

SUMMARY
Circulating levels of the gut microbe-derived metabolite trimethylamine-N-oxide (TMAO) have recently been linked to cardiovascular disease (CVD) risk. Here we performed transcriptional profiling in mouse models of altered reverse cholesterol transport (RCT), and serendipitously identified the TMAO-generating enzyme flavin monooxygenase 3 (FMO3) as a powerful modifier of cholesterol metabolism and RCT. Knockdown of FMO3 in cholesterol-fed mice alters biliary lipid secretion, blunts intestinal cholesterol absorption, and limits the production of hepatic oxysterols and cholesteryl esters. Furthermore, FMO3 knockdown stimulates basal and liver X receptor (LXR)-stimulated macrophage RCT, thereby improving cholesterol balance. Conversely, FMO3 knockdown exacerbates hepatic ER stress and inflammation in part by decreasing hepatic oxysterol levels and subsequent LXR activation. FMO3 is thus identified as a central integrator of hepatic cholesterol and triacylglycerol metabolism, inflammation, and ER stress. These studies suggest that the gut microbiota-driven TMA/FMO3/TMAO pathway is a key regulator of lipid metabolism and inflammation.
INTRODUCTION
Atherosclerosis and associated cardiovascular disease (CVD) remains the largest cause of mortality in developed countries (Go et al., 2013). Despite widespread use of statins, CVD-associated mortality and morbidity has been reduced by only ~30% (Go et al., 2013), demonstrating a clear need for better therapeutic strategies. Elevation of high density lipoprotein (HDL) function is thought to be an attractive therapeutic strategy (Rader and Tall, 2012). However, recent clinical trials (Boden et al., 2011; Schwartz et al. 2012) and Mendelian randomization studies (Voight et al., 2012) have failed to show clinical benefits of HDL cholesterol elevation, calling into question the role of HDL cholesterol as a surrogate marker of protection from atherosclerosis (Rader and Tall, 2012). Both proponents and critics alike of the “HDL hypothesis” agree on one thing – further studies are needed to understand the mechanism regulating the fundamental process of HDL-driven reverse cholesterol transport (RCT). The prevailing model for RCT is that cholesterol from the artery wall is delivered to the liver via HDL, from where it is then secreted into bile before leaving the body through the feces (Dietschy and Turley, 2002; Rader et al., 2009; Rosenson et al., 2012). However, we and others have recently demonstrated that RCT can also proceed through a non-biliary pathway known as transintestinal cholesterol excretion (TICE), which persists in both the surgical or genetic absence of biliary cholesterol secretion (Temel et al., 2010; Temel and Brown 2012; Brown et al., 2008; LeMay et al., 2013; van der Velde et al., 2007; van der Veen et al., 2009). Under normal physiologic conditions the hepatobiliary route predominates and TICE is a minor pathway, only contributing ~30% of the total cholesterol lost through the feces in mice (Temel and Brown 2012). However, pharmacologic activation of liver X receptor (LXR) can preferentially stimulate the non-biliary pathway to where TICE contributes greater than 60% of the total cholesterol lost through the feces (van der Veen et al., 2009). Although mechanisms regulating the classic hepatobiliary pathway of RCT have been well defined (Yu et al., 2002; Graf et al., 2003; Groen et al., 2008; Wiersma et al., 2009; Temel et al., 2007), almost no mechanistic information exists for the non-biliary TICE pathway (Brown and Temel 2012; Brufau et al., 2011).
We have previously described several mouse models where the non-biliary TICE pathway is either chronically (Temel et al., 2010) or acutely stimulated (Marshall et al., 2014). Here we have taken advantage of these mouse models as hypothesis generating tools. To identify regulators of TICE and RCT we performed transcriptional profiling in NPC1L1-liver-transgenic mice (which exhibit chronic TICE stimulation; Temel et al., 2010) and second generation acyl-CoA:cholesterol acyltransferase 2 (ACAT2) antisense oligonucleotide (ASO) treated mice (which exhibit acute TICE stimulation; Marshall et al., 2014). From this screening, we found that the hepatic expression of flavin-containing monooxygenase 3 (FMO3) was coordinately downregulated in mouse models of stimulated TICE. In parallel, independent studies have shown that plasma levels of FMO3’s product trimethylamine-N-oxide (TMAO) are highly predictive of atherosclerosis in humans, and TMAO is proatherogenic in mice (Wang et al., 2011; Wang et al., 2014; Tang et al., 2013; Koeth et al., 2013; Bennett et al., 2013; Brown and Hazen 2014). Additionally, we have recently shown that dietary supplementation with the FMO3 product TMAO inhibits macrophage RCT in vivo (Koeth et al. 2013). Here we follow up on these collective observations, and demonstrate that FMO3 knockdown stimulates non-biliary macrophage RCT and reorganizes whole body cholesterol balance. The current studies have also uncovered a previously unknown role for FMO3 in regulating hepatic liver X receptor (LXR) signaling, which controls endoplasmic reticulum (ER) stress and inflammation. Collectively, these observations identify the gut microbiota-driven TMA/FMO3/TMAO pathway as a key integrator of lipid metabolism and immune cell function, and specifically identify FMO3 as a key regulator of sterol balance and RCT, independent of its role in TMAO production. Given that this pathway has now been identified in two separate screens (work described here & metabolomics screening by Wang et al., 2011), we believe this pathway represents a central regulatory node for CVD pathogenesis.
RESULTS
Transcriptional Profiling Identifies FMO3 as a Regulator of RCT
To identify potential regulators of macrophage RCT, we performed microarray analysis in liver from two independent mouse models where TICE was either chronically (NPC1L1-liver-transgenic mice; Temel et al. 2010) or acutely (ACAT2 ASO treatment; Marshall et al., 2014) stimulated. With a stringent threshold (p<0.001) set for differentially expressed genes (DEGs), there were less than 100 DEGs within each array dataset (Figure 1A and 1B), and the vast majority of DEGs were not coordinately regulated in both cohorts (Table S1, Table S2, Table S3, and Table S4). In fact, there were only two genes that were coordinately regulated in both models of TICE augmentation (Figure 1C and 1D). The sole gene that was upregulated in both acute and chronic TICE mouse models was Gm10567, which is predicted to be non-coding RNA of unknown biological function (Figure 1C). The only gene that was coordinately downregulated in both TICE models was FMO3 (Figure 1D, TableS1, and Table S2). We subsequently utilized real time PCR to confirm that FMO3 mRNA expression was significantly reduced in both acute (ACAT2 ASO treated) and chronic (NPC1L1-liver-transgenic mice) TICE models (Figure 1E and 1F). Interestingly, FMO3 mRNA expression closely reflects the concentration of cholesterol in bile, given that FMO3 expression is significantly reduced in both NPC1L1-liver transgenic mice and in mice with diminished expression of the biliary cholesterol half transporter ABCG8 (data not shown). Given that the FMO3 enzymatic product TMAO has recently been associated with increased CVD risk in humans, and shown to both promote atherosclerosis and inhibit RCT in mice (Wang et al., 2011; Tang et al., 2013; Koeth et al., 2013; Bennett et al., 2013; Brown and Hazen 2014), we decided to further interrogate the primary role of FMO3 in cholesterol balance and RCT.
Figure 1. Transcriptional Profiling Defines FMO3 as a Regulator of RCT.
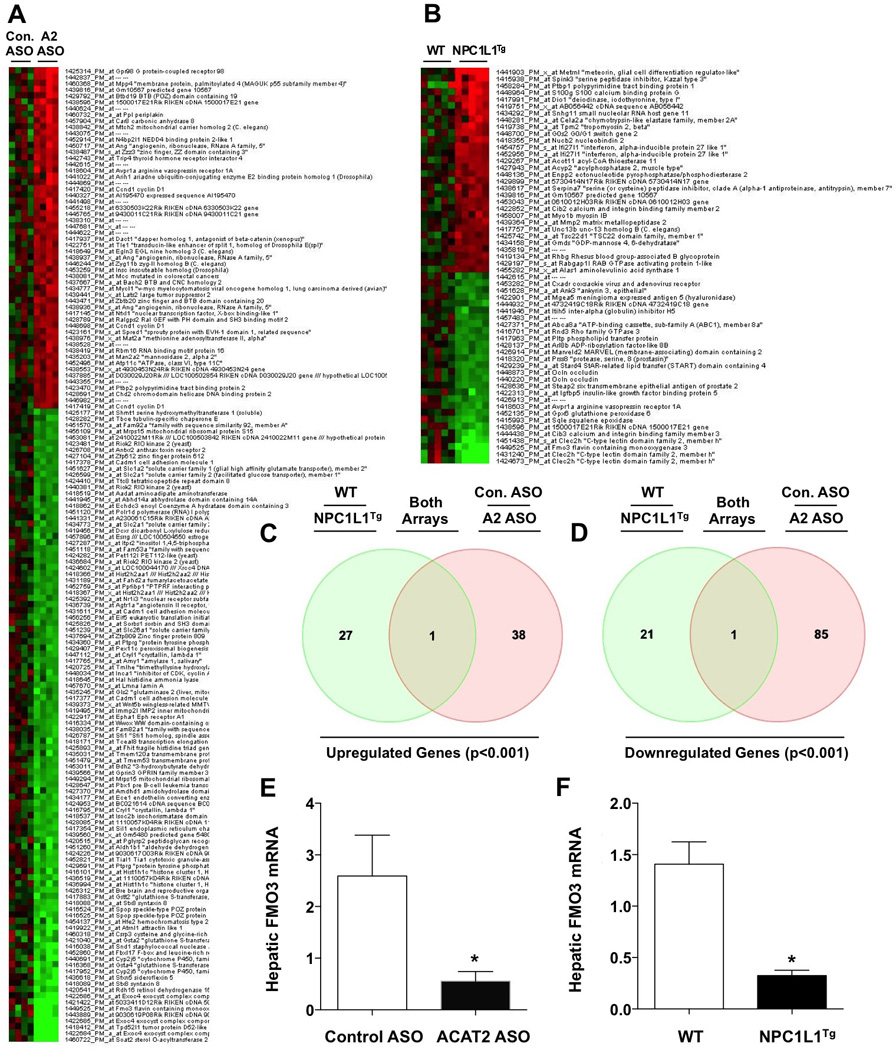
(A) Microarray analysis in acute TICE mouse model. Female C57BL/6 mice were treated with a non-targeting control ASO (Con. ASO) or an ASO targeting ACAT2 mRNA (A2 ASO) as previously described for 1 week (Marshall et al., 2014). Differentially expressed genes (DEGs) are shown as a heatmap (all genes shown have p<0.001, n = 4).
