

Abstract
Infection of gut-resident CD4+ memory T-cells during acute HIV and SIV infection is associated with rapid loss of these cells and damage to the epithelial barrier. Damage to the epithelial barrier allows translocation of microbial products from the intestinal lumen into the body. Immune activation caused by these microbial products has been associated with disease progression. Although microbial translocation has been demonstrated in SIV-infected nonhuman primates, the identity of translocating bacteria has not been determined. In this study we examined the communities of bacteria both within the GI tract and systemic tissues of both healthy and experimentally SIV-infected Asian macaques. While there were only modest changes in the GI tract-associated microbiome resulting from infection, there is substantial dysbiosis after administration of antiretrovirals. Analysis of bacterial DNA isolated from tissues of infected animals revealed a preference for the phylum Proteobacteria, suggesting that they preferentially translocate. Consistent with this finding, we observed increased metabolic activity of Proteobacterial species within the colonic lumen of SIV-infected animals. Overall these data provide insights into disease progression and suggest that therapies aimed at altering the composition and metabolic activity of the GI tract microbiome could benefit chronically-HIV infected individuals particularly those on antiretroviral therapies.
Introduction
Human Immunodeficiency Virus (HIV) infection in humans and Simian Immunodeficiency Virus (SIV) infection in Asian macaques leads to the development of chronic inflammation that persists even in antiretroviral (ARV)-treated individuals with undetectable plasma viral loads1. In ARV-treated, HIV-infected individuals the residual inflammation is associated with non-HIV comorbidities, including cardiovascular disease, neurologic disorders, cancers, and an overall increased mortality1,2. The importance of persistent chronic immune activation is highlighted by the fact that immune activation is a better predictor of untreated disease progression then either peripheral blood CD4+ T-cell count or viral load2,3.
Potential mechanisms driving inflammation include cytokine induced immune activation secondary to immunological response to HIV/SIV replication, subclinical co-infections such as cytomegalovirus and Epstein-Barr virus, and microbial products that translocate from the lumen of the intestine into peripheral circulation4–7. The gastrointestinal (GI) tract represents the largest mucosal organ in the body and carries a very large percentage of the body’s leukocytes. This concentration of immunological defense within the GI tract is likely due to the need to contain, and potentially respond to, the large microbial mass within the lumen.
In progressive HIV and SIV infections an important site of viral replication and early CD4+ T-cell depletion is within the GI tract lamina propria8. During the short period of the acute phase of infection massive numbers of CCR5+CD4+ T cells are infected which subsequently leads to cell death. The few remaining CD4+ T cells within the lamina propria are skewed away from producing IL17 and IL22, and epithelial cells become apoptotic with subsequent areas of focal damage to the epithelial barrier of the GI tract9–13. This damage to the barrier that separates the intestinal microbiota from the rest of the body allows translocation of microbial products into the lamina propria with subsequent systemic dissemination14. In turn, this microbial translocation is associated with increased immune activation14–16.
The intestinal microbiota is a complex community of bacteria. It is comprised of over 1000 species of bacteria and has roughly the same metabolic capacity as the liver. The makeup of the bacterial community varies along the length of the intestine and alterations in the composition of the microbiome, known as dysbiosis, have been associated with various disease states17–20. For example, decreased bacterial diversity and increased proportions of species of the phylum Proteobacteria have been associated with Crohn’s Disease and increased levels of species of the Firmicutes phylum are observed in obesity21–24.
Dysbiosis has also been demonstrated in HIV-infected individuals with an expansion of the species belonging to the Proteobacteria phylum25,26. Indeed, a disproportionate amount of Proteobacteria within the microbiome is a common occurrence in diseases manifest by inflammation within the GI tract. While dysbiosis may occur in HIV-infected individuals25–27, several studies have found that dysbiosis does not seem to be a common occurrence in progressive SIV-infection of Asian macaques28–31. Therefore, a clear picture of how the microbiota and virus interact with one another indirectly and how these interactions influence disease progression remain elusive.
Here we use experimental infection of Asian macaques (both pigtail macaques, PTM, and rhesus macaques, RM) with SIV to examine changes in the microbiome. High throughput sequencing techniques and quantitative PCR were used to query longitudinal changes in the composition and metabolic activity of the GI tract microbiome after infection and in response to treatment with ARVs. We then further explore the question of which bacteria translocate from the GI tract and account for the observed microbial products in the periphery.
Results
Alterations in the gut microbiome during SIV infection and anti-retroviral therapy
In order to examine potential changes in the GI tract microbiome resulting from SIV infection and ARV therapy we longitudinally sampled stool from 11 experimentally SIV-infected PTM that were subsequently ARV-treated with or without probiotics for 10 months, and sequenced variable regions 1 thru 3 of DNA coding for 16S ribosomal RNA by high throughput sequencing (Figure 1). Given that inflammation peaks coincident with acute SIV replication, we first sequenced DNA from the GI tract microbiome using DNA isolated from stool collected at days 0 and 21 following infection (Figure 1a–b, Supplementary Figure S1). Taxonomic assignments of stool bacterial DNA from individual uninfected animals and animals at the peak of acute infection revealed only slight changes in community makeup (Figure 1a–b), suggesting that significant dysbiosis of the GI tract microbiota may not occur as a result of inflammation ongoing within acute infection. In order to examine long-term consequences of SIV infection on the composition of the GI tract microbiome, we followed broad changes in community structure by qPCR using DNA extracted from longitudinal stool samples. PCR primers were selected to detect total bacterial 16S DNA or sequences specific to the three predominant bacterial phyla (Bacteroidetes, Firmicutes and Proteobacteria). Relative levels of each of these three phyla within the GI tract microbiome were followed longitudinally (Figure 1c–e). While the relative abundance of Bacteroidetes and Firmicutes did not following infection, the abundance of Proteobacteria – the least abundant of the phyla examined – changed over the course of infection. A statistically insignificant increase in Proteobacteria species in the stool was noted from pre-infection to acute infection (Day 0 to 21). Abundance then decreased into the chronic phase of infection to levels lower than the uninfected time point (1.28% to 0.05%, P<0.0001). More strikingly, initiation of ARVs noticeably perturbed the composition of the GI tract microbiota. Adminisitration of ARVs decreased the abundance of both Bacteroidetes and Firmicutes (45.4% to 33.46%, and 54.3% to 43.2%, P=0.055 respectively) while increasing the abundance of Proteobacteria (0.30% to 23.34%, P=0.0003). Despite the fact that ARV therapy was continued regularly in all animals, the GI tract microbiome returned to a state similar to pre-therapy levels within two weeks. These data support the notion that the microbiome is fairly plastic and has the capacity to normalize over time.
