

Abstract
Objective:
To investigate the effect of different natalizumab washout (WO) periods on recurrence of MRI and clinical disease activity in patients switching from natalizumab to fingolimod.
Methods:
In this multicenter, double-blind, placebo-controlled trial (TOFINGO), patients with relapsing-remitting multiple sclerosis (RRMS) were randomized 1:1:1 to 8-, 12-, or 16-week WO followed by fingolimod treatment over 32 weeks from last natalizumab infusion (LNI). Brain MRI was performed at baseline and weeks 8, 12, 16, 20, and 24.
Results:
Of 142 enrolled and randomized patients, 112 (78.9%) completed the study (8 weeks, n = 41/50; 12 weeks, n = 31/42; 16 weeks, n = 40/50). Number (95% confidence interval [CI]) of active (new/newly enlarged T2) lesions from LNI through 8 weeks of fingolimod treatment (primary outcome) was similar in the 8-week (2.1 [1.7–2.6]) and 12-week WO groups (1.7 [1.3–2.2]) and higher in the 16-week WO group (8.2 [7.3–9.1]). During the WO period only, the number (95% CI) of active lesions increased with increasing WO duration (8 weeks, 0.4 [0.2–0.6]; 12 weeks, 2.1 [1.6–2.6]; 16 weeks, 3.6 [3.0–4.2]). Over the 24 weeks from LNI, gadolinium-enhancing T1 lesion counts were lower in the 8-week WO group (14.1 [5.67–22.53]) than in the 12-week (21.3 [1.41–41.19]) or 16-week (18.5 [8.40–28.60]) WO groups. More patients were relapse-free in the 8-week (88%) and 12-week (91%) WO groups than the 16-week WO group (84%). Sixty-eight percent of patients experienced adverse events (mostly mild/moderate), with similar incidence across groups. No unusually severe relapses or opportunistic infections occurred.
Conclusions:
Initiating fingolimod therapy 8–12 weeks after natalizumab discontinuation is associated with a lower risk of MRI and clinical disease reactivation than initiation after 16-week WO.
Classification of evidence:
This study provides Class II evidence that for patients with RRMS switching from natalizumab to fingolimod, shorter natalizumab WO periods are associated with less MRI disease activity than are longer WO periods.
Natalizumab is an α4 integrin antagonist used for the treatment of patients with relapsing multiple sclerosis (MS). Its use, however, can be associated with progressive multifocal leukoencephalopathy (PML), an uncommon but severe and potentially fatal opportunistic brain infection caused by the JC virus (JCV), which most likely results from compromised brain immunosurveillance.1,2 In anti-JCV seropositive patients (approximately 55% of patients with MS),3 the risk of PML increases with duration of natalizumab treatment and with previous immunosuppressant use.1 Anti-JCV seropositive patients with a history of previous immunosuppressant use who have been receiving natalizumab for more than 2 years have a PML risk of up to 13 per 1,000 vs <1 per 1,000 for patients with none of these risk factors.1 Therefore, in patients found to be anti-JCV seropositive, a switch from natalizumab to another treatment is considered in order to mitigate the increased risk of PML.4–7 Other factors may also lead to a decision to discontinue natalizumab, for example, persistent anti-natalizumab antibodies, suboptimal efficacy, tolerability issues,1 or patient preference for an oral therapy. However, natalizumab discontinuation could lead to the rapid return of relapse activity and MRI lesion development to pretreatment levels.1,4,8,9
Fingolimod 0.5 mg is a once-daily oral treatment10 for relapsing forms of MS11,12 with superior efficacy to the first-line therapy interferon-β-1a IM with regards to relapse rate and MRI outcomes.13 Post hoc analyses of relapse data from an open-label observational study showed that fingolimod has a beneficial effect on relapse rates in patients who discontinued natalizumab before the study, suggesting that fingolimod provides disease control after natalizumab discontinuation.14 Indirect comparisons based on phase 3 data further support the use of fingolimod as a potentially suitable switch therapy from natalizumab in appropriate patients.15
This double-blind, randomized, parallel-group study aimed to establish the optimal timing for initiating fingolimod therapy following natalizumab discontinuation, which would avoid potentially harmful additive effects on immune surveillance and would still maintain appropriate disease control.
METHODS
Study oversight and design.
This 32-week, patient- and rater-blinded, randomized, multicenter, parallel-group study was originally planned to include about 600 patients with relapsing-remitting MS (RRMS) who received their last natalizumab treatment at randomization. The sponsor prematurely stopped enrollment owing to an internal decision; however, all 142 randomized patients were allowed to continue in the study as per protocol.
Standard protocol approvals, registrations, and patient consents.
Patients eligible for inclusion were aged 18–65 years with RRMS (diagnosed using the 2010 McDonald criteria)16 and an Expanded Disability Status Scale (EDSS) score of 0–6. All patients had received natalizumab treatment for at least 6 months immediately before study screening, but were considered for treatment discontinuation for at least one of the following reasons: treatment duration >2 years, positive JCV antibody status, pretreatment with immunosuppressive drugs, adverse events (AEs) including hypersensitivity reactions, presence of anti-natalizumab neutralizing antibodies, or any other valid medical reason.
The last natalizumab infusion (LNI) had to occur within 1 week of randomization. Key exclusion criteria were consistent with those described in the phase 3 fingolimod clinical trials.13,17 Patients who had previously been treated at any time with cladribine or mitoxantrone were excluded.
All patients gave written informed consent. The study (ClinicalTrials.gov identifier NCT01499667) involved 44 study centers, and was conducted in accordance with the ethical principles of the Declaration of Helsinki.18,19 The protocol and all amendments were approved by each study center's ethics committee.
Study randomization and masking.
The study consisted of 3 phases: screening, washout (WO) following the LNI, and treatment with fingolimod 0.5 mg once daily (figure e-1 on the Neurology® Web site at Neurology.org). During the WO phase, patients were randomized 1:1:1 to 1 of 3 different natalizumab WO/fingolimod treatment regimens: 8-week WO (no therapy for 8 weeks following LNI) followed by 24 weeks of treatment with oral fingolimod 0.5 mg; 12-week WO (no therapy for 8 weeks following LNI and placebo for 4 weeks) followed by 20 weeks of treatment with fingolimod 0.5 mg; or 16-week WO (no therapy for 8 weeks following LNI and placebo for 8 weeks) followed by 16 weeks of treatment with fingolimod 0.5 mg. Randomization was managed centrally using a validated automated system (Cenduit, Durham, NC) (details are provided in the supplementary material).
Study outcomes.
The objective of the study was to evaluate disease control, using MRI and clinical outcomes, during different periods of WO following discontinuation of natalizumab and also after switching to fingolimod treatment.