(B) Microarray analysis in chronic TICE mouse model. Female wild type (WT) or NPC1L1-liver transgenic mice (NPC1L1Tg; Temel et al., 2010) were maintained on a high cholesterol diet (0.2%, wt/wt) for 8 weeks. Differentially expressed genes (DEGs) are shown as a heatmap (all genes shown have p<0.001, n = 5).
(C) Venn Diagram showing coordinately upregulated genes in both microarray datasets. The shared gene indicated is Gm10567, and is of unknown function.
(D) Venn Diagram showing coordinately downregulated genes in both microarray datasets. The shared gene is FMO3.
(E) qPCR quantification of hepatic FMO3 mRNA levels in female control and ACAT2 ASO-treated mice (n=5).
(F) qPCR quantification of hepatic FMO3 mRNA levels in female WT and NPC1L1-liver transgenic mice (NPC1L1Tg) (n=5).
ASO-Mediated Knockdown of FMO3 Reorganizes Whole Body Cholesterol Balance
To examine the role of FMO3 in cholesterol balance, we utilized second generation antisense oligonucleotide (ASO) targeting (Crooke, 1997) in mice fed different levels of dietary cholesterol. FMO3 ASO treatment very effectively reduced hepatic FMO3 mRNA (Figure 2A) and protein (Figure 2B) expression, when compared to mice treated with a non-targeting control ASO. As expected, FMO3 knockdown significantly increased circulating levels of FMO3’s substrate trimethylamine (TMA) (Figure 2C), while reciprocally decreased circulating levels of FMO3’s product TMAO (Figure 2D). In parallel, FMO3 knockdown resulted in significant elevations of TMA and reciprocal decreases in TMAO in the liver (data not shown). FMO3 knockdown did not alter the overall health or growth of mice during the period of investigation (data not shown), but did result in a striking reorganization of cholesterol balance. First, FMO3 knockdown significantly reduced intestinal cholesterol absorption in mice fed a low cholesterol diet, and more modestly decreased cholesterol absorption in mice fed a high cholesterol diet (Figure 2E). Corresponding trends were seen in the mass amount of cholesterol lost in feces, where FMO3 knockdown significantly increased fecal neutral sterol loss in mice fed a low cholesterol diet, but the trend for increased fecal sterol loss failed to reach significance in FMO3 ASO treated mice fed a high cholesterol diet (Figure 2F). In both dietary conditions FMO3 knockdown significantly reduced hepatic cholesteryl ester levels (Figure 2G). However, hepatic free cholesterol levels were not altered by diet or ASO treatment (Figure 2H). FMO3 knockdown also reduced the biliary secretion of cholesterol (Figure 2I, Figure S1A, Figure S1B) and phospholipids (Figure S1E and S1F), without dramatically altering biliary bile acid levels (Figure S1C and S1D). FMO3 ASO-driven reductions in biliary phospholipid were accompanied by significant reductions in the hepatic expression of mdr2 (P-glycoprotein) (data not shown). Finally, FMO3 knockdown did not dramatically alter plasma lipid levels in low cholesterol-fed mice, but did cause a modest shift in plasma cholesterol distribution in high cholesterol-fed mice (Figure 2J–2P). In mice fed a high cholesterol diet, FMO3 knockdown significantly reduced very low density lipoprotein (VLDL) cholesterol levels (Figure 2L and 2M), while causing an elevation of low density lipoprotein (LDL) cholesterol levels (Figure 2L and 2N). In contrast, FMO3 knockdown did not alter levels of HDL cholesterol (Figure 2O) or plasma triglycerides (TG) (Figure 2P). Detergent block experiments demonstrated that FMO3 knockdown does not alter VLDL-TG secretion, but does blunt VLDL-CE secretion significantly (data not shown). Finally, FMO3 knockdown resulted in a significant reduction of the total bile acid pool size (Figure S2A), which is likely driven by diminished expression of the major bile acid synthetic enzymes Cyp7a1 and Cyp8b1 (Figure S2B and S2C). Collectively, these results show that FMO3 knockdown reorganizes cholesterol balance in a diet-specific manner, indicating a mechanistic link between FMO3 and cholesterol and bile acid metabolism.
Figure 2. ASO-Mediated Knockdown of FMO3 Reorganizes Whole Body Cholesterol Balance.
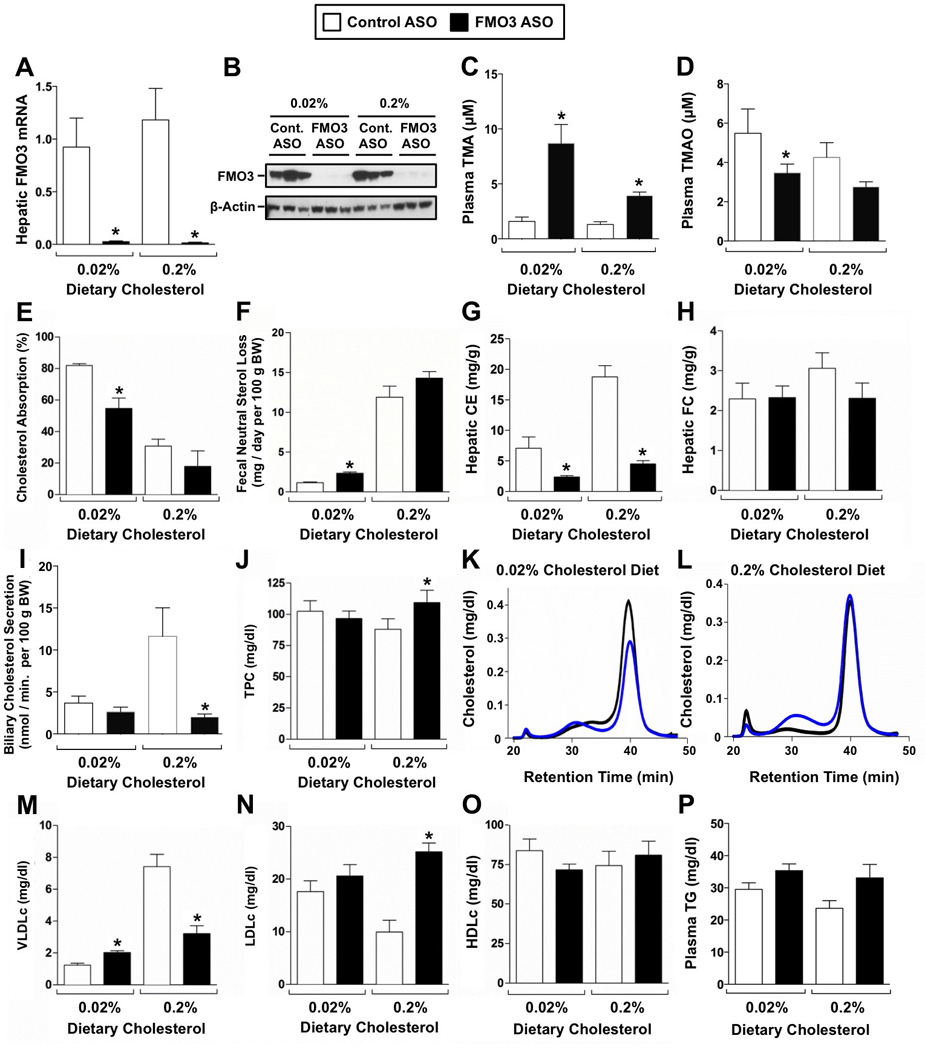
Female C57BL/6 mice were fed either a low (0.02%, wt/wt) or high (0.2%, wt/wt) cholesterol diet and treated with either a control (non-targeting) ASO or an ASO targeting FMO3 mRNA for 6 weeks.
(A) qPCR quantification of hepatic FMO3 mRNA levels.
(B) Western blot determination of FMO3 protein levels.
(C) Circulating levels of the FMO3 substrate trimethylamine (TMA).
(D) Circulating levels of the FMO3 product trimethylamine-N-oxide (TMAO).
(E) Fractional cholesterol absorption was determined using the dual fecal isotope method.
(F) Fecal neutral sterol excretion was determined by gas liquid chromatography.
(G) Hepatic cholesteryl ester (CE) levels.
(H) Hepatic free cholesterol (FC) levels.
(I) Biliary cholesterol secretion rate was measured following common bile duct cannulation.
(J) Total plasma cholesterol (TPC) levels.
(K) Cholesterol distribution of pooled plasma in mice fed a low cholesterol diet (n=5 per pool).
