

Abstract
“Friends come and go but enemies accumulate.”
Arthur Bloch
Mitochondrial networks in eukaryotic cells are maintained via regular cycles of degradation and biogenesis. These complex processes function in concert with one another to eliminate dysfunctional mitochondria in a specific and targeted manner and coordinate the biogenesis of new organelles. This review covers the two aspects of mitochondrial turnover, focusing on the main pathways and mechanisms involved. The review also summarizes the current methods and techniques for analyzing mitochondrial turnover in vivo and in vitro, from the whole animal proteome level to the level of single organelle.
Keywords: mitochondria, autophagy, mitophagy, mitochondrial biogenesis, cardiac
Introduction
The endosymbiotic event that occurred billions of years ago between the SAR11 clade Rickettsiales and the microorganism which accepted them up sparked the evolution of all eukaryotic life on the planet [1]. These bacteria, which over time transformed into organelles we now call mitochondria, play a crucial role in maintaining all cellular functions as the most efficient and abundant source of adenosine triphosphate (ATP), essential for meeting the energetic demands of eukaryotic cells. For example, in the heart, mitochondria are by far the most abundant organelle and the main source (∼90%) of ATP (via oxidative phosphorylation) required for contraction of the organ [2]. Furthermore, mitochondria regulate calcium stores [3], fuel utilization [4], intracellular signaling [5] and apoptosis [6]. Due to this wide spectrum of cellular functions that mitochondria are involved in, the organelles play a pivotal role in mediating cellular homeostasis. As a result, maintenance of a healthy population of mitochondria is essential for cell survival. This is especially important due to the fact that mitochondria in a cell produce ATP at differential rates, comprising a heterogeneous population [7]. Those organelles which are quiescent, inefficient at ATP production or producing high levels of reactive oxygen species (ROS) combine to contribute to an extremely heterogeneous network. Impaired mitochondrial quality control results in accumulation of damaged mitochondria that may release more reactive oxygen species [8], produce less ATP [9], have a lower threshold for cytochrome c release resulting in apoptosis [10], undergo mitochondrial permeability transition pore (MPTP) opening resulting in necrosis [11], or may release mitochondrial components (mtHSP60, oxidized mitochondrial DNA) into cytosol where its recognition by receptors for damage-associated molecular patterns (DAMP) activates inflammation [12]. Mitochondrial turnover is therefore an integral aspect of quality control in which dysfunctional mitochondria are selectively eliminated through autophagy (mitophagy) and replaced through expansion of preexisting mitochondria (biogenesis) [13]. In this review, we focus on the mechanics of mitochondrial turnover and the techniques currently employed to measure and monitor the dynamic network, constantly kept in balance by two arms of mitochondrial quality control: mitophagy and mitochondrial biogenesis.
Mitophagy
In order to maintain an efficient supply of energy to the cell, mitochondria must constantly perform maintenance on a molecular and network level. On a protein level, this is absolutely crucial due to the fact that oxidative phosphorylation (OX-PHOS) leads to oxidative damage to mitochondrial proteins over time [14]. Furthermore, the vast majority of mitochondrial proteins are encoded in the nucleus, and accumulation of respiratory chain subunits can lead to an imbalance in respiratory chain complexes. To address this problem, mitochondria contain a number of proteases responsible for targeted protein quality control (UPRmt, the mitochondrial unfolded protein response): most prominently by the Lon protease and other AAA+ family proteases (ATPases associated with a variety of cellular activities) [15]. These proteases degrade the superfluous subunits into small peptides and act like chaperones to extract unfolded proteins from the inner mitochondrial membrane and target them for degradation via the proteasome [16]. Although the mechanism of removal of these peptides from the mitochondria into the cytosol is unclear, in mammals the responsible transporter is believed to be ABCme10, a homolog of the C. elegans protein Haf1 which removes peptide fragments generated by the C. elegans protease ClpP [17]. The extruded peptides are recognized by transcription factors CHOP, C/EBPβ, and cJun/AP1, which upregulate the expression of UPRmt factors such as mitochondrial chaperone hsp60, ClpP, Yme1L1, and MPPβ, Tim17A, NDUFB2, and EndoG. [18]. Finally, in response to the unfolded mitochondrial proteins, PKR (double-stranded RNA-activated protein kinase) phosphorylates eIF2α and cJun, thereby suppressing the translation and import of nuclear-encoded mitochondrial proteins [19].
However, this targeted approach is not sufficient for rapid removal of whole dysfunctional mitochondria or a large number of damaged components. To accomplish this, cells employ the process of selective mitochondrial autophagy (mitophagy), which eliminates damaged and dysfunctional mitochondria permitting the synthesis of new mitochondrial components and their insertion into the remaining functional mitochondria. Using the heart as an example again, mitochondria turn over with a half-life of 14 days [20]. Rat cardiomyocytes have roughly 1000 mitochondria per cell, suggesting that under basal resting conditions, one mitochondrion per cell is replaced every 40 minutes. Intriguingly, a number of enzymes involved in oxidative phosphorylation (OX-PHOS) are expressed with diurnal variation, suggesting that mitochondrial turnover is regulated by the circadian cycle. This seems likely given that a period of fasting occurs during sleep; therefore mitochondrial elimination may alternate with regeneration on a daily basis in a manner resembling a sine wave, with 3-4% of the mitochondrial population replaced each day. However, this may be much higher: in the cultured mouse atrial cell line HL-1 subjected to nutrient deprivation, the number of mitochondria is reduced by 70% within 3.5 hours [21].
Eukaryotic cells generally rely on two processes to eliminate and recycle components to maintain homeostasis working in concert: the ubiquitin proteasome system (UPS) and autophagy. Autophagy is a cellular degradation system in eukaryotic cells that allows for the bulk recycling of unwanted cytoplasmic aggregate proteins or dysfunctional organelles in a lysosome-dependent manner [22]. In order to facilitate and initiate mitophagy, mitochondrial dynamics (fusion and fission) play a critical role in mitochondrial turnover, favoring fission and suppressing fusion, enabling engulfment by autophagosomes. Fission of reticulate mitochondria into smaller fragments is a crucial requirement for mitophagy to occur [23, 24]. The key regulator of this process is dynamin-related protein 1 (Drp1), a GTPase in the dynamin super family of proteins recruited to the mitochondria, which in concert with proteins Fission 1 (Fis1), mitochondrial fission factor (Mff), and mitochondrial dynamics proteins of 49 (MiD49) and 51 kDa (MiD51) is responsible for mitochondrial fragmentation [25-28]. While the importance of Fis1 as a fission partner of Drp1 is unclear and may be cell-type dependent, the role of the other three proteins appears essential: Mff assists in the assembly of Drp1 and MiD49 and MiD51 may play a regulatory role by recruiting Drp1 and maintaining it in inactive state until fission is required, stimulated by cell signaling [29]. Proteins that promote OMM fusion such as Mitofusin 1 and 2 (Mfn1 and 2) are ubiquitinated and eliminated by the UPS, while other proteins such as Optic atrophy protein 1 (OPA, regulator of fusion of the inner mitochondrial membrane), are degraded during mitophagy by the inner membrane zinc metalloprotease OMA1 and AAA proteases [30-32].
The best-characterized pathway of mitophagy depends on the recruitment of E3 ubiquitin ligase Parkin. Mitophagy in this context is triggered by cellular stresses such as ischemia, which trigger depolarization of the outer mitochondrial membrane (OMM) (best induced by utilizing chemical uncouplers of mitochondria such as carbonyl cyanide 4-(trifluoromethoxy) phenylhydrazone (FCCP) or carbonyl cyanide m-chlorophenylhydrazone (CCCP)) [33]. The depolarization results in stabilization of the serine/threonine kinase phosphatase and tensin homolog (PTEN)-induced kinase 1 (PINK1) on the outer mitochondrial membrane (OMM) and recruitment of the E3 ubiquitin ligase Parkin, which attaches ubiquitin moieties to OMM proteins [34-37]. The interplay between PINK1 and Parkin is a crucial step in mediating the clearance of dysfunctional mitochondria [38, 39].
PINK1 is constitutively made and continuously degraded by the mitochondria-specific proteases presenilin-associated rhomboid-like protein (PARL) and mitochondrial processing peptidase (MPP). These proteases are inactivated by the loss of membrane potential across the inner mitochondrial membrane through an as yet uncharacterized mechanism; this inactivation results in the accumulation of PINK1 on the OMM where it can phosphorylate the OMM proteins through its kinase domain which faces the cytosol and facilitates the targeting of said proteins to Parkin [40-42]. The targets of PINK1 include Parkin itself [43, 44], mitofusin 2 (Mfn2) [45], and mitochondrial rho 1 (MIRO) [46], a component of the microtubule-associated motor complex that anchors kinesin to mitochondria. More controversially, Voltage-dependent anion channel 1 (VDAC1) has been suggested to be a Parkin target essential for mitophagy [34], although this finding is in dispute [47]. Of those targets, Mfn2, which under basal conditions functions in mitochondrial fusion events and links endoplasmic reticulum to mitochondria, acts as a Parkin receptor during mitophagy following phosphorylation by PINK1, recruiting Parkin to the mitochondria where it ubiquitinates the OMM targets. Ubiquitination and proteasomal degradation of MIRO, Mfn2, and Mfn1 prevent the targeted mitochondria from rejoining the mitochondrial network through fusion [45, 46] [48-50]. Further, the presence of ubiquitin on the OMM proteins facilitates recruitment of autophagy adapter proteins such as neighbor of BRCA1 (NBR1) or sequestosome-1 (p62/SQSTM1). These adaptor proteins have an ubiquitin binding domain (UBA) and microtubule-associated protein 1 light chain 3 (LC3) interacting region (LIR) which serves as an anchor for the developing autophagosomal membrane in proximity to the tagged mitochondrion in a zipper-like process [51, 52]. Other possible actors in Parkin-dependent mitophagy include SMAD-specific E3 ubiquitin ligase 1 (SMURF1), although its E3 ubiquitin ligase function appears to be superfluous to its ability to facilitate mitophagy [53]. Yet another player in promoting Parkin-dependent mitophagy is Activating molecule in Beclin 1-regulated autophagy (Ambra1), which dissociates from mitochondrial Bcl-2 to bind Beclin1 to initiate autophagy [54, 55]. Ambra1 interacts with Parkin to promote mitophagy, but is not a substrate of Parkin [56].