The primary outcome was the number of active (new or newly enlarged) T2 lesions from the LNI, during WO, and through 8 weeks of fingolimod treatment, adjusted for the observation time (figure e-1).
The key secondary outcomes were the number of active (new or newly enlarged) T2 lesions from the LNI through to the end of the WO, and the number of active T2 lesions during the first 8 weeks of fingolimod treatment only (figure e-1). Other secondary outcomes (assessed up to week 24, the last MRI scan) included change in T2 lesion volume from baseline, gadolinium-enhancing (Gd+) lesion count, proportion of patients free from Gd+ lesions, proportion of patients free from active T2 lesions, proportion of patients free from relapses, and safety and tolerability parameters.
Assessments.
Disease control was assessed by analyzing MRI scans, MS relapses, and EDSS scores. MRI scans were obtained at baseline and at weeks 8, 12, 16, 20, and 24 and were examined by a central MRI reading center blinded to treatment allocation and clinical information. Active T2 lesions were counted by comparing the most recent MRI scan with the previous MRI scan (usually 4 weeks apart, except for weeks 0–8), not by comparison with the baseline scan. For periods of time involving multiple MRI scans, the sum of the active T2 lesion counts of each MRI was used. Gd+ lesions were counted at every scan. Patients were asked to report symptoms indicative of relapse, which had to be confirmed by an EDSS assessment in the study center within 7 days of onset. These, and the scheduled EDSS assessments at screening, week 16, and at the end of the study (week 32), were performed by a trained and certified independent evaluating physician blinded to the other data collected during the trial. Only confirmed relapses were included in analyses. In case of confirmed relapse or if MRI-related activity reached a prespecified threshold (a new lesion [Gd+ or new T2] >0.5 cm or ≥2 new lesions [Gd+ or new T2, regardless of size]), patients had the option to continue on current treatment or to be transitioned to open-label fingolimod or switched to a nonprotocol MS treatment.
AEs, serious AEs (SAEs), vital signs, and laboratory evaluations were monitored throughout the study. ECGs were obtained at baseline and at weeks 8, 12, and 16, and ophthalmologic examinations were performed at baseline and weeks 24 and 32. Satisfaction with and preference for treatments was assessed using the Treatment Satisfaction Questionnaire for Medication–9 (TSQM-9) and treatment preference questionnaire (details are provided in the supplementary material).
Statistical analysis.
Sample size calculations are reported in the supplementary material. For the primary and key secondary outcomes, a primary analysis was performed using a negative binomial regression model adjusted for each WO group, the total volume of T2 lesions at baseline, and the observation period. Owing to early termination of study enrollment and drop in sample size, the power to detect statistically significant differences in the number of active T2 lesions based on the negative binomial model across WO groups was reduced to 30%–40% of that originally planned. In addition, reported p values should be interpreted with caution because of the smaller than expected sample size. Summary statistics are provided for other outcomes. An exploratory outlier analysis of active T2 lesion counts was also performed; the 90th percentile (P90) of the number of active T2 lesions (only 10% of patients have greater than or equal to this number of active T2 lesions) was determined and the mean plus 2 times SD (mean + 2 SD) was calculated.
Kaplan-Meier plots were produced based on time to first event data, with an increase in active (new or newly enlarged) T2 lesions being considered as an event. The modified full analysis set was used for the primary outcome and included all patients who completed 8 weeks of fingolimod therapy and had an evaluable MRI scan at this time point. The analyses of the secondary variables were based on the full analysis set (FAS), which included all randomized patients, analyzed according to the WO group assigned at randomization. The safety set (SS) included all randomized patients, analyzed according to the WO group assigned on the basis of the closest match to the day on which the patient first received fingolimod. Patients with an actual WO period that fell exactly midway between the 8- and 12-week groups (i.e., 10 weeks) or the 12- and 16-week groups (i.e., 14 weeks) were assigned to the group with the longer WO time. The SS was used for analysis of all safety and tolerability variables.
Classification of evidence.
Given that, at the time of study initiation, there was no evidence-based guidance on the optimal length of the WO period between natalizumab discontinuation and fingolimod initiation, the TOFINGO study was designed to determine if different natalizumab WO periods had an effect on recurrence of MRI and clinical disease activity in patients switching from natalizumab to fingolimod. This study provides Class II evidence that for patients with RRMS switching from natalizumab to fingolimod, shorter natalizumab WO periods are associated with less MRI disease activity than longer WO periods.
RESULTS
The first patient visit took place in September 2011; the study was then terminated early in March 2012, with the final patient visit in November 2012.
Patient disposition.
A total of 142 patients were randomized and were analyzed as part of the FAS: 50, 42, and 50 patients to the 8-, 12-, and 16-week WO groups, respectively. Of these, 112 (78.9%) patients completed the study (41, 31, and 40 in the 3 groups, respectively) (figure 1). The most frequent reasons for study discontinuation reported by the investigators across the WO groups were administrative problems (n = 14 [9.9%]) and protocol deviations (n = 6 [4.2%]).
Figure 1. CONSORT flow diagram.

WO = washout.