(L) Cholesterol distribution of pooled plasma in mice fed a high cholesterol diet (n=5 per pool).
(M) Very low density lipoprotein cholesterol (VLDLc) levels.
(N) Low density lipoprotein cholesterol (LDLc) levels.
(O) High density lipoprotein cholesterol (HDLc) levels.
(P) Total plasma triglyceride (TG) levels.
All data represent the mean ± S.E.M. from 5–10 mice per group, * = significantly different from the control ASO group within each diet group (p<0.05).
FMO3 Coordinates Sterol-Sensing Transcription Factor Activation in the Liver
Cellular cholesterol levels are carefully sensed and regulated by at least two major transcriptional mechanisms involving sterol regulatory element-binding proteins (SREBPs) and liver X receptors (LXRs) (Tontonoz and Mangelsdorf, 2003; Brown and Goldstein, 1999). Interestingly, FMO3 knockdown results in reciprocal regulation of the SREBP and LXR pathways in mouse liver (Figure 3) in a diet-specific manner. In mice fed a low cholesterol diet, where endogenous cholesterol synthesis rates are high, FMO3 knockdown caused significant elevation in SREBP2 target genes including HMG-CoA reductase (Figure 3J), HMG-CoA synthase (Figure 3K), and squalene synthase (Figure 3L). In contrast, in mice fed a high cholesterol diet, where the liver is burdened with excess cholesterol, FMO3 knockdown caused a marked reduction in LXR target genes including ATP binding cassette transporters G5 (Figure 3A) and G8 (Figure 3B), lysophosphatidylcholine acyltransferase 3 (LPCAT3) (Figure 3D), SREBP1c (Figure 3F), fatty acid synthase (Figure 3G), and stearoyl-CoA desaturase 1 (Figure 3H). However, LXR alpha and ATP binding cassette transporter A1 (ABCA1) mRNA expression was not altered by FMO3 knockdown (Figure 3C, 3E). FMO3 ASO-driven alteration in LXR signaling can likely be explained by reduced expression of oxysterol synthetic enzymes Cyp27a1 and Cyp46a1 (data not shown) resulting in decreased availability of endogenous oxysterol ligands in the liver (Figure 3M–3R). Interestingly, circulating oxysterol levels were not similarly altered by FMO3 knockdown, with the exception of the minor oxysterol 25-hydroxycholesterol being slightly elevated (data not shown). In agreement with dampened LXR signaling, FMO3 knockdown strikingly decreased the expression of LXR target genes involved in de novo lipogenesis, and blunted hepatic steatosis (data not shown). Collectively, these data suggest that FMO3 knockdown depletes hepatic cholesterol stores to a level where SREBP2-driven transcription is activated (Figure 3I–3L), and LXR signaling is reciprocally repressed (Figure 3A–3H) due to diminished oxysterol abundance (Figure 3M–3R).
Figure 3. FMO3 Coordinates Sterol-Sensing Transcription Factor Activation in the Liver.
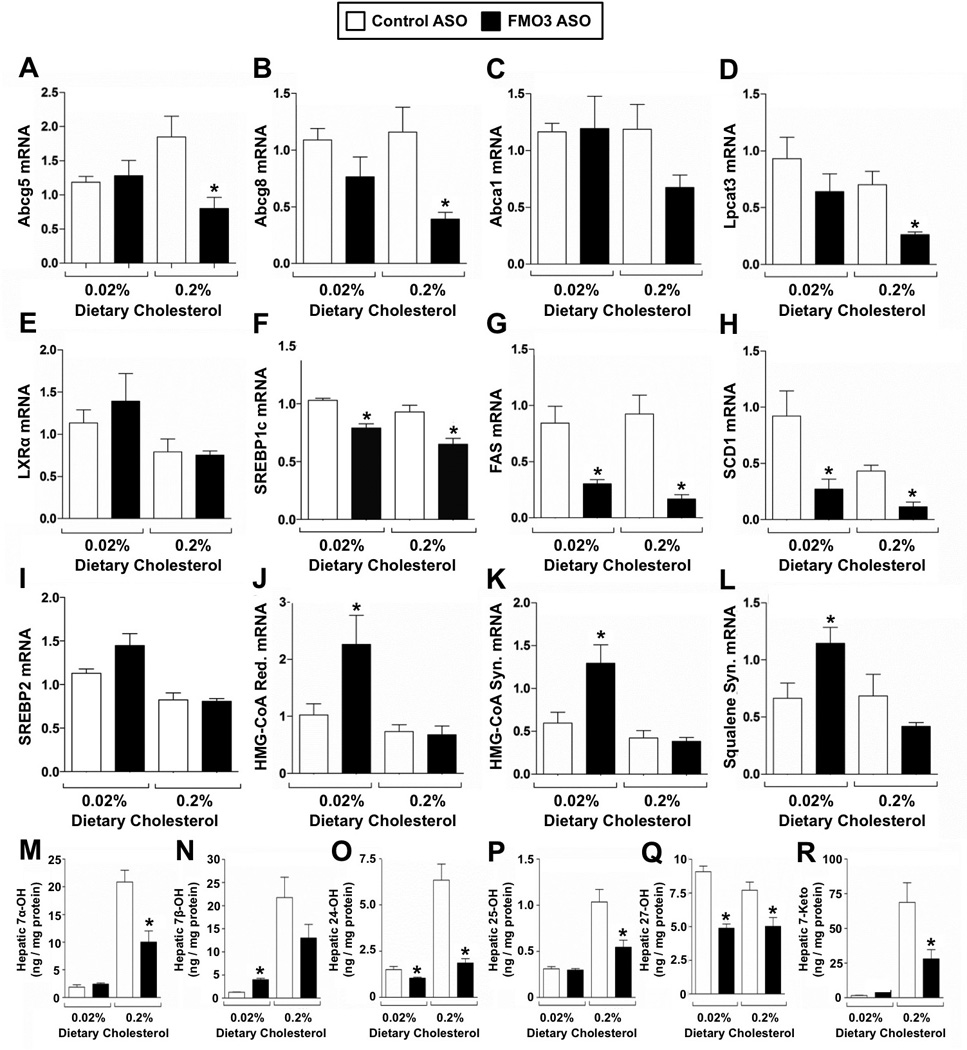
Female C57BL/6 mice were fed either a low (0.02%, wt/wt) or high (0.2%, wt/wt) cholesterol diet and treated with either a control (non-targeting) ASO or an ASO targeting FMO3 mRNA for 6 weeks.
(A–H) Expression of LXR and downstreatm target genes in the liver.
(I–L) Expression of SREBP2 and downstream target genes in the liver.
(M–R) Hepatic levels of oxysterols
All data represent the mean ± S.E.M. from 4–5 mice per group, * = significantly different from the control ASO group within each diet group (p<0.05).
FMO3 Knockdown Stimulates Non-Biliary Macrophage RCT
Given that FMO3 was coordinately downregulated in both acute and chronic models of TICE (Figure 1), and prior studies showed TMAO fed mice had significant reductions in macrophage RCT (Koeth et al., 2013), we wondered whether FMO3 might be linked mechanistically to biliary or non-biliary RCT. In our macrophage RCT studies, a subset of mice were treated with the LXR agonist T0901317, since it is known that LXR activation promotes macrophage RCT primarily by augmenting the non-biliary TICE pathway (van der Veen et al., 2009). FMO3 knockdown very modestly decreased total plasma cholesterol levels (Figure 4A). When treated with the LXR agonist T0901317, both control ASO- and FMO3 ASO-treated mice exhibited mild hypercholesterolemia, with the majority of the cholesterol elevation seen in large HDL particles (data not shown), as has been previously described (Grefhorst et al., 2002). Following [3H]-cholesterol labeled macrophage injection into the peritoneal cavity, the plasma [3H]-cholesterol accumulation was significantly lower in FMO3 inhibited mice, both in the vehicle and T0901317-treated groups (Figure 4B). This reduction in [3H]-cholesterol was largely due to a decrease in large HDL particles (Figure 4C), which tracked closely with the cholesterol mass distribution (data not shown). Importantly, in control ASO-treated mice there was an expected increase in LXR agonist-inducible biliary [3H]-cholesterol secretion (Figure 4G). In contrast, FMO3 ASO-treated mice had lower biliary [3H]-cholesterol recovery, both in the vehicle and T0901317-treated groups, and lacked the expected LXR agonist-inducible response for biliary cholesterol secretion (Figure 4G). Interestingly, FMO3 ASO-treated mice also lack T0901317-induced biliary [3H]-bile acid secretion (Figure 4H). Despite having reduced levels of biliary [3H]-cholesterol (Figure 4G), FMO3 knockdown mice had increased basal and LXR agonist-stimulated disposal of cholesterol into the feces, as measured by both [3H]-cholesterol recovery (Figure 4D) and mass fecal neutral sterol loss (Figure 4F). FMO3 knockdown also modestly increased fecal [3H]-bile acid recovery in both basal and LXR-stimulated conditions (Figure 4E). At the time of necropsy the hepatic recovery of macrophage derived [3H]- cholesterol in the form of [3H]-cholesterol (Figure 4I) and [3H]-bile acid (Figure 4J) was not altered by FMO3 knockdown. Likewise, intestinal recovery of macrophage derived [3H]-cholesterol in the form of [3H]-cholesterol (Figure 4K) and [3H]-bile acid (Figure 4L) was not altered by FMO3 knockdown, but there was a slight increase in intestinal [3H]-cholesterol in mice treated with T0901317 (Figure 4K). Interestingly, LXR activation significantly represses hepatic FMO3 mRNA (Figure 4M) and protein (Figure 4N). However, FMO3 knockdown did not dramatically alter canonical LXR-driven target gene expression in the liver and small intestine in these mice maintained on a low cholesterol diet (data not shown). Of particular relevance to RCT, FMO3 knockdown significantly reduced intestinal NPC1L1 expression (Figure 4O), which is in agreement with alterations in intestinal cholesterol absorption (Figure 2E). Collectively, these results demonstrate that FMO3 knockdown promotes both basal and LXR agonist-stimulated macrophage RCT (Figure 4D and 4F), while repressing biliary cholesterol levels (Figure 4G, S1A, and S1B) and intestinal cholesterol absorption (Figure 2E).