Recently, a number of groups have reported a novel mode of Parkin activation by PINK1, through the phosphorylation of ubiquitin at its Serine 65 residue (ubiquitinPhospho–Ser65) [57-59]. The E3 ubiquitin ligase function of Parkin is autoinhibited under basal conditions, and is activated by PINK1 via direct phosphorylation of the conserved Ser65 residue in the Ubl domain and the direct phosphorylation of ubiquitin. The studies point to a two-step activation mechanism of Parkin in which an initial phosphorylation of residue Ser65 by PINK1 within the Ubl domain following mitochondrial depolarization results in a conformational change that disrupts the interaction of said domain and the C-terminus of Parkin. This “open” conformation primes Parkin for optimal binding of ubiquitinPhospho–Ser65 that results in the active E3 form of the protein which then targets mitochondrial membrane proteins for proteasomal degradation.
In addition to the PINK1/Parkin/ubiquitin axis, Parkin-independent mitophagy can be initiated through atypical members of the Bcl-2 homology domain 3 (BH3) family members such as BCL2/adenovirus E1B 19 kDa protein-interacting protein 3 (BNIP3) and BCL2/adenovirus E1B 19 kDa protein-interacting protein 3-like protein (BNIP3L aka NIX). These proteins insert into the OMM and facilitate engulfment by the autophagosome through a LIR domain that can interact with LC3 isoforms, including gamma-aminobutyric acid receptor-associated protein (GABARAP) and GABARAP-like 1 (GABARAPL1) [60, 61]. However, Bnip3 can also work in conjunction with Parkin, by recruiting Drp1 and Parkin to the mitochondria to promote fission and mitophagy [62]. In hypoxic conditions mitophagy has been reported to be mediated by the OMM protein FUN14 domain containing 1 (FUNDC1) which contains a LIR [63] (Fig. 1).
Figure 1. Overview of Mitophagy.
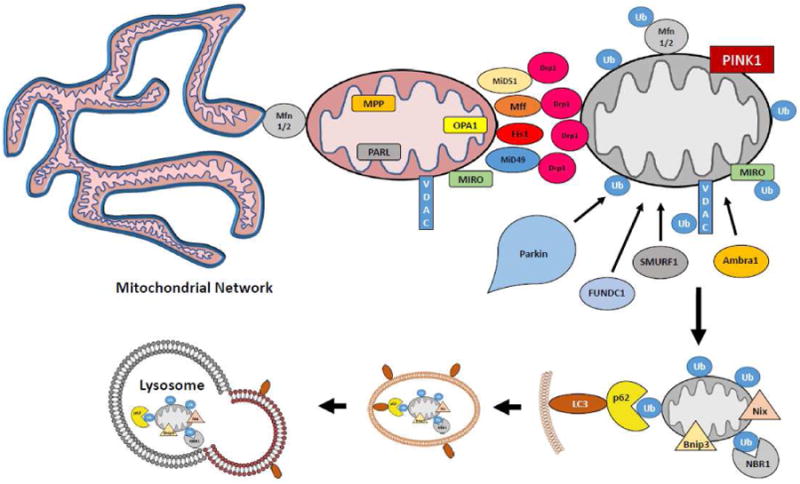
Inactivation of mitochondria-resident proteases in depolarized mitochondria leads to an accumulation of PINK1 on the outer mitochondrial membrane. The phosphorylation of outer membrane proteins such as Mfn1/2 leads to their ubiquitination by Parkin. The fission machinery separates the dysfunctional mitochondrion from the network, and the ubiquitinated proteins serve as binding sites for autophagy adaptor proteins such as p62. These proteins contain LC3 binding sites, leading to encapsulation of the organelle in an autophagic vesicle, which is delivered to the lysosome for degradation.
While mitophagy is responsible for bulk degradation of mitochondria, turnover of individual components may proceed at asynchronous rates through redistribution of components via fusion events, selective degradation of proteins via mitochondrial proteases, and proteasomal elimination of some OMM proteins. Even in the case of Parkin-dependent mitophagy, some outer membrane proteins are recycled through transfer to the ER [64]. Proteomic studies using heavy isotope labeling [65] revealed that proteins of the IMM turn over with rates that are similar to mitochondrial turnover based on historic radiolabeling studies [66, 67], suggesting that IMM proteins (primarily OXPHOS constituents) may be primarily cleared via mitophagy. This also corresponds to studies which showed that matrix and OMM were readily redistributed across the mitochondrial network when fusion and fission were intact; however, IMM constituents redistributed much more slowly [24].
Mitochondrial Biogenesis
Over the course of evolution, mitochondrial protein expression and translation was transposed to the DNA of the host cell, leaving a residual circular genome of 16 kb, which in mammals encodes only 13 protein subunits required for aerobic respiration. Furthermore, mitochondrial genomes replicate in a manner that is radically different from the mammalian DNA replication mechanisms, utilizing the “bootstrap” method in which processed transcripts (bootlaces) hybridize with the displaced parental strand as the replication fork advances [68]. This mechanism minimizes the occurrence and impact of single-strand breaks that threaten genome stability, limit the occurrence of replication-dependent deletions and insertions, and defend against invading elements such as viruses. Mitochondrial biogenesis, the second arm of mitochondrial turnover, therefore hinges on coordination of nuclear and mitochondrial-encoded gene expression.
The key transcription factor that directly and indirectly regulates the expression of mitochondrial nuclear-encoded proteins is the Peroxisome proliferator-activated receptor gamma coactivator-1 alpha (PGC-1α) [69]. First identified for the increase in its transcriptional activity in the context of thermogenesis and as the binding partner of peroxisome proliferator-activated receptor γ (PPARγ), PGC-1α is a member of the nuclear receptor superfamily, proteins responsible for assembling macrocomplexes into functional transcriptional machinery at specific DNA sequences [70, 71]. As such, PGC-1α is responsible for the main thrust of mitochondrial biogenesis in the nucleus by controlling the expression of nuclear respiratory factors 1 and 2 (NRF-1 and NRF-2), transcription factors which themselves control the expression of mitochondrial transcription factor A (Tfam) [72]. Tfam is a mitochondrial resident transcription factor responsible for expression of mitochondrial genes (tRNAs, rRNAs and 13 subunits of respiratory chain) from the heavy and light mitochondrial DNA (mtDNA) [73]. Several studies have also confirmed the central role of PGC-1α in mitochondrial biogenesis, by demonstrating that increased expression of PGC-1α in response to stimuli such as cold and exercise leads to increased expression of mitochondrial enzymes such as ATP synthetase ((β-subunit), COX (cytochrome c oxidase) subunits (COX II and COX IV) and δ-aminolevulinate synthase (δ-ALAS) [74, 75]. In all, overexpression of PGC-1α leads to upregulation of 151 genes that encode mitochondrial proteins involved in many metabolic functions of mitochondria such as fatty acid β-oxidation (FAO), tricarboxylic acid cycle and oxidative phosphorylation, as well as mitochondrial ribosomal machinery and mitochondrial membrane transport proteins [76]. In addition, the PGC-1α isoform PGC-1β is thought to control increased mitochondrial biogenesis and basal oxygen consumption, but is not induced by cold or exercise, suggesting alternate pathways to induction of mitochondrial biogenesis [77].
Beyond directing the transcription of respiratory chain mRNAs, PGC-1α also interacts with and increases the expression of downstream transcription factors such as peroxisome proliferator-activated receptors (PPARs), hormone receptors for estrogen and thyroid hormone, as well as ERRs (estrogen-related receptors) α and γ [78]. ERRs are a set of nuclear receptors that lack an associated ligand and are closely linked with mitochondrial biogenesis in general and PGC-1α in particular. [79] In fact, ERRs may be a key co-activator of PGC-1α activity, as the effect of over-expression of PGC-1α can be inhibited by siRNA targeted to ERRα, or mimicked by overexpression of ERRα, demonstrating the close relationship between PGC-1α activation and ERRα [76]. Another family of nuclear receptors that plays a role together with PGC-1α in mitochondrial biogenesis are the PPARs, which in heterodimerization with members of retinoid X receptor (RXR) drive the transcription of nuclear genes encoding mitochondrial FAO enzymes [80].
Cell signaling cascades are the driving force behind the activation of PGC-1α. Two pathways of transcriptional activation have been well characterized: through calcineurin A (CnA) and Ca2+/calmodulin-dependent protein kinase IV (CaMKIV). CnA interacts with and activates myocyte enhancer factors 2C and 2D (MEF2C and MEF2D), which regulate the transcription of PGC-1α directly [81, 82]. MEF2C and 2D are upregulated by PGC-1α, which results in a feed forward loop that allows PGC-1α to continue to drive its own upregulation [83]. CaMKIV activates PGC-1α by phosphorylating the transcription factor cAMP response element (CRE)-binding protein (CREB); once phosphorylated, CREB binds to promoter elements on the PGC-1α gene and strongly drives its expression [81]. The p38 mitogen-activated protein kinase (p38 MAPK) and AMP-dependent Kinase (AMPK) have also been implicated as regulators of PGC-1α expression. p38 MAPK activity is increased following exercise, leading to the activation of MEF2 and activating transcription factor 2 (ATF2), both of which upregulate the expression of PGC-1α [84, 85].
The activity of PGC-1α is further highly regulated by post-translational modifications: direct phosphorylation by signaling kinases and acetylation/deacetylation. A number of kinases have been implicated in control of PGC-1α activity: AMPK and Akt during starvation and p38 MAPK after endurance exercise [86-88]. The activation of AMPK in response to glucose depletion results in direct phosphorylation of PGC-1α on threonine-177 and serine-538, crucial for activation of PGC-1α-dependent transcription from the PGC-1α promoter [86]. In a similar fashion, p38 MAPK increases the activity of PGC-1α by directly phosphorylating threonine-262, serine-265, and threonine-268, resulting in stabilization of the protein and the disruption of the interaction between PGC-1α and its inhibitor p160MBP [87, 89]. Conversely, insulin inhibits the activity of PGC-1α through Akt, which can directly phosphorylate the serine-570 residue on PGC-1α, or indirectly through phosphorylation of the Clk2 kinase which then in turn phosphorylates the C-terminal serine- and threonine-rich regions of PGC-1α, decreasing its co-transcriptional activity [90, 91].
Other post-translational control of PGC-1α involves glycogen synthase kinase 3β (GSK3β), which has been shown to inhibit PGC-1α activity in response to acute oxidative stress by increasing its proteasomal degradation and inhibiting the activity of Sirt1, an NAD-dependent deacetylase thought to activate PGC-1α [92]. The deacetylation is crucial for the activation of PGC-1α, as the protein is heavily acetylated by acetyltransferase GCN5, inhibiting its activity and sequestering it in the nuclear foci rather than the promoter regions of its target genes [93]. Sirt1 activity is dependent upon the coenzyme nicotinamide adenine dinucleotide (NAD+), and it is highly sensitive to the changes in energy requirements of the cell; as the ratio of NAD+/NADH changes in response to fasting, exercise or redox stress, Sirt1 increases the deacetylation PGC-1α, resulting in an increase in transcription of PGC-1α targets, leading to mitochondrial biogenesis [94-96]. AMPK, which directly modulates the activity of PGC-1α by phosphorylation, may also control its activity indirectly by increasing NAD+ levels in the cell by fatty acid oxidation, increasing the activity of Sirt1 [96]. Ubiquitination and methylation have also been demonstrated to play a role in regulating the activity of PGC-1α in response to energy demands and oxidative stress, states that require mitochondrial biogenesis [97].