Figure 1. SIV and ARV treatment related changes in the GI tract microbiome.

The GI tract microbiome of experimentally SIV-infected PTM was followed longitudinally by analyzing 16S rDNA content of stool. a) Stool DNA from day 0 and 21 post infection was amplified by PCR for 454 pyrosequencing analysis. Bar graphs indicate the relative makeup of the bacterial community in individual animals at each timepoint. b) The average bacterial community makeup of all 11 animals shown in panel A at day 0 and 21. Longitudinal qPCR analysis of the relative amount of c) Bacteroidetes, d) Firmictues, e) Proteobacteria or f) Lactobacillus present in the stool. Red dashed vertical line indicates the start of ARV therapy.
We also studied longitudinal changes in some microbes thought to impart specific benefits to the host. Lactobacillus is a genus of Gram-positive facultative anaerobes from the phylum Firmicutes that has often been identified as an important member of the mammalian GI tract microbiome. Several Lactobacillus species are used, therapeutically, as probiotics. We therefore longitudinally examined the abundance of Lactobacillus in the fecal microbiome of PTM over the course of SIV infection and after administration of therapeutics (Figure 1f). The average abundance of Lactobacillus in the animals prior to SIV infection was 8.3%. However, SIV infection led to a drastic reduction in the level of Lactobacillus overtime (2.7% at day 48, P=0.003, and 1.8% at day 84 post infection, P=0.004). Interestingly, treatment for just one month led to a recovery of Lactobacillus (Figure 1f, P=0.03).
The modest changes to the composition of the microbiome we observed lead us to ask whether the metabolic activity of the microbiome might also be changing, and whether this activity might be masked by simply examining the composition of the microbiome. In order to measure metabolic activity we measured the levels of bacterial ribosomal RNA present in the stool since more metabolically active bacteria will produce higher levels of rRNA compared to less metabolically active species32–34. RNA was isolated from the same samples as above using Trizol. Reverse transcription was performed using a bacterial 16S-specific primer. Following reverse transcription the resultant cDNA was analyzed for the presence of Bacteroidetes, Firmicutes and Proteobacteria as above (Figure 2). Interestingly, we observed that the relative abundance of each phyla was vastly different when comparing RNA to DNA, suggesting that all bacterial phyla are not equally metabolically active. To determine the relative differences in metabolic activity we graphed the ratio of RNA/DNA for each bacterial phylum (Figure 2a). Examining these ratios revealed that Bacteroidetes were relatively inactive while Proteobacteria were highly active. This suggests that Proteobacteria, while small in abundance, are of incredible importance given their proclivity for metabolic activity. These data are consistent with published analyses of bacterial metabolism within the GI tract35. We then examined metabolic activity of the GI tract microbiome longitudinally after SIV infection and after ARV treatment (Figure 2b–d). As with the DNA analysis, the only change noted as a result of SIV infection was an increase in the activity of Proteobacteria. Moreover, similar to our studies of the DNA composition of the microbiota, ARV therapy induced sudden changes in the metabolic activity of all three phyla, which quickly normalized to pre-therapy levels. After administration of ARVs the relative abundance of Bacteroidetes RNA increased (7.62% to 26.13% P<0.001, Figure 2b), Firmicutes decreased (85.91% to 63.81%, P<0.001, Figure 2c) and Proteobacteria was increased (6.47% to 10.06%, P<0.001, Figure 2d).
Figure 2. Changes in the metabolic activity of the microbiome.

The metabolic activity of the GI tract microbiome of experimentally SIV-infected PTM was followed longitudinally by analyzing 16S RNA content of stool via RT-qPCR. a) The ratio of RNA/DNA for each of the three major phyla (Bacteroidetes, Firmicutes and Proteobacteria) for all animals prior to infection. Longitudinal RT-qPCR analysis of the relative metabolic activity of b) Bacteroidetes, c) Firmictues, d) Proteobacteria or e) Lactobacillus present in the stool. Red dashed vertical line indicates the start of ARV therapy.
We also examined the metabolic activity of fecal Lactobacillus species. The relative abundance of metabolically active Lactobacillus decreased following SIV infection (From 6.25% at baseline, to 4.85%, 1.25% and 0.28% over the course of infection, Figure 2e). Induction of therapy caused a brief restoration of Lactobacillus rRNA levels at day 98, but the effect was lost by day 105.
Abundance of Proteobacteria increases in the tissues of SIV infected animals
In order to determine if dysbioses observed in the composition and metabolic activity of the microbiome in SIV-infected macaques might influence the identity of microbes that translocate from the lumen of the intestines into systemic tissues, we isolated bacterial DNA from specific tissues and performed bacterial phyla-specific qPCR. Specifically, DNA was extracted from the feces, colon, liver, and mesenteric lymph node (MLN) of SIV-uninfected and chronically SIV-infected, ARV naïve, RM (Figures 3 and 4). Similar tissue samples were not available from untreated, SIV-infected PTM. However, consistent with what we had seen within the faecal microbiota of SIV-infected and uninfected PTM, examination of the stool-associated microbiome from the RM revealed no drastic dysbiosis attributed to SIV infection (Figure 3a). Similarly, specific analyses of the relative abundance of each phylum revealed no changes relating to infection within samples taken directly from the colon of these RM (Figure 3b).