Figure 4. FMO3 Knockdown Stimulates Non-Biliary Macrophage RCT.
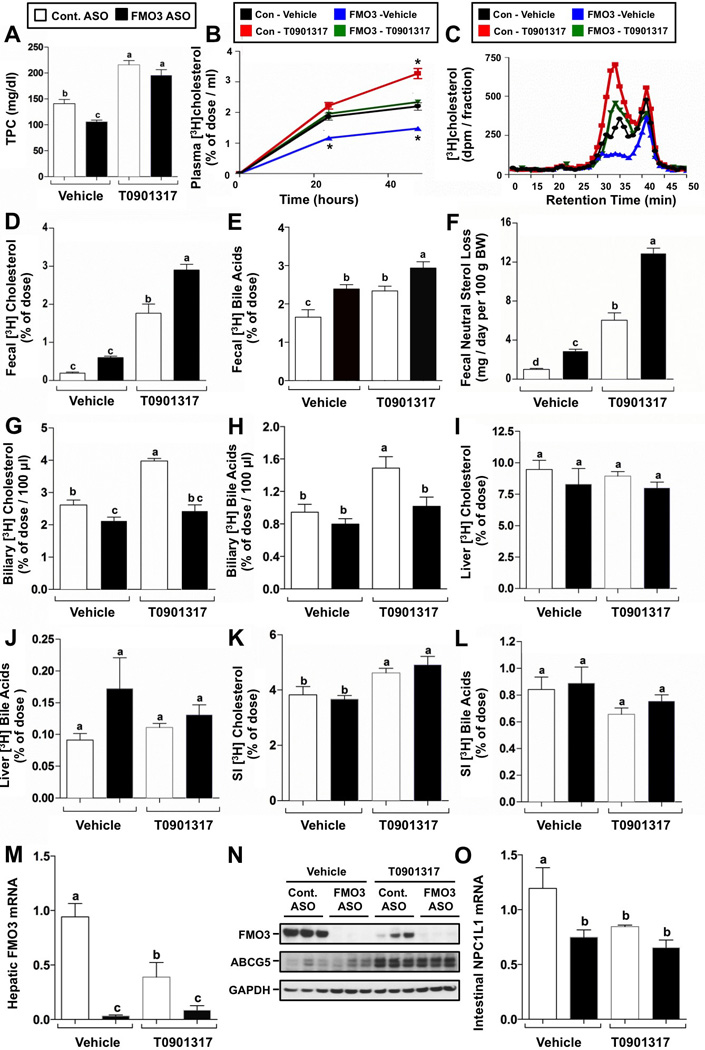
Female C57BL/6 mice were fed a low (0.02%, wt/wt) cholesterol diet and treated with either a control (non-targeting) ASO or an ASO targeting FMO3 mRNA for 6 weeks. During the last week of treatment, mice also were orally gavaged with either a vehicle or an exogenous LXR agonist (T0901317). During the last 48 hours, mice were singly housed, and macrophage RCT into different compartments was measured as described in the Experimental Procedures.
(A) Total plasma cholesterol (TPC) levels.
(B) Time course of [3H]cholesterol accumulation in plasma.
(C) [3H]cholesterol distribution of pooled plasma (n = 5 per pool).
(D) [3H]cholesterol recovery in the feces.
(E) [3H]bile acids recovery in the feces.
(F) Mass fecal neutral sterol excretion.
(G) [3H]cholesterol recovery in gall bladder bile.
(H) [3H]bile acids recovery in gall bladder bile.
(I) [3H]cholesterol recovery in the liver.
(J) [3H]bile acids recovery in the liver.
(K) [3H]cholesterol recovery in the small intestine (SI) wall.
(L) [3H]bile acids recovery in the small intestine (SI) wall.
(M) qPCR quantification of hepatic FMO3 mRNA levels.
(N) Western blot analysis of hepatic FMO3 protein levels.
(O) qPCR quantification of NPC1L1 mRNA levels in the duodenum.
All data represent the mean ± S.E.M. from 6–10 mice per group, and means not sharing a common superscript differ significantly, (p < 0.05).
FMO3 Knockdown Promotes Hepatic ER Stress and Inflammation by Dampening LXR Activation
LXR activation has previously been shown to dampen both inflammatory responses and ER stress by upregulating direct target genes such as LPCAT3 (Rong et al., 2013), and transrepressing proinflammatory genes such as iNOS and COX-2 (Joseph et al., 2003). Given that oxysterol ligand availability is lower in FMO3 knockdown mice, we hypothesized that this may promote hepatic inflammation and ER stress. Indeed, FMO3 knockdown caused marked infiltration of macrophages into the liver (Figure 5A and 5B) and increased the expression of macrophage-derived proinflammatory cytokines and chemokines (Figure 5C and 5D). Furthermore, FMO3 knockdown increased the expression of several genes linked to ER stress (ATF3, CHOP) (Figure 5C and 5D), and increased CHOP protein expression (Figure 5E). FMO3 knockdown also increased the activation of c-Src (Figure 5E), which has also recently been linked to saturated fatty acid-induced ER stress and inflammatory signaling (Holzer et al., 2011). Importantly, either providing endogenous LXR agonists (i.e feeding 0.2% cholesterol in the diet) or providing an exogenous LXR agonist (T0901317) blunted FMO3 ASO-driven hepatic inflammation (Figure 5B, 5C, and 5D), c-Src activation (Figure 5E), and ER stress (Figure 5C, 5D, and 5E). Collectively, these results suggest that FMO3 knockdown promotes hepatic inflammation and ER stress in part by diminishing LXR activity. Collectively, our results are consistent with hepatic FMO3 serving as a critical determinant of the well-known ability of LXR to reciprocally regulate lipid metabolism and inflammation (Joseph et al., 2003; Rong et al., 2013).
Figure 5. FMO3 Knockdown Promotes Hepatic Inflammation and ER Stress by Dampening LXR Activation.
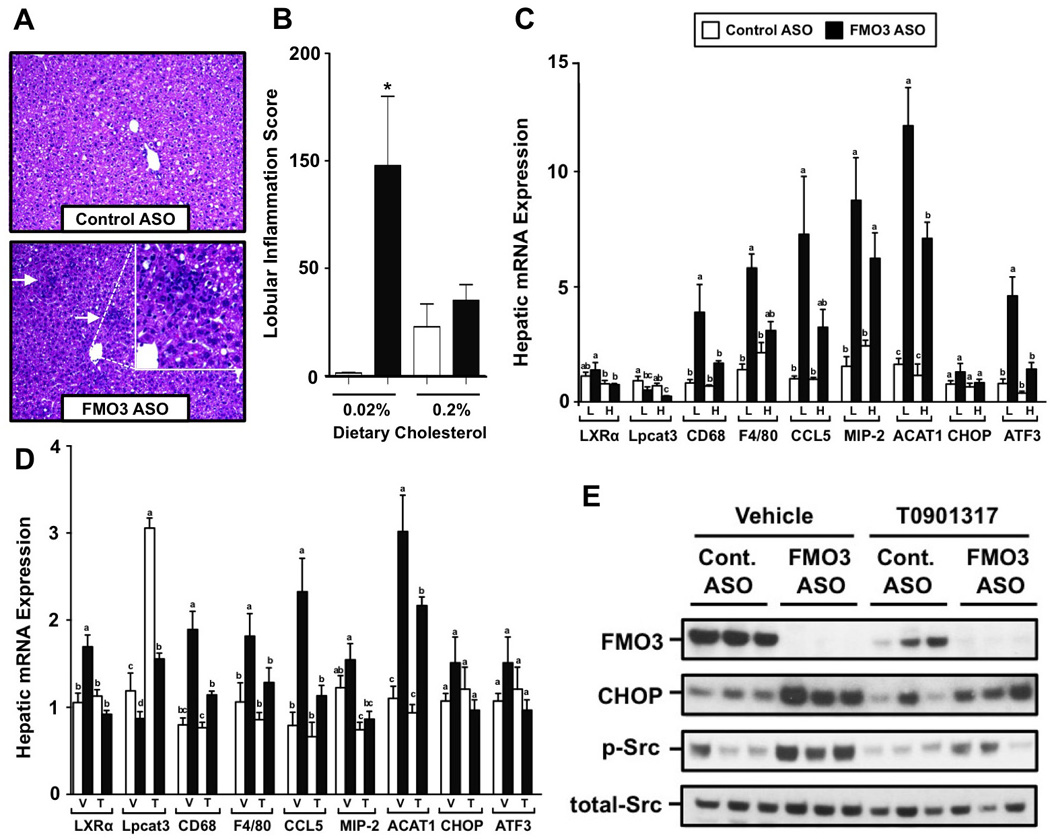
For dietary cholesterol-induced LXR activation (panels A–C), female C57BL/6 mice were fed either a low (0.02%, wt/wt) or high (0.2%, wt/wt) cholesterol diet and treated with either a control (non-targeting) ASO or an ASO targeting knockdown of FMO3 for 6 weeks. For exogenous ligand-induced LXR activation (panels D–E), female C57BL/6 mice were fed a low (0.02%, wt/wt) cholesterol diet and treated with either a control (non-targeting) ASO or an ASO targeting FMO3 mRNA for 6 weeks. During the last week of treatment, mice also were orally gavaged with either a vehicle or an exogenous LXR agonist (T0901317) as described in the macrophage RCT experiments in the Experimental Procedures.