Beyond transcription of nuclear and mitochondrial encoded messages, protein synthesis, import, and assembly must occur. The mRNAs in mitochondria are translated by the mitochondrial ribosomes in the matrix, while nuclear-encoded mRNAs are translated in polyribosomes closely associated with the mitochondrial outer membrane, facilitating co-translational import [98]. Protein import machinery is complex, and defects in the process, whether inborn or acquired, can lead to a range of diseases (reviewed in [99]). Assembly of OXPHOS subunits depends upon coordination of protein import and mitochondrial protein synthesis. An imbalance triggers the mitochondrial unfolded protein response which can culminate in mitophagy [100].
Mitochondrial biogenesis is intimately linked to mitophagy. mTOR, a central regulator of autophagy, is a serine/threonine kinase that directly controls mitochondrial biogenesis through activation of PGC-1α and its gene targets. mTOR also upregulates biogenesis through the mTOR complex 1 (mTORC1), which prevents the binding of the eukaryotic translation initiation factor 4E (eIF4E)-binding proteins (4E-BP) to their targets, stimulating the translation of nuclear encoded mitochondrial proteins including members of complex V, complex I, and Tfam [101, 102]. In addition to activation of biogenesis and mitophagy, repressors of biogenesis are important players controlling mitochondrial turnover. One of the more recently discovered direct repressors of PGC-1α is the protein Parkin-interacting substrate, PARIS (ZNF746) [103]. PARIS is a target of Parkin ubiquitin ligase, which controls the levels of Paris via the ubiquitin proteasome pathway. Accumulated PARIS in the nucleus leads to direct inactivation of PGC-1α transcription and inhibition of PGC-1α-dependent genes. Other repressors of mitochondrial biogenesis operate in less direct manner. Another repressor, nuclear co-repressor 1 (Ncor1) acts as a transcriptional repressor of PPARγ, PPARδ and ERR as well as an inhibitor of their activity by docking with histone deacetylases such as HDAC3 and SIRT1 thereby curbing the expression and activity of MEF2, PPARδ, and ERR and their downstream transcriptional targets involving mitochondrial oxidative metabolism [104] (Fig. 2).
Figure 2. Overview of Mitochondrial Biogenesis.
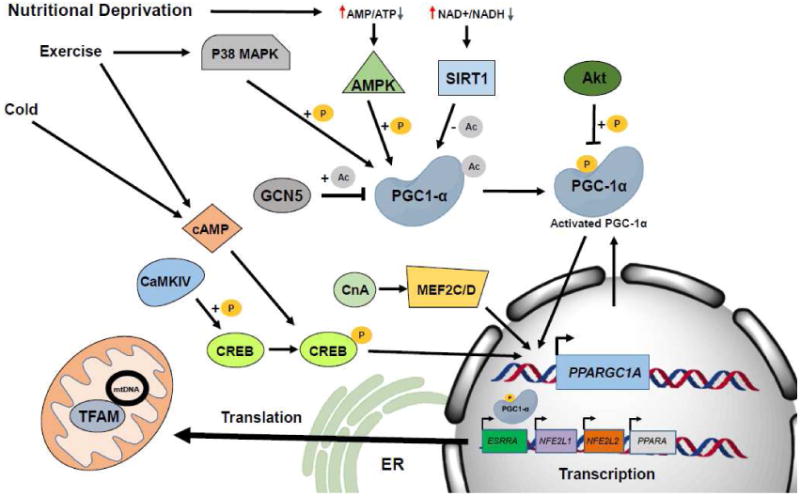
The bulk of biogenesis is controlled by the transcription factor PGC-1α. External stimuli such as cold, exercise or nutritional deprivation activate signaling cascades which lead to activation of PGC-1α. PGC-1α activates the transcription of other transcription factors such as NRF1, which are responsible for driving the transcription of nuclear-encoded mitochondrial proteins.
Mitochondrial Lipid Turnover
Mitochondria possess two lipid bilayer membrane structures, and the outer mitochondrial membrane (OMM) and inner mitochondrial membrane (IMM) differ radically in their composition. Much of the lipid for both membranes derives from the endoplasmic reticulum (ER). Importantly, phosphatidic acid derived from the ER is transferred to the inner leaflet of the IMM where it is enzymatically converted to cardiolipin [105]. Cardiolipin is essential to maintain the high degree of proton impermeability of the IMM, and defects in its biosynthesis and remodeling lead to severe mitochondrial defects and myocardial dysfunction [106, 107]. Additional lipids are synthesized in the ER and transported to the mitochondria by direct membrane interactions and by diverse shuttle systems. Of note, phosphatidylserine, synthesized in ER, is converted in IMM to phosphatidylethanolamine and exported back to ER for conversion to phosphatidylcholine [108]. Sphingolipids are synthesized in the ER but when ceramides are delivered to the OMM, they serve as important binding sites for LC3-II, thereby mediating mitophagy [109]. Mitochondrial membranes have also been suggested as a source for autophagosome membrane lipids, particularly as the mitochondria are the key site for synthesis of phosphatidylethanolamine needed for lipidation of LC3 [110, 111]. During mitophagy, most of the membrane lipids are degraded in the lysosome, liberating free fatty acids [112]; this process may be disrupted in the presence of lipid peroxides, leading to accumulation of lipofuscin [113].
Experimental Methods for Examining Mitochondrial Turnover
Pulse-chase labeling
Classic approaches to monitoring mitochondrial turnover involved the use of radioactive isotope labeling of mitochondrial proteins [67], phospholipids [114], and heme groups [66]. Studies of cardiac mitochondria revealed an average half-life of 17 days under steady state conditions [115]. This has been confirmed more recently by non-radioactive isotope labeling and proteomic analysis [65, 116]. Many of the mitochondrial inner membrane/matrix components exhibited coordinated turnover, suggesting autophagic degradation, whereas outer membrane components had more variable rates of turnover. Subcellular distribution (subsarcolemmal vs. interfibrillar) also influenced turnover rates [116].
Proteomics
Until quite recently, much of the work on the rate of mitochondrial turnover was performed through proteomic approaches. For example, Major et. al, made use of 2-D PAGE analysis to identify changes in protein abundance in mitochondria isolated from yeast grown on a nonfermentable carbon source and then incubated in a buffer containing an ATP-regenerating system [117]. Using this approach, the group was able to isolate and identify 60 proteins which exhibited a change by matrix-assisted laser desorption ionization-TOF (MALDI-TOF) mass spectrometry, covering roughly 11% of yeast mitochondrial proteins listed in the MitoP2 database. Further use of this technique allowed for identification of changes in mitochondrial protein levels when yeast cells were subjected to different temperature conditions. The authors were able to identify several groups of proteins with differential stability under stress conditions, and presented a mechanism by which mitochondrial turnover in yeast under conditions of stress involves mitochondrial resident proteases. The drawbacks to using 2-D PAGE are the limitations presented by the difficulty of resolving low-abundance or hydrophobic proteins, reproducibility and the low-throughput nature of the method [118].
An alternate, and more elegant approach to analyzing the mitochondrial turnover on a proteome-wide level has been developed by Kim et al. [65]. The approach makes use of the fact that heavy water (2H2O) intake is a safe, cost efficient and effective way to induce the incorporation of 2H into biomolecules. Combined with liquid-chromatography mass spectrometry (LC-MS), this technique allows for straightforward and accurate measurement of protein half-life in mouse organs. By combining the technique with post-collection data analysis, the group was able to observe kinetics of 458 liver and heart mitochondrial proteins, allowing for comparison of wholesale mitochondrial dynamics comparison between the two organs. The data revealed that mouse heart mitochondrial proteins had a much longer half-life (slower turnover) than their analogs in the liver (approximately four times higher). Deeper analysis of the data revealed that in both tissues, proteins responsible for protein folding had a much shorter half-life than those involved in redox reactions. In contrast, proteins responsible for biosynthesis had a much faster turnover rate in the heart than in the liver, suggesting tissue-specific turnover rates for mitochondria in mammals. Furthermore, the variable turnover rate in proteins from isolated mitochondria gives further support to the previously stated hypothesis, whereby mitochondria segregate dysfunctional proteins for clearance by autophagy during cycles of fission and fusion. The study also emphasizes the significant role that mitochondrial resident proteases must play in order to clear dysfunctional protein components and maintain homeostasis in the organelle. The drawback of this approach is the fact that in this case protein turnover is analyzed on a global level, from a population of mitochondria; furthermore, posttranslational modifications of mitochondrial proteins, which may be important in segregating newly-synthesized from older mitochondria, may be missed by this approach. On a more practical level, this approach requires access to stable isotopes and mass spectrometry equipment, and may be cost prohibitive for many laboratories.
Imaging mitophagy with GFP-LC3
Kim and Lemasters used hepatocytes from GFP-LC3-transgenic mice for confocal imaging of autophagosomes as they formed around mitochondria labeled with TMRM (tetramethylrhodamine methyl ester) [119]. In response to nutrient deprivation, autophagosomes formed around polarized mitochondria which retained the dye until the autophagosome fully enclosed the mitochondrion. This form of mitochondrial autophagy appears to be independent of mitochondrial membrane potential, in contrast to Parkin-dependent mitophagy, which is initiated by membrane depolarization and stabilization of PINK1 [37]. Use of mitochondrial colocalization with GFP-LC3 for a quantitative readout of mitophagy is challenging, because mitochondrial engulfment takes less than 10min, followed by loss of GFP-LC3 fluorescence soon after acidification. Catching such a dynamic process “in the act” makes quantitation difficult. A related imaging approach was used by Twig et al. [24] to examine mitophagy under steady state conditions in Ins1 cells. They noted that mitochondrial fission resulted in asymmetric daughter mitochondria; the depolarized mitochondrion was less likely to undergo subsequent fusion events but rather was engulfed by an autophagosome within 40 minutes. These contrasting observations of mitophagy may reflect different processes: one dependent on mitochondrial depolarization and PINK1 for Parkin and p62-dependent mitophagy, and another mediated by increased ROS as described by Scherz-Shouval et al. [120]. Bnip3, an adaptor for LC3 [121], is induced to dimerize by ROS [122] and may participate in starvation-induced mitophagy.
mt-Keima
An alternate approach to studying mitochondrial turnover involves utilizing mitochondria-targeted fluorescent proteins. A study by Katayama et al. utilized a coral fluorescent protein that is resistant to lysosomal proteases, Keima, which exhibits a green fluorescence at neutral pH and red fluorescence at acidic pH (inside autolysosomes) [123]. The authors fused the protein with a mitochondrial signal sequence to localize it to the inner membrane of the mitochondria (mt-Keima). By co-transfecting cells with mt-Keima and EGFP-Parkin, they were able to quantitatively observe discrete autophagic events following the induction of mitophagy by incubation with Carbonyl cyanide m-chlorophenyl hydrazone (CCCP) (mitochondrial membrane uncoupling agent) and oligomycin (ATP synthase inhibitor). Over a time course, the green fluorescence of mt-Keima shifted from green to red as mitochondria (or mitochondrial inner membrane components) were delivered to the lysosomes. Although a powerful tool for studying autophagy in general and mitophagy in particular, mt-Keima does not address the mitochondrial biogenesis arm of the turnover, nor does it provide information on the “age” of the particular mitochondria it is expressed in.