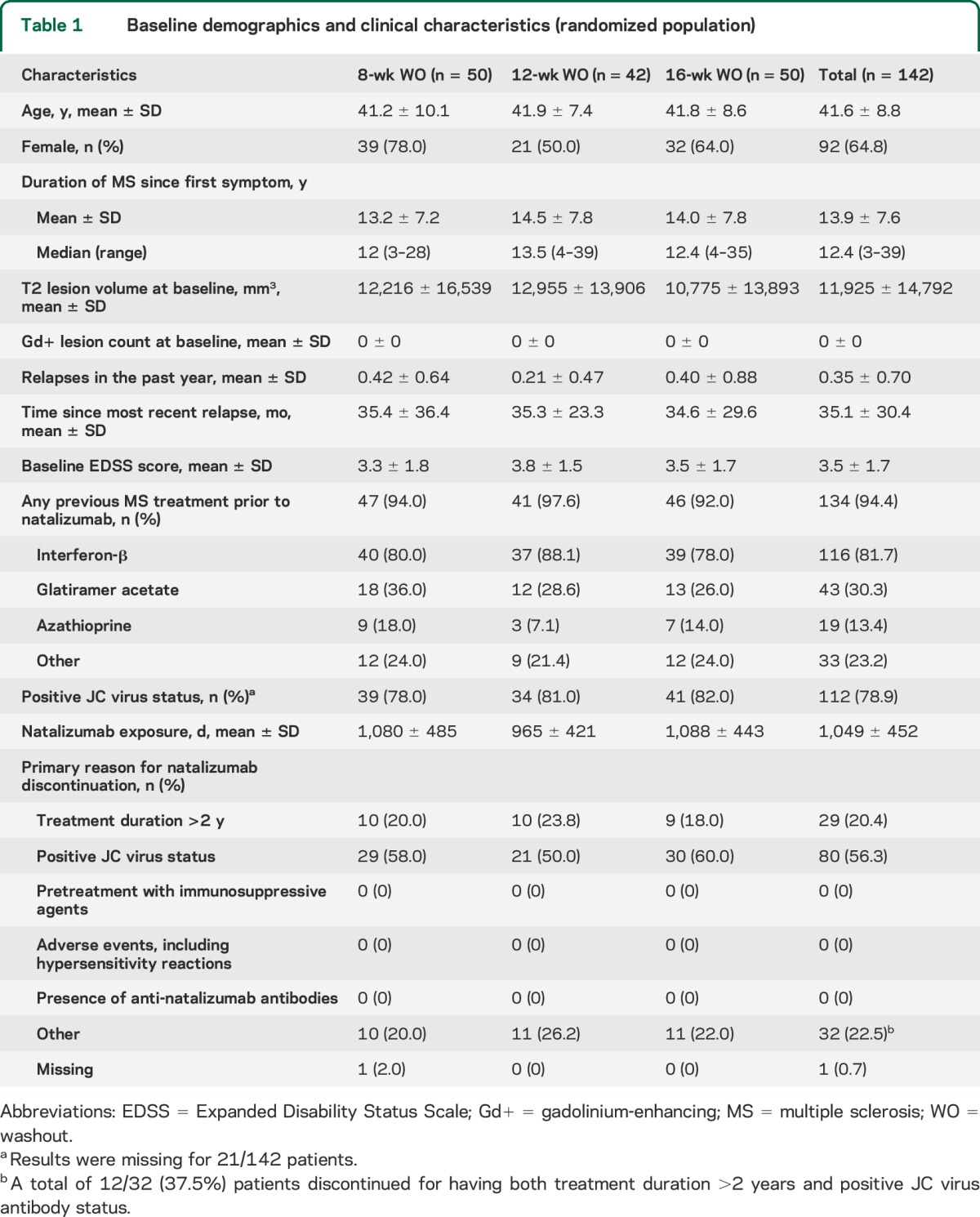
Given the relatively small sample size, baseline demographic and MS characteristics were reasonably well balanced between WO groups (table 1), with the exception of T2 lesion volume being lower in the 16-week WO group and relapses in the previous year and natalizumab exposure being lower in the 12-week WO group (all nonsignificant).
Table 1.
Baseline demographics and clinical characteristics (randomized population)
Primary outcome.
The mean number (adjusted for observation time) of active T2 lesions from the LNI through 8 weeks of fingolimod treatment was similar in the 8-week and 12-week WO groups and significantly higher in the 16-week WO than the 12-week WO group (p = 0.0393, figure 2A). Numerically more patients were free from active T2 lesions in the 8-week and 12-week WO groups, compared with those in the 16-week WO group based on Kaplan-Meier estimates for the time to first active T2 lesion (secondary outcome), which was shortest for the 8-week WO group (figure 2B).
Figure 2. Active T2 lesions.

(A) Number of active T2 lesions (sum of all active T2 lesions on MRI scans during specified time interval in each group) since last infusion of natalizumab for the washout (WO) period and 8 weeks of fingolimod therapy (adjusted for observation time). (B) Kaplan-Meier estimate of time to a new or newly enlarged T2 lesion (adjusted for observation time; modified full analysis set population). The number of new or newly enlarged T2 lesions (C) since the last natalizumab infusion for the WO period only and (D) for the first 8 weeks of fingolimod therapy only (adjusted for the observation time; full analysis set population). *Two-sided statistical significance at 0.05 level, calculated using a negative binomial regression model adjusted for WO group and baseline T2 lesion volume, with the logarithm of the observation period as the offset variable. **Difference was statistically significant for the 8-week vs the 12-week (p < 0.0001) and 16-week (p = 0.0013) WO groups for the first key secondary endpoint and for the second key secondary endpoint, for the 8-week vs the 16-week WO group (p = 0.0351). A 2-sided statistical significance at 0.05 level was indicated, calculated using the negative binomial regression model adjusted for WO group, baseline T2 lesion volume, and observation period. CI = confidence interval.
Key secondary outcomes.
During the WO period, the mean number of active T2 lesions (adjusted for observation time) (figure 2C) significantly increased with increasing duration of WO (8-week vs 12-week WO [p < 0.0001] and vs 16-week WO [p = 0.0013]).
During the first 8 weeks of fingolimod treatment, the mean number of active T2 lesions was numerically lower in the 8-week compared with the 12-week WO group and significantly lower in the 8-week than the 16-week WO group (p = 0.0351) (figure 2D). Similarly, more patients were free from active T2 lesions in the 8-week than in the 12- or 16-week WO groups (figure 2D).
Relapses.
Over the 24-week study period, the annualized relapse rate was highest for patients in the 16-week WO group and similar in the 8-week and 12-week WO groups (table 2). Accordingly, over the entire study a smaller proportion of patients (no significant difference) were relapse-free in the 16-week group vs the 8-week or 12-week WO groups. There was no clear pattern of relapse occurrence relative to the time since the last natalizumab dose.
Table 2.