Figure 3. Analysis of the colon associated microbiome in SIV infection.

DNA was extracted from the colon tissue of RM characterized as uninfected or SIV-infected. The relative abundance of Bacteroidetes, Firmicutes and Proteobacteria was determined by qPCR for 16S rDNA sequence. a) Bar graph showing the community structure of the colon microbiome for each of the animals. b) Dot plot showing the relative abundance of each phyla as in panel a.
Figure 4. Analysis of bacteria present in the mesenteric lymph node of SIV infected Rhesus macaques.

DNA was extracted from the MLN of RM characterized as uninfected or SIV infected. The relative abundance of Bacteroidetes, Firmicutes and Proteobacteria was determined by qPCR for 16S rDNA sequence. a) Bar graph showing the community structure of bacteria from the MLN for each of the animals. b) Dot plot showing the relative abundance of each phyla as in panel a.
Next we examined the relative abundance of these three bacterial phyla in MLN. This analysis revealed that bacterial DNA present within MLN was not, simply, a representation of bacterial DNA present within the colon. Rather, we found a different proportion of bacterial DNA in the MLN (Figure 4). In MLN there was a slightly increased level of Firmicutes compared to levels found within the colon and, perhaps more importantly, the levels of Proteobacteria present in the MLN increased after SIV infection. Indeed, a statistically significant change in the relative abundance of Proteobacteria present in the MLN was observed for chronically infected RM (7.04% in acute to 26.13% in chronically-infected animals P=0.012, Figure 4b). These data suggest that Proteobacteria may be preferentially translocating from the gut. To test this hypothesis more rigorously, we compared the relative abundance of Proteobacterial DNA amongst different tissues in our animals (Figure 5). This analysis confirmed that the abundance of Proteobacteria in the MLN of chronically infected animals was increased compared to the abundance in the gut (15.86% in the colon versus 26.13% in the MLN P=0.055). These data strongly suggest that the increased abundance of Proteobacteria may be due to an increase in translocation of these specific bacteria.
Figure 5. Abundance of tissue-associated Proteobacteria differs from the colon.
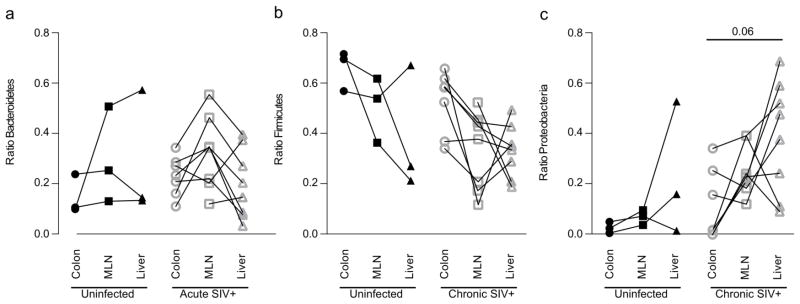
The relative abundance of a) Bacteroidetes, b) Firmicutes and c) Proteobacteria in the colon, liver and MLN of uninfected or SIV-infected RM was determined by qPCR for Proteobacteria 16S rDNA.
Specific species of bacteria are more likely to be found in peripheral tissues
To probe further the difference between the bacterial communities found within the GI tract and the species of translocating bacteria found in the tissues of SIV-infected animals we performed 454, high-throughput, pyrosequencing. DNA isolated from the colon, liver and MLN of uninfected or experimentally SIV-infected RM was amplified using 16s primers specifically labeled for 454 analysis. Amplified PCR products were then sequenced. Principal coordinates analysis (PCA) of the bacterial DNA isolated and amplified from different tissues suggested slight differences between the bacterial communities in the peripheral tissue as opposed to the colon (Figure 6a–b). Analysis of bacterial sequences from the liver and colon of SIV-infected animals revealed the presence of three distinct populations (Figure 6a, left panel). The majority of the colon samples fell within a single population; while the liver associated bacteria had a tendency to associate in the other two populations. Examination of the bacterial DNA in MLN of SIV-infected animals showed a clear separation from bacterial DNA in the colon (Figure 6b, left panel), revealing two populations separated by changes in principal component 1 with the majority of the colon samples falling in the leftmost population. Samples from the MLN tended to cluster to the right. Comparisons of bacterial communities in uninfected animals revealed no obvious separation (Figure 6a–b, right panels).
Figure 6. Specific families of proteobacteria translocate from the gut.

DNA extracted from the colon, liver and MLN of uninfected and experimentally SIV-infected RM was amplified using bacterial 16S primers tagged for 454 pyro-sequencing analysis. a) Principal component analysis showing differences in the bacterial communities of the colon (blue) and liver (red) in SIV-infected (left panel) and uninfected (right panel) animals. b) Principal component analysis showing differences in the bacterial communities of the colon (blue) and MLN (green) in SIV-infected (left panel) and uninfected (right panel) animals. c) Metastats analysis was performed to determine which bacteria were present at significantly different abundance in either the liver or MLN compared to the colon of all RM or PTM used in the study. The graph shows bacterial families that are significantly under represented (blue) or over represented (red) in the tissues of each animal group.
The PCA data confirm that the translocating bacteria are not simply a representation of GI tract luminal bacteria. To identify more closely what might drive these differences we looked at the bacterial taxa present in each of the tissues of these RM and also included several SIV-infected PTM (animals from Figures 1–2) that had been treated. We performed a metastats analysis to describe which OTUs are differentially represented between liver or MLN as compared to colon (Figure 6c). Examination at the family level taxonomy assignments of OTUs revealed a propensity for nine families of bacteria to be under-represented in the RM liver or MLN as compared to the colon (Blue colored samples). Included in these were both the Helicobacter and Campylobacter species of Proteobacteria. Ten families of bacteria were identified as being over-represented in the RM MLN and liver including: two different alpha Proteobacteria, three beta Proteobacteria, one gamma Proteobacteria as well as Staphylococcus and Streptococcus species. These studies confirmed the earlier qPCR analysis, which suggested that Proteobacteria was over-represented in the MLN of SIV-infected animals. The comparison to PTM on suppressive ARVs suggests that treatment of the virus does not reverse this phenomenon.