MitoTimer (inducible expression)
In order to study mitochondrial turnover at the level of a single mitochondrion, our laboratory has developed the MitoTimer protein. Timer is a mutant of DsRed fluorescent protein developed by Terskikh et al. [124]. The Timer protein transitions from green fluorescence to a more stable red conformation as it matures over a span of 48 hours. Furthermore, the protein is very stable under physiological conditions, insensitive to variations in ionic strength, and changes in pH between 7.0 and 8.0. Timer or Timer-fusion proteins are widely used to monitor cellular processes including gene expression, intracellular protein recycling, cell survival, and the kinetics of viral infection [125-128]. We fused the Timer protein with the N-terminal mitochondrial signal sequence from the cytochrome c oxidase subunit VIII (COX8) to target the protein to the inner membrane of the mitochondria. The protein can be monitored as it matures from green to red over 48 hours (Fig. 3A), and it is retained in the mitochondria for a number of days (presumably corresponding to mitochondrial half-life). We cloned MitoTimer into a construct with a tetracycline-inducible promoter in conjunction with the reverse tetracycline transactivator (rtTA) to allow for synchronized and time-restricted expression of MitoTimer. A short (2 hours) pulse with tetracycline, followed by removal of any residual tetracycline, is sufficient to achieve MitoTimer expression across the mitochondrial population of the cell. In order for mitochondrial fusion to occur, mitochondria must maintain a high membrane potential [24] and express mitofusin-1 or -2 (Mfn1/2) [129]. A study by Ferree et al. utilized MitoTimer to show that it is evenly distributed across the mitochondrial network in wild type cells under basal conditions; in contrast, mouse embryonic fibroblasts (MEFs) derived from Mfn1/2 double knockout mice displayed a heterogeneous distribution of green vs. red MitoTimer, demonstrating that fusion events enable mixing of old and new proteins within the mitochondrial network [129]. High membrane potential is required for mitochondrial protein import [130]; thus MitoTimer will not be imported into dysfunctional mitochondria with low membrane potential [131].
Figure 3. MitoTimer.
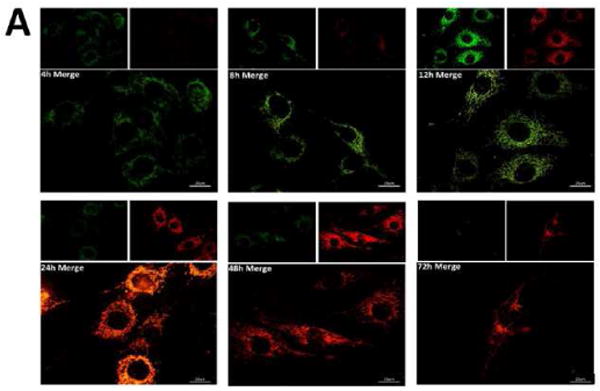
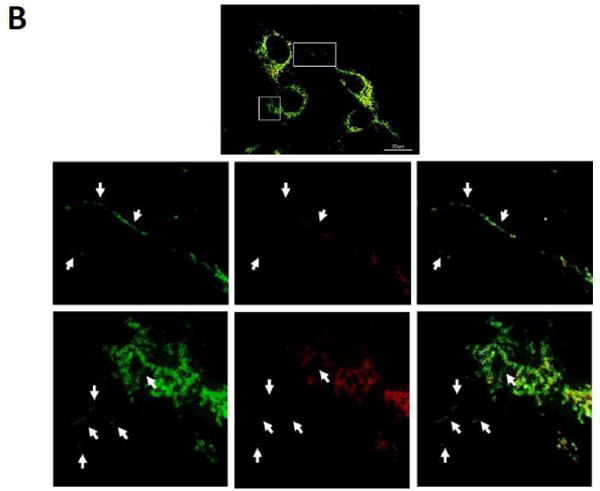
(A) C2C12 cells expressing Tet-inducible MitoTimer, exposed to Dox for 1 h, and then imaged at 4, 8, 12, 24, 48 and 72 h. Merged images of green and red channels are shown; smaller insets show the expression levels of MitoTimer green and MitoTimer red fluorescence. (B) Heterogeneous expression of MitoTimer fluorescence, indicating mitochondria with differential rates of import.
Previous work demonstrated that the rate of diffusional exchange of contents across the mitochondrial network is fastest for outer membrane constituents, intermediate for matrix contents, and slowest for inner membrane proteins [132]. Consistent with evidence indicating that turnover of mitochondrial inner membrane and matrix proteins depends upon mitophagy [133, 134], inhibition of autophagy enhanced the accumulation of red (older) MitoTimer [129], demonstrating the utility of the construct for the assessment of mitophagy.
Biogenesis could be assessed after a second Dox pulse to induce another round of expression, (readily evident as green MitoTimer), and which can reveal mitochondrial subpopulations that are more actively engaged in protein import, a hallmark of mitochondrial biogenesis. Interestingly, MitoTimer import during biogenesis in C2C12 cells is not uniform, but is enriched in a perinuclear zone (Fig. 3B). While this may be simply due to mRNA proximity, it is possible that the subpopulation of mitochondria enriched for green MitoTimer are specialized for mitochondrial regeneration. A perinuclear distribution of newly-synthesized MitoTimer was also noted in neuronal cells, while mature MitoTimer predominated in the distal portion of the neurites [131]. This finding is consistent with previous work suggesting that autophagosomes initiate distally in neurons (near the oldest mitochondria) and then migrate towards the soma, where they fuse with lysosomes [135].
MitoTimer (constitutive expression)
It is also possible to obtain information about mitochondrial turnover by expressing MitoTimer in a constitutive, rather than a conditional manner, in animal models. By expressing constitutive MitoTimer in Drosophila heart tube, Laker et al. demonstrated that exercise training resulted in an increase in the number of green mitochondria, corresponding to increased mitochondrial biogenesis [136]. Furthermore, the authors demonstrated the utility of MitoTimer for monitoring mitochondrial turnover in mice, where MitoTimer delivered by electroporation was constitutively expressed in skeletal muscle. The ratio of red to green MitoTimer in the skeletal muscle was increased in mice on a high fat diet compared to normal chow-fed animals, consistent with loss of lysosomal function needed for mitophagy [137]. Suppression of autophagy results in accumulation of mitochondria with lower membrane potential and increased oxidative damage [138]. Conversely, Laker et al. observed an increase in the green:red ratio of MitoTimer in mice subjected to exercise training, even in the presence of a high fat diet [136]. Exercise upregulates autophagy (enhancing clearance of older mitochondria) [139] and also stimulates mitochondrial biogenesis [140], providing two drivers for the green-shifted ratio of MitoTimer in their model.
Conclusions
Derangements of mitochondrial turnover are recognized in a growing number of disorders. The field is poised for important new discoveries, with the advent of new technologies including deuterium oxide labeling with high resolution mass spectrometry analyses in humans [141] and the ability to image mitophagy and biogenesis in cells and tissues. Medicine, which has traditionally focused on organ pathophysiology, may well have to adjust its focus to include organelle pathology as an aspect of human disease.
Highlights.
