responses are strictly dependent on D1 receptor activation or are restricted to D1-expressing neurons [6–8], in the present study we used only neurons responding to application of D1 agonist SKF83959 (10 μM) with an increase in [Ca2+]i [26, 27] (Fig. 1b); accordingly, these neurons were considered D1-positive, signaling via Gq-coupled pathways [26]. When incubated with Ca2+-free saline, neuronal Ca2+ response to application of SKF83959 was reduced from 197 ± 4.6 nM (n = 581 cells in Ca2+-containing saline) to 96 ± 3.6 nM (n = 6 cells in Ca2+-free medium) and further, largely abolished by presence of IP3R inhibitors xestospongin C (XeC, 10 μM, 15 min) and 2-aminoethoxydiphenyl borate (2-APB, 100 μM, 15 min) − Δ[Ca2+]i = 7 ± 2.1 nM (n = 6, Fig. 1b). This indicates that indeed, in the responsive neurons, SKF83959 promotes Ca2+ mobilization from intracellular stores via IP3Rs, supporting the activity of a Gq-coupled pathway.
s3.2. Cocaine enhances IP3-dependent Ca2+ mobilization via σ1R activation
In D1-expressing neurons incubated with Ca2+-containing saline, microinjection of cocaine (100 μM, final concentration inside the cell) did not elicit an increase in [Ca2+]i, the effect being similar to that produced by microinjection of control buffer (Fig. 1c); Δ[Ca2+]i was 28 ± 4.7 nM, and the area under curve of the Ca2+ response (A.U.C.) was 33.8 ± 4.4 nM x min for cocaine (n = 6 D1-positive nAcc neurons), while for control vehicle the effects measured 21 ± 4.2 nM and 36.5 ± 3.7 nM x min (n = 6), respectively (Fig. 1c).
In an additional series of experiments, we tested the effect of intracellular administration cocaine on IP3-induced Ca2+ response in D1-positive accumbens neurons. To evaluate the effect of cocaine co-injection on the Ca2+ mobilization triggered by IP3, additional experiments were carried out in Ca2+-free saline, in order to prevent interference with any Ca2+ entry mechanism. At 20 nM, IP3 microinjection into D1-positive nAcc neurons produced a Ca2+ response of 276 ± 2.8 nM (n = 6 cells), while 10 μM cocaine co-injected with 20 nM IP3 triggered a significantly enhanced effect, measuring 364 ± 3.6 nM (n = 6 cells, Fig. 2a). To establish a concentration-response curve, increasing concentrations of IP3 (1–60 nM) were injected either alone or in combination with 10 μM cocaine (n = 5 to 6 cells for each concentration tested). The two concentration-response curves are presented in Fig. 2b; cocaine significantly shifted the IP3 concentration-response curve to the left, diminishing the EC50 for IP3 from 22.4 nM (when administered alone) to 17.8 nM (when co-administered with cocaine).
Figure 2. Cocaine shifts to the left the IP3-induced concentration-Ca2+ response curve via σ1R.
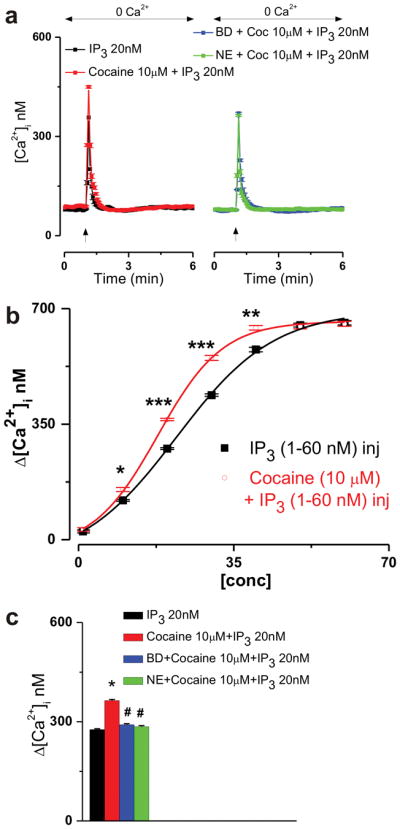
a, Averaged Ca2+ responses elicited by IP3 (20 nM) microinjection alone or in combination with cocaine (10 μM) or by IP3 (20 nM) and cocaine (10 μM) coinjection upon σ1R blockade with either BD-1063 (BD, 10 μM) or NE-100 (NE, 3 μM). b, Concentration-response curves indicating the effect of IP3 (1, 10, 20, 30, 40, 50 and 60 nM) microinjection into D1-expressing nAcc neurons (incubated in Ca2+-free saline) when injected alone (black) or in combination with 10 μM cocaine (red); P < 0.005(*), P <0.001(**) and P < 0.00001(***); comparison of the two data sets yielded statistical significance for the two fits: F = 5.1715; P = 0.0378 < 0.05. c, Comparison of the Ca2+ increases triggered by treatments mentioned in a; P < 0.00001 compared with IP3 (*) or to combined cocaine and IP3 microinjection (#).
Blocking σ1Rs with either the selective and prototypical σ1R antagonist BD-1063 [28] (10 μM, 20 min) or with NE-100 (3 μM, 20 min), another selective σ1R inhibitor [29, 30], was sufficient to prevent cocaine-induced augmentation of IP3-mediated Ca2+ mobilization: Δ[Ca2+]i were 291 ± 3.3 nM (BD-1063 pretreatment, n = 6 cells) and 285 ± 3.2 nM (NE-100 pretreatment, n = 6) compared with 364 ± 3.6 nM (10 μM cocaine co-injected with 20 nM IP3, no antagonists, n = 6) and 276 ± 2.8 nM (20 nM IP3 alone, n = 6 cells, Fig. 2a, c).
Next, we evaluated the effect of cocaine microinjection on the IP3-induced Ca2+ mobilization in Ca2+-containing saline-incubated D1-positive neurons. IP3 (20 nM) microinjection alone robustly elevated [Ca2+]i by 332 ± 4.8 nM (A.U.C. of 111 ± 4.4 nM x min, n = 6 cells), while in the presence of 10 μM co-injected cocaine, the effect was greatly enhanced, measuring 576 ± 5.3 nM in amplitude (A.U.C. of 234 ± 6.1 nM x min, n = 6) (Fig. 3a, b). Incubation of neurons with BD-1063 (10 μM, 20 min) reduced the Ca2+ response to co-injected cocaine and IP3 to that of IP3 alone (Δ [Ca2+]i was 337 ± 5.8 nM, A.U.C. was 104 ± 3.3 nM x min, n = 6, Fig. 3a, b), indicating that the cocaine-triggered augmentation was σ1R-mediated. This conclusion is supported by the inability of BD-1063 to reduce the effect IP3 microinjection alone (Δ [Ca2+]i was 342 ± 5.2 nM, A.U.C. was 114 ± 4.8 nM x min, Fig. 3a, b). To further strengthen our findings, we evaluated whether the σ1R antagonist NE-100 would block the cocaine-mediated enhancement of IP3-induced Ca2+ response: indeed, similar to the effects seen in presence of extracellular Ca2+, 20 min pretreatment of neurons with 3 μM NE-100 resulted in a significant reduction of the Ca2+ increase promoted by IP3 and cocaine co-injection, the response measuring 341 ± 5.2 nM in amplitude and having an A.U.C. of 108 ± 4.8 nM x min (Fig. 3a, b). Noteworthy, σ1R blockade by either BD-1063 or NE-100 had largely identical diminishing effect on the response triggered by combined cocaine and IP3 administration, both in the presence and in the absence of extracellular Ca2+.