Possible sources of translocating Proteobacteria
There are at least two explanations for the increased abundance of Proteobacteria species within the liver and MLN of SIV-infected Asian macaques. One possibility is that bacteria are not translocating from the colon, where the percentage of Proteobacteria is relatively low, but are instead translocating from the small intestine where Proteobacterial species are more abundant (Figure 7a). The second is that the species seen in the liver and MLN are more capable of translocating across the damaged colonic epithelium into tissues within the body or better equipped to replicate in the tissue. To address this possibility directly, we used laser capture microdissection in combination with staining against bacterial antigens within paraffin-embedded colon tissue from SIV-infected RM to identify and sample areas of the lamina propria that contained translocating bacteria (Figure 7b). DNA was extracted from isolated tissue regions corresponding to areas of observed translocation or from bacteria within the lumen of the colon from two SIV-infected RM. qPCR analysis revealed that translocating bacteria proximal to the lumen of the colon were enriched for Proteobacterial species (Figure 7c). Although we cannot rule out that Proteobacteria may also be translocating from the small intestine, this experiment shows clear evidence for preferential translocation from the colon itself, which harbors the vast majority of the human microbiome36.
Figure 7. Evidence for translocation of Proteobacteria from the colon.

a) DNA was extracted from luminal swabs taken from the duodenum, jejunum, ileum, cecum and rectum from four uninfected RM. Relative abundance of Bacteroidetes, Firmicutes and Proteobacteria was determined by qPCR for 16S rDNA. b) To examine the makeup of bacteria present in the lamina propria of experimentally SIV-infected RM laser capture microdissection was performed on slide mounted colon tissue that had been stained with antibodies against bacterial lipopolysaccharide. Colored regions indicate samples taken from the lumen (blue), lamina propria (pink) and control (yellow). c) DNA was isolated from pooled samples from the laser capture and the relative abundance of Bacteroidetes, Firmicutes and Proteobacteria was determined for samples from the lumen or sites of translocation in the lamina propria by qPCR.
Correlation between proteobacteria translocation and immune activation
Microbial translocation into the peripheral tissue has the capability of inducing immune activation, irrespective of the identity of bacterial species. The translocation of Proteobacteria represents an even greater potential for activation as these bacteria are often thought to contain a large number of potentially pathogenic species. To assess the potential for Proteobacteria to be driving immune activation we isolated cells from the MLN of the RM in our cohort and used flow cytometry to examine the percentage of activated CD4+ T-cells (Figure 8a). As expected, SIV+ RM showed a higher percentage of HLA-DR+CD4+ T cells than uninfected controls (data not shown). Interestingly, the percentages of HLA-DR+CD4+ T cells correlated positively with the relative amount of Proteobacteria found in the same tissue (Figure 8a, P=0.037). We also examined potential relationships between plasma viral load and Proteobacteria within MLN (Figure 8b), which revealed no correlation. These data show that while Proteobacterial translocation seems unrelated to viral load, that is may be related to the overall level of immune activation.
Figure 8. Correlation of proteobacteria translocation and activation of CD4+ T-cells.
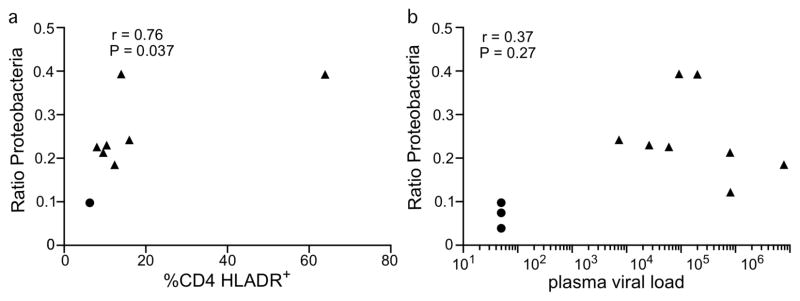
a) T cells from the MLN of RM were examined by flow cytometry to examine the percentage of CD4+ T-cells expressing the activation marker HLA-DR. Percentage of CD4+ T-cells expressing HLA-DR was plotted against the relative amount of Proteobacteria found in the MLN. b) Viral loads were determined at the time of necropsy and charted versus relative amount of Proteobacteria.
Discussion
Study of the microbiome has revealed a relationship between the composition of the body’s commensal flora and several disease states17–21,23. Examination of the microbiome is of potential importance in the study of progressive HIV and SIV infections. These viruses quickly decimate the GI tract-resident CD4+ T-cell population. This depletion is associated with, and may be related causally to, translocation of intestinal products from the lumen into the lamina propria and then systemically14,28. Microbial translocation is associated with increased immune activation seen in progressive SIV and HIV infections2,3. In this study we examined changes in the composition of the GI tract microbiome longitudinally in experimentally SIV-infected Asian macaques and also examined the character of the bacterial populations that were translocating from the GI tract into tissues. Use of non-human primates for this work allowed us to control for factors that might confound human studies such as diet, lifestyle and use of pharmaceutics.
