

Abstract
We used high throughput sequencing to examine the structure and composition of the T cell receptor β chain in Common Variable Immune Deficiency (CVID). TCRβ CDR3 regions were amplified and sequenced from genomic DNA of 44 adult CVID subjects and 22 healthy adults, using a high-throughput multiplex PCR. CVID TCRs had significantly less junctional diversity, fewer n-nucleotide insertions and deletions, and completely lacked a population of highly modified TCRs, with 13 or more V-gene nucleotide deletions, seen in healthy controls. The CVID CDR3 sequences were significantly more clonal than control DNA, and displayed unique V gene usage. Despite reduced junctional diversity, increased clonality and similar infectious exposures, DNA of CVID subjects shared fewer TCR sequences as compared to controls. These abnormalities are pervasive, found in out-of-frame sequences and thus independent of selection and were not associated with specific clinical complications. These data support an inherent T cell defect in CVID.
Keywords: Common Variable Immune Deficiency, T cell receptor, High throughput sequencing, Junctional diversity, VDJ recombination, Clonality, Clone sharing, EBV, CMV, Adult
1. Introduction
Common Variable Immune Deficiency (CVID) is usually categorized as a B cell defect, based on the most prominent characteristics of hypogammaglobulinemia and loss of functional antibody [1]. In the majority of cases the complex genetic factors leading to this syndrome remain unknown [2]. The CVID syndrome includes a wide clinical heterogeneity which, aside from bacterial infections, includes increased incidence of autoimmune cytopenias, granulomatous and lymphocytic lung disease, enteropathy, and lymphoid hyper-plasia and lymphoma, suggesting intrinsic defects of cellular immunity [3–5]. In addressing the T cell compartment, we and others have described T lymphopenia, loss of CD4+ T cells, defective proliferation to specific and non-specific activators [6, 7], reduced numbers of T cell receptor excision circles (TRECs), and, perhaps as a result, a deficiency of thymic emigrants and circulating naïve T cells [8–10]. The multiple T cell defects described suggests that the CVID syndrome has characteristics of a combined immune deficiency. While the T cell receptors in CVID have not been directly examined, older studies using spectratyping suggested a contraction of the T cell repertoire in some subjects [7, 11]. Methods such as spectratyping, however, fail to capture the complexity and diversity of the T cell repertoire, and are at best semi-quantitative.
Here we used high throughput sequencing to examine directly the structure and diversity of the T cell repertoire in a large cohort of subjects with CVID, providing quantitative sequence information about the structure of variable (V), diversity (D), and joining (J) regions of the β chain of the T cell receptor (TCR), and allowing determination of the structure of V(D)J junctions. This segment constitutes the third complementary determining region (CDR3), the most variable portion of the TCR, and thus reflects the breadth of T cell antigen-binding capacity. Examining junctional diversity, genetic recombination, clonality, and CDR3 structure, we demonstrate that in comparison to control DNA, TCR sequences in CVID have fewer deletions from germline sequences, have fewer n-nucleotide additions, are significantly more clonal, lack a highly modified population of TCRs found in healthy donor blood, and demonstrate a globally constricted T cell repertoire.
2. Methods
2.1. Demographics and clinical characteristics
Enrolled subjects were diagnosed with CVID based on standard criteria including reduced serum IgG, IgA, and/or IgM by two or more confidence intervals below the normal range and specific antibody deficiency [12]. Peripheral blood DNA samples of 44 CVID subjects, 15 females and 29 males of ages 9 to 64 years with a mean of 40, and 22 healthy adult volunteers, 12 females and 10 males of ages 23 to 66 years with a mean of 34, were examined. Both patients and controls were predominantly of Caucasian heritage. Clinical features of patients and immunological characteristics are described in Online Repository Tables S1 and S2. All CVID subjects were receiving replacement immunoglobulin at the time of study participation for at least one year; none were ill or on immunomodulatory or immunosuppressive medications at the time of blood collection. Eight patients were known to have TACI mutations, C104R (4), A181E (2), R72H (1) and V220 (1). The Institutional Review Board of Mount Sinai Hospital approved this study, and written informed consent was obtained from all patients.
2.2. TCRβ sequencing and analysis
Peripheral blood mononuclear cells were isolated from heparinized whole blood samples (5 to 8 ml) by centrifugation of diluted blood over Hypaque 1077 (Sigma-Aldrich), or Ficoll-Paque (GE Healthcare, Uppsala, Sweden). Genomic DNA was isolated from these cells via column purification (Qiagen, Valencia, CA) or centrifugation (5 Prime, Gaithersburg, MD). Equivalent amounts of control and CVID DNA were used for sequencing. A 60 bp sequence of the rearranged TCRβ CDR3 region was amplified and sequenced for all samples using the immunoSEQ™ assay, a bias corrected multiplex PCR and high-throughput assay for the re-arranged DNA of T cells (Adaptive Biotechnologies) [13]. Each unique sequence represents a clone. For each clone, the nucleotide sequence, the V, D, and J gene usage, as well as copy number and frequency were determined. Clonality was calculated from Shannon's Entropy as previously described [14]. The predicted amino acid sequence of the productive sequences was determined. The mean CDR3 Kyte–Doolittle hydropathy index was interpolated from this sequence. Additional methods are in the Online Repository.