Figure 3. σ1R-mediated enhancement by microinjected cocaine of the IP3-induced [Ca2+]i increase.

a, Averaged tracings of the Ca2+ responses elicited by intracellular microinjection of IP3 (20 nM) alone or in presence of σ1R antagonist BD-1063 (BD, 10 μM); or cocaine (C, 10μM) and IP3 (20 nM) in absence, or presence of either BD (10 μM) or NE-100 (NE, 3 μM); the fast Ca2+ chelator BAPTA-AM (200 μM); or TRPC blocker SKF96365 (SKF, 2 μM). b, Comparison of the amplitudes and areas under curve (A.U.C.) of the Ca2+ responses; P < 0.00001 compared with IP3 alone (*), with combined cocaine and IP3 microinjection (#), or with all other treatment groups (+). c, Fura-2 AM fluorescence ratios (340 nm/380 nm) of cultured nAcc neurons before and after D1 agonist SKF83959 (10 μM), after washing of SKF83959 and after microinjection of indicated compounds in absence and presence of the antagonist pretreatment indicated in the right side; cold colors indicate low levels of [Ca2+]i; hot colors indicate high levels of [Ca2+]i; fluorescence scale (0–2) is magnified in each panel showing the effect of injected compounds.
In presence of the fast Ca2+ chelator BAPTA-AM (200 μM, 30 min incubation), combined intracellular administration of cocaine and IP3 produced a small and insignificant response, measuring 49 ± 4.1 nM in amplitude and with an A.U.C. of 14 ± 3.3 nM x min (n = 6, Fig. 3a, b). In presence of SKF96365 (2 μM), that blocks receptor- and store-operated Ca2+ entry via transient receptor potential canonical (TRPC) channels [31, 32], the cocaine-induced potentiation was abolished (Δ[Ca2+]i was 268 ± 6.4 nM, A.U.C. of 76 ± 4.6 nM x min, n = 6, Fig. 3a, b). Fig. 3c depicts representative examples of changes in 340 nm/380 nm Fura-2 fluorescence ratio of nAcc neurons in presence of D1 agonist SKF83959, followed (after washing of SKF83959) by intracellular microinjection of IP3 alone or IP3 and cocaine, in absence and presence of the indicated antagonists.
Next, we tested whether cocaine might amplify Ca2+ responses induced by another intracellular Ca2+ release mediator, cyclic ADP ribose (cADPR), which acts on ryanodine receptors [33]. Because cADPR has also been involved in the activation of TRPM2 (also known as TRPC7 or LTRPC2) [34, 35], to avoid any TRPC-mediated effect, experiments were performed in absence of extracellular Ca2+. In cells incubated with Ca2+-free saline, cADPR (20 μM) injection elevated [Ca2+]i by 174 ± 2.8 nM (A.U.C. of 52 ± 1.3 nM x min, n = 6 cells Fig. 4a, b). Cocaine (10 μM) and cADPR (20 μM) co-injection induced an similar response to that of cADPR alone, measuring 171 ± 2.6 nM in amplitude (A.U.C. of 54 ± 1.1 nM x min, n = 6, Fig. 4a, b), while microinjection of a higher concentration of cADPR (50 μM) induced a proportionally higher response, of 629 ± 3.7 nM (A.U.C. of 326 ± 2.8 nM x min, n = 6 cells, Fig. 4a, b), indicating that 20 μM cADPR produced a submaximal effect. Thus, cocaine does not enhance cADPR-mediated Ca2+ signaling in nAcc neurons.
Figure 4. Cocaine does not enhance cADPR or OAG-mediated Ca2+ signaling in nAcc neurons.

a, Averaged Ca2+ tracings indicating the response of Ca2+-free saline-incubated neurons to microinjection of 20 μM cADPR alone or co-injected with 10 μM cocaine or to microinjection of 50 μM cADPR. b, Comparison of the amplitudes and areas under curve of the Ca2+ responses to treatments indicated in a; P < 0.00001 compared to basal Ca2+ levels (*) or to the effect of 20 μM cADPR (**). c, Averaged Ca2+ tracings corresponding to the effects produced by OAG (75 μM) in Ca2+-free and Ca2+-containing saline, in absence and presence of cocaine (10 μM), or to the effect of 100 μM OAG in Ca2+-negative and Ca2+-positive conditions. d, Comparison of the effects in Ca2+-containing saline induced by treatments indicated in d; P < 0.00001 compared to basal Ca2+ levels (*) or to the effect of 75 μM OAG in presence of extracellular Ca2+ (**).
Since diacylglycerol (DAG) has been reported to promote TRPC activation downstream of phospholipase C [36], we tested the effect of the membrane-permeable DAG analogue 1-oleoyl-2-acetyl-sn-glycerol (OAG) on D1-expressing nAcc neurons. Application of OAG (75 μM) in cells incubated with Ca2+-free saline elicited no effect (Fig. 4c), while addition of Ca2+ to the extracellular medium unmasked a response measuring 251 ± 7.8 nM (A.U.C. of 1183 ± 9.3 nM x min, n = 12 neurons, Fig. 4c, d). A likewise effect was produced by 75 μM OAG in cells treated with 10 μM cocaine: no response in absence of extracellular Ca2+ and an [Ca2+]i elevation by 258 ± 6.7 nM (A.U.C. of 1164 ± 9.7 nM x min, n = 12, Fig. 4c, d) upon changing to Ca2+-containing saline. Since absence of a cocaine-enhancing effect may occur as a consequence of the employment of a concentration of OAG eliciting a maximal response, we tested the effect of 100 μM OAG and noted an increase in [Ca2+]i by 383 ± 11.8 nM upon Ca2+ addition (A.U.C. of 1847 ± 10.2 nM x min, n = 12, Fig. 4c, d), significantly higher than that produced by 75 μM OAG.
In D1-positive nAcc neurons, the TRPC antagonist SKF96365 (2 μM) did not block the Ca2+ elevation in response to KCl (30 mM) (Fig. 5), which produces depolarization-induced activation of voltage-gated Ca2+ channels. Thus, as we have previously reported [25], at 2 μM SKF96365 has no antagonistic effect on voltage-gated Ca2+ channels.