Longitudinal analysis of the GI tract microbiome via analysis of stool-associated DNA revealed modest changes to the composition over the time of infection. Specifically, the abundance of the phylum Proteobacteria and the genus Lactobacillus decreased following infection. A recent study by Dillon et al. failed to find any significant change in a single time point study of the stool of HIV-1-infected patients26. Examination of colonic biopsies suggested that in HIV-1-infected individuals the GI tract microbiome experiences an increase in the abundance of Proteobacteria26,37. However, colon tissue associated samples may be more indicative of adhering and translocating bacteria than the overall microbiome. Another study showed a correlation between high levels of Lactobacillus and retention of CD4+ T cells in HIV-1-infected individuals38. This is of particular interest when paired with our finding that SIV infection of macaques actually decreases the abundance of Lactobacillus and that this trend is partially reversed by anti-retroviral therapy, suggesting a possible causal effect. Indeed, multiple studies have suggested a positive role for inclusion of Lactobacillus containing probiotics in HIV therapy31,39–41.
In our studies examining both DNA and RNA levels we saw the greatest change in the microbiome in immediate response to the initiation of ARV therapy. This is perhaps unsurprising as a commonly reported side effect of ARV use in humans is diarrhea, consistent with the idea that ARV treatment can cause dysbiosis. It is, however, noteworthy because of the interplay between the immune system and the microbiome in HIV/SIV infection. We cannot tell from these data whether ARV induced changes in the microbial community may alter the host immune response to infection or alter viral replication, but changes in the microbiota should be considered in future studies. That the relative proportions of the measured bacteria begin to return to pre-therapy levels within weeks is also interesting. This suggests that although ARVs can alter the microbiome, they are likely not having any persistent anti-microbial effects. Our data are consistent with the a previous analysis of the stool microbiome of ARV-treated HIV-infected individuals which also suggested no long-term changes driven by ARVs31. However, restoration to a state similar to pre-infection might require prolonged courses of therapy42.
Ample evidence from multiple labs demonstrates that microbial translocation occurs following intestinal inflammation in SIV and HIV infection. However, the identity of translocating microbes, and the mechanisms underlying microbial translocation have remained somewhat elusive. There are at least two possibilities relating to the identities of translocating microbial products: 1) the intestinal contents passively leak from the damaged GI tract and; 2) only certain bacteria are capable of translocation. Analysis of bacterial DNA in the liver and MLN of chronically SIV-infected Asian macaques showed a preference for the accumulation of Proteobacteria (Figures 4–6). A preference for Proteobacteria was seen even relatively early in the process of systemic translocation as laser capture analysis of translocating bacteria revealed an increased abundance of Proteobacteria as compared to the luminal contents (Figure 7). These results fit with those published by Dillon et al. who showed an increase in Proteobacteria in colon biopsy samples taken from HIV infected individuals and may be attributed to the increased metabolic activity we observe among proteobacter species and the motile and facultative anaerobic nature of many proteobacter species. Moreover, sampling the colon endoscopically may be more indicative of bacteria that have successfully breached the mucus layer, and are adhering and translocating into the lamina propria. Consistent with the hypothesis that the proclivity of Proteobacteria to translocate to influence immunity, we observed a correlation within the MLN between activated CD4+ T cells and the proportion of Proteobacteria (Figure 8). Overall, these data suggest that modulation of the microbiome, and identification of bacterial species which translocate, will aid in development of therapeutic interventions aimed at restoring this dysbiosis and are consistent with several studies demonstrating that probiotic supplementation of ARVs lead to decreased systemic inflammation 43–45.
Methods
Animals
Eight rhesus macaques (Macaca mulatta) were infected intravenously with 3000 TCID50 of SIVmac239 and were sacrificed during the chronic phase of infection (>4 weeks). Three healthy uninfected control animals were sacrificed as controls. At necropsy tissue samples were harvested from the colon, mesenteric lymph nodes and liver and flash frozen in liquid nitrogen for later DNA extraction.
Twelve pigtail macaques (Macaca nemestrina) were infected intravenously with 3000 TCID50 of SIVmac239. Blood and stool were sampled longitudinally over the course of infection. Ninety-one days after infection we began a treatment regimen of daily doses of PMPA (20–30 mg/kg), FTC (30 mg/kg), L812 (240 mg) Additionally, six of the animals were treated with daily doses of the probiotic VSL #3 and received five doses of IL-21 at one week intervals starting at day 98 post infection. PTM were used in this portion of the study as our prior experienced indicates these animals to be more receptive to oral medications compared to RM. Set point and treatment viral loads are shown in Supplementary Figure S1.
Animals were housed and cared in accordance with American Association for Accreditation of Laboratory Animal Care standards in AAALAC accredited facilities, and all animal procedures were performed according to protocols approved by the Institutional Animal Care and Use Committees of the National Institute of Allergy and Infectious Diseases.
Isolation of Nucleic Acids
DNA was isolated from 180–220 mg samples of stool using the QIAmp DNA Stool Kit (Qiagen) following the manufacturers instructions. RNA was isolated from stool using Trizol (Life Technologies) following the manufacturers protocol with the addition of two to three additional phenol:chloroform extractions following the initial phase separation. DNA was isolated from tissues using the DNeasy Blood and Tissue Kit (Qiagen) following the manufacturers instructions. DNA and RNA was extracted from luminal swabs by tandem use of the RNeasy Mini and DNeasy Blood and Tissue Kits (Qiagen). Nucleic acids were washed off of the luminal swabs using buffer RLT with beta-mercaptoethanol from the RNeasy kit. This volume was then used to extract RNA following the manufacturer’s protocol. Flow through from the binding step and RW1 buffer was step was saved and combined for use in DNA extraction. Collected flow-through was combined and precipitated with ethanol. The resulting pellet was treated with buffer ATL plus proteinase K from the DNeasy kit and used for DNA extraction. DNA was extracted from laser capture microdissection samples by phenol:chloroform extraction followed by ethanol precipitation.