2.3. Statistical methods
Statistical analyses were performed and graphs made using the R statistical programming language (version 2.15.2) and GraphPad Prism (version 5.01). A p-value of <0.05 was regarded as significant. V, D, or J family and gene usage was compared using a two-way ANOVA with Tukey HSD multiple comparisons test. Unpaired t-tests were used for comparison of normally distributed numerical data, while nonparametric data were assessed with Wilcoxon tests. Pearson coefficients were calculated to test correlation. Normality was established using histograms and the Shapiro–Wilk test. Clustering analysis of V genes was performed using the hclust function in R. The number of sequences using each V gene was scaled for each individual. The Manhattan distance was calculated, and the complete clustering algorithm was used to determine clustering.
3. Results
3.1. T-cell receptors in CVID have reduced junctional diversity
Aggregate effects of thymic selection and peripheral expansion shape the Vβ repertoire. During VDJ recombination, the addition and deletion of nucleotides can result in non-productive sequences that are carried in genomic DNA if the second locus is successfully rearranged. From the CDR3 sequences obtained from CVID and control DNA samples, we determined whether the sequence was productive (in frame) or non-productive (out-of-frame or containing stop codons). While equal amounts of input DNA were used in each case, controls had a higher number of unique sequences per sample (average of 81,820) than CVID subjects (average of 31,547 unique sequences per sample); however, both were in the range expected for peripheral blood DNA of healthy adults [15]. (Two CVID samples with low numbers of sequences (<10,000) were excluded.) Our results, initially unexpected, showed that the CVID samples contained significantly smaller proportion of non-productive sequences (15.9% ± 0.32% n = 42) as compared to control samples (17.2% ± 0.27% n = 22, p = 0.01). Since T cell V(D)J rearrangement inevitably alters the CDR3 sequence by deletion of templated germline bases and insertion of non-templated bases into the Vβ–Dβ and Dβ–Jβ junctions, we compared the mean number of deletions and insertions in unique CVID TCRβ CDR3 sequences to those from control DNA. The results showed that CVID CDR3 sequences were in fact closer to germline in configuration, with fewer deletions or insertions, possibly contributing to the decreased frequency of out-of-frame sequences. The mean number of CDR3 deletions (from V, D, and J genes) in CVID samples was 15.0 ± 0.04 bases, while for controls the mean number of deletions was 15.90 ± 0.17 bases (Fig. 1a; p-value < 0.0001 and Online Repository Fig. S1a; p-value < 0.0001). (We examined the number of deletions from Vβ, 5′ and 3′ ends of Dβ and Jβ. Significantly fewer deletions from the Jβ and 5′ end of Dβ, Online Repository Fig. S2, were primarily responsible for the fewer total deletions.) The mean number of CDR3 n-nucleotide insertions (V–D and D–J) was also reduced for CVID at 7.7 ± 0.04 bases, as compared to control samples with a mean of 8.7 ± 0.13 inserted bases (Fig. 1b, p-value < 0.0001; Online Repository Fig. S1b; p = 0.01). As CVID sequences had both fewer deletions and insertions, the net median CDR3 length was similar to that of control DNA (CVID 40.5 ± 0.05 bases and control subjects 40.5 ± 0.06). Since the out-frame sequences are not shaped by selection processes, we also compared the frequencies of insertions and deletions in unique sequences of this type. Unlike productive sequences, the predicted CDR3 lengths of non-productive sequences were significantly different (CVID 43.22 ± 0.069 bases; healthy controls 42.97 ± 0.083 bases; p = 0.02). CVID samples had an average of 15.1 ± 0.08 deletions and 11.4 ± 0.13 insertions in stop-terminated sequences, and 14.5 ± 0.05 deletions and 10.0 ± 0.06 insertions in frame shift mutated CDR3s. In contrast, control TCR sequences with stop codons had mean 16.2 ± 0.16 deletions and 12.5 ± 0.13 insertions, and those with frame shifts had 16.2 ± 0.16 deletions and 11.0 ± 0.13 insertions (ANOVA, p < 0.001). Thus both in frame and out-of-frame sequences had significantly fewer nucleotide modifications in CVID samples, suggesting that CVID T cell progenitors have intrinsic defects in their recombination events. Similar analysis of the total repertoire did not show the differences between the groups in the number of deletions (Online Repository Fig. S3a). This was due to a relative abundance of T cells with fewer deletions in the total repertoire. Reciprocal changes were seen in insertions (Online Repository Fig. S3b).
Figure 1.

Fewer VDJ deletions and n-nucleotide insertions in CVID CDR3 sequences compared to normal controls. a) The sum of the numbers of deletions from Vβ, Dβ, and Jβ sequences were calculated for each sequence. The mean number of deletions (in bases) for each patient (red square) or control (blue filled circle) is shown (y-axis). The horizontal bar represents the mean of each group and the whiskers represent the standard error of the mean. b) The sum of the number of insertions between Vβ and Dβ, and Dβ and Jβ sequences were calculated for each sequence. The mean number of insertions (in bases) for each patient (red square) or control (blue filled circle) is shown (y-axis). The horizontal bar represents the mean of each group and the whiskers represent the standard error of the mean. p-Values of t-test are indicated. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
3.2. CVID CDR3 sequences lack a population of highly modified TCRs
To better characterize the differences between CVID and control TCRs, we next analyzed deletions at the V locus, a more sensitive measure than total deletions. In control subjects, the distribution of deletions from the V gene exhibited a bimodal curve (Fig. 2a). While the majority of clones had less than 13 V-region gene deletions, 0.11% of clones were characterized by highly modified sequences, with up to 29 deletions (median 15) in this region. On the other hand, the maximum number of deletions in V genes for any CVID sample was 12. Control TCRs from T cells with these highly modified sequences contained a unique amino acid composition (Online Repository Fig. S4) with lower hydrophobicity scores (Fig. 2b), a population absent in subjects with CVID who are missing this portion of the normal T cell repertoire.