Figure 5. SKF96365 (2 μM) is devoid of inhibitory activity at voltage-gated Ca2+ channels in nAcc neurons.
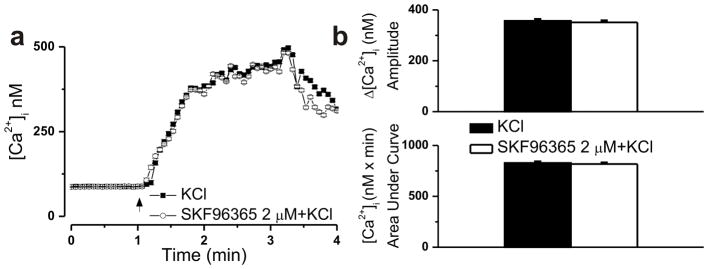
a, Averaged Ca2+ responses induced by 30 mM KCl in absence and presence of SKF96365 (2 μM). b, The amplitudes of the Ca2+ increases produced by KCl measured 357 ± 5.6 nM (n = 31 cells) in absence and 351 ± 5.9 nM (n = 37) in presence of SKF96365; the areas under curve were 828 ± 11.3nM x min and 817 ± 10.9 nM x min, respectively.
3.3. Cocaine produces σ1R-mediated potentiation of the Ca2+ response triggered by ATP
Next, we examined the effect of cocaine on ATP, an IP3-generating molecule, in D1-expressing accumbens neurons. To confirm that ATP mobilizes IP3-sensitive Ca2+ stores in nAcc neurons, we first examined the effect of extracellular application of ATP (20 μM) on [Ca2+]i in Ca2+-free saline, in the absence and presence of inhibitors of endoplasmic reticulum and lysosomal Ca2+ release channels. In the absence of extracellular Ca2+, ATP (20 μM) elevated [Ca2+]i of nAcc neurons by 278 ± 3.6 nM (A.U.C. of 78.8 ± 3.1 nM x min, n = 9 cells, Fig. 6a, b). Blockade of IP3Rs with XeC (10 μM, 15 min) and 2-APB (100 μM, 15 min), but not inhibition of ryanodine receptors with ryanodine (Ry, 10 μM, 1h) or of lysosomal NAADP-sensitive two pore channels with Ned-19 (5 μM, 15 min) [37], abolished the Ca2+ response of nAcc neurons to ATP, indicating that it was IP3R-mediated (Fig. 6a). In the presence of these blockers, ATP increased [Ca2+]i by 23 ± 2.7 nM (XeC + 2-APB; A.U.C. of 5.6 ± 1.8 nM x min, n = 9 cells), by 269 ± 3.1 nM (Ry; A.U.C. of 74.7 ± 3.8 nM x min, n = 9) and by 274 ± 4.9 nM (Ned-19; A.U.C. of 77.3 ± 3.7 nM x min, n = 9), respectively (Fig. 6b).
Figure 6. Cocaine potentiates the Ca2+ response of nAcc neurons to IP3-generating molecules.

a–b, ATP promotes Ca2+ mobilization from IP3-sensitive pools in accumbens neurons: a, Averaged tracings of the Ca2+ responses elicited by ATP (20 μM) in Ca2+-free saline, in absence or presence of IP3R blockers xestospongin C (XeC) and 2-aminoethoxydiphenyl borate (2-APB); ryanodine receptor blocker ryanodine (Ry); or lysosomal two-pore channel inhibitor Ned-19. b, Comparison of the amplitudes and the areas under curve (A.U.C.) of the Ca2+ increases produced by the indicated treatments; *P < 0.00001 compared with all other treatment groups. c–d, Cocaine produces σ1R-dependent potentiation of the ATP-induced Ca2+ elevation in D1-expressing accumbens neurons: c, Averaged tracings of the Ca2+ responses induced by bath application of ATP (20 μM) or cocaine (C, 10 μM) and ATP (20 μM), in naïve neurons or neurons incubated with either σ1R antagonist BD-1063 (BD, 10 μM); or Ca2+ chelator BAPTA-AM (200 μM); or TRPC blocker SKF96365 (SKF, 2 μM). d, Comparison of the amplitudes and the areas under curve (A.U.C.) of the Ca2+ responses produced by the indicated treatments; P < 0.00001 compared with ATP alone (*), with combined cocaine and ATP administration (#), or with all other treatment groups (+).
In Ca2+-containing saline, application of ATP (20 μM) alone elevated [Ca2+]i of D1-positive neurons by 377 ± 4.8 nM (A.U.C. of 108 ± 4.4 nM x min, n = 29 cells, Fig. 6c, d). Combined administration of cocaine (10 μM) and ATP (20 μM) produced a greatly potentiated increase in [Ca2+]i, measuring 613 ± 8.6 nM in amplitude and with an A.U.C. of 247 ± 4.3 nM x min (n = 47 cells, Fig. 6c, d). The cocaine-induced enhancement was completely abrogated by pretreatment of cells with σ1R antagonist BD-1063 (10 μM, 20 min) or with the TRPC blocker SKF96365 (2 μM, 20 min), when the Ca2+ response was similar to that promoted by ATP alone; Δ[Ca2+]i was 364 ± 6.1 nM (A.U.C. of 103 ± 4.6 nM x min, n = 31) in the case of BD-1063 and 272 ± 5.7 nM (A.U.C. of 79 ± 5.2 nM x min, n = 28) in the case of SKF96365 (Fig. 6c, d). In presence of BAPTA-AM (200 μM, 30 min), co-administration of cocaine and ATP no longer elicited a significant increase in [Ca2+]i (amplitude of 23 ± 3.5 nM, A.U.C. of 15 ± 2.3 nM x min, n = 42, Fig. 6c, d).
3.4. Intracellular microinjection of cocaine amplifies IP3-induced depolarization via σ1Rs
The mean resting potential of dissociated D1-positive nAcc neurons was −71.6 ± 0.02 mV (n = 258 cells depolarizing in response to SKF83959 in the preliminary screen test). Microinjection of cocaine or control vehicle had no effect on neuronal membrane potential; ΔVm were −1.2 ± 0.34 mV (n = 6) and −0.9 ± 0.47 mV (n = 6), respectively, (Fig. 7a, b). Intracellular administration of IP3 (20 nM) produced a slight and rather transient depolarization of 5.12 ± 0.42 mV, which in the presence of co-injected cocaine (10 μM) was converted to a greatly enhanced and more prolonged response, measuring 9.77 ± 0.53 mV in amplitude (Fig. 7c, d). The cocaine-dependent component of the effect was abolished upon blocking σ1Rs with BD-1063 (10 μM, 20 min pretreatment), when the combined administration of IP3 and cocaine depolarized neuronal membrane potential by 5.53 ± 0.39 mV (n = 6), similar to IP3 injection alone (Fig. 7c, d). In presence of the Ca2+ chelator BAPTA-AM (200 μM, 30 min) or of TRPC inhibitor SKF96365 (2 μM, 20 min), the IP3 and cocaine-triggered depolarization were lost, the changes in resting membrane potential measuring 0.79 ± 0.62 mV (n = 6) and 1.34 ± 0.47 mV (n = 6), respectively (Fig. 7c, d).