Clinical disease activity
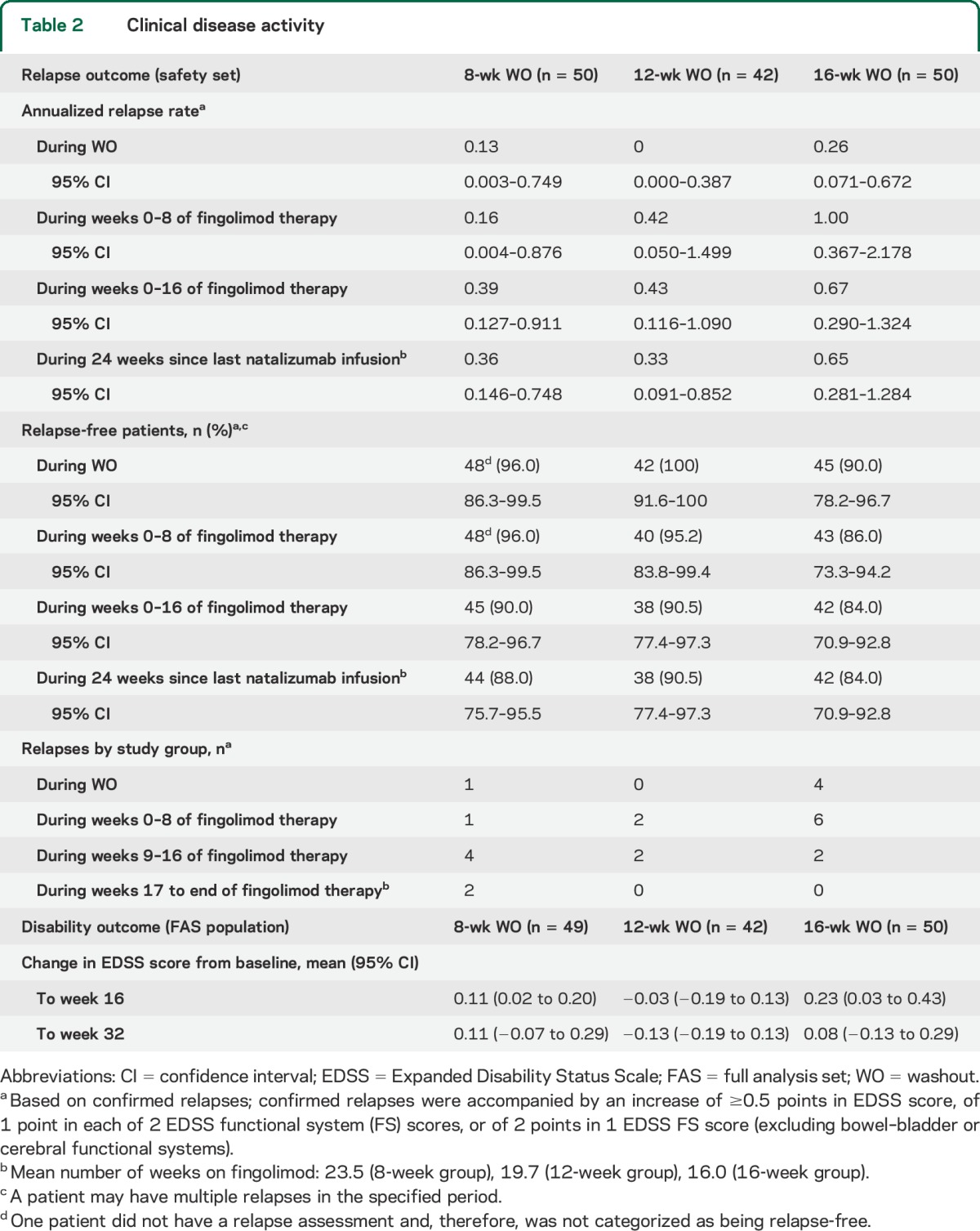
During the WO period, 1 patient in the 16-week group reported a relapse, classified by the investigator as a moderate AE, and received IV methylprednisolone. During fingolimod therapy, 1 relapse was reported as a moderate AE in the 12-week group. In the 16-week group, 3 relapses were reported as AEs in 3 patients; one was classed as a moderate AE and 2 patients were hospitalized (SAEs). Across groups, all relapsed patients were treated with IV methylprednisolone.
Other secondary outcomes.
Active T2 lesion counts and proportion of patients free from active T2 lesions over 24 weeks are summarized in table 3. Up to week 24, patients in the 8-week WO group had numerically lower mean and median number of Gd+ T1 lesions (not significant) and proportionately more patients were free from Gd+ lesions than in the 12- or 16-week WO groups (table 3). Active T2 lesion counts per observation interval (during the first 8 weeks of WO and every 4 weeks thereafter) are summarized in table e-1.
Table 3.
MRI disease activity at week 24 (FAS population)
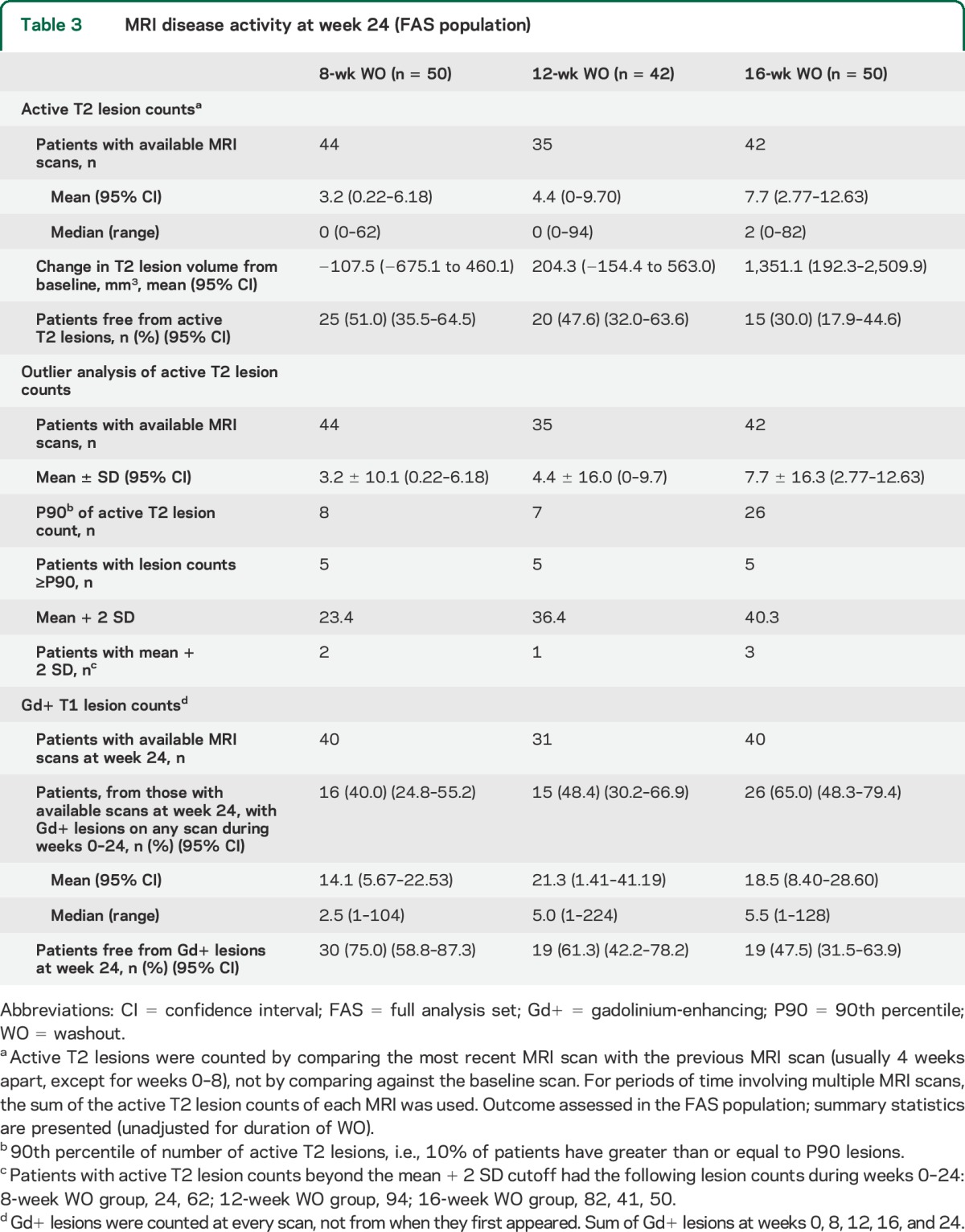
A small number of patients had high active T2 lesion counts (table 3); of the 121 patients in the FAS, 15 patients were at or above the P90 in terms of a cumulative number of active T2 lesions over 24 weeks, with the greatest number of active T2 lesions found in patients in the 16-week WO group. Six of these 15 patients had active T2 lesion counts that were greater than the mean + 2 SD (table 3). Interestingly, 5 of these patients relapsed during the study: 2 patients in the 8-week WO group at study day 91 and day 105 (34 and 49 days since starting fingolimod, respectively), and 3 patients in the 16-week WO group at study days 113, 138, and 139 (1, 26, and 14 days since starting fingolimod, respectively). MRI activity preceded or coincided with relapses in all cases and all relapses were mild/moderate in severity; all patients recovered either partially or completely.