Mitochondria come and mitochondria go, always from the one befo'
Knowledge grows, we can refine, since new tools have come on line
MitoTimer and mKeima give a lovely glow, and mass spectrometry with D2O
Reveal biogenesis and mitophagy as the key to organismal longevity
Acknowledgments
The authors gratefully acknowledge the support of NIH P01 HL112730 (to RAG) and the Dorothy and E. Phillip Lyon Chair in Molecular Cardiology in honor of Clarence M. Agress, MD (to RAG).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Literature Cited
- 1.Thrash JC, Boyd A, Huggett MJ, Grote J, Carini P, Yoder RJ, Robbertse B, Spatafora JW, Rappe MS, Giovannoni SJ. Phylogenomic evidence for a common ancestor of mitochondria and the SAR11 clade. Scientific reports. 2011;1:13. doi: 10.1038/srep00013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Murphy E, Steenbergen C. Preconditioning: the mitochondrial connection. Annual review of physiology. 2007;69:51–67. doi: 10.1146/annurev.physiol.69.031905.163645. [DOI] [PubMed] [Google Scholar]
- 3.Duchen MR. Mitochondria and calcium: from cell signalling to cell death. The Journal of Physiology. 2000;529:57–68. doi: 10.1111/j.1469-7793.2000.00057.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Liesa M, Shirihai Orian S. Mitochondrial Dynamics in the Regulation of Nutrient Utilization and Energy Expenditure. Cell Metabolism. 2013;17:491–506. doi: 10.1016/j.cmet.2013.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tait SW, Green DR. Mitochondria and cell signalling. Journal of cell science. 2012;125:807–815. doi: 10.1242/jcs.099234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pradelli LA, Beneteau M, Ricci JE. Mitochondrial control of caspase-dependent and -independent cell death. Cellular and molecular life sciences : CMLS. 2010;67:1589–1597. doi: 10.1007/s00018-010-0285-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kuznetsov AV, Margreiter R. Heterogeneity of Mitochondria and Mitochondrial Function within Cells as Another Level of Mitochondrial Complexity. International Journal of Molecular Sciences. 2009;10:1911–1929. doi: 10.3390/ijms10041911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Song M, Chen Y, Gong G, Murphy E, Rabinovitch PS, Dorn GW., 2nd Super-suppression of mitochondrial reactive oxygen species signaling impairs compensatory autophagy in primary mitophagic cardiomyopathy. Circulation research. 2014;115:348–353. doi: 10.1161/CIRCRESAHA.115.304384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kim Nam C, Tresse E, Kolaitis RM, Molliex A, Thomas Ruth E, Alami Nael H, Wang B, Joshi A, Smith Rebecca B, Ritson Gillian P, Winborn Brett J, Moore J, Lee JY, Yao TP, Pallanck L, Kundu M, Taylor JP. VCP Is Essential for Mitochondrial Quality Control by PINK1/Parkin and this Function Is Impaired by VCP Mutations. Neuron. 2013;78:65–80. doi: 10.1016/j.neuron.2013.02.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Winklhofer KF. Parkin and mitochondrial quality control: toward assembling the puzzle. Trends in Cell Biology. 2014;24:332–341. doi: 10.1016/j.tcb.2014.01.001. [DOI] [PubMed] [Google Scholar]
- 11.Kim I, Rodriguez-Enriquez S, Lemasters JJ. Minireview: Selective Degradation of Mitochondria by Mitophagy. Archives of biochemistry and biophysics. 2007;462:245–253. doi: 10.1016/j.abb.2007.03.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Krysko DV, Agostinis P, Krysko O, Garg AD, Bachert C, Lambrecht BN, Vandenabeele P. Emerging role of damage-associated molecular patterns derived from mitochondria in inflammation. Trends in immunology. 2011;32:157–164. doi: 10.1016/j.it.2011.01.005. [DOI] [PubMed] [Google Scholar]
- 13.Andres AM, Stotland A, Queliconi BB, Gottlieb RA. A time to reap, a time to sow: Mitophagy and biogenesis in cardiac pathophysiology. Journal of molecular and cellular cardiology. 2014 doi: 10.1016/j.yjmcc.2014.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kirkinezos IG, Moraes CT. Reactive oxygen species and mitochondrial diseases. Semin Cell Dev Biol. 2001;12:449–457. doi: 10.1006/scdb.2001.0282. [DOI] [PubMed] [Google Scholar]
- 15.Bender T, Lewrenz I, Franken S, Baitzel C, Voos W. Mitochondrial enzymes are protected from stress-induced aggregation by mitochondrial chaperones and the Pim1/LON protease. Mol Biol Cell. 22:541–554. doi: 10.1091/mbc.E10-08-0718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Leonhard K, Guiard B, Pellecchia G, Tzagoloff A, Neupert W, Langer T. Membrane protein degradation by AAA proteases in mitochondria: extraction of substrates from either membrane surface. Molecular cell. 2000;5:629–638. doi: 10.1016/s1097-2765(00)80242-7. [DOI] [PubMed] [Google Scholar]
- 17.Haynes CM, Yang Y, Blais SP, Neubert TA, Ron D. The matrix peptide exporter HAF-1 signals a mitochondrial UPR by activating the transcription factor ZC376.7 in C. elegans. Mol Cell. 2010;37:529–540. doi: 10.1016/j.molcel.2010.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Aldridge JE, Horibe T, Hoogenraad NJ. Discovery of genes activated by the mitochondrial unfolded protein response (mtUPR) and cognate promoter elements. PLoS One. 2007;2:e874. doi: 10.1371/journal.pone.0000874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Haynes CM, Ron D. The mitochondrial UPR - protecting organelle protein homeostasis. Journal of cell science. 2010;123:3849–3855. doi: 10.1242/jcs.075119. [DOI] [PubMed] [Google Scholar]
- 20.Menzies RA, Gold PH. The Turnover of Mitochondria in a Variety of Tissues of Young Adult and Aged Rats. J Biol Chem. 1971;246:2425–2429. [PubMed] [Google Scholar]
- 21.Carreira RS, Lee Y, Ghochani M, Gustafsson AB, Gottlieb RA. Cyclophilin D is required for mitochondrial removal by autophagy in cardiac cells. Autophagy. 2010;6 doi: 10.4161/auto.6.4.11553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mizushima N, Levine B, Cuervo AM, Klionsky DJ. Autophagy fights disease through cellular self-digestion. Nature. 2008;451:1069–1075. doi: 10.1038/nature06639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tanaka A, Cleland MM, Xu S, Narendra DP, Suen DF, Karbowski M, Youle RJ. Proteasome and p97 mediate mitophagy and degradation of mitofusins induced by Parkin. J CellBiol. 2010;191:1367–1380. doi: 10.1083/jcb.201007013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Twig G, Elorza A, Molina AJ, Mohamed H, Wikstrom JD, Walzer G, Stiles L, Haigh SE, Katz S, Las G, Alroy J, Wu M, Py BF, Yuan J, Deeney JT, Corkey BE, Shirihai OS. Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. EMBO J. 2008;27:433–446. doi: 10.1038/sj.emboj.7601963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yoon Y, Krueger EW, Oswald BJ, McNiven MA. The mitochondrial protein hFis1 regulates mitochondrial fission in mammalian cells through an interaction with the dynamin-like protein DLP1. Mol Cell Biol. 2003;23:5409–5420. doi: 10.1128/MCB.23.15.5409-5420.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Otera H, Wang C, Cleland MM, Setoguchi K, Yokota S, Youle RJ, Mihara K. Mff is an essential factor for mitochondrial recruitment of Drp1 during mitochondrial fission in mammalian cells. The Journal of cell biology. 2010;191:1141–1158. doi: 10.1083/jcb.201007152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Palmer CS, Osellame LD, Laine D, Koutsopoulos OS, Frazier AE, Ryan MT. MiD49 and MiD51, new components of the mitochondrial fission machinery. EMBO reports. 2011;12:565–573. doi: 10.1038/embor.2011.54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Smirnova E, Griparic L, Shurland DL, van der Bliek AM. Dynamin-related protein Drp1 is required for mitochondrial division in mammalian cells. Mol Biol Cell. 2001;12:2245–2256. doi: 10.1091/mbc.12.8.2245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Loson OC, Song Z, Chen H, Chan DC. Fis1, Mff, MiD49, and MiD51 mediate Drp1 recruitment in mitochondrial fission. Molecular biology of the cell. 2013;24:659–667. doi: 10.1091/mbc.E12-10-0721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.MacVicar TD, Lane JD. Impaired OMA1-dependent cleavage of OPA1 and reduced DRP1 fission activity combine to prevent mitophagy in cells that are dependent on oxidative phosphorylation. Journal of cell science. 2014;127:2313–2325. doi: 10.1242/jcs.144337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kaser M, Kambacheld M, Kisters-Woike B, Langer T. Oma1, a novel membrane-bound metallopeptidase in mitochondria with activities overlapping with the m-AAA protease. J Biol Chem. 2003;278:46414–46423. doi: 10.1074/jbc.M305584200. [DOI] [PubMed] [Google Scholar]
- 32.McBride H, Soubannier V. Mitochondrial function: OMA1 and OPA1, the grandmasters of mitochondrial health. Current biology : CB. 2010;20:R274–276. doi: 10.1016/j.cub.2010.02.011. [DOI] [PubMed] [Google Scholar]
- 33.Huang C, Andres AM, Ratliff EP, Hernandez G, Lee P, Gottlieb RA. Preconditioning involves selective mitophagy mediated by Parkin and p62/SQSTM1. PLoS One. 2011;6:e20975. doi: 10.1371/journal.pone.0020975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Geisler S, Holmstrom KM, Skujat D, Fiesel FC, Rothfuss OC, Kahle PJ, Springer W. PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nature cellbiology. 2010;12:119–131. doi: 10.1038/ncb2012. [DOI] [PubMed] [Google Scholar]
- 35.Matsuda N, Sato S, Shiba K, Okatsu K, Saisho K, Gautier CA, Sou YS, Saiki S, Kawajiri S, Sato F, Kimura M, Komatsu M, Hattori N, Tanaka K. PINK1 stabilized by mitochondrial depolarization recruits Parkin to damaged mitochondria and activates latent Parkin for mitophagy. J Cell Biol. 2010;189:211–221. doi: 10.1083/jcb.200910140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Narendra D, Tanaka A, Suen DF, Youle RJ. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J Cell Biol. 2008;183:795–803. doi: 10.1083/jcb.200809125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Narendra DP, Jin SM, Tanaka A, Suen DF, Gautier CA, Shen J, Cookson MR, Youle RJ. PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. PLoS Biol. 2010;8:e1000298. doi: 10.1371/journal.pbio.1000298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Clark IE, Dodson MW, Jiang C, Cao JH, Huh JR, Seol JH, Yoo SJ, Hay BA, Guo M. Drosophila pink1 is required for mitochondrial function and interacts genetically with parkin. Nature. 2006;441:1162–1166. doi: 10.1038/nature04779. [DOI] [PubMed] [Google Scholar]
