

Abstract
AIM: This study investigated the mechanisms of protection afforded by the proton pump inhibitor lansoprazole against gastric injury induced by different non-steroidal anti-inflammatory drugs (NSAIDs) in rats.
METHODS: Male Sprague-Dawley rats were orally treated with indomethacin (100 µmol/kg), diclofenac (60 µmol/kg), piroxicam (150 µmol/kg) or ketoprofen (150 µmol/kg). Thirty minutes before NSAIDs, animals were orally treated with lansoprazole 18 or 90 µmol/kg. Four hours after the end of treatments, the following parameters were assessed: gastric mucosal PGE2, malondialdehyde (MDA), myeloperoxidase (MPO) or non-proteic sulfhydryl compounds (GSH) levels; reverse transcription-polymerase chain reaction (RT-PCR) of mucosal COX-2 mRNA; gastric acid secretion in pylorus-ligated animals; in vitro effects of lansoprazole (1-300 µmol/L) on the oxidation of low density lipoproteins (LDLs) induced by copper sulphate.
RESULTS: All NSAIDs elicited mucosal necrotic lesions which were associated with neutrophil infiltration and reduction of PGE2 levels. Increments of MPO and MDA contents, as well as a decrease in GSH levels were detected in the gastric mucosa of indomethacin- or piroxicam-treated animals. Indomethacin enhanced mucosal cyclooxygenase-2 expression, while not affecting cyclooxygenase-1. At the oral dose of 18 µmol/kg lansoprazole partly counteracted diclofenac-induced mucosal damage, whereas at 90 µmol/kg it markedly prevented injuries evoked by all test NSAIDs. Lansoprazole at 90 µmol/kg reversed also the effects of NSAIDs on MPO, MDA and GSH mucosal contents, without interfering with the decrease in PGE2 levels or indomethacin-induced cyclooxygenase-2 expression. However, both lansoprazole doses markedly inhibited acid secretion in pylorus-ligated rats. Lansoprazole concentration-dependently reduced the oxidation of LDLs in vitro.
CONCLUSION: These results suggest that, besides the inhibition of acid secretion, lansoprazole protection against NSAID-induced gastric damage depends on a reduction in mucosal oxidative injury, which is also responsible for an increment of sulfhydryl radical bioavailability. It is also suggested that lansoprazole does not influence the down-regulation of gastric prostaglandin production associated with NSAID treatment.
Keywords: Lansoprazole, Gastric injury, Non-steroidal anti-inflammatory drugs
INTRODUCTION
Non-steroidal anti-inflammatory drugs (NSAIDs) are among the most commonly prescribed drugs due to their high efficacy in the treatment of pain, fever and inflammation. However, the use of these drugs is associated with the occurrence of adverse digestive effects, consisting most notably of gastric erosions, ulceration, bleeding and perforation[1]. It is widely accepted that both the beneficial and detrimental effects of NSAIDs are attributable to their ability to inhibit prostaglandin synthesis through a direct blockade of cyclooxygenase (COX)[2]. In particular, the sequelae of suppressed prostaglandin production, which may contribute to the pathogenesis of gastroduodenal ulceration, include a decrease in mucus synthesis and secretion of mucus, an inhibition of bicarbonate secretion, a reduction of mucosal blood flow, an alteration of microvascular structure and an increase in acid and pepsinogen secretion[1,3].
The pathogenesis of NSAID-induced gastrointestinal damage may also depend on prostaglandin-independent mechanisms, such as uncoupling of oxidative phosphorylation, alterations of mucosal cell turnover as well as neutrophil activation followed by enhanced endothelial adhesion[1,4]. These mechanisms, in combination with those related to prostaglandin suppression, lead to microvessel occlusion and subsequent hyperproduction of reactive oxygen metabolites. Such substances are then able to induce oxidative tissue injury which seems to play a prominent role in the development of mucosal ulceration caused by NSAIDs[3,5].
The impairment of mucosal defence allows gastric acid to cause injury. Therefore, gastric acid is regarded as a pivotal step in the pathogenesis of gastroduodenal ulceration, and acid suppression is viewed as a main option in the treatment of NSAID-induced mucosal damage[6]. For these reasons, proton pump inhibitors (PPIs), including lansoprazole and other related compounds, have been investigated in clinical studies in patients under treatment with NSAIDs, and proven to be effective in both prevention and healing of NSAID-induced gastric lesions[7]. Indeed, PPIs can markedly reduce gastric acidity through the blockade of H+, K+-ATPase, the enzyme responsible for the final step in the secretion of hydrochloric acid by parietal cells[8]. However, there is also evidence to suggest that these drugs can activate gastric protective mechanisms independent of acid reduction[9,10]. For instance, in previous studies omeprazole and lansoprazole prevented gastric injury elicited by acidified ethanol or hemorrhagic shock through an enhancement of mucosal defence[9].