(A) H&E stained liver sections (200× magnification) from female C57BL/6 mice fed a low (0.02%, wt/wt) cholesterol diet and treated with either a control ASO or FMO3 ASO for 6 weeks. Arrows indicate areas of localized immune cell infiltration.
(B) Hepatic lobular inflammation score from pathological report; Data represent the mean ± S.E.M. from 4 mice per group, * = significantly different from the control ASO group within each diet group (p<0.05).
(C) qPCR quantification of genes involved in inflammation and ER stress in the liver of mice fed a low (L) or high (H) cholesterol diet.
(D) qPCR quantification of genes involved in inflammation and ER stress in the liver of vehicle (V) or T0901317-treated (T) mice.
(E) Western blot analysis of FMO3, CHOP, total Src, and activated Src (p-SrcTyr416); n=3 individual animals shown per group
Data represent the mean ± S.E.M. from 4–8 mice per group, and means not sharing a common superscript differ significantly, (p < 0.05). * = significantly different from the control ASO group within each diet group (p<0.05).
FMO3 Gain of Function Reciprocally Reorganizes Cholesterol Balance and Hepatic Inflammation
To confirm the specificity of our results with ASO-mediated knockdown of FMO3, we performed parallel gain of function experiments by means of adenoviral-mediated gene delivery (Figure S3). Administration of FMO3 adenovirus resulted in an 18-fold increase in hepatic FMO3 mRNA levels (Figure S3A). Hepatic overexpression of FMO3 resulted in modest but significant reductions in fecal neutral sterol loss (Figure S3B), while more substantially increasing biliary cholesterol loss (Figure S3C). Furthermore, FMO3 overexpression resulted in a ~20% increase in total plasma cholesterol levels (Figure S3D). In further agreement with loss of function data, FMO3 overexpression significantly reduced the hepatic expression of genes involved in inflammation (CD68, F4/80) and ER stress (ATF3) (Figure S3E, S3F, and S3G). FMO3 overexpression also increased expression of the LXR target genes ABCA1 (Figure S3H) and LPCAT3 (Figure S3I). These data further support the notion that FMO3 is a central regulator of cholesterol balance and hepatic inflammatory responses, given that both gain and loss of function impacts the same biochemical pathways.
FMO3 Knockdown Regulates Intestinal Cholesterol Balance in a Gut Microbe-Dependent Fashion
FMO3 has many potential endogenous and xenobiotic substrates (Cashman and Zhang, 2006; Cashman 2008) that could be involved in its ability to regulate cholesterol balance and inflammation. However, we know that the FMO3 substrate TMA and product TMAO are generated solely from gut microbe-dependent metabolism of dietary methylamine nutrients (Wang et al., 2011; Koeth et al., 2013). To determine the involvement of gut microbial metabolites in the ability of FMO3 ASOs to reorganize cholesterol balance and hepatic inflammation we suppressed gut microbial communities using a poorly absorbed cocktail of antibiotics. After only one week of treatment, levels of both the FMO3 substrate TMA and its product TMAO were barely detectable in antibiotic treated mice (Figure S4A, S4B). Interestingly, antibiotic-mediated suppression of gut microbes completely blocked the ability of FMO3 ASO treatment to reduce intestinal cholesterol absorption (Figure S4C) and elevate fecal neutral sterol loss (Figure S4D). However, antibiotic treatment did not significantly alter the ability of FMO3 ASO treatment to increase hepatic CD68 expression (Figure S4F), decrease hepatic FAS expression (Figure S4G), or decrease hepatic Cyp8b1 expression (Figure S4H). Collectively, these results suggest that the ability of FMO3 ASO treatment to alter intestinal cholesterol absorption and fecal neutral sterol excretion likely relies on gut microbial metabolites, of which TMA is a strong candidate, but FMO3 ASO-driven effects on hepatic inflammation, fatty acid synthesis, and bile acid synthesis likely arise from another non-microbial source.
The Ability of FMO3 ASOs to Reorganize Lipid Metabolism and Inflammation Does not Arise from Diminished TMAO Levels
Given that TMAO has been linked to CVD risk, and the fact that FMO3 ASO treatment reduces TMAO levels in the circulation and liver (Figure 2D, data not shown), we wanted to determine the involvement of TMAO in the lipid and inflammatory phenotypes seen with FMO3 knockdown. To achieve this we provided TMAO as a dietary supplement in control and FMO3 ASO treated mice. As expected FMO3 knockdown reduced circulating TMAO levels, but dietary TMAO supplementation effectively blocked this effect (Figure S4J). Surprisingly, dietary TMAO supplementation in FMO3 inhibited mice resulted in a marked accumulation of plasma TMA (~80-fold above control levels), likely arising from bacterial conversion of TMAO to TMA (Ansaldi et al., 2007) coupled with diminished hepatic conversion of TMA into TMAO by FMO3. Despite the ability of dietary TMAO to rescue circulating plasma TMAO levels in FMO3 ASO treated mice (Figure S4J), this did not alter the ability of FMO3 ASO treatment to alter cholesterol balance (Figure S4K, S4L) or hepatic gene expression (Figure S4M–S4P). Interestingly, in this TMAO add back experiment, the levels of circulating TMA are positively correlated (R2=0.45, P<0.0001) with the amount of fecal neutral sterol loss (data not shown), indicating a potential role for TMA as a gut microbe-derived signal regulating cholesterol balance. Collectively, these data strongly suggest that although chronic elevation of TMAO can be proatherogenic (Wang et al., 2011, Koeth et al., 2013), TMAO is not likely involved in the ability of FMO3 inhibitors to reorganize cholesterol balance and hepatic inflammation.
DISCUSSION
Now identified in two independent screens of altered RCT or CVD risk (Figure 1; Wang et al., 2011), the gut microbe-driven TMA/FMO3/TMAO pathway is increasingly being pursued for the treatment or prevention of CVD. However, this pathway exhibits many levels of complexity including dietary inputs (Wang et al., 2011; Koeth et al., 2013), requirement of bacterial metabolism (Brown and Hazen 2014), and complex hormonal regulation of the FMO enzyme family (Bennett et al. 2013). Furthermore, TMAO feeding (Wang et al., 2011; Koeth et al., 2013) and FMO3 knockdown (data shown here) exert some consistent but many divergent effects on cholesterol and bile acid metabolism across multiple tissues (Figure S5). Although FMO3 and TMAO are directly biochemically connected, our studies suggest that the FMO3 enzyme and the TMAO product likely impact lipid metabolism and inflammation through distinct mechanisms. Therefore it will become increasingly important to differentiate the ability of FMO3 itself versus TMAO to alter the pathogenesis of CVD. The major findings of the current study include the following: (1) transcriptional profiling links FMO3 to RCT, (2) FMO3 knockdown reorganizes multiple processes determining cholesterol balance, (3) FMO3 is a negative regulator of non-biliary reverse cholesterol transport, (4) FMO3 regulates LXR activity to impact hepatic inflammation and ER stress, (5) FMO3 gain of function reciprocally reorganizes intestinal cholesterol balance and dampens hepatic inflammation, (6) the reorganization of cholesterol balance seen with FMO3 knockdown does not involve TMAO, yet can be blocked by antibiotic-mediated suppression of gut microbes, and (7) the hepatic inflammation seen with FMO3 knockdown does not likely involve either TMA or TMAO because it is not suppressed by antibiotics or dietary TMAO. Collectively, these observations identify transcriptional control of FMO3 as an important regulatory switch by which cholesterol balance and hepatic inflammatory responses are integrated.
Given that normalization of circulating TMAO levels in FMO3 inhibited mice does not rescue the abnormal lipid and inflammatory phenotype, it is likely that other FMO3 substrates or products are involved. An obvious candidate would be the accumulation of the FMO3 substrate TMA in FMO3 ASO treated mice. Two pieces of evidence support that TMA may be involved in the ability of FMO3 to regulate cholesterol balance: 1) antibiotic treatment, a condition that causes disappearance of circulating TMA, blocks the ability of FMO3 ASO treatment to blunt intestinal cholesterol absorption and increase fecal cholesterol loss (Figure S4C, S4D), and 2) Circulating TMA levels significantly correlate with the level of fecal neutral sterol loss (data not shown). Although these data suggest a potential role for TMA in regulating cholesterol balance, there is no similar support for TMA being involved in the hepatic inflammation seen in FMO3 inhibited mice given that antibiotic treatment does not reverse FMO3 ASO-driven hepatic inflammation. Furthermore, the ability of FMO3 knockdown to suppress genes involved in hepatic lipogenesis (FAS, ACC1, SCD1) does not involve the TMA/TMAO axis, given that antibiotic treatment or dietary TMAO supplementation do not reverse FMO3 ASO-driven repression of these genes (Figure S4). Collectively, these data suggest that the ability of FMO3 inhibitors to alter cholesterol balance, inflammation, and ER stress likely involves several molecular mechanisms including gut microbial metabolism (alterations in cholesterol absorption and fecal neutral sterol loss) and gut microbe-independent mechanisms (alterations in hepatic inflammation, ER stress, and lipogenesis). Therefore, it will become increasingly important to identify and consider all potential substrates and products (both natural products and xenobiotics) of FMO3 that have the potential to reorganize these diverse phenotypes. Identification of such FMO3 regulated substrates and products could create untapped therapeutic opportunities for lipid- or inflammation-driven diseases.