- 39.Park J, Lee SB, Lee S, Kim Y, Song S, Kim S, Bae E, Kim J, Shong M, Kim JM, Chung J. Mitochondrial dysfunction in Drosophila PINK1 mutants is complemented by parkin. Nature. 2006;441:1157–1161. doi: 10.1038/nature04788. [DOI] [PubMed] [Google Scholar]
- 40.Greene AW, Grenier K, Aguileta MA, Muise S, Farazifard R, Haque ME, McBride HM, Park DS, Fon EA. Mitochondrial processing peptidase regulates PINK1 processing, import and Parkin recruitment. EMBO Rep. 2012;13:378–385. doi: 10.1038/embor.2012.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jin SM, Lazarou M, Wang C, Kane LA, Narendra DP, Youle RJ. Mitochondrial membrane potential regulates PINK1 import and proteolytic destabilization by PARL. J Cell Biol. 2010;191:933–942. doi: 10.1083/jcb.201008084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhou C, Huang Y, Shao Y, May J, Prou D, Perier C, Dauer W, Schon EA, Przedborski S. The kinase domain of mitochondrial PINK1 faces the cytoplasm. Proc Natl Acad Sci U S A. 2008;105:12022–12027. doi: 10.1073/pnas.0802814105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Iguchi M, Kujuro Y, Okatsu K, Koyano F, Kosako H, Kimura M, Suzuki N, Uchiyama S, Tanaka K, Matsuda N. Parkin-catalyzed ubiquitin-ester transfer is triggered by PINK1-dependent phosphorylation. J Biol Chem. 2013;288:22019–22032. doi: 10.1074/jbc.M113.467530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lazarou M, Jin SM, Kane LA, Youle RJ. Role of PINK1 binding to the TOM complex and alternate intracellular membranes in recruitment and activation of the E3 ligase Parkin. Developmental cell. 2012;22:320–333. doi: 10.1016/j.devcel.2011.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chen Y, Dorn GW., 2nd PINK1-phosphorylated mitofusin 2 is a Parkin receptor for culling damaged mitochondria. Science. 2013;340:471–475. doi: 10.1126/science.1231031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wang X, Winter D, Ashrafi G, Schlehe J, Wong YL, Selkoe D, Rice S, Steen J, LaVoie MJ, Schwarz TL. PINK1 and Parkin target Miro for phosphorylation and degradation to arrest mitochondrial motility. Cell. 2011;147:893–906. doi: 10.1016/j.cell.2011.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Narendra D, Kane LA, Hauser DN, Fearnley IM, Youle RJ. p62/SQSTM1 is required for Parkin-induced mitochondrial clustering but not mitophagy; VDAC1 is dispensable for both. Autophagy. 2010;6:1090–1106. doi: 10.4161/auto.6.8.13426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gegg ME, Cooper JM, Chau KY, Rojo M, Schapira AH, Taanman JW. Mitofusin 1 and mitofusin 2 are ubiquitinated in a PINK1/parkin-dependent manner upon induction of mitophagy. Hum Mol Genet. 2010;19:4861–4870. doi: 10.1093/hmg/ddq419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Glauser L, Sonnay S, Stafa K, Moore DJ. Parkin promotes the ubiquitination and degradation of the mitochondrial fusion factor mitofusin 1. J Neurochem. 2011;118:636–645. doi: 10.1111/j.1471-4159.2011.07318.x. [DOI] [PubMed] [Google Scholar]
- 50.Poole AC, Thomas RE, Yu S, Vincow ES, Pallanck L. The mitochondrial fusion-promoting factor mitofusin is a substrate of the PINK1/parkin pathway. PLoS One. 2010;5:e10054. doi: 10.1371/journal.pone.0010054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kirkin V, Lamark T, Sou YS, Bjorkoy G, Nunn JL, Bruun JA, Shvets E, McEwan DG, Clausen TH, Wild P, Bilusic I, Theurillat JP, Overvatn A, Ishii T, Elazar Z, Komatsu M, Dikic I, Johansen T. A role for NBR1 in autophagosomal degradation of ubiquitinated substrates. Mol Cell. 2009;33:505–516. doi: 10.1016/j.molcel.2009.01.020. [DOI] [PubMed] [Google Scholar]
- 52.Pankiv S, Clausen TH, Lamark T, Brech A, Bruun JA, Outzen H, Overvatn A, Bjorkoy G, Johansen T. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J Biol Chem. 2007;282:24131–24145. doi: 10.1074/jbc.M702824200. [DOI] [PubMed] [Google Scholar]
- 53.Orvedahl A, Sumpter R, Jr, Xiao G, Ng A, Zou Z, Tang Y, Narimatsu M, Gilpin C, Sun Q, Roth M, Forst CV, Wrana JL, Zhang YE, Luby-Phelps K, Xavier RJ, Xie Y, Levine B. Image-based genome-wide siRNA screen identifies selective autophagy factors. Nature. 2011;480:113–117. doi: 10.1038/nature10546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Fimia GM, Stoykova A, Romagnoli A, Giunta L, Di Bartolomeo S, Nardacci R, Corazzari M, Fuoco C, Ucar A, Schwartz P, Gruss P, Piacentini M, Chowdhury K, Cecconi F. Ambra1 regulates autophagy and development of the nervous system. Nature. 2007;447:1121–1125. doi: 10.1038/nature05925. [DOI] [PubMed] [Google Scholar]
- 55.Strappazzon F, Vietri-Rudan M, Campello S, Nazio F, Florenzano F, Fimia GM, Piacentini M, Levine B, Cecconi F. Mitochondrial BCL-2 inhibits AMBRA1-induced autophagy. EMBO J. 2011;30:1195–1208. doi: 10.1038/emboj.2011.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Van Humbeeck C, Cornelissen T, Hofkens H, Mandemakers W, Gevaert K, De Strooper B, Vandenberghe W. Parkin interacts with Ambra1 to induce mitophagy. J Neurosci. 2011;31:10249–10261. doi: 10.1523/JNEUROSCI.1917-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kazlauskaite A, Kelly V, Johnson C, Baillie C, Hastie CJ, Peggie M, Macartney T, Woodroof HI, Alessi DR, Pedrioli PG, Muqit MM. Phosphorylation of Parkin at Serine65 is essential for activation: elaboration of a Miro1 substrate-based assay of Parkin E3 ligase activity. Open biology. 2014;4:130213. doi: 10.1098/rsob.130213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Koyano F, Okatsu K, Kosako H, Tamura Y, Go E, Kimura M, Kimura Y, Tsuchiya H, Yoshihara H, Hirokawa T, Endo T, Fon EA, Trempe JF, Saeki Y, Tanaka K, Matsuda N. Ubiquitin is phosphorylated by PINK1 to activate parkin. Nature. 2014;510:162–166. doi: 10.1038/nature13392. [DOI] [PubMed] [Google Scholar]
- 59.Kane LA, Lazarou M, Fogel AI, Li Y, Yamano K, Sarraf SA, Banerjee S, Youle RJ. PINK1 phosphorylates ubiquitin to activate Parkin E3 ubiquitin ligase activity. The Journal of cell biology. 2014;205:143–153. doi: 10.1083/jcb.201402104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Chakrama FZ, Seguin-Py S, Le Grand JN, Fraichard A, Delage-Mourroux R, Despouy G, Perez V, Jouvenot M, Boyer-Guittaut M. GABARAPL1 (GEC1) associates with autophagic vesicles. Autophagy. 2010;6:495–505. doi: 10.4161/auto.6.4.11819. [DOI] [PubMed] [Google Scholar]
- 61.Novak I, Kirkin V, McEwan DG, Zhang J, Wild P, Rozenknop A, Rogov V, Lohr F, Popovic D, Occhipinti A, Reichert AS, Terzic J, Dotsch V, Ney PA, Dikic I. Nix is a selective autophagy receptor for mitochondrial clearance. EMBO Rep. 2010;11:45–51. doi: 10.1038/embor.2009.256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lee Y, Lee HY, Hanna RA, Gustafsson AB. Mitochondrial autophagy by Bnip3 involves Drp1-mediated mitochondrial fission and recruitment of Parkin in cardiac myocytes. Am J Physiol Heart Circ Physiol. 2011;301:H1924–1931. doi: 10.1152/ajpheart.00368.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Liu L, Feng D, Chen G, Chen M, Zheng Q, Song P, Ma Q, Zhu C, Wang R, Qi W, Huang L, Xue P, Li B, Wang X, Jin H, Wang J, Yang F, Liu P, Zhu Y, Sui S, Chen Q. Mitochondrial outer-membrane protein FUNDC1 mediates hypoxia-induced mitophagy in mammalian cells. Nat Cell Biol. 2012;14:177–185. doi: 10.1038/ncb2422. [DOI] [PubMed] [Google Scholar]
- 64.Saita S, Shirane M, Nakayama KI. Selective escape of proteins from the mitochondria during mitophagy. Nature communications. 2013;4:1410. doi: 10.1038/ncomms2400. [DOI] [PubMed] [Google Scholar]
- 65.Kim TY, Wang D, Kim AK, Lau E, Lin AJ, Liem DA, Zhang J, Zong NC, Lam MP, Ping P. Metabolic Labeling Reveals Proteome Dynamics of Mouse Mitochondria. Mol Cell Proteomics. 2012 doi: 10.1074/mcp.M112.021162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Aschenbrenner B, Druyan R, Albin R, Rabinowitz M. Haem a cytochrome c and total protein turnover in mitochondria from rat heart and liver. Biochem J. 1970;119:157–160. doi: 10.1042/bj1190157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lipsky NG, Pedersen PL. Mitochondrial turnover in animal cells. Half-lives of mitochondria and mitochondrial subfractions of rat liver based on [14C]bicarbonate incorporation. J Biol Chem. 1981;256:8652–8657. [PubMed] [Google Scholar]
- 68.Holt IJ, Jacobs HT. Unique features of DNA replication in mitochondria: A functional and evolutionary perspective. BioEssays : news and reviews in molecular, cellular and developmental biology. 2014 doi: 10.1002/bies.201400052. [DOI] [PubMed] [Google Scholar]
- 69.Wu Z, Puigserver P, Spiegelman BM. Transcriptional activation of adipogenesis. Curr Opin Cell Biol. 1999;11:689–694. doi: 10.1016/s0955-0674(99)00037-x. [DOI] [PubMed] [Google Scholar]
- 70.Puigserver P, Wu Z, Park CW, Graves R, Wright M, Spiegelman BM. A cold-inducible coactivator of nuclear receptors linked to adaptive thermogenesis. Cell. 1998;92:829–839. doi: 10.1016/s0092-8674(00)81410-5. [DOI] [PubMed] [Google Scholar]
- 71.Xiao X, Wang P, Chou KC. Recent progresses in identifying nuclear receptors and their families. Current topics in medicinal chemistry. 2013;13:1192–1200. doi: 10.2174/15680266113139990006. [DOI] [PubMed] [Google Scholar]