Figure 7. Intracellular microinjection of cocaine enhances IP3-induced depolarization in the nAcc.

a–b, Cocaine microinjection alone does not modify neuronal membrane potential: a, Representative recordings of the resting membrane potential of D1-expressing nAcc neurons treated intracellularly with either control vehicle or cocaine (C, 10 μM, final intracellular concentration). b, Neither control vehicle microinjection or cocaine microinjection did produce a significant change in neuronal membrane potential. c-d, Cocaine potentiates the IP3-dependent depolarization of D1-expressing accumbens neurons: c, Characteristic changes in resting membrane potential of neurons microinjected with either IP3 (20 nM) or IP3 (20 nM) and cocaine (C, 10 μM) in absence and presence of bath-applied σ1R inhibitor BD-1063 (BD, 10 μM), of the Ca2+ chelator BAPTA-AM (200 μM) or of TRPC blocker SKF96365 (SKF, 2 μM). d, Comparison of the amplitudes of the depolarizations induced by treatment conditions described in c; P < 0.00001 compared with IP3 injection alone (*), with combined cocaine and IP3 microinjection (#), or with all other treatment groups (+).
3.5. Cocaine enhances ATP-elicited depolarization of D1-positive nAcc neurons
Cocaine produced a likewise augmentation of the amplitude and duration of the depolarization promoted by the IP3-generating molecule ATP (Fig. 8a). ATP (20 μM) depolarized D1-expressing nAcc neurons by 6.71 ± 0.39 mV (n = 36) in absence and by 11.63 ± 0.54 (n = 52 cells) in presence of co-applied cocaine (10 μM) (Fig. 8b). Pretreatment of cells with σ1R blocker BD-1063 (10 μM, 20 min) virtually eliminated the cocaine-mediated amplification, as neurons depolarized only by 7.38 ± 0.47 mV (n = 46) when ATP and cocaine were co-applied in this condition (Fig. 8a, b). The depolarization triggered by combined administration of cocaine and ATP measured 5.68 ± 0.53 mV (n = 39) when neurons were preincubated with the fast Ca2+ chelator BAPTA-AM (200 μM, 30 min) and 5.92 ± 0.41 mV (n = 43) upon pretreatment with TRPC blocker SKF96365 (2 μM, 20 min) (Fig. 8a, b). These results support a contribution of the [Ca2+]i elevation and of cation entry via TRPC in the mechanism of cocaine-induced enhancement of ATP effects in D1-positive nAcc neurons.
Figure 8. Cocaine enhances the ATP-induced depolarization.
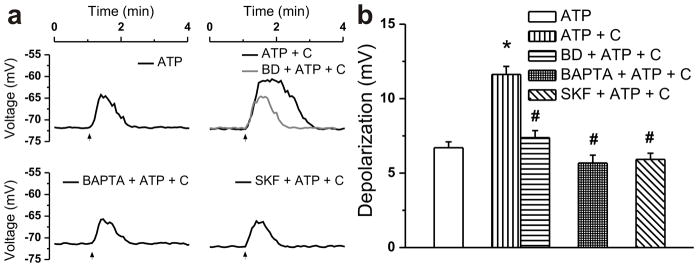
a, Typical recordings of membrane potential modifications of D1-expressing accumbens neurons treated extracellularly with ATP (20 μM) or ATP (20 μM) and cocaine (C, 10 μM) in absence and presence of the σ1R inhibitor BD-1063 (BD, 10 μM), of the Ca2+ chelator BAPTA-AM (200 μM) or of the TRPC blocker SKF96365 (SKF, 2 μM). b, Comparison of the amplitudes of the depolarizations induced by treatments described in a; P < 0.00001 compared with ATP alone (*) or with combined cocaine and ATP administration (#).
3.6. Blockade of σ1Rs or TRPC in the nAcc reverses cocaine-induced hyperlocomotion and sensitization in vivo
The role of σ1Rs and TRPC in the nAcc in cocaine-induced hyperactivity was investigated in the rat. To this end, the ability of the selective σ1R antagonist BD-1063 (80 μg/0.5 μl, intra-nAcc), and the TRPC blocker SKF96365 (20 μg/0.5 μl, intra-nAcc), to attenuate acute cocaine (15 mg/kg ip)-induced hyperlocomotion was tested. Two-way ANOVA with repeated measures over days revealed a statistically significant main effect of day (F5,202 = 22.821, p < 0.001), a significant treatment main effect (F5, 202 = 13.994, p < 0.001) and no treatment × day interaction (F25,202 = 1.296, p= 0.175). Post test analysis revealed that administration of cocaine following aCSF infusion produced a significant increase in ambulatory activity on Days 2–5 compared with aCSF + vehicle administration (Fig. 8b; aC vs aV; p < 0.05). Pretreatment with BD-1063 or SKF96365 into the accumbens significantly inhibited cocaine-induced locomotion on Days 2–5 (aC vs BC or SC, p<0.05). Infusion of BD-1063 or SKF96365 alone (prior to a saline injection) did not alter locomotion when compared to the levels of activity after aCSF infusion and saline injection (Fig. 8b, BV or SV vs aV; p > 0.05). On day 5, activity of the SKF96365 + cocaine group was different from the three vehicle control groups (SC vs aV/BV/SV; post hoc p < 0.05), but significantly lower than the cocaine alone group (SC vs aV; p < 0.05).
Locomotor sensitization occurs as the result of repeated cocaine administration. The ability of the BD-1063 and SKF96365 to block the development of locomotor sensitization to repeated cocaine was investigated. Seven days following the five daily treatments described above, all rats were challenged with cocaine (15 mg/kg ip) on Day 12 without further intracranial infusions and activity recorded. Rats receiving daily intra-accumbens aCSF plus cocaine for 5 days showed significantly greater ambulatory response to the cocaine challenge on Day 12 than did rats receiving daily intra-accumbens aCSF plus saline, demonstrating locomotor sensitization (Fig. 9b; aC vs aV, p < 0.05). Activity following the cocaine challenge of the groups receiving daily intra-accumbens BD-1063 or SKF96365 plus cocaine was significantly lower than those receiving aCSF plus cocaine (BC/SC vs aC, p < 0.05). These data demonstrate that cocaine-induced hyperactivity and sensitization can be blocked by pretreatment with the σ1R antagonist BD-1063 or TRPC blocker SFK96365.