PCR Primers
Pan 16s: 785F GGACTACGGATTAGATACCCTGGTAGTCC, 919R CTTGTGCGGGTCCCCGTCAAT
Bacteroidetes 16s: BactF285 GGTTCTGAGAGGAGGTCCC, Bact UniR338 GCTGCCTCCCGTAGGAGT
Firmicutes 16s: Firm934F GGAGYATGTGGTTTAATTCGAAGCA, Firm1060R AGCTGACGACAACCATGCAC
Proteobacteria 16s: Pro 1080gF TCGTCAGCTCGTGTYGTGA, Pro g1202R CGTAAGGGCCATGATG
454 Pyro-sequencing
For sequencing of 16S rDNA amplicon libraries were prepared from sample DNA using Accuprime High Fidelity Taq polymerase (Invitrogen) and universal primers flanking variable regions V1 (primer 27F; AGAGTTTGATCCTGGCTCAG) and V3 (primer 534R; ATTACCGCGGCTGCTGG). For each sample, the universal primers were tagged with unique sequences (“barcodes”) to allow for multiplexing/demultiplexing PCR products were then purified using the Agencourt Ampure XP Kit (Beckman Counter Genomics) and quantitated using the QuantIT dsDNA High-Sensitivity Assay Kit (Invitrogen). Approximately equivalent amounts of each PCR product were then pooled and purified with a Qiagen minElute column (Qiagen) into 30 μl TE buffer prior to sequencing on a 454 FLX instrument using Titanium chemistry. Raw sequences of the V1-V3 hypervariable regions of the 16S rRNA gene were processed and analyzed using Mothur v.1.31.246. Sequences were trimmed of primer and barcode sequences (primer differences allowed, 2 bp, barcodes, 1 bp) to a minimum length of 200bp and denoised using the Mothur implementation of PyroNoise47. Sequences were aligned using the SILVA database, and chimeras removed using UCHIME48. The taxonomic assignment was determined using the RDP Classifier included in Mothur. Operational Taxonomic Units (OTUs) were defined at an identity cutoff of 97%. Metastats49 was used to detect differentially abundant OTUs controlling the false discovery rate (<0.05). To visualize clustering of communities among samples, distance matrices were generated using the Yue and Clayton theta (relative abundance) measures of dissimilarity and used in PCoA plots.
Laser Capture Micro-dissection
Serials sections (7 μm) were cut, with the first and third sections being placed on glass slides for immunohistochemistry analysis and the second and fourth placed on Acturus ® PEN Membrane Frame slides (Applied Biosystems LCM0521) for laser capture microdissection (LCM). Immunohistochemistry for bacterial products was performed on the first and third slides using a biotin-free polymer approach (Golden Bridge International, Inc.), which were dewaxed and rehydrated with double-distilled H2O. Antigen retrieval was performed by heating sections in 1X DIVA Decloacker reagent (Biocare Medical) in a pressure cooker set at 122°C for 30 s followed by cooling to room temperature. Nonspecific Ig-binding sites were blocked with Blocking buffer (TBS containing 0.05% Tween-20 and 0.25% casein) for 30 min at room temperature. Escherischia coli Ab (1:10,000; DAKO) was diluted in Blocking buffer and incubated overnight at 4°C. Slides were washed in 1x TBS with 0.05% Tween-20 and endogenous peroxidases blocked using 1.5% (v/v) H2O2 in TBS (pH 7.4) for 5 min. Slides were incubated with Rabbit Polink-1 HRP for 30 min at room temperature, washed and incubated with Impact™ DAB (3,3-diaminobenzidine; Vector Laboratories) to develop the staining. The slides were washed in ddH2O, counterstained with hematoxylin, mounted in Permount (Fisher Scientific), and scanned at high magnification (x200) using the ScanScope CS System (Aperio Technologies) yielding high-resolution data from the entire tissue section.
After the regions of interest (ROI) were determined and mapped on the digital Aperio scans of each tissue, the laser capture microdissection was performed on the specified ROI using an Arcturus XT, Nikon TE2000 Microscope system. The targeted areas (translocation-positive, Translocation-negative area and lumen) were captured on 3 different caps (CapSure Macro LCM Caps, Applied Biosystems) for each animal. We determined the optimal laser setting for LCM of colon tissue was 0.5% transmission, UV cutting speed 270, UV Cut length 400micron, IR spot spacing 60 and 3 IR spots per cut length. The IR capture spot size was set to produce a spot 15um in diameter at a power of 70 mW for 24 duration. The caps carrying the tissue were placed in a GeneAmp tube (Applied Biosystems, N8010611) that contained 50 ul of Proteinase K (Acturus Picopure DNA Extraction Kit, Applied Biosystems). Each tube was inverted to allow total immersion of the tissue in the PK solution and incubated at 65°C for 20 hours. The samples were then stored at −20°C until processed for qPCR.
T-cell Analysis
Multicolor flow cytometric analysis was performed on isolated cells according to standard procedures using human mAbs that cross-react with PTM and RM. Cells were stained with Aqua Live/Dead amine dye (Invitrogen) to identify dead cells. Predetermined optimal concentrations were then used of the following antibodies: anti-CD3-Alexa700 (clone SP34-2, BDPharmigen), anti-CD8 PacBlue (clone RPA-T8, BDPharmigen), anti-CD4 PE-Cy5.5 (clone OKT4, eBioscience), anti-CD4 PE-Cy5.5 (clone L200, BDPharmigen), anti-Ki67 FITC (clone B26, BDPharmigen), anti-CCR5 PE (clone 3A9, BDPharmigen), anti-CD28 ECD (clone 28.2, Beckman Coulter), anti-CD95 PE-Cy5 (clone DX2, BDPharmigen), anti-CD14 APC (clone M5E2, BDPharmigen), All samples were permeabilized and fixed using CytoFix/Perm Kit (BDPharmigen) and intracellularly stained to detect Ki67. The acquired data were anlalyzed using FlowJo software (Treestar).
Statistical Analyses
All data was analyzed using Prism v6.0 (GraphPad Software). Differences in populations were determined by performing the non-parametric Mann-Whitney test. Correlation was determined using the Spearman text. In both cases a p-value of less than 0.05 was considered significant.