At present, a large variety of endogenous factors, including prostaglandins, growth factors, digestive hormones, sensory peptides, nitric oxide, and sulfhydryl compounds, has been implicated in the mechanisms through which gastric mucosa counteracts ulcerative stimuli[11], and some of these factors have been proposed to account for the gastroprotective effects of PPIs[9,10]. Nevertheless, the possible contribution of acid-independent mechanisms to the protective effects of these drugs against NSAID-induced gastric injury remains to be clarified. Overall, the present study was designed to: (1) examine the influence of lansoprazole on gastric mucosal damage induced by different NSAIDs; (2) evaluate to what extent factors and mechanisms related to gastric mucosal defence are involved in the protective actions of lansoprazole; (3) assess whether lansoprazole is endowed with direct antioxidant properties.
MATERIALS AND METHODS
Animals and experimental design
Albino male Wistar rats, with 200-220 g body weight, were used throughout the study. They were fed standard laboratory chow and tap water ad libitum and were not used for at least one week after their delivery to the laboratory. The animals were housed, four in a cage, in temperature-controlled rooms on a 12-h light cycle at 22-24°C and 50-60% relative humidity. They were deprived of food 24 h before the experiment. However, free access to water ad libitum was allowed until 1 h before the beginning of experiments. Experiments were performed in accordance with the provisions of the European Community (EC) Council Directive 86-609, recognized and adopted by the Italian Government.
In experiments on gastroprotection, lansoprazole (18 or 90 µmol/kg) or its vehicle was administered by intragastric gavage. The following NSAIDs, which are associated with moderate to high levels of risk for gastric injury in clinical settings[4], were then administered by intragastric gavage at doses known to induce gastric lesions in rats: indomethacin (100 µmol/kg[12]), diclofenac (60 µmol/kg[13]), piroxicam (150 µmol/kg[14]), and ketoprofen (150 µmol/kg[15]). In all cases, a 30 min interval was allowed between lansoprazole and NSAID administration. Four hours after treatment with NSAIDs, the animals were sacrificed by cervical dislocation, their stomachs were rapidly removed and processed for the evaluation of the following parameters: (1) histomorphometric analysis of gastric mucosal damage; (2) quantitative estimation of mucosal inflammatory activity; (3) assay of malondialdehyde (MDA), non-proteic sulfhydryl compounds and prostaglandin E2 (PGE2) in the mucosal layer; (4) assessment of mucosal expression of COX isoforms (COX-1, COX-2).
In an additional set of experiments, the effects of lansoprazole (18 or 90 µmol/kg) or its vehicle were assayed on gastric acid secretion in rats subjected to pylorus ligation. Dose levels of lansoprazole were selected on the basis of previous studies in rat models of gastric injury, showing that this PPI exerted maximal protective actions at 90 µmol/kg[9].
Animals subjected to NSAID-induced gastric mucosal damage
Histomorphometric evaluation of gastric mucosal damage The histomorphometric estimation of gastric mucosal damage was carried out as previously described[16]. Briefly, the stomach was opened along the greater curvature, gently washed with saline (154 mmol/L NaCl), pinned upon a cork plate with the mucosal surface turned upwards, and fixed in 10% formalin buffered with phosphate for 24 h at 4°C. Each stomach was dissected in parallel strips perpendicular to the lesser curvature at a distance of 2 mm. The strips from each stomach were sequentially placed on a glass slide and oriented with the side of each strip distal to the pylorus upwards. A solution of melted 3% agar was gently poured on the strips and quickly cooled at 4°C to induce solidification. The agar block was then removed from the glass slide, dehydrated, and embedded in paraffin wax. Three micrometers thick paraffin sections were cut using a microtome and stained with hematoxylin and eosin. Sections were examined by light microscopy and the length of both total and damaged mucosa was evaluated by means of a micrometric scale. The lesion index was estimated as the length fraction of damaged mucosa over the total length of mucosa, and expressed in percentage values.