4. Discussion
Emerging evidence points to IP3 involvement both in σ1R-mediated signaling and in cocaine-promoted responses. σ1R activation potentiates not only IP3R-mediated Ca2+ release from the ER [24], but also IP3 formation [38, 39]. IP3R blockade at central levels inhibits cocaine-induced place preference in mice [40]. Cocaine binds to σ1Rs with a 2 μM affinity [9]. In vivo studies have shown that pharmacologically relevant doses of cocaine produce striatal levels of the drug in a low μM range [41, 42]. For instance, it has been reported that after systemic administration to mice of a 10 mg/kg dose of cocaine, a peak value for cocaine of 2.6 μg/g (~7.6 – 8.5 μM) at 5 min was seen in the brain, whereas after a 25 mg/kg the peak value was 6.7 μg/g (~19.7 – 22.1 μM) [41]; moreover, the cocaine concentrations in the brain were always higher than those in the plasma, with an average brain/plasma ratio of 7 [41].
We report here that, although ineffective by itself at concentrations up to 100 μM, at 10 μM cocaine strongly enhances IP3-induced Ca2+ responses in D1-expressing nAcc neurons by activating intracellularly-located σ1Rs, significantly shifting the IP3 concentration-response curve to the left. Our finding is supported by previous indications that in NG108 cells, cocaine promotes σ1R-dependent increase in IP3R3 sensitivity for IP3, further translated into augmented Ca2+ efflux from the ER in the presence of IP3-generating mediators such as bradykinin [10, 43]. Accordingly, we found that the Ca2+ response induced by ATP (which clearly mobilized only IP3-sensitive intracellular Ca2+ stores in our paradigm) was similarly enhanced by cocaine via σ1Rs. Conversely, cocaine had no effect on the ryanodine receptor-mediated Ca2+ release.
We note two potential implications of our findings. On the one hand, cocaine is a dopamine transporter blocker, producing indirect activation of phospholipase C β-coupled D1-like receptors, which results in IP3 accumulation in the striatum [44]. Thus, one putative mechanism of cocaine-induced activation of D1-positive neurons in vivo would include two steps: a dopamine-dependent increase in IP3 levels, likely associated with an increase in [Ca2+]i, followed by a dopamine-independent, cocaine-directed activation of σ1Rs, resulting in potentiation of the initial Ca2+ response.
On the other hand, increased levels of ATP synthase β-chain have been found in the nAcc of cocaine-overdose victims [45], suggesting that cocaine may elicit local ATP elevation in this brain region. σ1Rs promote ATP production via increased mitochondrial Ca2+ uptake [46] and potentiate ATP-triggered Ca2+ mobilization [47]. ATP induces IP3-mediated Ca2+ release by activating Gq-coupled P2Y(1) receptors, which are expressed by neurons in the nAcc [48]. In view of our present findings, once ATP levels in the nAcc increase, cocaine is expected to directly activate D1-positive neurons, by σ1R-dependently enhancing ATP effects.
Another important result of our study is that cocaine-triggered enhancement of IP3- or ATP-mediated Ca2+ elevation was proportionally translated into a σ1R-dependent augmentation of IP3- or ATP-induced depolarization of D1-expressing nAcc neurons. Experiments using the fast Ca2+ chelator BAPTA-AM clearly indicate contingency of the depolarization on [Ca2+]i increase.
Changes in resting membrane potential determine the ease with which excitatory synaptic inputs bring the membrane potential closer to the threshold for action potential firing. In vivo, the membrane potential of D1-positive medium spiny neurons oscillates between “down-states” characterized by highly negative values (ranging from −75 to −85 mV) and “up-states” consisting in periodic plateau depolarizations (Vm ~ −55 mV) triggered by convergent, temporally coherent excitatory synaptic inputs [49, 50]. Accordingly, we found that the resting membrane potential of dissociated D1-positive neurons was ~ −72 mV. Given that dopamine depolarizes D1-expressing neurons and that under conditions of cocaine administration synaptic dopamine levels are increased, we propose that cocaine-induced σ1R activation favors the D1 pathway in the nAcc. Indeed, we found that cocaine-mediated locomotor hyperactivity and sensitization is significantly attenuated by prior administration of a σ1R antagonist into the rat nAcc.
IP3-dependent Ca2+ signaling has been shown to be involved in the activation of TRPC [25, 51]. TRPC subtypes 4 and 5 are expressed in the rodent nAcc at low and moderate levels, respectively [52], and have been implicated in cocaine-triggered motivation/reward-directed behaviors [53, 54]. Rats lacking a functional trpc4 gene showed reduced cocaine self-administration [54]; mice with selective trpc5 knock-down in the forebrain exhibited increased cocaine self-administration on the first day of testing, although were similar to wild-type mice during the maintenance phase of self-administration [53]. We report here that TRPC are critical for cocaine-triggered amplification of IP3/ATP-dependent Ca2+ responses and membrane potential changes in D1-expressing nAcc neurons. Importantly, cocaine does not enhance OAG-mediated Ca2+ signaling, indication that IP3 is a critical molecule in the pathway linking cocaine and TRPC. Moreover, the TRPC-involving route in the nAcc mediates cocaine-induced hyperlocomotion and locomotor sensitization, as indicated by our experiments using intra-accumbal delivery of a TRPC blocker.
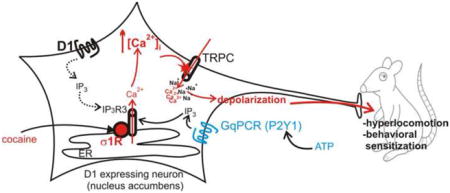
Thus, we propose a new mechanism by which cocaine stimulates or maintains D1-positive neurons in active state: σ1R activation at the ER, resulting in dissociation from inhibitory proteins [10, 11, 43] and consequent increases in IP3R3 sensitivity for IP3 [10, 43]. This in turn correlates with increased Ca2+ efflux from the ER, which promotes TRPC opening [25, 51] and cation entry, resulting in further [Ca2+]i increase and membrane depolarization, converging to locomotor hyperactivity and sensitization (Fig. 10).
Figure 10. Mechanism of cocaine-induced activation of D1-expressing nAcc neurons.
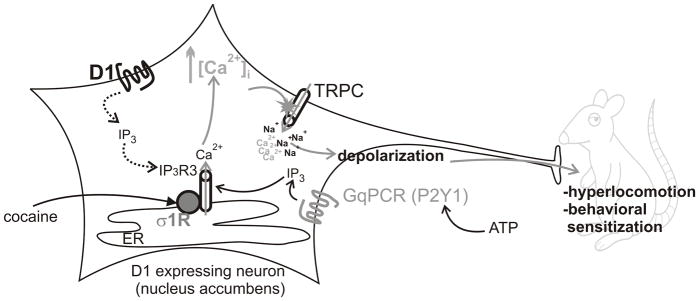
Cocaine activates endoplasmic reticulum (ER)-located σ1Rs and potentiates Ca2+ release from the ER via IP3 receptors type 3 (IP3 R3) promoted by GPCR agonists (Gq-coupled, such as ATP). The increase in cytosolic Ca2+ triggers activation of TRPC and additional Ca2+ entry, as well as Na2+ entry, followed by depolarization and activation of these neurons, triggering hyperlocomotion and behavioral sensitization in vivo.