Our discovery of FMO3 as a coordinately downregulated gene in acute and chronic TICE models (Figure 1) lead us to hypothesize that FMO3 may be a key integrator of biliary and non-biliary RCT. To our surprise, FMO3 knockdown actually phenocopies our chronic TICE mouse model (NPC1L1-liver-transgenic mice), which displays severely reduced biliary cholesterol levels with normal fecal cholesterol loss (Temel et al. 2010). Much like NPC1L1- liver-transgenic mice (Temel et al., 2010), FMO3 inhibited mice seem to preferentially utilize the non-biliary TICE pathway for fecal disposal of cholesterol (Figure 4). In particular, FMO3 ASO treatment strongly enhances LXR agonist-driven macrophage RCT into the feces (Figure 4D), while at the same time preventing LXR agonist-driven increases in biliary cholesterol (Figure 4G). This observation is important because LXR agonists preferentially stimulate the non-biliary TICE pathway (van der Veen et al., 2009), further supporting FMO3 as a negative regulator of TICE. It is interesting to note that treatment with the LXR agonist T0901317 in control ASO treated mice significantly reduces hepatic FMO3 mRNA (Figure 4M) and FMO3 protein (Figure 4N). These data demonstrate that now in three independent conditions where TICE is stimulated (van der Veen et al., 2009; Temel et al., 2010; Marshall et al., 2014), FMO3 is transcriptionally repressed (Figure 1, 4M, 4N). Collectively, these findings demonstrate that FMO3 gene expression is coordinately repressed in several mouse models of augmented TICE, and FMO3 knockdown promotes basal and LXR agonist-stimulated non-biliary RCT (Figure 4). However, it is important to point out that since multiple steps of cholesterol balance are altered by FMO3 knockdown, it is difficult to interpret which step predominates to promote RCT, and whether the reorganization of cholesterol balance seen in FMO3 knockdown mice is derived simply from alterations in TICE. For instance, by inhibiting cholesterol absorption (Figure 2E) FMO3 knockdown could indirectly impact both biliary and non-biliary macrophage RCT rates (Sehayek and Hazen, 2008; Jakulj et al. 2010). Therefore, although FMO3 knockdown does clearly reorganize biliary and non-biliary RCT pathway towards TICE predominance, it also alters multiple steps of forward and reverse cholesterol transport culminating in a net negative cholesterol balance.
It has long been known that a complex reciprocal relationship exists between many lipid metabolic and inflammatory pathways. This antagonism, collectively known as transrepression, is orchestrated by crosstalk between several nuclear hormone receptors (GR, PPARα, PPARγ, PPARδ, LXRα, NURR1, etc.) and the proinflammatory master transcription factor NF-κB (Glass and Saijo, 2010; Joseph et al., 2003; Khovidhunkit et al., 2003). Activation of diverse nuclear hormone receptors (GR, PPARα, PPARγ, PPARδ, LXRα, NURR1, etc.) can transrepress T cell- and macrophage-driven inflammatory responses orchestrated by NF-κB, while in a reciprocal manner activation of NF-κB-driven proinflammatory signaling effectively blunts nuclear hormone receptor signaling (Glass and Saijo, 2010; Joseph et al., 2003; Khovidhunkit et al., 2003). Relevant to our work, LXR activation can strongly suppress NF-κB-driven cytokine and chemokine responses to a multitude of inflammatory stimuli (Joseph et al., 2003; Glass and Saijo, 2010). This transrepression pathway likely underlies the hepatic inflammation and ER stress seen with FMO3 knockdown (Figure 5). Recently, it was demonstrated that a direct transcriptional target of LXR (LPCAT3) mediates a large part of this transrepressive pathway (Rong et al., 2013). Here we show that FMO3 activity is a major determinant of both LXR activation (Figure 3) and downstream anti-inflammatory responses in the liver. Our data suggest that the TMA/FMO3/TMAO pathway is a previously underappreciated regulator of LXR activity, which has broad implications in sterol balance and inflammatory processes.
Given that the link between the metaorganismal TMAO pathway and CVD has only been established in the last three years, many additional studies are required to gain insights into where therapeutic interventions should be targeted. The studies described here along with previous reports linking TMAO to CVD risk in humans (Wang et al., 2011; Wang et al., 2014; Tang et al., 2013; Koeth et al., 2013) provide compelling evidence that the TMA/FMO3/TMAO pathway is a central regulatory pathway that deserves further exploration. However, the ability of TMAO to promote atherosclerosis (Wang et al., 2011; Wang et al., 2014; Tang et al., 2013; Koeth et al., 2013) may be mutually exclusive from the ability of FMO3 inhibitors to reorganize cholesterol balance and hepatic inflammation. Our studies highlight the necessity to understand the repertoire of substrates that can be utilized by FMO3, and also open the possibility that FMO3 may have regulatory functions distinct from its enzymatic activity. Given that FMO3 knockdown and gain of function reciprocally reorganize cholesterol balance, inflammation, and ER stress, FMO3 is uniquely positioned among the FMO family of enzymes to impact human disease. However, further studies are warranted to determine whether this pathway can be exploited pharmacologically in lipid- or inflammatory-driven disease. In particular identification of FMO3 substrates and products that play a primary role in promoting non-biliary macrophage RCT, without increasing hepatic inflammation, would be an attractive strategy for innovative cholesterol lowering drugs in the post statin era. Advancement in our understanding of the enzymology of FMO3 is thus predicted to not only be informative in xenobiotic toxicology studies, but also may provide insights into therapeutic strategies for the treatment or prevention of atherosclerosis.
EXPERIMENTAL PROCEDURES
Animal Studies
The acute and chronic TICE mouse models used for screening purposes have been previously described (Marshall et al., 2014; Temel et al., 2010). For ASO-mediated knockdown of FMO3, female C57BL/6 mice were either maintained on low (0.02%) or high (0.2%) cholesterol diet for a period of 6 weeks, and simultaneously injected with control (non-targeting) or FMO3 targeting ASOs biweekly (25 mg/kg BW) as previously described (Marshall et al. 2014). Macrophage RCT experiments were done as previously described (Temel et al., 2010). All methods used for lipid biochemistry (quantification of cholesterol, oxysterols, bile acids, and phospholipids) have been previously described (Marshall et al., 2014; Koeth et al., 2013; Thomas et al., 2013). All mice were maintained in an American Association for Accreditation of Laboratory Animal Care-approved animal facility, and all experimental protocols were approved by the Institutional Animal Care and Use Committee at Wake Forest University School of Medicine and the Cleveland Clinic Lerner Research Institute.
RNA and Protein Methods
Tissue RNA extraction, real time PCR, and microarray analyses were performed as previously described (Thomas et al., 2013; Marshall et al., 2014). Immunoblotting was conducted as previously described (Thomas et al., 2013). A detailed description of RNA and protein methods is available in the online supplementary methods section.
Statistical Analysis
Most data are expressed as the mean ± S.E.M., and were analyzed using either a one-way or two-way analysis of variance (ANOVA) followed by Student’s t tests for post hoc analysis using JMP version 5.0.12 software (SAS Institute, Cary, NC). For microarray analysis we used empirical Bayes method implemented in R package limma (Smyth 2004).
Supplementary Material
ACKNOWLEDGEMENTS
This work was supported by National Institutes of Health and Office of Dietary Supplements grants: R00 HL096166 (J.M.B.), R01 HL122283 (J.M.B.), R01 HL103866 (S.L.H.), P20 HL113452 (S.L.H.), and U54 GM069338 (H.A.B.). Further support was provided by the Cleveland Clinic Foundation General Clinical Research Center of the Cleveland Clinic/Case Western Reserve University CTSA (1UL1RR024989). S.L.H is also partially supported by a gift from the Leonard Krieger Fund. Authors thank Stephen Milne for assistance with the glycerophospholipid mass spectrometry analysis. Some of the mass spectrometry studies were performed in the Lerner Research Institute Mass Spectrometry Core, with instruments partially supported by a Center of Innovation Award by AB Sciex.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
CONFLICTS OF INTEREST
M.W., D.M.S., A.C.B., D.F., A.D.G., A.L.B., S.M., A.M., R.C.S., J.S., X.R., T.d.A.V., J.C., P.T.I., D.S.M, H.A.B., X.L., P.P., P.T., A.J.L., R.E.T., and J.M.B. have no conflicts of interest to declare. S.L.H. and Z.W. are named as co-inventors on pending and issued patents held by the Cleveland Clinic relating to cardiovascular diagnostics and therapeutics, and have the rights to receive royalty payments for inventions or discoveries related to cardiovascular diagnostics. S.L.H. reports he has been paid as a consultant by the following companies: Cleveland Heart Lab, Inc., Esperion, Liposciences Inc., and Procter & Gamble. S.L.H. also reports he has received research funds from Cleveland Heart Lab, Esperion, Liposciences Inc., Proctor & Gamble, Roche, and Takeda. Richard Lee, Rosanne Crooke, and Mark Graham are employees at Isis Pharmaceuticals, Inc. (Carlsbad, CA).
SUPPLEMENTAL INFORMATION
Supplemental Information Includes Supplemental Experimental Procedures, 4 supplementary tables, and 5 supplementary figures which can be found with this article online.