Figure 2.
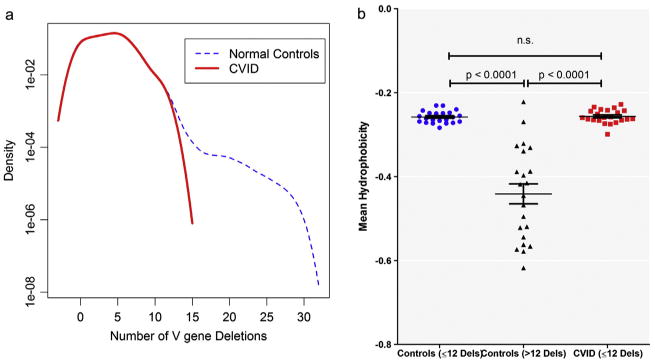
CVID TCR sequences lack a unique population of TCRs characterized by more than 12 deletions from the V gene. a) A density curve of V-deletions in all unique clones in healthy controls (blue dashed line) or CVID (red bold line). The number of deletions is displayed on the x-axis while frequency is expressed on a log scale on the y-axis. b) Mean Kyte–Doolittle hydropathy index (y-axis) for control DNA sequences with ≤12 deletions (blue filled circles), >12 deletions (black triangles) and CVID sequences (red squares), all of which had ≤12 Vβ deletions. A negative number indicates hydrophilicity and a positive number indicates hydrophobicity. t-Test p-values are indicated. n.s. denotes statistically not significant. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
3.3. Increased clonality in CVID
Since there were significantly fewer insertions and deletions in CVID, we examined the degree of clonality, based on the normalized reciprocal of Shannon's Entropy. Independent of the number of sequences examined, CVID subjects had increased TCRβ clonality as compared to healthy controls (t-test, p-value = 0.018) (Fig. 3a). This was a feature most characteristic of about a third of subjects, and bore no relationship to the age of the subject, or to the presence or absence of autoimmunity, lymphoid hyperplasia, interstitial or granulomatous lung disease, splenomegaly, previous splenectomy, enteropathy, previously treated B-cell lymphoma or TACI polymorphisms (Table 1). To examine what fraction of the total repertoire was occupied by the largest clones, we sorted the sequences in each sample by copy number. In healthy controls, the top 1% largest clones occupied 20.03% ± 1.56% of the repertoire, whereas for CVID samples, the top 1% of clones occupied a significantly larger fraction at 33.16% ± 2.56% (t-test with Welch's correction for unequal variances, p < 0.0001) (Fig. 3b). There was no correlation between clonality and the number of productive clones (r = −0.20, p = 0.7295), nor to the number of sequences examined, as clonality in each case was normalized to the number of productive sequences.
Figure 3.

CVID TCR sequences have increased clonality compared to healthy controls. a) Clonality of CVID (red square) and control DNA (blue filled circle) is shown. Horizontal line represents mean and whiskers represent standard error of mean. The groups were compared by t-test and the p-value is indicated. b) All productive clones were ranked by their normalized copy number. The sum of the copy numbers of the top 1% of clones was calculated. The figure depicts the sum of the copy number as a percent of the normalized copy number of all productive sequences on the y-axis and group on the x-axis (CVID – red square; controls – blue filled circle). The groups were compared by t-test and the p-value is indicated. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
Table 1.
CVID complications and clonality.
| Present, mean ± SEM, n | Absent, mean ± SEM, n | p-Value* | |
|---|---|---|---|
| Autoimmunity | 0.19 ± 0.023, n = 18 | 0.18 ± 0.033, n = 26 | 0.84 |
| B cell lymphoma | 0.12 ± 0.028, n = 4 | 0.19 ± 0.021, n = 40 | 0.08 |
| Splenomegaly | 0.21 ± 0.037, n = 19 | 0.16 ± 0.019, n = 25 | 0.19 |
| Interstitial lung disease | 0.22 ± 0.035, n = 20 | 0.15 ± 0.019, n = 24 | 0.10 |
| Enteropathy | 0.13 ± 0.043, n = 5 | 0.19 ± 0.021, n = 39 | 0.32 |
| Autoimmune cytopenias | 0.18 ± 0.026, n = 18 | 0.18 ± 0.028, n = 26 | 0.95 |
| TACI mutations | 0.20 ± 0.05, n = 8 | 0.18 ± 0.02, n = 36 | 0.73 |
Unpaired t-test.