Previous studies examining cocaine-induced σ1R activation noted a potential cocaine-mediated translocation of σ1Rs from the ER to the plasma membrane of cells in striatal slices, where σ1Rs interact with a D1-D1 homomer [22] or a heteromer of D1 and H3 histamine receptor [55], thus facilitating D1-mediated effects. Another σ1R-dependent effect of cocaine involves formation of higher order oligomers between σ1R and D2 receptors in mouse striatal slices and inhibition of signaling via D2 [23]. These plasma membrane-initiated mechanisms were clearly demonstrated in transfected cells treated with 15–30 μM cocaine, while their potential occurrence in brain striatal slices was apparent at higher cocaine concentrations (150 μM) meant to allow diffusion into the tissue [22, 23, 55]. Another study identified a cocaine-induced association of σ1R and K+ channels at the plasmelemma of nAcc shell neurons with implications for cocaine-promoted changes in neuronal excitability and associated behavioral sensitization [56]. Our study is the first indication of σ1R-directed effects of cocaine occurring upon their intracellular activation in nAcc neurons, and not upon translocation at the plasma membrane. Moreover, we identify a previously unknown mechanism of cocaine action in D1-expressing nAcc neurons, involving TRPC activation, providing a putative explanation of the in vivo results here shown, as well as those of others [54].
Highlights.
Cocaine enhances IP3-mediated Ca2+ signaling via sigma-1-receptor (σ1R)
This induces TRP canonical (TRPC) activation and depolarization of accumbal neurons
Intra-accumbal σ1R/TRPC blockade reduces cocaine-hyperlocomotion/sensitization.
We show behavioral relevance of a novel cocaine-triggered pathway in the accumbens.
Acknowledgments
This work was supported by the NIH grants HL090804 (to EB), DA035926 (to MEA), P30DA013429 (to EMU).
Abbreviations The abbreviations used are
- aCSF
artificial cerebrospinal fluid
- ANOVA
analysis of variance
- A.U.C
area under curve
- BAPTA
1,2-Bis(2-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid tetrakis
- [Ca2+]i
intracellular Ca2+ concentration
- DiBAC4(3)
bis-(1,3-dibutylbarbituric acid) trimethine oxonol
- ER
endoplasmic reticulum
- HBSS
Hank’s balanced salt solution
- IP3
inositol 1,4,5-trisphoshate
- IP3R
IP3 receptor
- nAcc
nucleus accumbens
- σ1R
sigma-1 receptor
- TRPC
transient receptor potential canonical channels
Footnotes
Author contributions: J.L.B., E.D., G.C.B., P.Z., G.Y. and E.B. performed experiments; J.L.B., E.D., G.C.B., P.Z., G.Y., M.E.A., E.M.U. and E.B. analyzed data; E.B. and E.M.U. designed experiments; E.D. and J.L.B. drafted the manuscript; E.B., M.E.A. and E.M.U. finalized the manuscript; all authors read and approved the final version of the manuscript.
The authors declare no competing financial interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Carroll FI, Howell LL, Kuhar MJ. Pharmacotherapies for treatment of cocaine abuse: preclinical aspects. J Med Chem. 1999;42:2721–36. doi: 10.1021/jm9706729. [DOI] [PubMed] [Google Scholar]
- 2.Gorelick DA, Gardner EL, Xi ZX. Agents in development for the management of cocaine abuse. Drugs. 2004;64:1547–73. doi: 10.2165/00003495-200464140-00004. [DOI] [PubMed] [Google Scholar]
- 3.Rush CR, Stoops WW. Agonist replacement therapy for cocaine dependence: a translational review. Future Med Chem. 2012;4:245–65. doi: 10.4155/fmc.11.184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chen R, Tilley MR, Wei H, Zhou F, Zhou FM, Ching S, Quan N, Stephens RL, Hill ER, Nottoli T, Han DD, Gu HH. Abolished cocaine reward in mice with a cocaine-insensitive dopamine transporter. Proc Natl Acad Sci U S A. 2006;103:9333–8. doi: 10.1073/pnas.0600905103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Giros B, Jaber M, Jones SR, Wightman RM, Caron MG. Hyperlocomotion and indifference to cocaine and amphetamine in mice lacking the dopamine transporter. Nature. 1996;379:606–12. doi: 10.1038/379606a0. [DOI] [PubMed] [Google Scholar]
- 6.Anderson SM, Pierce RC. Cocaine-induced alterations in dopamine receptor signaling: implications for reinforcement and reinstatement. Pharmacol Ther. 2005;106:389–403. doi: 10.1016/j.pharmthera.2004.12.004. [DOI] [PubMed] [Google Scholar]
- 7.Bertran-Gonzalez J, Bosch C, Maroteaux M, Matamales M, Herve D, Valjent E, Girault JA. Opposing patterns of signaling activation in dopamine D1 and D2 receptor-expressing striatal neurons in response to cocaine and haloperidol. J Neurosci. 2008;28:5671–85. doi: 10.1523/JNEUROSCI.1039-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Xu M, Hu XT, Cooper DC, Moratalla R, Graybiel AM, White FJ, Tonegawa S. Elimination of cocaine-induced hyperactivity and dopamine-mediated neurophysiological effects in dopamine D1 receptor mutant mice. Cell. 1994;79:945–55. doi: 10.1016/0092-8674(94)90026-4. [DOI] [PubMed] [Google Scholar]
- 9.Sharkey J, Glen KA, Wolfe S, Kuhar MJ. Cocaine binding at sigma receptors. Eur J Pharmacol. 1988;149:171–4. doi: 10.1016/0014-2999(88)90058-1. [DOI] [PubMed] [Google Scholar]
- 10.Hayashi T, Su TP. Regulating ankyrin dynamics: Roles of sigma-1 receptors. Proc Natl Acad Sci U S A. 2001;98:491–6. doi: 10.1073/pnas.98.2.491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Su TP, Hayashi T, Maurice T, Buch S, Ruoho AE. The sigma-1 receptor chaperone as an inter-organelle signaling modulator. Trends Pharmacol Sci. 2010;31:557–66. doi: 10.1016/j.tips.2010.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Katz JL, Su TP, Hiranita T, Hayashi T, Tanda G, Kopajtic T, Tsai SY. A Role for Sigma Receptors in Stimulant Self Administration and Addiction. Pharmaceuticals (Basel) 2011;4:880–914. doi: 10.3390/ph4060880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Maurice T, Su TP. The pharmacology of sigma-1 receptors. Pharmacol Ther. 2009;124:195–206. doi: 10.1016/j.pharmthera.2009.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Alonso G, Phan V, Guillemain I, Saunier M, Legrand A, Anoal M, Maurice T. Immunocytochemical localization of the sigma(1) receptor in the adult rat central nervous system. Neuroscience. 2000;97:155–70. doi: 10.1016/s0306-4522(00)00014-2. [DOI] [PubMed] [Google Scholar]