Determination of mucosal myeloperoxidase content Myeloperoxidase (MPO) was assayed as previously reported by Pacheco et al[17] and assumed as a quantitative index to estimate the degree of mucosal infiltration by polymorp-honuclear cells elicited by treatment with test NSAIDs. Briefly, specimens of mucosa were rapidly scraped from the underlying tissue layers of gastric wall using two glass slides kept cold on ice. Mucosal samples (300 mg) were homogenized 3 times (30 s each) at 4°C with a polytron homogenizer (Cole Parmer Homogenizer, Vernon Hills, IL, USA) in 1 mL of ice-cold 50 mmol/L phosphate buffer (pH 6.0) containing 0.5% of hexadecyltrime-thylammonium bromide to prevent the pseudoperoxidase activity of hemoglobin as well as to solubilize membrane-bound MPO. The homogenate was sonicated for 10 s, frozen-thawed 3 times and spun by centrifugation for 20 min at 18 000 g. The supernatant was then recovered and used for determination of MPO concentration by means of enzyme-linked immunosorbent assay (ELISA) (Bioxytech, Oxis International Inc., Portland, OR, USA). All samples were assayed within 2 d after collection. The results were expressed as ng of MPO per 100 mg of gastric mucosa.
Quantitative evaluation of mucosal oxidative damage MDA concentrations in samples of gastric mucosa were determined to obtain quantitative estimations of membrane lipid peroxidation evoked by test NSAIDs. For this purpose, specimens of mucosa were scraped from the underlying tissue layers of gastric wall using two glass slides kept cold on ice. The mucosa was weighed, minced by forceps, homogenized in 2 mL of cold buffer (Tris-HCl 20 mmol/L, pH 7.4) using a polytron homogenizer (Cole Parmer Homogenizer), and spun by centrifugation at 1 500 g for 10 min at 4°C. Aliquots of supernatants were then used for subsequent assay procedures. Mucosal MDA concentrations were estimated by colorimetric assay (Calbiochem-Novabiochem Corporation, San Diego, CA, USA), and the results were expressed as nmol of MDA per milligram of gastric mucosa.
Assay of sulfhydryl compounds in gastric mucosa The concentrations of reduced glutathione (GSH) in specimens of gastric mucosa were determined to estimate the tissue content of non-proteic sulfhydryl compounds[18]. For this purpose, samples of mucosa were scraped from the underlying layers of gastric wall using two glass slides kept cold on ice. The scraped mucosa was weighed, minced by forceps, homogenized in 2 mL of cold buffer [0.4 mol/L 2-(N-morpholino)-ethanesulfonic acid, 0.1 mol/L phosphate, and 2 mmol/L ethylendiaminotetracetic acid (EDTA), pH 6] using a polytron homogenizer (Cole Parmer Homogenizer), and spun by centrifugation at 10 000 r/min for 15 min at 4°C. Aliquots of supernatants were deproteinated by a solution containing 1.25 mol/L metaphosphoric acid and 4 mol/L triethanolamine, to avoid interferences due to particulate components or proteic sulfhydryl groups, and then used for the subsequent assay procedures. Gastric mucosal levels of GSH were determined by enzyme colorimetric assay (Cayman Chemicals, Ann Arbor, MI, USA), and the results were expressed as nmol of GSH per milligram of gastric mucosa.
Assay of PGE2 in gastric mucosa
Enzyme immunoassay of PGE2 in gastric mucosa was performed by means of a commercial kit (Cayman Chemical Company, Ann Arbor, MI, USA). Briefly, specimens of mucosa were rapidly scraped from the underlying tissue layers of gastric wall using two glass slides kept cold on ice. The mucosa was weighed, minced by forceps, and homogenized in 1 mL of cold phosphate buffer (PBS 0.1 mol/L, pH 7.4, containing 1 mmol/L EDTA and 10 µmol/L indomethacin) per gram of tissue using a polytron homogenizer (Cole Parmer Homogenizer, Vernon Hills, IL, USA). The resulting homogenate was added with an equal volume of absolute ethanol, and stirred with vortex. After 5-min incubation at room temperature, the homogenate was spun by centrifugation at 1 500 r/min for 10 min at 4°C. The supernatant was added with 1 mol/L HCl until pH 4. Before the assay, samples were subjected to purification by means of superclean LC-18 SPE columns (Sigma Co., St. Louis, MO, USA). For this purpose, 0.5 mL of sample was added to 2 mL of ethanol and vortexed. After incubation at room temperature for 5 min, the sample was spun by centrifugation at 3 000 r/min for 10 min. The supernatant was then removed and applied to the LC-18 SPE column which was previously activated with 5 mL of methanol and then with 5 mL of ultrapure water. The column was subsequently washed with 5 mL of ultrapure water and 5 mL of hexane. PGE2 was then eluted with 5 mL of ethyl acetate containing 1% methanol. The eluted ethyl acetate fractions were collected and evaporated to dryness with nitrogen. Aliquots were then used for subsequent enzyme immunoassay. PGE2 concentration was expressed as nanogram per gram of mucosal tissue.