3.4. Clonality is related to the absolute T cell count
TCRβ clonality in CVID samples was highly correlated with absolute T cell counts in peripheral blood (Pearson coefficient of correlation, r = 0.67, p < 0.001, Fig. 4). Previous work using spectratyping suggested that a few CVID subjects with higher numbers of CD8+ T cells had more evidence of clonality [11]. While there was a positive correlation with absolute CD8+ T cell counts for the group, (r = 0.66, p < 0.001), this was due to 5 patients with reduced CD4/8 ratios and higher CD8 T cell counts (as shown in Online Repository Fig. S5). Excluding these samples eliminated this correlation (r = −0.33, r2 = 0.11, p = 0.16).
Figure 4.

Clonality correlates with absolute T cell count in peripheral blood. Clonality is depicted on the y-axis absolute CD3+ T cell count on the x-axis. The linear regression line is shown and coefficient of correlation, r, indicated on the figure. Clonality of control samples and respective laboratory ranges for normal are indicated in the crosshatched area.
3.5. CVID subjects share fewer clones than normal controls
Prior studies have demonstrated that any two healthy individuals share approximately 10,000 CDR3 amino acid sequences, independent of the degree of HLA similarity [15]. To compare sharing of clones amongst CVID subjects and healthy controls, we randomly resampled the repertories of these cohorts separately, 12 samples at a time, for 100 rounds, to determine how many individuals in each cohort shared a TCR, as defined by amino acid sequence. Despite increased clonality, CVID subjects shared fewer TCRβ clones as compared to healthy controls (Fig. 5; p < 0.0001). In fact, a significantly larger portion of the repertoire of CVID subjects (85.2 ± 0.46) was not shared with other members of this group, as compared to healthy controls (83.3 ± 0.21; p-value = 0.0007, Fig. S6). As the highly-shared sequences in control samples did not include a preponderance of sequences with large numbers of n-nucleotide insertions, the lack of shared clones in CVID could not be explained by the loss of this highly modified population in patients.
Figure 5.

CVID TCR sequences share fewer clones compared to normal controls. CVID and control DNA were iteratively sampled, 12 individuals at a time for 100 repeats. The amino acid sequences of CDR3 regions of productive clones were compared separately amongst CVID patients (red squares) or healthy controls (blue filled circles). The number of individuals that share each clone in a sample of DNA was determined. The mean number of clones shared amongst 2 to 12 individuals was then determined. The number of individuals who share a clone is designated on the x-axis. The mean number of clones shared amongst patients or controls is depicted on the y-axis on a log10 scale. Error bars represent the 95% confidence interval. Kolmogorov–Smirnov test p-values are indicated. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
For normal controls, sharing of TCR sequences has been partly attributed to T cell activation by ubiquitous viral antigens such as EBV and CMV [16]. To determine if clonal sharing in controls could be attributed to an expansion of these common anti-viral sequences, and to determine if such clones could also be found in CVID samples, we interrogated both data sets for 70 EBV and 245 CMV TCR sequences previously published [16–20]. DNA of 11 of the 22 controls contained EBV related clones, and 10 of 22 contained CMV related TCR sequences. These sequences were found in proportionally fewer CVID subjects, however, with 10 of 42 containing EBV TCR sequences and 5 of 42 containing CMV TCR sequences (Table 2).
Table 2.
CVID DNA was less likely to have known EBV and CMV specific TCR sequences.
| EBV | CMV | |||||
|---|---|---|---|---|---|---|
|
|
|
|||||
| Controls | CVID | p-Value | Controls | CVID | p-Value | |
| Viral clone containing samples | 12/22 | 11/42 | 0.03* | 10/22 | 5/42 | 0.003* |
| Clones/sample | 3.3 ± 0.9 | 3.9 ± 1.3 | 0.72† | 1.7 ± 0.5 | 1 ± 0.0 | 0.34† |
| Mean normalized frequency | 0.0027 ± 0.0006 | 0.0057 ± 0.0016 | 0.09† | 0.016 ± 0.015 | 0.0002 ± 3.9 × 105 | 0.31 † |
χ2 test.
t-Test.
3.6. Altered TCRVβ gene usage
As the construction of the CVID CRD3 demonstrated less deviation from germline sequences, greater degrees of clonality, and a reduction in shared sequences, we examined the use of V gene segments in CVID compared to control DNA. For this, we calculated the proportion of sequences belonging to a specific V gene family, for both unique productive and nonproductive sequences. For productive sequences, the usage of the different V genes was significantly different between the groups (ANOVA with Tukey HSD correction for multiple comparisons p = 0.023). V-genes TRBV12-3/4, TRBV18-0, TRBV19-0, TRBV2-0, TRBV24-1, TRBV29-1, TRBV3-1, TRBV7-2, TRBV7-6, TRBV7-9, and TRBV9-0 were significantly overrepresented in CVID, whereas TRBV10-1, TRBV10-2, TRBV20-1, TRBV12-5, TRBV30-0, TRBV5-1, TRBV6-1, TRBV6-2/3, TRBV6-4, TRBV6-7, and TRBV6-8 were significantly underrepresented (Fig. 6a). Non-biased clustering analyses of V region use demonstrated that DNA samples from patients and controls could be entirely segregated (Fig. 6b). Jβ genes can be grouped into Jβ clusters 1 and 2. We noted that CVID patients favored cluster 1 genes TRBJ1-1 (CVID 12.2% ± 0.51%, controls 9.2% ± 0.84%, p = 0.002) and TRBJ1-5 (CVID 6.6% ± 0.64%, controls 4.4% ± 0.19%, p = 0.018) with a reciprocal decrease in cluster 2 genes TRBJ2-3 (CVID 12.0% ± 0.99%, controls 15.5% ± 0.54%, p = 0.017), TRBJ2-4 (CVID 2.3% ± 0.16%, controls 2.9% ± 0.10%, p = 0.024) and TRBJ2-6 (CVID 2.1% ± 0.17%, controls 3.4% ± 0.17%, p b 0.0001). The usage of the cluster 1 gene, TRBJ1-4 (CVID 3.7% ± 0.23%, controls 4.7% ± 0.24%, p = 0.006) was also decreased in CVID (Fig. S7). Vβ and Jβ usage did not segregate based on the presence of autoimmune or lymphoproliferative complications (Table E4).