Author Contributions
J.M.B. planned the project, designed experiments, analyzed data, and wrote the manuscript; J.M.B., R.E.T., and S.L.H. design experiments and provided useful discussion directing the project; M.W., D.M.S., A.C.B., D.F., A.D.G., A.L.B., S.M., A.M., R.C.S., Z.W., J.S., and J.C. conducted mouse experiments, analyzed data, and aided in manuscript preparation; R.G.L., R.M.C., and M.J.G. provided antisense oligonucleotides and valuable discussion; I.M., V.L., C.H., U.T., and R.Z. performed all in vitro enzyme assays and provided valuable discussion; X.L. provided histopathological reports; X.R., T.dA.V., D.M.S, P.T. and A.J.L. performed adenoviral overexpression studies in mice; Z.W., S.L.H., P.P., P.T.I., D.S.M. and H.A.B. performed mass spectrometric analyses and provided critical insights for these studies; All authors were involved in the editing of the final manuscript.
REFERENCES
- Altmann SW, Davis HR, Jr, Zhu LJ, Yao X, Hoos LM, Tetzloff G, Iyer SP, Maguire M, Golovko A, Zeng M, Wang L, Murgolo N, Graziano MP. Niemann-pick C1 like 1 protein is critical for intestinal cholesterol absorption. Science. 2004;20:1201–1204. doi: 10.1126/science.1093131. [DOI] [PubMed] [Google Scholar]
- Ansaldi M, Theraulaz L, Baraquet C, Panis G, Mejean V. Aerobic TMAO respiration in Escherichia coli. Mol. Microbiol. 2007;66:484–494. doi: 10.1111/j.1365-2958.2007.05936.x. [DOI] [PubMed] [Google Scholar]
- Bennett BJ, de Aguiar Vallim TQ, Wang Z, Shih DM, Meng Y, Gregory J, Allayee H, Lee R, Graham M, Crooke R, et al. Trimethylamine-N-oxide, a metabolite associated with atherosclerosis, exhibits complex genetic and dietary regulation. Cell Metab. 2013;17:49–60. doi: 10.1016/j.cmet.2012.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boden WE, Probstfield JL, Anderson T, Chaitman BR, Desvignes-Nickens P, Koprowicz K, McBride R, Teo K, Weintraub W, Zhao XQ, et al. AIM-HIGH investigators. Niacin in patients with low HDL cholesterol levels receiving intensive statin therapy. N. Engl. J. Med. 2011;365:2255–2267. doi: 10.1056/NEJMoa1107579. [DOI] [PubMed] [Google Scholar]
- Brown JM, Hazen SL. Metaorganismal nutrient metabolism as a basis of cardiovascular disease. Curr. Opin. Lipidol. 2014;25:48–53. doi: 10.1097/MOL.0000000000000036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown MS, Goldstein JL. A proteolytic pathway that controls the cholesterol content of membranes, cells, and blood. Proc. Natl. Acad. Sci. USA. 1999;96:11041–11048. doi: 10.1073/pnas.96.20.11041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brufau G, Groen AK, Kuipers F. Reverse cholesterol transport revisited: contribution of biliary versus intestinal cholesterol excretion. Arterioscler. Thromb. Vasc. Biol. 2011;31:1726–1733. doi: 10.1161/ATVBAHA.108.181206. [DOI] [PubMed] [Google Scholar]
- Busby MG, Fischer L, da Costa KA, Thompson D, Mar MH, Zeisel SH. Choline- and betaine-defined diets for use in clinical research and for the management of trimethylamiuria. J. Am. Diet. Assoc. 2004;104:1836–1845. doi: 10.1016/j.jada.2004.09.027. [DOI] [PubMed] [Google Scholar]
- Cashman JR, Zhang J. Human flavin-containing monooxyge- nases. Annu. Rev. Pharmacol. Toxicol. 2006;46:65–100. doi: 10.1146/annurev.pharmtox.46.120604.141043. [DOI] [PubMed] [Google Scholar]
- Cashman JR. Role of flavin-monooxygenase in drug development. Expert Opin. Drug Metab. Toxicol. 2008;4:1507–1521. doi: 10.1517/17425250802522188. [DOI] [PubMed] [Google Scholar]
- Craciun S, Balskus EP. Microbial conversion of choline to trimethylamine requires a glycyl radical enzyme. Proc. Natl. Acad. Sci. USA. 2012;109:21307–21312. doi: 10.1073/pnas.1215689109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crooke ST. Advances in understanding the pharmacological properties of antisense oligonucleotides. Adv. Pharmacol. 1997;40:1–49. doi: 10.1016/s1054-3589(08)60136-2. [DOI] [PubMed] [Google Scholar]
- Dietschy JM, Turley SD. Control of cholesterol turnover in the mouse. J. Biol. Chem. 2002;277:3801–3804. doi: 10.1074/jbc.R100057200. [DOI] [PubMed] [Google Scholar]
- Glass CK, Saijo K. Nuclear receptor transrepression pathways that regulate inflammation in macrophages and T cells. Nat. Rev. Immunol. 2010;10:365–376. doi: 10.1038/nri2748. [DOI] [PubMed] [Google Scholar]
- Go AS, Mozaffarian D, Roger VL, Benjamin EJ, Berry JD, Borden WB, Bravata DM, Dai S, Ford ES, Fox CS, et al. Executive summary: heart disease and stroke statistics – 2013 update: a report from the American Heart Association. Circulation. 2013;127:143–152. doi: 10.1161/CIR.0b013e318282ab8f. [DOI] [PubMed] [Google Scholar]
- Graf GA, Yu L, Li WP, Gerard R, Tuma PL, Cohen JC, Hobbs HH. ABCG5 and ABCG8 are obligate heterodimers for protein trafficking and biliary cholesterol excretion. J. Biol. Chem. 2003;278:48275–48282. doi: 10.1074/jbc.M310223200. [DOI] [PubMed] [Google Scholar]
- Grefhorst A, Elzinga BM, Voshol PJ, Plosch T, Kok T, Bloks VW, van der Sluijs FH, Havekes LM, Romijn JA, Verkade HJ, Kuipers F. Stimulation of lipogenesis by pharmacological activation of the liver X receptor leads to production of large, triglyceride-rich very low density lipoprotein particles. J. Biol. Chem. 2002;277:34182–34190. doi: 10.1074/jbc.M204887200. [DOI] [PubMed] [Google Scholar]
- Groen A, Kunne C, Jongsma G, van den Oever K, Mok KS, Petruzzelli M, Vrins C, Bull L, Paulusma CC, Oude Elferink RP. Abcg5/8 independent biliary cholesterol excretion in Atp8b1-deficient mice. Gastroenterology. 2008;134:2091–2100. doi: 10.1053/j.gastro.2008.02.097. [DOI] [PubMed] [Google Scholar]
- Holzer RG, Park EJ, Li N, Tran H, Chen M, Choi C, Solinas G, Karin M. Saturated fatty acids induce c-Src clustering within membrane microdomains, leading to JNK activation. Cell. 2011;147:173–184. doi: 10.1016/j.cell.2011.08.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jakulj L, Vissers MN, van Roomen CP, van der Veen JN, Vrins CL, Kunne C, Stellaard F, Kastelein JJ, Groen AK. Ezetimibe stimulates faecal neutral sterol excretion depending on abcg8 function in mice. FEBS Lett. 2010;584:3625–3628. doi: 10.1016/j.febslet.2010.07.035. [DOI] [PubMed] [Google Scholar]
- Joseph SB, Castrillo A, Laffitte BA, Mangelsdorf DJ, Tontonoz P. Reciprocal regulation of inflammation and lipid metabolism by liver X receptors. Nat. Med. 2003;9:213–229. doi: 10.1038/nm820. [DOI] [PubMed] [Google Scholar]
- Khovidhunkit W, Moser AH, Shigenaga JK, Grunfeld C, Feingold Endotoxin down-regulates ABCG5 and ABCG8 in mouse liver and ABCA1 and ABCG1 in J774 murine macrophages: differential role of LXR. J. Lipid Res. 2003;44:1728–1736. doi: 10.1194/jlr.M300100-JLR200. [DOI] [PubMed] [Google Scholar]
- Koeth RA, Wang Z, Levison BS, Buffa JA, Org E, Sheehy BT, Britt EB, Fu X, Wu Y, Li L, et al. Intestinal microbiota metabolism of L-carnitine, a nutrient in red meat, promotes atherosclerosis. Nat. Med. 2013;19:576–585. doi: 10.1038/nm.3145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le May C, Berger JM, Lespine A, Pilot B, Prieur X, Letessier E, Hussain MM, Collet X, Cariou B, Costet P. Transintestinal cholesterol excretion is an active metabolic process modulated by PCSK9 and statin involving ABCB1. Arterioscler. Thromb. Vasc. Biol. 2013;33:1484–1493. doi: 10.1161/ATVBAHA.112.300263. [DOI] [PubMed] [Google Scholar]
- Li Q, Korzan WJ, Ferrero DM, Chang RB, Roy DS, Buchi M, Lemon JK, Kaur AW, Stowers L, Fendt M, Liberles SD. Synchronous evolution of an odor biosynthesis pathway and behavioral response. Curr. Biol. 2013;23:11–20. doi: 10.1016/j.cub.2012.10.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li-Hawkins J, Gafvels M, Olin M, Lund EG, Andersson U, Schuster G, Bjorkhem I, Russell DW, Eggertsen G. Cholic acid mediates negative feedback regulation of bile acid synthesis in mice. J. Clin. Invest. 2002;110:1191–1200. doi: 10.1172/JCI16309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshall SM, Kelley KL, Davis MA, Wilson MD, Lee RG, Crooke RM, Graham MJ, Rudel LL, Brown JM, Temel RE. Acute sterol O-acyltransferase (SOAT2) knockdown rapidly mobilizes hepatic cholesterol for fecal excretion. PLos One. 2014;9:e98953. doi: 10.1371/journal.pone.0098953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rader DJ, Tall AR. The not-so-simply HDL story: Is it time to revise the HDL cholesterol hypothesis? Nat. Med. 2012;18:1344–1346. doi: 10.1038/nm.2937. [DOI] [PubMed] [Google Scholar]