There was no significant difference in mean EDSS scores between treatment groups at week 32 (table 2). Treatment preference questionnaire and TSQM-9 results are presented in table e-2.
Safety.
AEs during the study, including both the WO and fingolimod treatment periods, were reported for 67.6% of patients; the overall proportions of patients with AEs were the same in the 8-week and 16-week WO groups, and lower in the 12-week WO group. In particular, more AEs were reported during the longer vs shorter WO periods, and during fingolimod therapy, the number of reported AEs was highest for the group that had received fingolimod for the longest duration (table 4). The most frequent AEs reported in at least 5% of patients during the WO period, regardless of WO group, were headache, sinusitis, depression, and dizziness (table 4). During fingolimod treatment, infection incidence was greater in the 8-week WO group (n/N = 15/50 patients; 30.0%) than in the 12-week (n/N = 6/42; 14.3%) or 16-week (n/N = 7/50; 14.0%) groups. In particular, a higher incidence of nasopharyngitis (n = 6, 12%) and urinary tract infections (n = 3, 6%) were reported in the 8-week group than in the 12-week (n = 1, 1% for both) or 16-week groups (n = 2, 4%; n = 0, respectively) (table 4), which accounted for the majority of the infections reported during fingolimod therapy. The majority of infections were mild and the occurrence did not show any particular temporal pattern (data on file); there was no spike in infection frequency during the first 8 weeks of treatment in the 8-week group when natalizumab may not have fully washed out.
Table 4.
Adverse events reported in ≥5% of patients during WO and fingolimod treatment and serious adverse eventsa during WO and fingolimod treatment (safety set)
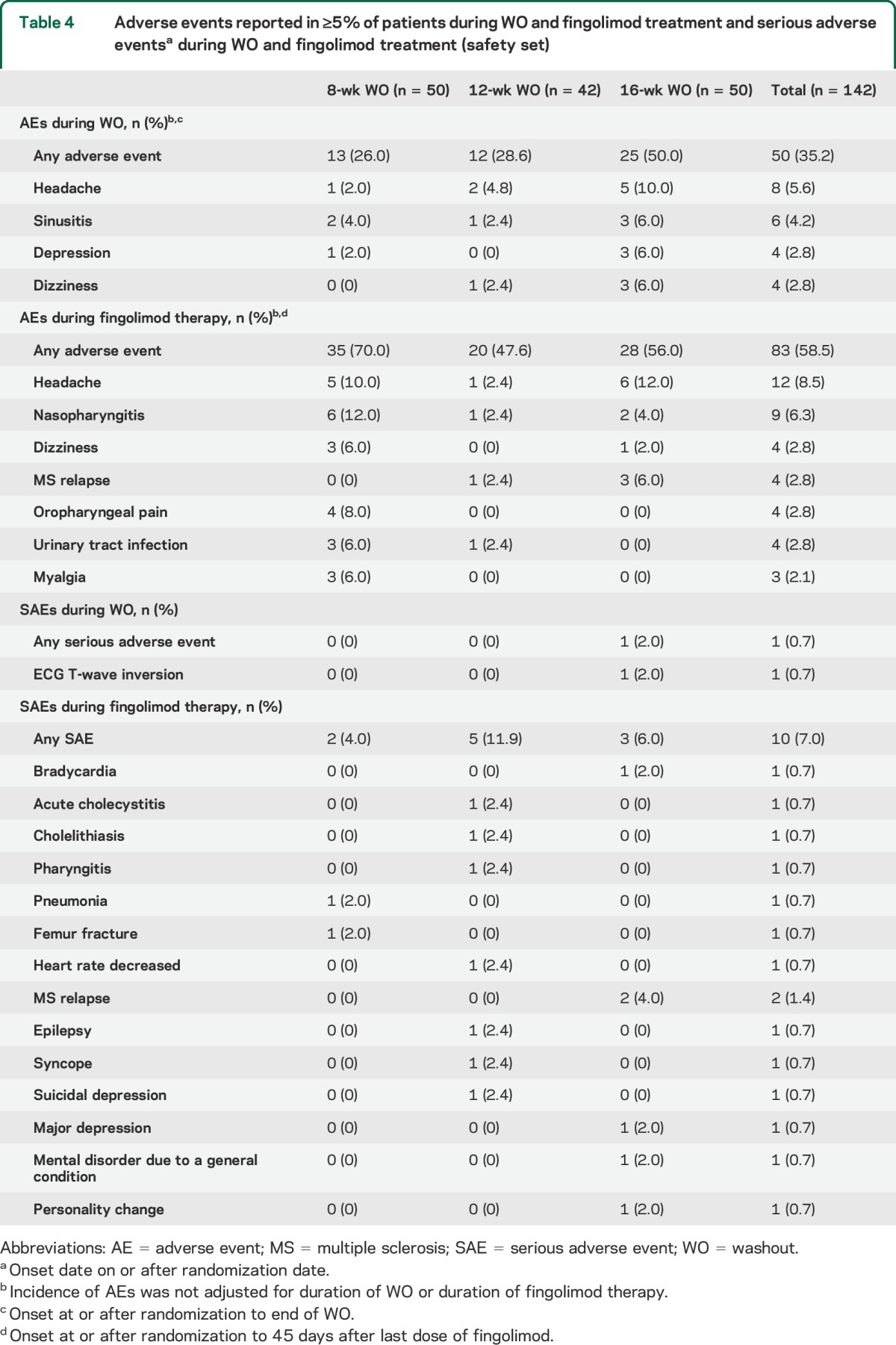
There were no deaths during the study. During the WO period, 1 patient in the 12-week group (dizziness) and 1 patient in the 16-week group (ECG T-wave inversion) discontinued owing to AEs; the latter was also reported as a SAE due to hospitalization of the patient. During the fingolimod treatment period, 2 patients discontinued fingolimod (12-week group, cholelithiasis; 16-week group, hepatic enzyme increase); SAEs were reported for 10 patients: 2 patients in the 8-week, 5 patients in the 12-week, and 3 patients in the 16-week WO groups (table 4). None of the SAEs was reported as severe. No systemic opportunistic infections or PML cases were reported. There were no clinically significant differences between the WO groups in laboratory and vital sign parameters during the WO and fingolimod treatment periods (data not shown).