Figure 6.
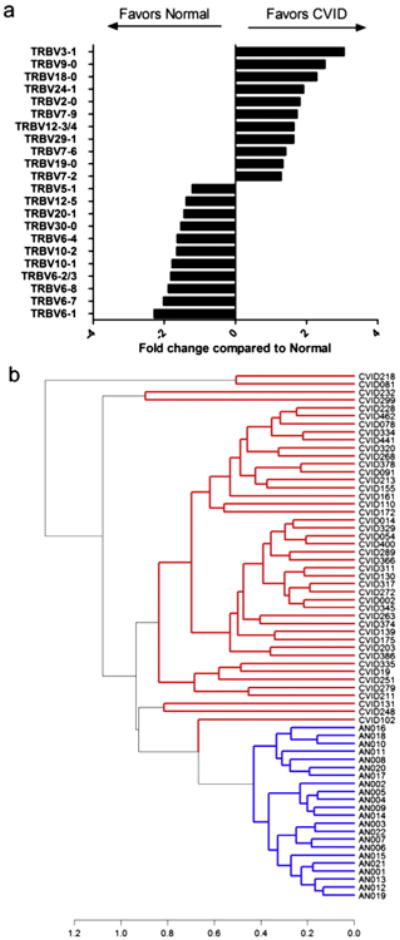
CVID DNA contains divergent Vβ families as compared to control DNA. a) The average fraction of sequences utilizing each V gene was calculated for CVID patients and controls. Ratios of CVID:normal were determined for V genes where significant differences were present. Ratios less than 1 are depicted as their negative reciprocal for ease of comparison. b) Non-biased clustering of scaled V gene distribution for the 42 CVID and 22 control subjects was performed using Manhattan distance and complete clustering algorithms. CVID DNA samples, shown in red, are completely segregated from control DNA, shown in blue. Clustering distance is indicated on the x-axis. Sample numbers are indicated on the right prefaced by CVID or AN (adult normal) for controls. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
3.7. Amino acid consequences
Both the gene preference and the junctional diversity impact the resulting amino acid sequence of the CDR3. These determine the charge and hydrophobicity of the peptide binding pocket and may affect the peptides recognized in the context of MHC. We found that the mean number of charged residues was significantly fewer in CVID sequences [1.994 ± 0.006, p < 0.0001] compared to controls [2.042 ± 0.006] (Fig. S8). To identify the determinants of this difference we modeled the number of charged residues with group (CVID or control), Vβ gene, Jβ gene, and number of insertions as the independent variables. Group (CVID or control) was a significant determinant of the number of charged residues (ANOVA with Tukey HSD correction for multiple comparisons, all p values < 0.0001), as were the choices of V and J genes and number of insertions (p values < 0.0001). Similarly, group assignment was a significant independent determinant of mean hydrophobicity.
To better understand changes in charge and hydrophobicity, we examined the distribution of these characteristics at each amino acid position for the modal CDR length of 15 amino acids. Significant differences in charge were seen at residues1, 4 to6 and 9 to 12(Fig. S9a) and in hydrophobicity at residues 1 to 5, 13 and 15 (Fig. S9b), indicating alterations in CDR3 physiochemical properties in CVID.
4. Discussion
A number of studies have demonstrated that the CVID T cell compartment suffers from numerical [8–11, 21, 22] and functional impairments [6, 7, 23–25]. While early studies have suggested a diminished diversity of the TCRβ repertoire based on spectratyping in a few subjects [7, 11], TCRβ sequences of large number of adult CVID subjects have not been examined in depth. Using high throughput sequencing we examined CVID TCRs of peripheral blood cells for CDR3 structure and diversity, and found that the CVID CDR3 repertoire is strikingly deficient in junctional modifications in V–D and D–J segments, resulting in sequences that are more similar to the germline configuration. As lengths of TCRα and β CDR3 are highly constrained [26], there was a proportional lack of deletions and insertions in CVID sequences. This was not the case in non-productive sequences where on average longer CDR3 lengths were observed in the CVID group. The presence of sequences with fewer n-nucleotides could result from either a central defect or from extra-thymic selection processes in the periphery. When analyzing unique sequences, each clone had equal weight in this analysis and demonstrated the lack of junctional modifications in CVID patients. As non-productive CVID CDR3 sequences also had fewer n-nucleotides, an intrinsic defect in the recombination process itself is more likely than defective peripheral selection. In the total repertoire this lack of junctional modifications was less apparent because peripheral expansion of T cells bearing TCRβ with fewer modifications in the periphery previously reported to occur in the normal population [27] was not seen in the CVID samples. As the CDR3 loop of TCRβ chain contributes the main contacts with antigenic peptides, reduced junctional diversity could lead to a contracted repertoire, which might explain the loss of circulating antigen-specific T cells previously observed in immunized CVID subjects [28]. While the CDR3 TCR Vβ repertoire of control subjects contained a unique population of highly modified sequences (more than 12 Vβ deletions and insertions), clones of this sort, characterized by a unique amino acid composition and greater hydrophilicity, were completely absent in CVID DNA samples.