- 15.Gundlach AL, Largent BL, Snyder SH. Autoradiographic localization of sigma receptor binding sites in guinea pig and rat central nervous system with (+)3H-3-(3-hydroxyphenyl)-N-(1-propyl)piperidine. J Neurosci. 1986;6:1757–70. doi: 10.1523/JNEUROSCI.06-06-01757.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Matsumoto RR. Targeting sigma receptors: novel medication development for drug abuse and addiction. Expert Rev Clin Pharmacol. 2009;2:351–8. doi: 10.1586/ecp.09.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Matsumoto RR, Liu Y, Lerner M, Howard EW, Brackett DJ. Sigma receptors: potential medications development target for anti-cocaine agents. Eur J Pharmacol. 2003;469:1–12. doi: 10.1016/s0014-2999(03)01723-0. [DOI] [PubMed] [Google Scholar]
- 18.Hiranita T, Mereu M, Soto PL, Tanda G, Katz JL. Self-administration of cocaine induces dopamine-independent self-administration of sigma agonists. Neuropsychopharmacology. 2013;38:605–15. doi: 10.1038/npp.2012.224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hiranita T, Soto PL, Tanda G, Katz JL. Reinforcing effects of sigma-receptor agonists in rats trained to self-administer cocaine. J Pharmacol Exp Ther. 2010;332:515–24. doi: 10.1124/jpet.109.159236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Romieu P, Martin-Fardon R, Maurice T. Involvement of the sigma1 receptor in the cocaine-induced conditioned place preference. Neuroreport. 2000;11:2885–8. doi: 10.1097/00001756-200009110-00011. [DOI] [PubMed] [Google Scholar]
- 21.Romieu P, Phan VL, Martin-Fardon R, Maurice T. Involvement of the sigma(1) receptor in cocaine-induced conditioned place preference: possible dependence on dopamine uptake blockade. Neuropsychopharmacology. 2002;26:444–55. doi: 10.1016/S0893-133X(01)00391-8. [DOI] [PubMed] [Google Scholar]
- 22.Navarro G, Moreno E, Aymerich M, Marcellino D, McCormick PJ, Mallol J, Cortes A, Casado V, Canela EI, Ortiz J, Fuxe K, Lluis C, Ferre S, Franco R. Direct involvement of sigma-1 receptors in the dopamine D1 receptor-mediated effects of cocaine. Proc Natl Acad Sci U S A. 2010;107:18676–81. doi: 10.1073/pnas.1008911107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Navarro G, Moreno E, Bonaventura J, Brugarolas M, Farre D, Aguinaga D, Mallol J, Cortes A, Casado V, Lluis C, Ferre S, Franco R, Canela E, McCormick PJ. Cocaine inhibits dopamine D2 receptor signaling via sigma-1-D2 receptor heteromers. PLoS One. 2013;8:e61245. doi: 10.1371/journal.pone.0061245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hayashi T, Su TP. Sigma-1 receptor chaperones at the ER-mitochondrion interface regulate Ca(2+) signaling and cell survival. Cell. 2007;131:596–610. doi: 10.1016/j.cell.2007.08.036. [DOI] [PubMed] [Google Scholar]
- 25.Deliu E, Brailoiu GC, Eguchi S, Hoffman NE, Rabinowitz JE, Tilley DG, Madesh M, Koch WJ, Brailoiu E. Direct evidence of intracrine angiotensin II signaling in neurons. Am J Physiol Cell Physiol. 2014;306:C736–44. doi: 10.1152/ajpcell.00131.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chun LS, Free RB, Doyle TB, Huang XP, Rankin ML, Sibley DR. D1-D2 dopamine receptor synergy promotes calcium signaling via multiple mechanisms. Mol Pharmacol. 2013;84:190–200. doi: 10.1124/mol.113.085175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jin LQ, Goswami S, Cai G, Zhen X, Friedman E. SKF83959 selectively regulates phosphatidylinositol-linked D1 dopamine receptors in rat brain. J Neurochem. 2003;85:378–86. doi: 10.1046/j.1471-4159.2003.01698.x. [DOI] [PubMed] [Google Scholar]
- 28.Matsumoto RR, Bowen WD, Tom MA, Vo VN, Truong DD, De Costa BR. Characterization of two novel sigma receptor ligands: antidystonic effects in rats suggest sigma receptor antagonism. Eur J Pharmacol. 1995;280:301–10. doi: 10.1016/0014-2999(95)00208-3. [DOI] [PubMed] [Google Scholar]
- 29.Berardi F, Ferorelli S, Colabufo NA, Leopoldo M, Perrone R, Tortorella V. A multireceptorial binding reinvestigation on an extended class of sigma ligands: N-[omega-(indan-1-yl and tetralin-1-yl)alkyl] derivatives of 3,3-dimethylpiperidine reveal high affinities towards sigma1 and EBP sites. Bioorg Med Chem. 2001;9:1325–35. doi: 10.1016/s0968-0896(01)00011-6. [DOI] [PubMed] [Google Scholar]
- 30.Chaki S, Okuyama S, Ogawa S, Tanaka M, Muramatsu M, Nakazato A, Tomisawa K. Solubilization and characterization of binding sites for [3H]NE-100, a novel and potent sigma 1 ligand, from guinea pig brain. Life Sci. 1996;59:1331–40. doi: 10.1016/0024-3205(96)00458-4. [DOI] [PubMed] [Google Scholar]
- 31.Clapham DE, Julius D, Montell C, Schultz G. International Union of Pharmacology. XLIX. Nomenclature and structure-function relationships of transient receptor potential channels. Pharmacol Rev. 2005;57:427–50. doi: 10.1124/pr.57.4.6. [DOI] [PubMed] [Google Scholar]
- 32.Zhu X, Jiang M, Birnbaumer L. Receptor-activated Ca2+ influx via human Trp3 stably expressed in human embryonic kidney (HEK)293 cells. Evidence for a non-capacitative Ca2+ entry. J Biol Chem. 1998;273:133–42. doi: 10.1074/jbc.273.1.133. [DOI] [PubMed] [Google Scholar]
- 33.Guse AH. Calcium mobilizing second messengers derived from NAD. Biochim Biophys Acta. 2014 doi: 10.1016/j.bbapap.2014.12.015. [DOI] [PubMed] [Google Scholar]