Supplementary Material
Acknowledgments
The authors would like to thank Julie Segre and Clayton Deming for significant technical assistance and the execution of the 454 sequencing. We would like to thank CLIC/BBC for advice and helpful discussions. Funding for this study was provided in part by the Division of Intramural Research/NIAID/NIH.
Footnotes
Disclosure The authors declare that they have no conflict of interest.
The content of this publication does not necessarily reflect the views or policies of DHHS, nor does the mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government.
Bibliography
- 1.Rodger AJ, et al. Activation and coagulation biomarkers are independent predictors of the development of opportunistic disease in patients with HIV infection. The Journal of infectious diseases. 2009;200:973–983. doi: 10.1086/605447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sandler NG, et al. Plasma Levels of Soluble CD14 Independently Predict Mortality in HIV Infection. The Journal of infectious diseases. 2011;203:780–790. doi: 10.1093/infdis/jiq118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Marchetti G, et al. Microbial translocation predicts disease progression of HIV-infected antiretroviral-naive patients with high CD4+ cell count. Aids. 2011 doi: 10.1097/QAD.0b013e3283471d10. [DOI] [PubMed] [Google Scholar]
- 4.Brenchley JM, et al. Microbial translocation is a cause of systemic immune activation in chronic HIV infection. Nat Med. 2006;12:1365–1371. doi: 10.1038/nm1511. [DOI] [PubMed] [Google Scholar]
- 5.Gordon SN, et al. Disruption of intestinal CD4+ T cell homeostasis is a key marker of systemic CD4+ T cell activation in HIV-infected individuals. J Immunol. 2010;185:5169–5179. doi: 10.4049/jimmunol.1001801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jacobson MA, et al. Human herpesvirus replication and abnormal CD8+ T cell activation and low CD4+ T cell counts in antiretroviral-suppressed HIV-infected patients. PloS one. 2009;4:e5277. doi: 10.1371/journal.pone.0005277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kedzierska K, Crowe SM. Cytokines and HIV-1: interactions and clinical implications. Antivir Chem Chemother. 2001;12:133–150. doi: 10.1177/095632020101200301. [DOI] [PubMed] [Google Scholar]
- 8.Brenchley JM, et al. CD4+ T cell depletion during all stages of HIV disease occurs predominantly in the gastrointestinal tract. J Exp Med. 2004;200:749–759. doi: 10.1084/jem.20040874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Klatt NR, et al. Loss of mucosal CD103+ DCs and IL-17+ and IL-22+ lymphocytes is associated with mucosal damage in SIV infection. Mucosal immunology. 2012;5:646–657. doi: 10.1038/mi.2012.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Klatt NR, Brenchley JM. Th17 cell dynamics in HIV infection. Curr Opin HIV AIDS. 2010;5:135–140. doi: 10.1097/COH.0b013e3283364846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Heise C, et al. Human immunodeficiency virus infection of enterocytes and mononuclear cells in human jejunal mucosa. Gastroenterology. 1991;100:1521–1527. doi: 10.1016/0016-5085(91)90648-5. [DOI] [PubMed] [Google Scholar]
- 12.Ullrich R, et al. Mucosal HIV infection. Pathobiology. 1998;66:145–150. doi: 10.1159/000028012. [DOI] [PubMed] [Google Scholar]
- 13.Epple HJ, et al. Acute HIV infection induces mucosal infiltration with CD4+ and CD8+ T cells, epithelial apoptosis, and a mucosal barrier defect. Gastroenterology. 2010;139:1289–1300. doi: 10.1053/j.gastro.2010.06.065. [DOI] [PubMed] [Google Scholar]
- 14.Estes JD, et al. Damaged intestinal epithelial integrity linked to microbial translocation in pathogenic simian immunodeficiency virus infections. PLoS pathogens. 2010;6:e1001052. doi: 10.1371/journal.ppat.1001052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Klatt NR, et al. Compromised gastrointestinal integrity in pigtail macaques is associated with increased microbial translocation, immune activation, and IL-17 production in the absence of SIV infection. Mucosal immunology. 2010;3:387–398. doi: 10.1038/mi.2010.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pandrea I, et al. Cutting edge: Experimentally induced immune activation in natural hosts of simian immunodeficiency virus induces significant increases in viral replication and CD4+ T cell depletion. J Immunol. 2008;181:6687–6691. doi: 10.4049/jimmunol.181.10.6687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Larsen N, et al. Gut microbiota in human adults with type 2 diabetes differs from non-diabetic adults. PloS one. 2010;5:e9085. doi: 10.1371/journal.pone.0009085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Abu-Shanab A, Quigley EM. The role of the gut microbiota in nonalcoholic fatty liver disease. Nat Rev Gastroenterol Hepatol. 2010;7:691–701. doi: 10.1038/nrgastro.2010.172. [DOI] [PubMed] [Google Scholar]
- 19.Caradonna L, et al. Enteric bacteria, lipopolysaccharides and related cytokines in inflammatory bowel disease: biological and clinical significance. J Endotoxin Res. 2000;6:205–214. [PubMed] [Google Scholar]
- 20.Chang JY, et al. Decreased diversity of the fecal Microbiome in recurrent Clostridium difficile-associated diarrhea. The Journal of infectious diseases. 2008;197:435–438. doi: 10.1086/525047. [DOI] [PubMed] [Google Scholar]