TCR diversity is commonly measured as the inverse of the estimated clonality of the sample. Here we show that clonality of the TCRβ repertoire is clearly increased in CVID, independent of subject gender or age, and unrelated to the presence or absence of autoimmunity, inflammatory conditions, or lymphoid hyperplasia, conditions associated with oligoclonal T cell populations in other studies [29, 30]. We also did not find any evidence of clonal T cell expansions in patients who had previously treated B cell lymphoma, previous splenectomy or TACI polymorphisms. Taking all subjects with “lymphoproliferation” together also did not reveal a correlation with clonality. However, the accuracy of these correlations is limited by patient heterogeneity. It is notable that B cell oligoclonality in CVID, not indicative of lymphoma, can also be found in blood or tissues, as previously published [31].
One explanation for expanded T cell clonal populations in CVID might be homeostatic expansion, not unlikely in the setting of lymphopenia and reduced thymic output [32, 33]. Homeostatic proliferation would be consistent with our finding that clonality was closely correlated with the number of CD3 T cells in CVID subjects. For five subjects, clonality was also associated with increased numbers of CD8 T cells, as noted in an older study [11]. This association may be due to the expansion of activated CD8+ T cells in these subjects, resulting in relative paucity of naïve CD4+ T cells [8, 34]. While Vβ usage is known to differ between CD4+ and CD8+ T cells, Vβ usage for these CVID subjects did not cluster independently. Direct sequence analysis of repertoires of T cell subpopulations would be needed to clarify which differences led to the increased clonality observed in these subjects. Homeostatic proliferation is less able to explain the alterations in Vβ usage seen between CVID and controls, however, as the Vβ divergences were found in both productive and non-productive sequences. Instead, we suspect that the V region selection bias is intrinsic to thymic development in CVID, and not due to secondary selection or lineage commitment.
While we are not able to exclude the possibility that oligoclonality in CVID could be attributed to recurrent or chronic infections causing a selective expansion of some clones, we sought TCR sequences associated with recognizable T-cell activation in response to the ubiquitous viral infections EBV and CMV, both known to incite clonal expansions in healthy subjects. While evidence of increased CMV and EBV responsive T cells has been reported in CVID [35, 36], the CDR3 sequences associated with these viruses in normal individuals were actually less prevalent in CVID samples as compared to controls. This does not exclude the possibility that CVID DNA may contain T cell sequences related to these viruses as other unrecognized sequences may be still be present. In context of the overall reductions in clonal sharing in CVID, however, any anti-viral TCR sequences, if present, are more likely to be unique to each individual.
The biologic meaning of a repertoire with diminished junctional diversity, loss of highly modified clones and clonal expansions in CVID is unclear, but suggests an intrinsic contraction of the T cell repertoire, starting at the thymic level. While enhanced TCR clonality was notable in CVID DNA, clonal sharing, a feature normally observed between healthy individuals [15], was significantly reduced in CVID sequences. Both of these outcomes could possibly arise due to defects in TCRβ nucleotide insertions and deletions, leaving CVID subjects less capable of generating the full repertoire of sequences and leaving each to respond to shared microbial challenges with unique sequences. The intrinsic inability to expand T cell clones following stimulation, a known defect in this immune disorder, would further contribute to this deficit [25]. In agreement, the TCR repertoire defects found in CVID are similar tothe reduced T cell CDR3 diversity, expanded sequences closer to germline configuration, and skewed Vβ usage found in congenital immune deficiencies with the hallmark of abnormal T cell development such as severe combined immune deficiency, MHC class II defects, Omenn syndrome, and DiGeorge syndrome [37–42].
The TCR abnormalities in CVID are pervasive and do not appear to be associated with specific clinical complications, further supporting an inherent T cell defect in this disease. Gathered evidence thus builds a narrative of a combined immune defect rather than primary humoral failure for this immune defect.
Supplementary Material
Mean number of deletions when analyzing unique or total repertoires.
Normalized mean frequency of TCRS grouped by number of deletions for CVID (red) and healthy controls (blue)
Acknowledgments
We would like to thank Lin Radigan for her meticulous purification of peripheral blood DNA without which this study would not have been possible. We would like to thank Dr Harlan Robins and Adaptive Biotech Inc. for their generous support for sequencing and informatics.
This work was supported in part through the computational resources and staff expertise provided by the Department of Scientific Computing at the Icahn School of Medicine at Mount Sinai. This work was supported by grants from the National Institutes of Health, AI 1061093, AI-0860037, AI-1048693, T32-GM007280, the Jeffrey Modell Foundation, and the David S Gottesman Immunology Chair.
Footnotes
Supplementary data to this article can be found online at http://dx.doi.org/10.1016/j.clim.2015.01.002.
Conflict of interest statement: The authors declare that there are no conflicts of interest.