DISCUSSION
Currently, there is no evidence-based guidance on the optimal length of the WO period between natalizumab discontinuation and fingolimod initiation. In the EU label, caution is recommended for the initiation of fingolimod therapy within 2–3 months of natalizumab discontinuation.12 In this randomized, patient- and rater-blinded, parallel-group study, we assessed systematically the effects of variable WO periods between discontinuation of natalizumab and the initiation of fingolimod treatment. Generally, a shorter WO period was associated with less MS disease activity. WO periods of 8 weeks or 12 weeks were associated with less MRI and clinical disease activity compared with the 16-week WO, in line with reports showing the return of MS disease activity 16–28 weeks after natalizumab discontinuation6,9,20 and previous uncontrolled studies showing that initiating fingolimod later than 12 weeks after the LNI was associated with higher initial MRI and clinical disease activity.5–7,9,21,22 Although there was no difference between the 8-week and 12-week WO groups for the primary outcome, evidence from secondary endpoints on disease activity favored 8-week WO over longer WO periods.
Analysis of active T2 lesion outlier patients showed that lesions either preceded or coincided with relapses for all 6 patients with counts outside the mean + 2 SD; however, this analysis is limited by the fact that MRI scans were performed only at set study visits, so lesion activity may not have been captured at the time of onset.
Our data suggest that, for optimal disease control, fingolimod treatment should be initiated between 8 and 12 weeks after discontinuation of natalizumab infusions. Recently, a period of 4–8 weeks has been recommended in order to reduce the likelihood of disease reactivation.7,23 Nonetheless, the potential risk of additive effects on the immune system should be considered within the first 3 months since natalizumab discontinuation, given the long elimination half-life of natalizumab and also the prolonged effect of natalizumab on immunosurveillance.12,24 There is an overlap between natalizumab and fingolimod treatment in the 8-week WO group due to the half-life of natalizumab. However, treatment with fingolimod has a differential effect on T and B cells, and can preserve immunosurveillance because immunocompetent regulatory and memory cells are free to circulate throughout the body, including the CSF.25,26 It has recently been shown that fingolimod-treated patients exhibited increased regulatory B-cell frequencies in the CSF.27 Although vigilance for serious infections such as PML is warranted following natalizumab discontinuation, current experience has thus far not indicated an increased risk of PML attributable to fingolimod treatment,28 and no severe or systemic opportunistic infections occurred in any of the fingolimod study groups reported up to now.13,17,29,30 Although we observed a higher overall incidence of infections in the 8-week WO group, these were largely attributable to the higher incidence of nasopharyngitis and urinary tract infections. The majority of infections were mild or moderate in severity and an increased incidence during the first few weeks of fingolimod treatment, when natalizumab may not have fully washed out, was reassuringly not observed (data on file).
In this short-term study, treatment was generally well tolerated irrespective of the WO period; the incidence of SAEs and discontinuations due to AEs were similar across the 3 WO groups. Of note, there were no unusually severe relapses reported in this study. Severe relapses have been reported in a small number of patients switching from natalizumab to fingolimod in clinical practice settings and have been interpreted as the result of disease reactivation before the onset of action of fingolimod.31
There were limitations to this study, primarily related to the lower than initially planned number of patients enrolled after the decision to stop recruitment. This study was therefore too small to allow full exploration of the risks of a short WO period in terms of serious infections. However, despite the sample size, the study results were analyzed as stipulated in the original study protocol. Although the power for the analysis was decreased, the numerical trends and proportions are consistent and indicative.
Overall, findings from the primary endpoint of this blinded, controlled study suggest that initiation of fingolimod therapy 8–12 weeks after discontinuation of natalizumab infusions is more effective in controlling disease reactivation than if it is initiated after a 16-week WO. Evidence from secondary endpoints on disease activity favors 8-week over longer WO periods. Additional observational or controlled studies are needed to determine if shorter than 8-week WO intervals would further improve disease control with reasonable risks.
Supplementary Material
ACKNOWLEDGMENT
The authors thank the patients who participated in the study and the study site personnel.
GLOSSARY
- AE
adverse event
- EDSS
Expanded Disability Status Scale
- FAS
full analysis set
- Gd+
gadolinium-enhancing
- JCV
JC virus
- LNI
last natalizumab infusion
- MS
multiple sclerosis
- P90
90th percentile
- PML
progressive multifocal leukoencephalopathy
- RRMS
relapsing-remitting multiple sclerosis
- SAE
serious adverse event
- SS
safety set
- TSQM-9
Treatment Satisfaction Questionnaire for Medication–9
- WO
washout
Footnotes
Editorial, page 14
Supplemental data at Neurology.org
Contributor Information
Collaborators: Helmut Butzkueven, Richard Macdonell, Eva-Maria Maida, Veronika Ticha, Marta Vachova, Eva Meluzinova, Mikko Kallela, Heinz Wiendl, Kerstin Hellwig, Mathias Maeurer, Johannes Boehringer, Wilfried Lueer, Hans-Juergen von Giesen, Arnfin Bergmann, Andreas Bitsch, Thomas Brosch, Peter Emrich, Klaus Gehring, Michael Lang, Gerd Reifschneider, Gerhard Roth, Eugen Schlegel, Sylke Schlemilch-Paschen, Erik Strauss, Joerg Osterhage, Andreas Link, Andreas Steinbrecher, Nikolaos Vlaikidis, Antonios Tavernarakis, Nikolaos Fakas, Marinos Dalakas, Athanasios Kyritsis, Magdolna Simo, Gyula Panczel, Ron Milo, Adi Vaknin, Giancarlo Comi, Luigi Maria Grimaldi, Xavier Montalban, Guillermo Izquierdo, Oscar Fernandez Fernandez, Antonio Escartin Siquier, Exuperio Diez, and Ludwig Kappos
AUTHOR CONTRIBUTIONS
The study was designed by the sponsor, Novartis Pharma AG, in collaboration with the steering committee. Data were collected by the investigators and analyzed by the sponsor. Novartis contributed to the interpretation of the study. All authors had full access to the data and had final responsibility for the contents and decision to submit for publication. L. Kappos: design and conceptualization of the study, analysis and interpretation of the data, drafting and revising the manuscript. E.-W. Radue: design and conceptualization of the study, analysis and interpretation of the data, and drafting and revising the manuscript. G. Comi: design and conceptualization of the study, analysis and interpretation of the data, and drafting and revising the manuscript. X. Montalban: design and conceptualization of the study, analysis and interpretation of the data, and drafting and revising the manuscript. H. Butzkueven: design and conceptualization of the study, analysis and interpretation of the data, and drafting and revising the manuscript. H. Wiendl: design and conceptualization of the study, analysis and interpretation of the data, and drafting and revising the manuscript. G. Giovannoni: design and conceptualization of the study, analysis and interpretation of the data, and drafting and revising the manuscript. H.-P. Hartung: analysis and interpretation of the data and revising the manuscript. T. Derfuss: analysis and interpretation of the data and revising the manuscript. Y. Naegelin: design and conceptualization of the study, analysis and interpretation of the data, and drafting and revising the manuscript. T. Sprenger: design and conceptualization of the study, analysis and interpretation of the data, and drafting and revising the manuscript. N. Mueller-Lenke: design and conceptualization of the study, analysis and interpretation of the data, and drafting and revising the manuscript. S. Griffiths: drafting and revising the manuscript. P. von Rosenstiel: interpretation of the data and revising the manuscript. R. Gottschalk: design and conceptualization of the study, analysis and interpretation of the data, and revising the manuscript. Y. Zhang: design and conceptualization of the study, statistical analysis and interpretation of the data, and revising the manuscript. F. Dahlke: design and conceptualization of the study, analysis and interpretation of the data, and revising the manuscript. D. Tomic: design and conceptualization of the study, analysis and interpretation of the data, and drafting and revising the manuscript.