- 34.Kolisek M, Beck A, Fleig A, Penner R. Cyclic ADP-ribose and hydrogen peroxide synergize with ADP-ribose in the activation of TRPM2 channels. Mol Cell. 2005;18:61–9. doi: 10.1016/j.molcel.2005.02.033. [DOI] [PubMed] [Google Scholar]
- 35.Togashi K, Hara Y, Tominaga T, Higashi T, Konishi Y, Mori Y, Tominaga M. TRPM2 activation by cyclic ADP-ribose at body temperature is involved in insulin secretion. EMBO J. 2006;25:1804–15. doi: 10.1038/sj.emboj.7601083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Venkatachalam K, Montell C. TRP channels. Annu Rev Biochem. 2007;76:387–417. doi: 10.1146/annurev.biochem.75.103004.142819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Naylor E, Arredouani A, Vasudevan SR, Lewis AM, Parkesh R, Mizote A, Rosen D, Thomas JM, Izumi M, Ganesan A, Galione A, Churchill GC. Identification of a chemical probe for NAADP by virtual screening. Nat Chem Biol. 2009;5:220–6. doi: 10.1038/nchembio.150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Novakova M, Ela C, Bowen WD, Hasin Y, Eilam Y. Highly selective sigma receptor ligands elevate inositol 1,4,5-trisphosphate production in rat cardiac myocytes. Eur J Pharmacol. 1998;353:315–27. doi: 10.1016/s0014-2999(98)00398-7. [DOI] [PubMed] [Google Scholar]
- 39.Tsai SY, Hayashi T, Mori T, Su TP. Sigma-1 receptor chaperones and diseases. Cent Nerv Syst Agents Med Chem. 2009;9:184–9. doi: 10.2174/1871524910909030184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kurokawa K, Mizuno K, Shibasaki M, Ohkuma S. Regulation of type 1 inositol 1,4,5-triphosphate receptor by dopamine receptors in cocaine-induced place conditioning. Synapse. 2012;66:180–6. doi: 10.1002/syn.20997. [DOI] [PubMed] [Google Scholar]
- 41.Benuck M, Lajtha A, Reith ME. Pharmacokinetics of systemically administered cocaine and locomotor stimulation in mice. J Pharmacol Exp Ther. 1987;243:144–9. [PubMed] [Google Scholar]
- 42.Pettit HO, Pan HT, Parsons LH, Justice JB., Jr Extracellular concentrations of cocaine and dopamine are enhanced during chronic cocaine administration. J Neurochem. 1990;55:798–804. doi: 10.1111/j.1471-4159.1990.tb04562.x. [DOI] [PubMed] [Google Scholar]
- 43.Su TP, Hayashi T. Cocaine affects the dynamics of cytoskeletal proteins via sigma(1) receptors. Trends Pharmacol Sci. 2001;22:456–8. doi: 10.1016/s0165-6147(00)01740-5. [DOI] [PubMed] [Google Scholar]
- 44.Medvedev IO, Ramsey AJ, Masoud ST, Bermejo MK, Urs N, Sotnikova TD, Beaulieu JM, Gainetdinov RR, Salahpour A. D1 dopamine receptor coupling to PLCbeta regulates forward locomotion in mice. J Neurosci. 2013;33:18125–33. doi: 10.1523/JNEUROSCI.2382-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tannu N, Mash DC, Hemby SE. Cytosolic proteomic alterations in the nucleus accumbens of cocaine overdose victims. Mol Psychiatry. 2007;12:55–73. doi: 10.1038/sj.mp.4001914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Shioda N, Ishikawa K, Tagashira H, Ishizuka T, Yawo H, Fukunaga K. Expression of a truncated form of the endoplasmic reticulum chaperone protein, sigma1 receptor, promotes mitochondrial energy depletion and apoptosis. J Biol Chem. 2012;287:23318–31. doi: 10.1074/jbc.M112.349142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wu Z, Bowen WD. Role of sigma-1 receptor C-terminal segment in inositol 1,4,5-trisphosphate receptor activation: constitutive enhancement of calcium signaling in MCF-7 tumor cells. J Biol Chem. 2008;283:28198–215. doi: 10.1074/jbc.M802099200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Franke H, Kittner H, Grosche J, Illes P. Enhanced P2Y1 receptor expression in the brain after sensitisation with d-amphetamine. Psychopharmacology (Berl) 2003;167:187–94. doi: 10.1007/s00213-002-1386-6. [DOI] [PubMed] [Google Scholar]
- 49.Wilson CJ. The generation of natural firing patterns in neostriatal neurons. Prog Brain Res. 1993;99:277–97. doi: 10.1016/s0079-6123(08)61352-7. [DOI] [PubMed] [Google Scholar]
- 50.Wilson CJ, Kawaguchi Y. The origins of two-state spontaneous membrane potential fluctuations of neostriatal spiny neurons. J Neurosci. 1996;16:2397–410. doi: 10.1523/JNEUROSCI.16-07-02397.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Clapham DE. TRP channels as cellular sensors. Nature. 2003;426:517–24. doi: 10.1038/nature02196. [DOI] [PubMed] [Google Scholar]
- 52.Fowler MA, Sidiropoulou K, Ozkan ED, Phillips CW, Cooper DC. Corticolimbic expression of TRPC4 and TRPC5 channels in the rodent brain. PLoS One. 2007;2:e573. doi: 10.1371/journal.pone.0000573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Pomrenze MB, Baratta MV, Rasmus KC, Cadle BA, Nakamura S, Birnbaumer L, Cooper DC. Cocaine self-administration in mice with forebrain knock-down of trpc5 ion channels. F1000Res. 2013;2:53. doi: 10.12688/f1000research.2-53.v1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Rasmus KC, O’Neill CE, Bachtell RK, Cooper DC. Cocaine self-administration in rats lacking a functional trpc4 gene. F1000Res. 2013;2:110. doi: 10.12688/f1000research.2-110.v1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Moreno E, Moreno-Delgado D, Navarro G, Hoffmann HM, Fuentes S, Rosell-Vilar S, Gasperini P, Rodriguez-Ruiz M, Medrano M, Mallol J, Cortes A, Casado V, Lluis C, Ferre S, Ortiz J, Canela E, McCormick PJ. Cocaine disrupts histamine H3 receptor modulation of dopamine D1 receptor signaling: sigma1-D1-H3 receptor complexes as key targets for reducing cocaine’s effects. J Neurosci. 2014;34:3545–58. doi: 10.1523/JNEUROSCI.4147-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kourrich S, Hayashi T, Chuang JY, Tsai SY, Su TP, Bonci A. Dynamic interaction between sigma-1 receptor and Kv1.2 shapes neuronal and behavioral responses to cocaine. Cell. 2013;152:236–47. doi: 10.1016/j.cell.2012.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]