- Rader DJ, Alexander ET, Weibel GL, Billheimer J, Rothblat GH. The role of reverse cholesterol transport in animals and humans and relationship to atherosclerosis. J. Lipid Res. 2009;50(Suppl.):S189–S194. doi: 10.1194/jlr.R800088-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rong X, Albert CJ, Hong C, Duerr MA, Chamberlain BT, Tarling EJ, Ito A, Gao J, Wang B, Edwards PA, et al. LXRs regulate ER stress and inflammation through dynamic modulation of membrane phospholipid composition. Cell Metab. 2013;18:685–697. doi: 10.1016/j.cmet.2013.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenson RS, Brewer HB, Jr, Davidson WS, Fayad ZA, Fuster V, Goldstein J, Hellerstein M, Jiang XC, Phillips MC, Rader DJ, et al. Cholesterol efflux and atheroprotection: advancing the concept of reverse cholesterol transport. Circulation. 2012;125:1905–1919. doi: 10.1161/CIRCULATIONAHA.111.066589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryu SD, Kang JH, Yi HG, Nahm CH, Park CS. Hepatic flavin-containing monooxygenase activity attenuated by cGMP-independent nitric oxide-mediated mRNA destabilization. Biochem. Biophys. Res. Commun. 2004a;324:409–416. doi: 10.1016/j.bbrc.2004.09.065. [DOI] [PubMed] [Google Scholar]
- Ryu SD, Yi HG, Cha YN, Kang JH, Kang JS, Jeon YC, Park HK, Yu TM, Lee JN, Park CS. Flavin-containing monooxygenase activity can be inhibited by nitric oxide-mediated S-nitrosylation. Life Sci. 2004b;75:2559–2572. doi: 10.1016/j.lfs.2004.05.018. [DOI] [PubMed] [Google Scholar]
- Schwartz GG, Olsson AG, Abt M, Ballantyne CM, Barter PJ, Brumm J, Chaitman BR, Holme IM, Kallend D, Leiter LA, et al. Effect of dalcetrapib in patients with a recent acute coronary syndrome. N. Engl. J. Med. 2012;367:2089–2099. doi: 10.1056/NEJMoa1206797. [DOI] [PubMed] [Google Scholar]
- Schwarz M, Russell DW, Dietshcy JM, Turley SD. Marked reduction in bile acid synthesis in cholesterol 7alpha-hydroxylase-deficient mice does not lead to diminished tissue cholesterol turnover or to hypercholesterolemia. J. Lipid Res. 1998;39:1833–1843. [PubMed] [Google Scholar]
- Sehayek E, Hazen SL. Cholesterol absorption from the intestine is a major determinant of reverse cholesterol transport from peripheriphal tissue macrophages. Arterioscler. Thromb. Vasc. Biol. 2008;28:1296–1297. doi: 10.1161/ATVBAHA.108.165803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smyth GK. Linear models and empirical Bayes methods for assessing differential expression in microarray experiments. Stat. Appl. Genet. Mol. Biol. 2004;3 doi: 10.2202/1544-6115.1027. article 3. [DOI] [PubMed] [Google Scholar]
- Tang WH, Wang Z, Levison BS, Koeth RA, Britt EB, Fu X, Wu Y, Hazen SL. Intestinal microbial metabolism of phosphatidylcholine and cardiovascular risk. N. Engl. J. Med. 2013;368:1575–1584. doi: 10.1056/NEJMoa1109400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Temel RE, Tang W, Ma Y, Rudel LL, Willingham MC, Ioannou YA, Davies JP, Nilsson LM, Yu L. Hepatic Niemann-Pick C1-like 1 regulates biliary cholesterol concentrations and is a target of ezetimibe. J. Clin. Invest. 2007;117:1968–1978. doi: 10.1172/JCI30060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Temel RE, Sawyer JK, Yu L, Lord C, Degirolamo C, McDaniel A, Marshall S, Wang N, Shah R, Rudel LL, Brown JM. Biliary sterol secretion is not required for macrophage reverse cholesterol transport. Cell Metab. 2010;12:96–102. doi: 10.1016/j.cmet.2010.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Temel RE, Brown JM. Biliary and nonbiliary contributions to reverse cholesterol transport. Curr. Opin. Lipidol. 2012;23:85–90. doi: 10.1097/MOL.0b013e3283508c21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas G, Better JL, Lord CC, Brown AL, Marshall S, Ferguson D, Sawyer J, Davis MA, Melchior JT, Blume LC, et al. The serine hydrolase ABHD6 is a critical regulator of the metabolic syndrome. Cell Rep. 2013;5:508–520. doi: 10.1016/j.celrep.2013.08.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tontonoz P, Mangelsdorf DJ. Liver X receptor signaling pathways in cardiovascular disease. Mol. Endocrinol. 2003;17:985–993. doi: 10.1210/me.2003-0061. [DOI] [PubMed] [Google Scholar]
- van der Velde AE, Vrins CL, van den Oever K, Kunne C, Oude Elferink RP, Kuipers F, Groen AK. Direct intestinal cholesterol secretion contributes significantly to total fecal neutral sterol excretion in mice. Gastroenterology. 2007;133:967–975. doi: 10.1053/j.gastro.2007.06.019. [DOI] [PubMed] [Google Scholar]
- van der Veen JN, van Dijk TH, Vrins CL, van Meer H, Havinga R, Bijsterveld K, Tietge UJ, Groen AK, Kuipers F. Activation of liver X receptor stimulates transintestinal excretion of plasma cholesterol. J. Biol. Chem. 2009;284:19211–19219. doi: 10.1074/jbc.M109.014860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voight BF, Peloso GM, Orho-Melander M, Frikke-Schmidt T, Barbalic M, Jensen MK, Hindy G, Holm H, Ding EL, Johnson T, et al. Plasma HDL cholesterol and risk of myocardial infarction: a mendelian randomisation study. Lancet. 2012;380:572–580. doi: 10.1016/S0140-6736(12)60312-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voshol PJ, Havinga R, Wolters H, Ottenhoff R, Princen HM, Oude Elferink RP, Groen AK, Kuipers F. Reduced plasma cholesterol and increased fecal sterol loss in multidrug resistance gene 2 P-glycoprotien-deficient mice. Gastroenterology. 1998;114:1024–1034. doi: 10.1016/s0016-5085(98)70323-3. [DOI] [PubMed] [Google Scholar]
- Wallrabenstein I, Kuklan J, Weber L, Zborala S, Werner M, Altmuller J, Becker C, Schmidt A, Hatt H, Hummel T, Gisselmann G. Human trace amine-associated receptor TAAR5 can be activated by trimethylamine. PLoS One. 2013;9:e54950. doi: 10.1371/journal.pone.0054950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Klipfell E, Bennett BJ, Koeth R, Levison BS, Dugar B, Feldstein AE, Britt EB, Fu X, Chung YM, et al. Gut flora metabolism of phosphatidylcholine promotes cardiovascular disease. Nature. 2011;472:57–63. doi: 10.1038/nature09922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Tang WH, Buffa JA, Fu X, Britt EB, Koeth RA, Levison BS, Fan Y, Wu Y, Hazen SL. Prognostic value of choline and betaine depends on intestinal microbiota-generated metabolite trimethylamine-N-oxide. Eur. Heart J. 2014;35:904–910. doi: 10.1093/eurheartj/ehu002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiersma H, Gatti A, Nijstad N, Oude Elferink RP, Kuipers F, Tietge UJ. Scavenger receptor class B type I mediates biliary cholesterol secretion independent of ATP-binding cassette transporter g5/g8 in mice. Hepatology. 2009;50:1263–1272. doi: 10.1002/hep.23112. [DOI] [PubMed] [Google Scholar]
- Temel RE, Tang W, Ma Y, Rudel LL, Willingham MC, Ioannou YA, Davies JP, Nilsson LM, Yu L. Hepatic Niemann-Pick C1-like 1 regulates biliary cholesterol concentrations and is a target of ezetimibe. J. Clin. Invest. 2007;117:1968–1978. doi: 10.1172/JCI30060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yancey PH, Clark ME, Hand SC, Bowlus RD, Somero GN. Living with water stress: evolution of osmolyte systems. Science. 1982;217:1214–1222. doi: 10.1126/science.7112124. [DOI] [PubMed] [Google Scholar]
- Yu L, Hammer RE, Li-Hawkins J, Von Bergmann K, Lutjohann D, Cohen J, Hobbs HH. Disruption of Abcg5 and Abcg8 in mice reveals their crucial role in biliary cholesterol secretion. Proc. Natl. Acad. Sci. USA. 2002;99:16237–16242. doi: 10.1073/pnas.252582399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Chaluvadi MR, Reddy R, Motika MS, Richardson TA, Cashman JR, Morgan ET. Hepatic flavin-containing monooxygenase gene regulation in different mouse inflammation models. Drug Metab. Dispos. 2009;37:462–468. doi: 10.1124/dmd.108.025338. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.