STUDY FUNDING
The study was sponsored by Novartis Pharma AG.
DISCLOSURE
L. Kappos Institution (University Hospital Basel) has received the following in the last 3 years, used exclusively for research support: steering committee, advisory board, and consultancy fees from Actelion, Addex, Bayer Health Care, Biogen, Biotica, Genzyme, Lilly, Merck, Mitsubishi, Novartis, Ono Pharma, Pfizer, Receptos, Sanofi-Aventis, Santhera, Siemens, Teva, UCB, and Xenoport; speaker fees from Bayer Health Care, Biogen, Merck, Novartis, Sanofi-Aventis, and Teva; support of educational activities from Bayer Health Care, Biogen, CSL Behring, Genzyme, Merck, Novartis, Sanofi, and Teva; royalties from Neurostatus Systems GmbH; and grants from Bayer Health Care, Biogen, Merck, Novartis, Roche, Swiss MS Society, the Swiss National Research Foundation, and the European Union and Roche Research Foundations. E.-W. Radue has received honoraria for serving as speaker at scientific meetings and consultant for Bayer Schering, Biogen Idec, Merck Serono, and Novartis. He has received financial support for research activities from Actelion, Basilea Pharmaceutica, Biogen Idec, Merck Serono, and Novartis. G. Comi has received consulting fees for participating in advisory boards from Actelion Pharmaceuticals, Bayer Schering Pharma, GeNeuro, Merck Serono, Novartis, Sanofi-Aventis, and Teva Pharmaceutical Industries, and lecture fees from Bayer Schering Pharma, Biogen-Dompé, Merck Serono, Novartis, Sanofi-Aventis, Serono Symposia International Foundation, and Teva Pharmaceutical Industries. X. Montalban has received honoraria and travel expenses for speaking at scientific meetings, has been a steering committee member of clinical trials, or participated in advisory boards of clinical trials in the past years with Almirall, Bayer Schering Pharma, Biogen Idec, EMD Serono, Genentech, Genzyme, Novartis, Sanofi-Aventis, and Teva Pharmaceutical Industries. H. Butzkueven has received compensation for serving as a consultant or speaker for or has received research support from Biogen Idec, Merck Serono, Novartis, and Sanofi-Aventis; he is on the editorial boards of Multiple Sclerosis Journal and Multiple Sclerosis and Related Disorders; and he has received project grants from the National Health and Medical Research Council, Australian Research Council Linkage Grant RG, and National MS Society (USA). H. Wiendl has received compensation for serving as a consultant or speaker for, or has received research support from, Bayer Schering Pharma, Biogen Idec/Elan Corporation, Merck Serono, Novartis, Novo Nordisk, Sanofi-Aventis, and Teva Pharmaceutical Industries. G. Giovannoni has received compensation for serving as a consultant or speaker for, or has received research support from, AbbVie, Bayer Schering Healthcare, Biogen Idec, Canbex, Eisai, Elan, Five Prime Therapeutics, Genzyme, Genentech, GlaxoSmithKline, Ironwood Pharmaceuticals, Merck-Serono, Novartis, Pfizer, Roche, Sanofi-Aventis, Synthon BV, Teva Pharmaceutical Industries, UCB, and Vertex Pharmaceuticals. H.-P. Hartung has received speaking fees and honoraria for consulting from Bayer Healthcare, Biogen Idec, GeNeuro, Genzyme, EMD Serono, Novartis, Octapharma, Opexa, Receptos, Roche, Teva Pharmaceutical Industries, and Sanofi-Aventis. T. Derfuss served on advisory boards for Bayer Schering Pharma, Biogen Idec, GeNeuro, Genzyme, Merck-Serono GmbH, Mitsubishi Pharma, Novartis Pharma, and Teva Pharmaceutical Industries. He received travel support from Bayer Schering Pharma, Biogen Idec, Genzyme, and Merck Serono GmbH. He received research and/or unrestricted grants from Biogen Idec, the European Union, the German Research Foundation, Merck Serono GmbH, Novartis Pharma, the Swiss National Foundation, and the Swiss MS Society. Y. Naegelin reports no disclosures relevant to the manuscript. T. Sprenger has received no personal compensation. The University Hospital Basel, as his employer, received compensation for serving on scientific advisory boards from Actelion, ATI, Biogen Idec, Genzyme, and Novartis. N. Mueller-Lenke has received compensation for serving as a consultant or speaker for, or has received research support from, Biogen Idec, Merck-Serono, and Novartis. S. Griffiths is an employee of Oxford PharmaGenesis Ltd., funded by Novartis Pharma AG. P. von Rosenstiel is a Novartis employee. R. Gottschalk is a Novartis employee. Y. Zhang is a Novartis employee. F. Dahlke is a Novartis employee. D. Tomic is a Novartis employee. Go to Neurology.org for full disclosures.
REFERENCES
- 1.FDA. Tysabri US prescribing information. Revised December 2013. Available at: http://www.accessdata.fda.gov/drugsatfda_docs/label/2013/125104s840s847s889lbl.pdf. Accessed February 12, 2014.
- 2.Bloomgren G, Richman S, Hotermans C, et al. Risk of natalizumab-associated progressive multifocal leukoencephalopathy. N Engl J Med 2012;366:1870–1880. [DOI] [PubMed] [Google Scholar]
- 3.FDA. Tysabri US prescribing information. Revised May 2013. Available at: http://www.accessdata.fda.gov/drugsatfda_docs/label/2013/125104s813lbl.pdf. Accessed November 22, 2013.