- 72.Virbasius JV, Scarpulla RC. Activation of the human mitochondrial transcription factor A gene by nuclear respiratory factors: a potential regulatory link between nuclear and mitochondrial gene expression in organelle biogenesis. Proc Natl Acad Sci U S A. 1994;91:1309–1313. doi: 10.1073/pnas.91.4.1309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Fisher RP, Clayton DA. A transcription factor required for promoter recognition by human mitochondrial RNA polymerase. Accurate initiation at the heavy- and light-strand promoters dissected and reconstituted in vitro. J Biol Chem. 1985;260:11330–11338. [PubMed] [Google Scholar]
- 74.Wu Z, Puigserver P, Andersson U, Zhang C, Adelmant G, Mootha V, Troy A, Cinti S, Lowell B, Scarpulla RC, Spiegelman BM. Mechanisms controlling mitochondrial biogenesis and respiration through the thermogenic coactivator PGC-1. Cell. 1999;98:115–124. doi: 10.1016/S0092-8674(00)80611-X. [DOI] [PubMed] [Google Scholar]
- 75.Baar K, Wende AR, Jones TE, Marison M, Nolte LA, Chen M, Kelly DP, Holloszy JO. Adaptations of skeletal muscle to exercise: rapid increase in the transcriptional coactivator PGC-1. FASEB J. 2002;16:1879–1886. doi: 10.1096/fj.02-0367com. [DOI] [PubMed] [Google Scholar]
- 76.Schreiber SN, Emter R, Hock MB, Knutti D, Cardenas J, Podvinec M, Oakeley EJ, Kralli A. The estrogen-related receptor alpha (ERRalpha) functions in PPARgamma coactivator 1alpha (PGC-1alpha)-induced mitochondrial biogenesis. Proc Natl Acad Sci U S A. 2004;101:6472–6477. doi: 10.1073/pnas.0308686101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Meirhaeghe A, Crowley V, Lenaghan C, Lelliott C, Green K, Stewart A, Hart K, Schinner S, Sethi JK, Yeo G, Brand MD, Cortright RN, O'Rahilly S, Montague C, Vidal-Puig AJ. Characterization of the human, mouse and rat PGC1 beta (peroxisome-proliferator-activated receptor-gamma co-activator 1 beta) gene in vitro and in vivo. Biochem J. 2003;373:155–165. doi: 10.1042/BJ20030200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Ventura-Clapier R, Garnier A, Veksler V. Transcriptional control of mitochondrial biogenesis: the central role of PGC-1alpha. Cardiovasc Res. 2008;79:208–217. doi: 10.1093/cvr/cvn098. [DOI] [PubMed] [Google Scholar]
- 79.Giguere V. Transcriptional control of energy homeostasis by the estrogen-related receptors. Endocrine reviews. 2008;29:677–696. doi: 10.1210/er.2008-0017. [DOI] [PubMed] [Google Scholar]
- 80.Madrazo JA, Kelly DP. The PPAR trio: regulators of myocardial energy metabolism in health and disease. J Mol Cell Cardiol. 2008;44:968–975. doi: 10.1016/j.yjmcc.2008.03.021. [DOI] [PubMed] [Google Scholar]
- 81.Herzig S, Long F, Jhala US, Hedrick S, Quinn R, Bauer A, Rudolph D, Schutz G, Yoon C, Puigserver P, Spiegelman B, Montminy M. CREB regulates hepatic gluconeogenesis through the coactivator PGC-1. Nature. 2001;413:179–183. doi: 10.1038/35093131. [DOI] [PubMed] [Google Scholar]
- 82.Handschin C, Rhee J, Lin J, Tarr PT, Spiegelman BM. An autoregulatory loop controls peroxisome proliferator-activated receptor gamma coactivator 1alpha expression in muscle. Proc Natl Acad Sci U S A. 2003;100:7111–7116. doi: 10.1073/pnas.1232352100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Lin J, Wu H, Tarr PT, Zhang CY, Wu Z, Boss O, Michael LF, Puigserver P, Isotani E, Olson EN, Lowell BB, Bassel-Duby R, Spiegelman BM. Transcriptional co-activator PGC-1 alpha drives the formation of slow-twitch muscle fibres. Nature. 2002;418:797–801. doi: 10.1038/nature00904. [DOI] [PubMed] [Google Scholar]
- 84.Akimoto T, Pohnert SC, Li P, Zhang M, Gumbs C, Rosenberg PB, Williams RS, Yan Z. Exercise stimulates Pgc-1alpha transcription in skeletal muscle through activation of the p38 MAPK pathway. J Biol Chem. 2005;280:19587–19593. doi: 10.1074/jbc.M408862200. [DOI] [PubMed] [Google Scholar]
- 85.Zhao M, New L, Kravchenko VV, Kato Y, Gram H, di Padova F, Olson EN, Ulevitch RJ, Han J. Regulation of the MEF2 family of transcription factors by p38. Mol Cell Biol. 1999;19:21–30. doi: 10.1128/mcb.19.1.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Jager S, Handschin C, St-Pierre J, Spiegelman BM. AMP-activated protein kinase (AMPK) action in skeletal muscle via direct phosphorylation of PGC-1alpha. Proc Natl Acad Sci U S A. 2007;104:12017–12022. doi: 10.1073/pnas.0705070104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Puigserver P, Rhee J, Lin J, Wu Z, Yoon JC, Zhang CY, Krauss S, Mootha VK, Lowell BB, Spiegelman BM. Cytokine stimulation of energy expenditure through p38 MAP kinase activation of PPARgamma coactivator-1. Mol Cell. 2001;8:971–982. doi: 10.1016/s1097-2765(01)00390-2. [DOI] [PubMed] [Google Scholar]
- 88.Zong H, Ren JM, Young LH, Pypaert M, Mu J, Birnbaum MJ, Shulman GI. AMP kinase is required for mitochondrial biogenesis in skeletal muscle in response to chronic energy deprivation. Proc Natl Acad Sci U S A. 2002;99:15983–15987. doi: 10.1073/pnas.252625599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Sano M, Tokudome S, Shimizu N, Yoshikawa N, Ogawa C, Shirakawa K, Endo J, Katayama T, Yuasa S, Ieda M, Makino S, Hattori F, Tanaka H, Fukuda K. Intramolecular control of protein stability, subnuclear compartmentalization, and coactivator function of peroxisome proliferator-activated receptor gamma coactivator 1alpha. J Biol Chem. 2007;282:25970–25980. doi: 10.1074/jbc.M703634200. [DOI] [PubMed] [Google Scholar]
- 90.Li X, Monks B, Ge Q, Birnbaum MJ. Akt/PKB regulates hepatic metabolism by directly inhibiting PGC-1alpha transcription coactivator. Nature. 2007;447:1012–1016. doi: 10.1038/nature05861. [DOI] [PubMed] [Google Scholar]
- 91.Rodgers JT, Haas W, Gygi SP, Puigserver P. Cdc2-like kinase 2 is an insulin-regulated suppressor of hepatic gluconeogenesis. Cell Metab. 2010;11:23–34. doi: 10.1016/j.cmet.2009.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Anderson RM, Barger JL, Edwards MG, Braun KH, O'Connor CE, Prolla TA, Weindruch R. Dynamic regulation of PGC-1alpha localization and turnover implicates mitochondrial adaptation in calorie restriction and the stress response. Aging Cell. 2008;7:101–111. doi: 10.1111/j.1474-9726.2007.00357.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Lerin C, Rodgers JT, Kalume DE, Kim SH, Pandey A, Puigserver P. GCN5 acetyltransferase complex controls glucose metabolism through transcriptional repression of PGC-1alpha. Cell Metab. 2006;3:429–438. doi: 10.1016/j.cmet.2006.04.013. [DOI] [PubMed] [Google Scholar]
- 94.Houtkooper RH, Canto C, Wanders RJ, Auwerx J. The secret life of NAD+: an old metabolite controlling new metabolic signaling pathways. Endocrine reviews. 2010;31:194–223. doi: 10.1210/er.2009-0026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Gerhart-Hines Z, Rodgers JT, Bare O, Lerin C, Kim SH, Mostoslavsky R, Alt FW, Wu Z, Puigserver P. Metabolic control of muscle mitochondrial function and fatty acid oxidation through SIRT1/PGC-1alpha. EMBO J. 2007;26:1913–1923. doi: 10.1038/sj.emboj.7601633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Canto C, Gerhart-Hines Z, Feige JN, Lagouge M, Noriega L, Milne JC, Elliott PJ, Puigserver P, Auwerx J. AMPK regulates energy expenditure by modulating NAD+ metabolism and SIRT1 activity. Nature. 2009;458:1056–1060. doi: 10.1038/nature07813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Fernandez-Marcos PJ, Auwerx J. Regulation of PGC-1alpha, a nodal regulator of mitochondrial biogenesis. The American journal of clinical nutrition. 2011;93:884S–890. doi: 10.3945/ajcn.110.001917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Ahmed AU, Fisher PR. Import of nuclear-encoded mitochondrial proteins: a cotranslational perspective. International review of cell and molecular biology. 2009;273:49–68. doi: 10.1016/S1937-6448(08)01802-9. [DOI] [PubMed] [Google Scholar]
- 99.MacKenzie JA, Payne RM. Mitochondrial protein import and human health and disease. Biochim Biophys Acta. 2007;1772:509–523. doi: 10.1016/j.bbadis.2006.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Mottis A, Jovaisaite V, Auwerx J. The mitochondrial unfolded protein response in mammalian physiology. Mamm Genome. 2014 doi: 10.1007/s00335-014-9525-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Morita M, Gravel SP, Chenard V, Sikstrom K, Zheng L, Alain T, Gandin V, Avizonis D, Arguello M, Zakaria C, McLaughlan S, Nouet Y, Pause A, Pollak M, Gottlieb E, Larsson O, St-Pierre J, Topisirovic I, Sonenberg N. mTORC1 controls mitochondrial activity and biogenesis through 4E-BP-dependent translational regulation. Cell Metab. 2013;18:698–711. doi: 10.1016/j.cmet.2013.10.001. [DOI] [PubMed] [Google Scholar]
- 102.Cunningham JT, Rodgers JT, Arlow DH, Vazquez F, Mootha VK, Puigserver P. mTOR controls mitochondrial oxidative function through a YY1-PGC-1alpha transcriptional complex. Nature. 2007;450:736–740. doi: 10.1038/nature06322. [DOI] [PubMed] [Google Scholar]
- 103.Shin JH, Ko HS, Kang H, Lee Y, Lee YI, Pletinkova O, Troconso JC, Dawson VL, Dawson TM. PARIS (ZNF746) repression of PGC-1alpha contributes to neurodegeneration in Parkinson's disease. Cell. 2011;144:689–702. doi: 10.1016/j.cell.2011.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Yamamoto H, Williams EG, Mouchiroud L, Canto C, Fan W, Downes M, Heligon C, Barish GD, Desvergne B, Evans RM, Schoonjans K, Auwerx J. NCoR1 is a conserved physiological modulator of muscle mass and oxidative function. Cell. 2011;147:827–839. doi: 10.1016/j.cell.2011.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Claypool SM, Koehler CM. The complexity of cardiolipin in health and disease. Trends in Biochemical Sciences. 2012;37:32–41. doi: 10.1016/j.tibs.2011.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Schlame M, Kelley RI, Feigenbaum A, Towbin JA, Heerdt PM, Schieble T, Wanders RJ, DiMauro S, Blanck TJ. Phospholipid abnormalities in children with Barth syndrome. J Am Coll Cardiol. 2003;42:1994–1999. doi: 10.1016/j.jacc.2003.06.015. [DOI] [PubMed] [Google Scholar]