References
- 1.Al-Herz W, et al. Primary immunodeficiency diseases: an update on the classification from the International Union of Immunological Societies Expert Committee for Primary Immunodeficiency. Front Immunol. 2014;5:162. doi: 10.3389/fimmu.2014.00162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Warnatz K, Voll RE. Pathogenesis of autoimmunity in common variable immunodeficiency. Front Immunol. 2012;3:210. doi: 10.3389/fimmu.2012.00210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chapel H, et al. Common variable immunodeficiency disorders: division into distinct clinical phenotypes. Blood. 2008;112(2):277–286. doi: 10.1182/blood-2007-11-124545. [DOI] [PubMed] [Google Scholar]
- 4.Resnick ES, et al. Morbidity and mortality in common variable immune deficiency over 4 decades. Blood. 2012;119(7):1650–1657. doi: 10.1182/blood-2011-09-377945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gathmann B, et al. Clinical picture and treatment of 2212 patients with common variable immunodeficiency. J Allergy Clin Immunol. 2014 doi: 10.1016/j.jaci.2013.12.1077. [DOI] [PubMed] [Google Scholar]
- 6.Spickett GP, Webster AD, Farrant J. Cellular abnormalities in common variable immunodeficiency. Immunodefic Rev. 1990;2(3):199–219. [PubMed] [Google Scholar]
- 7.Giovannetti A, et al. Unravelling the complexity of T cell abnormalities in common variable immunodeficiency. J Immunol. 2007;178(6):3932–3943. doi: 10.4049/jimmunol.178.6.3932. [DOI] [PubMed] [Google Scholar]
- 8.Bateman E, et al. T cell phenotypes in patients with common variable immunodeficiency disorders: associations with clinical phenotypes in comparison with other groups with recurrent infections. Clin Exp Immunol. 2012;170(2):202–211. doi: 10.1111/j.1365-2249.2012.04643.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Guazzi V, et al. Assessment of thymic output in common variable immunodeficiency patients by evaluation of T cell receptor excision circles. Clin Exp Immunol. 2002;129(2):346–353. doi: 10.1046/j.1365-2249.2002.01893.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kamae C, et al. Common variable immunodeficiency classification by quantifying T-cell receptor and immunoglobulin κ-deleting recombination excision circles. J Allergy Clin Immunol. 2013;131(5):1437–4000000. doi: 10.1016/j.jaci.2012.10.059. [DOI] [PubMed] [Google Scholar]
- 11.Serrano D, et al. Characterization of the T cell receptor repertoire in patients with common variable immunodeficiency: oligoclonal expansion of CD8(+) T cells. Clin Immunol. 2000;97(3):248–258. doi: 10.1006/clim.2000.4941. [DOI] [PubMed] [Google Scholar]
- 12.Al-Herz W, et al. Primary immunodeficiency diseases: an update on the classification from the International Union of Immunological Societies Expert Committee for Primary Immunodeficiency. Front Immunol. 2011;2:54. doi: 10.3389/fimmu.2011.00054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Carlson CS, et al. Using synthetic templates to design an unbiased multiplex PCR assay. Nat Commun. 2013;4:2680. doi: 10.1038/ncomms3680. [DOI] [PubMed] [Google Scholar]
- 14.Sherwood AM, et al. Tumor-infiltrating lymphocytes in colorectal tumors display a diversity of T cell receptor sequences that differ from the T cells in adjacent mucosal tissue. Cancer Immunol Immunother. 2013;62(9):1453–1461. doi: 10.1007/s00262-013-1446-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Robins H, et al. Overlap and effective size of the human CD8+ T cell receptor repertoire. Sci Transl Med. 2010;2(47):47ra64. doi: 10.1126/scitranslmed.3001442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Venturi V, et al. TCR beta-chain sharing in human CD8+ T cell responses to cytomegalovirus and EBV. J Immunol. 2008;181(11):7853–7862. doi: 10.4049/jimmunol.181.11.7853. [DOI] [PubMed] [Google Scholar]
- 17.Price DA, et al. Avidity for antigen shapes clonal dominance in CD8+ T cell populations specific for persistent DNA viruses. J Exp Med. 2005;202(10):1349–1361. doi: 10.1084/jem.20051357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Iancu EM, et al. Clonotype selection and composition of human CD8 T cells specific for persistent herpes viruses varies with differentiation but is stable over time. J Immunol. 2009;183(1):319–331. doi: 10.4049/jimmunol.0803647. [DOI] [PubMed] [Google Scholar]
- 19.Miconnet I, Marrau A, Pantaleo G. TCR Repertoire Diversity in Virus-specific CD8 T-cells. 2010 doi: 10.4049/jimmunol.1003309. [DOI] [PubMed] [Google Scholar]
- 20.Miconnet I, et al. Large TCR diversity of virus-specific CD8 T cells provides the mechanistic basis for massive TCR renewal after antigen exposure. J Immunol. 2011;186(12):7039–7049. doi: 10.4049/jimmunol.1003309. [DOI] [PubMed] [Google Scholar]
- 21.Serana F, et al. Thymic and bone marrow output in patients with common variable immunodeficiency. J Clin Immunol. 2011;31(4):540–549. doi: 10.1007/s10875-011-9526-6. [DOI] [PubMed] [Google Scholar]
- 22.Arumugakani G, Wood P, Carter C. Frequency of Treg cells is reduced in CVID patients with autoimmunity and splenomegaly and is associated with expanded CD21lo B lymphocytes. J Clin Immunol. 2010;30(2):292–300. doi: 10.1007/s10875-009-9351-3. [DOI] [PubMed] [Google Scholar]