- 4.Martinelli V, Colombo B, Dalla Costa G, et al. Recurrent disease-activity rebound in a patient with multiple sclerosis after natalizumab discontinuations for pregnancy planning. Mult Scler Epub 2013 Jun 17. [DOI] [PubMed]
- 5.O'Connor PW, Goodman A, Kappos L, et al. Disease activity return during natalizumab treatment interruption in patients with multiple sclerosis. Neurology 2011;76:1858–1865. [DOI] [PubMed] [Google Scholar]
- 6.Cohen M, Maillart E, Tourbah A, et al. Switching from natalizumab to fingolimod in multiple sclerosis: a French prospective study. JAMA Neurol 2014;71:436–441. [DOI] [PubMed] [Google Scholar]
- 7.Jokubaitis VG, Li V, Kalincik T, et al. Fingolimod after natalizumab and the risk of short-term relapse. Neurology 2014;82:1204–1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gueguen A, Roux P, Deschamps R, et al. Abnormal inflammatory activity returns after natalizumab cessation in multiple sclerosis. J Neurol Neurosurg Psychiatry 2014;85:1038–1040. [DOI] [PubMed] [Google Scholar]
- 9.Fox RJ, Cree BA, De Seze J, et al. MS disease activity in RESTORE: a randomized 24-week natalizumab treatment interruption study. Neurology 2014;82:1491–1498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Brinkmann V, Billich A, Baumruker T, et al. Fingolimod (FTY720): discovery and development of an oral drug to treat multiple sclerosis. Nat Rev Drug Discov 2010;9:883–897. [DOI] [PubMed] [Google Scholar]
- 11.Novartis Pharmaceuticals Corp. Gilenya US Prescribing Information. Revised May 2012. Available at: http://www.pharma.us.novartis.com/product/pi/pdf/gilenya.pdf. Accessed April 16, 2014. [Google Scholar]
- 12.European Medicines Agency. Summary of product characteristics. Updated June 18, 2012. Available at: http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Product_Information/human/002202/WC500104528.pdf. Accessed April 16, 2014.
- 13.Cohen JA, Barkhof F, Comi G, et al. Oral fingolimod or intramuscular interferon for relapsing multiple sclerosis. N Engl J Med 2010;362:402–415. [DOI] [PubMed] [Google Scholar]
- 14.Comi G, Gold R, Dahlke F, et al. Relapses in patients treated with fingolimod after previous exposure to natalizumab. Mult Scler 2015;21:786–790. 10.1177/1352458514549404. [DOI] [PubMed] [Google Scholar]
- 15.Nixon RM, Bergvall N, Sfikas N, et al. Indirect comparisons of oral fingolimod versus natalizumab on measures of disease freedom in patients with multiple sclerosis, based on results from FREEDOMS and AFFIRM. Poster presented at AAN 2013, San Diego, CA, 2013; P01.209.
- 16.Polman CH, Reingold SC, Banwell B, et al. Diagnostic criteria for multiple sclerosis: 2010 revisions to the McDonald criteria. Ann Neurol 2011;69:292–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kappos L, Radue EW, O'Connor P, et al. A placebo-controlled trial of oral fingolimod in relapsing multiple sclerosis. N Engl J Med 2010;362:387–401. [DOI] [PubMed] [Google Scholar]
- 18.International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use. ICH Harmonised Tripartite Guideline. Guideline for Good Clinical Practice. Available at: http://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Efficacy/E6/E6_R1__Guideline.pdf. Accessed May 18, 2015. [Google Scholar]
- 19.World Medical Association. Declaration of Helsinki: Ethical Principles for Medical Research Involving Human Subjects. Available at: http://www.wma.net/en/30publications/10policies/b3/index.html. Accessed October 4, 2013. [Google Scholar]
- 20.Kappos L, Bates D, Edan G, et al. Natalizumab treatment for multiple sclerosis: updated recommendations for patient selection and monitoring. Lancet Neurol 2011;10:745–758. [DOI] [PubMed] [Google Scholar]
- 21.Havla J, Tackenberg B, Hellwig K, et al. Fingolimod reduces recurrence of disease activity after natalizumab withdrawal in multiple sclerosis. J Neurol 2013;260:1382–1387. [DOI] [PubMed] [Google Scholar]
- 22.Sempere AP, Martin-Medina P, Berenguer-Ruiz L, et al. Switching from natalizumab to fingolimod: an observational study. Acta Neurol Scand 2013;128:e6–e10. [DOI] [PubMed] [Google Scholar]
- 23.de Seze J, Ongagna JC, Collongues N, et al. Reduction of the washout time between natalizumab and fingolimod. Mult Scler 2013;19:1248. [DOI] [PubMed] [Google Scholar]
- 24.Stüve O, Marra CM, Jerome KR, et al. Immune surveillance in multiple sclerosis patients treated with natalizumab. Ann Neurol 2006;59:743–747. [DOI] [PubMed] [Google Scholar]
- 25.Mehling M, Brinkmann V, Antel J, et al. FTY720 therapy exerts differential effects on T cell subsets in multiple sclerosis. Neurology 2008;71:1261–1267. [DOI] [PubMed] [Google Scholar]
- 26.Claes N, Dhaeze T, Fraussen J, et al. Compositional changes of B and T Cell subtypes during fingolimod treatment in multiple sclerosis patients: a 12-month follow-up study. PLoS One 2014;9:e111115. 10.1371/journal.pone.0111115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Grützke B, Hucke S, Wildemann B, et al. Fingolimod treatment promotes regulatory B-cell phenotype and function in multiple sclerosis. J Neuroimmunol 2014;275:42–43. [Google Scholar]
- 28.Putzki N, Clifford DB, Bischof D, et al. Characteristics of PML cases in multiple sclerosis patients switching to fingolimod from natalizumab. Mult Scler 2014;Abstract FC3.1. [Google Scholar]
- 29.Calabresi PA, Radue EW, Goodin D, et al. Safety and efficacy of fingolimod in patients with relapsing-remitting multiple sclerosis (FREEDOMS II): a double-blind, randomised, placebo-controlled, phase 3 trial. Lancet Neurol 2014;13:545–556. [DOI] [PubMed] [Google Scholar]
- 30.Kappos L, Cohen J, Collins W, et al. Fingolimod in relapsing multiple sclerosis: an integrated analysis of safety findings. Mult Scler Relat Disord 2014;3:494–504. [DOI] [PubMed] [Google Scholar]
- 31.Centonze D, Rossi S, Rinaldi F, Gallo P. Severe relapses under fingolimod treatment prescribed after natalizumab. Neurology 2012;79:2004–2005. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.