- 21.Harper PH, Lee EC, Kettlewell MG, Bennett MK, Jewell DP. Role of the faecal stream in the maintenance of Crohn’s colitis. Gut. 1985;26:279–284. doi: 10.1136/gut.26.3.279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Manichanh C, et al. Reduced diversity of faecal microbiota in Crohn’s disease revealed by a metagenomic approach. Gut. 2006;55:205–211. doi: 10.1136/gut.2005.073817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Turnbaugh PJ, Backhed F, Fulton L, Gordon JI. Diet-induced obesity is linked to marked but reversible alterations in the mouse distal gut microbiome. Cell host & microbe. 2008;3:213–223. doi: 10.1016/j.chom.2008.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Turnbaugh PJ, et al. A core gut microbiome in obese and lean twins. Nature. 2009;457:480–484. doi: 10.1038/nature07540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mutlu EA, et al. A compositional look at the human gastrointestinal microbiome and immune activation parameters in HIV infected subjects. PLoS pathogens. 2014;10:e1003829. doi: 10.1371/journal.ppat.1003829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dillon SM, et al. An altered intestinal mucosal microbiome in HIV-1 infection is associated with mucosal and systemic immune activation and endotoxemia. Mucosal immunology. 2014 doi: 10.1038/mi.2013.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gori A, et al. Early impairment of gut function and gut flora supporting a role for alteration of gastrointestinal mucosa in human immunodeficiency virus pathogenesis. J Clin Microbiol. 2008;46:757–758. doi: 10.1128/JCM.01729-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Klatt NR, Funderburg NT, Brenchley JM. Microbial translocation, immune activation, and HIV disease. Trends Microbiol. 2013;21:6–13. doi: 10.1016/j.tim.2012.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Handley SA, et al. Pathogenic simian immunodeficiency virus infection is associated with expansion of the enteric virome. Cell. 2012;151:253–266. doi: 10.1016/j.cell.2012.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.McKenna P, et al. The macaque gut microbiome in health, lentiviral infection, and chronic enterocolitis. PLoS pathogens. 2008;4:e20. doi: 10.1371/journal.ppat.0040020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Klatt NR, et al. Probiotic/prebiotic supplementation of antiretrovirals improves gastrointestinal immunity in SIV-infected macaques. J Clin Invest. 2013 doi: 10.1172/JCI66227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Karner M, Fuhrman JA. Determination of Active Marine Bacterioplankton: a Comparison of Universal 16S rRNA Probes, Autoradiography, and Nucleoid Staining. Applied and environmental microbiology. 1997;63:1208–1213. doi: 10.1128/aem.63.4.1208-1213.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kemp PF, Lee S, Laroche J. Estimating the growth rate of slowly growing marine bacteria from RNA content. Applied and environmental microbiology. 1993;59:2594–2601. doi: 10.1128/aem.59.8.2594-2601.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Faith JJ, McNulty NP, Rey FE, Gordon JI. Predicting a human gut microbiota’s response to diet in gnotobiotic mice. Science. 2011;333:101–104. doi: 10.1126/science.1206025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Maurice CF, Haiser HJ, Turnbaugh PJ. Xenobiotics shape the physiology and gene expression of the active human gut microbiome. Cell. 2013;152:39–50. doi: 10.1016/j.cell.2012.10.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Simon GL, Gorbach SL. Intestinal flora in health and disease. Gastroenterology. 1984;86:174–193. [PubMed] [Google Scholar]
- 37.Vujkovic-Cvijin I, et al. Dysbiosis of the gut microbiota is associated with hiv disease progression and tryptophan catabolism. Sci Transl Med. 2013;5:193ra191. doi: 10.1126/scitranslmed.3006438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Perez-Santiago J, et al. Gut Lactobacillales are associated with higher CD4 and less microbial translocation during HIV infection. Aids. 2013;27:1921–1931. doi: 10.1097/qad.0b013e3283611816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hummelen R, et al. Effect of 25 weeks probiotic supplementation on immune function of HIV patients. Gut Microbes. 2011;2:80–85. doi: 10.4161/gmic.2.2.15787. [DOI] [PubMed] [Google Scholar]
- 40.Irvine SL, et al. Probiotic yogurt consumption is associated with an increase of CD4 count among people living with HIV/AIDS. J Clin Gastroenterol. 2010;44:e201–205. doi: 10.1097/MCG.0b013e3181d8fba8. [DOI] [PubMed] [Google Scholar]
- 41.Schunter M, et al. Randomized pilot trial of a synbiotic dietary supplement in chronic HIV-1 infection. BMC Complement Altern Med. 2012;12:84. doi: 10.1186/1472-6882-12-84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lozupone CA, et al. Alterations in the gut microbiota associated with HIV-1 infection. Cell host & microbe. 2013;14:329–339. doi: 10.1016/j.chom.2013.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Heilpern D, Szilagyi A. Manipulation of intestinal microbial flora for therapeutic benefit in inflammatory bowel diseases: review of clinical trials of probiotics, pre-biotics and synbiotics. Reviews on recent clinical trials. 2008;3:167–184. doi: 10.2174/157488708785700302. [DOI] [PubMed] [Google Scholar]
- 44.Madsen KL. The use of probiotics in gastrointestinal disease. Canadian journal of gastroenterology = Journal canadien de gastroenterologie. 2001;15:817–822. doi: 10.1155/2001/690741. [DOI] [PubMed] [Google Scholar]
- 45.Marteau PR, de Vrese M, Cellier CJ, Schrezenmeir J. Protection from gastrointestinal diseases with the use of probiotics. The American journal of clinical nutrition. 2001;73:430S–436S. doi: 10.1093/ajcn/73.2.430s. [DOI] [PubMed] [Google Scholar]
- 46.Schloss PD, et al. Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Applied and environmental microbiology. 2009;75:7537–7541. doi: 10.1128/AEM.01541-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Quince C, Lanzen A, Davenport RJ, Turnbaugh PJ. Removing noise from pyrosequenced amplicons. BMC bioinformatics. 2011;12:38. doi: 10.1186/1471-2105-12-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Edgar RC, Haas BJ, Clemente JC, Quince C, Knight R. UCHIME improves sensitivity and speed of chimera detection. Bioinformatics. 2011;27:2194–2200. doi: 10.1093/bioinformatics/btr381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.White JR, Nagarajan N, Pop M. Statistical methods for detecting differentially abundant features in clinical metagenomic samples. PLoS computational biology. 2009;5:e1000352. doi: 10.1371/journal.pcbi.1000352. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.