- 23.Thon V, et al. Defective integration of activating signals derived from the T cell receptor (TCR) and costimulatory molecules in both CD4+ and CD8+ T lymphocytes of common variable immunodeficiency (CVID) patients. Clin Exp Immunol. 2007;110(2):174–181. doi: 10.1111/j.1365-2249.1997.tb08314.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Di Renzo M, et al. Enhanced T cell apoptosis in common variable immunodeficiency: negative role of the fas/fasligand system and of the Bcl-2 family proteins and possible role of TNF-RS. Clin Exp Immunol. 2001;125(1):117–122. doi: 10.1046/j.1365-2249.2001.01560.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.North M, Webster A, Farrant J. Defects in proliferative responses of T cells from patients with common variable immunodeficiency on direct activation of protein kinase C. Clin Exp Immunol. 2008;85(2):198–201. doi: 10.1111/j.1365-2249.1991.tb05704.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rock EP, et al. CDR3 length in antigen-specific immune receptors. J Exp Med. 1994;179(1):323–328. doi: 10.1084/jem.179.1.323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Robins HS, et al. Comprehensive assessment of T-cell receptor beta-chain diversity in alphabeta T cells. Blood. 2009;114(19):4099–4107. doi: 10.1182/blood-2009-04-217604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Funauchi M, et al. Defects in antigen-driven lymphocyte responses in common variable immunodeficiency (CVID) are due to a reduction in the number of antigen-specific CD4+ T cells. Clin Exp Immunol. 1995;101(1):82–88. doi: 10.1111/j.1365-2249.1995.tb02281.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jones J, et al. Human autoimmunity after lymphocyte depletion is caused by homeostatic T-cell proliferation. Proc Natl Acad Sci U S A. 2013;110(50):20200–20205. doi: 10.1073/pnas.1313654110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Qi Q, et al. Mechanisms shaping the naive T cell repertoire in the elderly — thymic involution or peripheral homeostatic3 proliferation? Exp Gerontol. 2014 doi: 10.1016/j.exger.2014.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sander CA, et al. Lymphoproliferative lesions in patients with common variable immunodeficiency syndrome. Am J Surg Pathol. 1992;16(12):1170–1182. doi: 10.1097/00000478-199212000-00004. [DOI] [PubMed] [Google Scholar]
- 32.Hogan T, et al. Clonally diverse T cell homeostasis is maintained by a common program of cell-cycle control. J Immunol. 2013;190(8):3985–3993. doi: 10.4049/jimmunol.1203213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cassani B, et al. Homeostatic expansion of autoreactive immunoglobulin-secreting cells in the Rag2 mouse model of Omenn syndrome. J Exp Med. 2010;207(7):1525–1540. doi: 10.1084/jem.20091928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kuntz M, et al. Analysis of bulk and virus-specific CD8+ T cells reveals advanced differentiation of CD8+ T cells in patients with common variable immunodeficiency. Clin Immunol. 2011;141(2):177–186. doi: 10.1016/j.clim.2011.07.006. [DOI] [PubMed] [Google Scholar]
- 35.Raeiszadeh M, et al. The T cell response to persistent herpes virus infections in common variable immunodeficiency. Clin Exp Immunol. 2006;146(2):234–242. doi: 10.1111/j.1365-2249.2006.03209.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Marashi SM, et al. Inflammation in common variable immunodeficiency is associated with a distinct CD8(+) response to cytomegalovirus. J Allergy Clin Immunol. 2011;127(6):1385–1393(e4). doi: 10.1016/j.jaci.2011.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Minegishi Y, et al. Analysis of the CDR3 region of the rearranged IgH chain genes in patients with severe combined immunodeficiency and severe lymphopenia. J Immunol. 1996;156(12):4666–4671. [PubMed] [Google Scholar]
- 38.Sarzotti-Kelsoe M, et al. Thymic output, T-cell diversity, and T-cell function in long-term human SCID chimeras. Blood. 2009;114(7):1445–1453. doi: 10.1182/blood-2009-01-199323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lev A, et al. Characterizing T cells in SCID patients presenting with reactive or residual T lymphocytes. Clin Dev Immunol. 2012;2012:261470. doi: 10.1155/2012/261470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ciupe SM, et al. Quantification of total T-cell receptor diversity by flow cytometry and spectratyping. BMC Immunol. 2013;14:35. doi: 10.1186/1471-2172-14-35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lev A, et al. Thymic function in MHC class II-deficient patients. J Allergy Clin Immunol. 2013;131(3):831–839. doi: 10.1016/j.jaci.2012.10.040. [DOI] [PubMed] [Google Scholar]
- 42.Yu X, et al. Human syndromes of immunodeficiency and dysregulation are characterized by distinct defects in T-cell receptor repertoire development. J Allergy Clin Immunol. 2014;133(4):1109–1115(e14). doi: 10.1016/j.jaci.2013.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Mean number of deletions when analyzing unique or total repertoires.
Normalized mean frequency of TCRS grouped by number of deletions for CVID (red) and healthy controls (blue)