- 107.Acehan D, Vaz F, Houtkooper RH, James J, Moore V, Tokunaga C, Kulik W, Wansapura J, Toth MJ, Strauss A, Khuchua Z. Cardiac and skeletal muscle defects in a mouse model of human Barth syndrome. J Biol Chem. 2011;286:899–908. doi: 10.1074/jbc.M110.171439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Vance JE, Tasseva G. Formation and function of phosphatidylserine and phosphatidylethanolamine in mammalian cells. Biochim Biophys Acta. 2013;1831:543–554. doi: 10.1016/j.bbalip.2012.08.016. [DOI] [PubMed] [Google Scholar]
- 109.Sentelle RD, Senkal CE, Jiang W, Ponnusamy S, Gencer S, Selvam SP, Ramshesh VK, Peterson YK, Lemasters JJ, Szulc ZM, Bielawski J, Ogretmen B. Ceramide targets autophagosomes to mitochondria and induces lethal mitophagy. Nature chemical biology. 2012;8:831–838. doi: 10.1038/nchembio.1059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Rambold AS, Lippincott-Schwartz J. Starved cells use mitochondria for autophagosome biogenesis. Cell Cycle. 2010;9:3633–3634. doi: 10.4161/cc.9.18.13170. [DOI] [PubMed] [Google Scholar]
- 111.Luo S, Chen Q, Cebollero E, Xing D. Mitochondria: one of the origins for autophagosomal membranes? Mitochondrion. 2009;9:227–231. doi: 10.1016/j.mito.2009.04.004. [DOI] [PubMed] [Google Scholar]
- 112.Lettieri Barbato D, Tatulli G, Aquilano K, Ciriolo MR. FoxO1 controls lysosomal acid lipase in adipocytes: implication of lipophagy during nutrient restriction and metformin treatment. Cell death & disease. 2013;4:e861. doi: 10.1038/cddis.2013.404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Yin D, Yuan X, Brunk UT. Test-tube simulated lipofuscinogenesis. Effect of oxidative stress on autophagocytotic degradation. Mechanisms of ageing and development. 1995;81:37–50. doi: 10.1016/0047-6374(94)01580-f. [DOI] [PubMed] [Google Scholar]
- 114.Bailey E, Taylor CB, Bartley W. Turnover of mitochondrial components of normal and essential fatty acid-deficient rats. The Biochemical journal. 1967;104:1026–1032. doi: 10.1042/bj1041026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Menzies RA, Gold PH. The Turnover of Mitochondria in a Variety of Tissues of Young Adult and Aged Rats. Journal of Biological Chemistry. 1971;246:2425–2429. [PubMed] [Google Scholar]
- 116.Kasumov T, Dabkowski ER, Shekar KC, Li L, Ribeiro RF, Jr, Walsh K, Previs SF, Sadygov RG, Willard B, Stanley WC. Assessment of cardiac proteome dynamics with heavy water: slower protein synthesis rates in interfibrillar than subsarcolemmal mitochondria. Am J Physiol Heart Circ Physiol. 2013;304:H1201–1214. doi: 10.1152/ajpheart.00933.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Major T, von Janowsky B, Ruppert T, Mogk A, Voos W. Proteomic analysis of mitochondrial protein turnover: identification of novel substrate proteins of the matrix protease pim1. Molecular and cellular biology. 2006;26:762–776. doi: 10.1128/MCB.26.3.762-776.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Lopez MF, Melov S. Applied proteomics: mitochondrial proteins and effect on function. Circulation research. 2002;90:380–389. doi: 10.1161/hh0402.105757. [DOI] [PubMed] [Google Scholar]
- 119.Kim I, Lemasters JJ. Mitochondrial Degradation by Autophagy (Mitophagy) in GFP-LC3 Transgenic Hepatocytes during Nutrient Deprivation. Am J Physiol Cell Physiol. 2010 doi: 10.1152/ajpcell.00056.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Scherz-Shouval R, Shvets E, Fass E, Shorer H, Gil L, Elazar Z. Reactive oxygen species are essential for autophagy and specifically regulate the activity of Atg4. Embo J. 2007;26:1749–1760. doi: 10.1038/sj.emboj.7601623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Hanna RA, Quinsay MN, Orogo AM, Giang K, Rikka S, Gustafsson AB. Microtubule-associated protein 1 light chain 3 (LC3) interacts with Bnip3 protein to selectively remove endoplasmic reticulum and mitochondria via autophagy. J Biol Chem. 2012;287:19094–19104. doi: 10.1074/jbc.M111.322933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Kubli DA, Quinsay MN, Huang C, Lee Y, Gustafsson AB. Bnip3 functions as a mitochondrial sensor of oxidative stress during myocardial ischemia and reperfusion. Am J Physiol Heart Circ Physiol. 2008;295:H2025–2031. doi: 10.1152/ajpheart.00552.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Katayama H, Kogure T, Mizushima N, Yoshimori T, Miyawaki A. A sensitive and quantitative technique for detecting autophagic events based on lysosomal delivery. Chemistry & biology. 2011;18:1042–1052. doi: 10.1016/j.chembiol.2011.05.013. [DOI] [PubMed] [Google Scholar]
- 124.Terskikh A, Fradkov A, Ermakova G, Zaraisky A, Tan P, Kajava AV, Zhao X, Lukyanov S, Matz M, Kim S, Weissman I, Siebert P. “Fluorescent timer”: protein that changes color with time. Science. 2000;290:1585–1588. doi: 10.1126/science.290.5496.1585. [DOI] [PubMed] [Google Scholar]
- 125.Loson OC, Ha CM, Parpura V. Age-dependent spatial segregation of synaptobrevin 2-containing vesicles in astrocytes. J Neurochem. 2011 doi: 10.1111/j.1471-4159.2010.07018.x. [DOI] [PubMed] [Google Scholar]
- 126.Kozel BA, Rongish BJ, Czirok A, Zach J, Little CD, Davis EC, Knutsen RH, Wagenseil JE, Levy MA, Mecham RP. Elastic fiber formation: a dynamic view of extracellular matrix assembly using timer reporters. J Cell Physiol. 2006;207:87–96. doi: 10.1002/jcp.20546. [DOI] [PubMed] [Google Scholar]
- 127.Verkhusha VV, Otsuna H, Awasaki T, Oda H, Tsukita S, Ito K. An enhanced mutant of red fluorescent protein DsRed for double labeling and developmental timer of neural fiber bundle formation. J Biol Chem. 2001;276:29621–29624. doi: 10.1074/jbc.C100200200. [DOI] [PubMed] [Google Scholar]
- 128.Robinson SM, Tsueng G, Sin J, Mangale V, Rahawi S, McIntyre LL, Williams W, Kha N, Cruz C, Hancock BM, Nguyen DP, Sayen MR, Hilton BJ, Doran KS, Segall AM, Wolkowicz R, Cornell CT, Whitton JL, Gottlieb RA, Feuer R. Coxsackievirus B exits the host cell in shed microvesicles displaying autophagosomal markers. PLoS pathogens. 2014;10:e1004045. doi: 10.1371/journal.ppat.1004045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Ferree AW, Trudeau K, Zik E, Benador IY, Twig G, Gottlieb RA, Shirihai OS. MitoTimer probe reveals the impact of autophagy, fusion and motility on subcellular distribution of young and old mitochondrial protein and on relative mitochondrial protein age. Autophagy. 2013;9:1887–1896. doi: 10.4161/auto.26503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Neupert W, Herrmann JM. Translocation of Proteins into Mitochondria. Annual Review of Biochemistry. 2007;76:723–749. doi: 10.1146/annurev.biochem.76.052705.163409. [DOI] [PubMed] [Google Scholar]
- 131.Hernandez G, Thornton C, Stotland A, Lui D, Sin J, Ramil J, Magee N, Andres A, Quarato G, Carreira RS, Sayen MR, Wolkowicz R, Gottlieb RA. MitoTimer: A novel tool for monitoring mitochondrial turnover. Autophagy. 2013;9:1852–1861. doi: 10.4161/auto.26501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Wikstrom JD, Twig G, Shirihai OS. What can mitochondrial heterogeneity tell us about mitochondrial dynamics and autophagy? Int J Biochem Cell Biol. 2009;41:1914–1927. doi: 10.1016/j.biocel.2009.06.006. [DOI] [PubMed] [Google Scholar]
- 133.Lipsky NG, Pedersen PL. Perturbation by clofibrate of mitochondrial levels in animal cells. Implications for a model of mitochondrial genesis. J Biol Chem. 1982;257:1473–1481. [PubMed] [Google Scholar]
- 134.Grisolia S, Knecht E, Hernandez-Yago J, Wallace R. Turnover and degradation of mitochondria and their proteins. Ciba Foundation symposium. 1979:167–188. doi: 10.1002/9780470720585.ch11. [DOI] [PubMed] [Google Scholar]
- 135.Maday S, Wallace KE, Holzbaur EL. Autophagosomes initiate distally and mature during transport toward the cell soma in primary neurons. J Cell Biol. 2012;196:407–417. doi: 10.1083/jcb.201106120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Laker RC, Xu P, Ryall KA, Sujkowski A, Kenwood BM, Chain KH, Zhang M, Royal MA, Hoehn KL, Driscoll M, Adler PN, Wessells RJ, Saucerman JJ, Yan Z. A novel MitoTimer reporter gene for mitochondrial content, structure, stress and damage in vivo. J Biol Chem. 2014;289:12005–12015. doi: 10.1074/jbc.M113.530527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Rodriguez-Navarro JA, Kaushik S, Koga H, Dall'Armi C, Shui G, Wenk MR, Di Paolo G, Cuervo AM. Inhibitory effect of dietary lipids on chaperone-mediated autophagy. Proc Natl Acad Sci U S A. 2012;109:E705–714. doi: 10.1073/pnas.1113036109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Wikstrom JD, Katzman SM, Mohamed H, Twig G, Graf SA, Heart E, Molina AJA, Corkey BE, de Vargas LM, Danial NN, Collins S, Shirihai OS. {beta}-Cell Mitochondria Exhibit Membrane Potential Heterogeneity That Can Be Altered by Stimulatory or Toxic Fuel Levels. Diabetes. 2007;56:2569–2578. doi: 10.2337/db06-0757. [DOI] [PubMed] [Google Scholar]
- 139.Grumati P, Coletto L, Schiavinato A, Castagnaro S, Bertaggia E, Sandri M, Bonaldo P. Physical exercise stimulates autophagy in normal skeletal muscles but is detrimental for collagen VI-deficient muscles. Autophagy. 2011;7:1415–1423. doi: 10.4161/auto.7.12.17877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Wang L, Mascher H, Psilander N, Blomstrand E, Sahlin K. Resistance exercise enhances the molecular signaling of mitochondrial biogenesis induced by endurance exercise in human skeletal muscle. Journal of applied physiology (Bethesda, Md : 1985) 2011;111:1335–1344. doi: 10.1152/japplphysiol.00086.2011. [DOI] [PubMed] [Google Scholar]
- 141.Lam MP, Wang D, Lau E, Liem DA, Kim AK, Ng DC, Liang X, Bleakley BJ, Liu C, Tabaraki JD, Cadeiras M, Wang Y, Deng MC, Ping P. Protein kinetic signatures of the remodeling heart following isoproterenol stimulation. J Clin Invest. 2014;124:1734–1744. doi: 10.1172/JCI73787. [DOI] [PMC free article] [PubMed] [Google Scholar]