Western blot assay of COX-1 and COX-2 in gastric mucosa
Specimens of mucosa were scraped from the underlying tissue layers of gastric wall using two glass slides kept cold on ice. Mucosal samples were weighed and homogenized in lysis buffer containing: HEPES 10 mmol/L, NaCl 30 mmol/L, EDTA 0.2 mmol/L, phenylmethylsulphonyl fluoride 2 mmol/L, leupeptin 10 µg/mL, aprotinin 10 µg/mL, sodium fluoride 1 mmol/L, sodium orthovanadate 1 mmol/L, glycerol 2%, MgCl2 0.3 mmol/L, and Triton-X 100 1%, using the polytron homogenizer. Mucosal homogenates were spun by centrifugation at 20 000 g for 15 min at 4°C, and the resulting supernatants were then separated from pellets and stored at -80°C. Protein concentration was determined in each sample by the Bradford method (Protein Assay kit, Bio-Rad, Hercules, CA, USA). To perform Western blot analysis of COX-1 and COX-2, equivalent amounts of protein lysates (50 µg) were separated by electrophoresis on sodium dodecylsulfate polyacrylamide gel (8%) and transferred onto a nitrocellulose membrane. The blots were then blocked overnight with 5% non-fat dried milk in phosphate buffered saline, and incubated overnight at room temperature with goat polyclonal antiserum raised against rat COX-1 or COX-2 (dilution 1:1000). After repeated washings with 0.1% Tween-20 in Tris-buffered saline, a peroxidase-conjugated rabbit anti-goat antibody (dilution 1:10000) was added for 1 h at room temperature. After repeated washings with 0.1% Tween-20 in Tris-buffered saline, the immunoreactive bands were visualized by enhanced chemiluminescence (ECL, Amersham Biosciences Europe GmbH, Cologno Monzese, Italy). The relative expression of COX-1 or COX-2 was quantified by densitometric analysis with NIH Image computer program (Scion Corporation, USA).
Animals subjected to pylorus ligation
Pylorus ligation was carried out as previously described[19]. Briefly, during a short anesthesia with diethyl ether, the abdomen was opened by a midline laparotomy and the duodenum was exteriorized. Then, lansoprazole (18 or 90 µmol/kg) or its vehicle was directly injected into the distal portion of duodenum by means of a 25-gauge needle, the pylorus was ligated, the abdominal incision was closed with clips, and the animals were allowed to recover from anesthesia for 10 min. Two hours after pylorus ligation, the esophageal-gastric junction was ligated, and the whole stomach was excised. The gastric content was emptied, carefully collected in graduated centrifuge tubes, and spun by centrifugation at 3 000 r/min for 10 min. Samples with more than 0.5 mL of sediment were discarded. The level of acidity was measured by automatic potentiometric titration to pH 7.0 with 0.01 mol/L NaOH, using an Autotitrator pH Meter PHM82 (Radiometer, Copenhagen, Denmark), and evaluated as H+ output. The effect of lansoprazole was expressed as µEqH+/2 h.
In vitro assay of antioxidant activity of lansoprazole
To evaluate whether lansoprazole is endowed with direct antioxidant properties, the drug was incubated in a reaction mixture where human native low density lipoproteins (LDLs) were subjected to oxidation upon exposure to CuSO4 at 37°C for 120 min, in accordance with a standardized method[20]. Lansoprazole was assayed at concentrations ranging from 1 to 300 µmol/L. The reaction mixture consisted of 2 mL of a phosphate buffer (10 mmol/L KH2PO4/K2HPO4, pH 5.3) containing LDLs 150 µg/mL and CuSO4 1 µmol/L. At the end of incubation, the oxidative reaction was stopped by addition of buthyldihy-droxytoluene 0.5 mol/L in acetonitrile, and the extent of LDL oxidation was estimated by measurement of 8-iso-prostaglandin F2α (8-iso-PGF2α) concentrations. For this purpose, 100-µL aliquots of the reaction mixture were used to assay 8-iso-PGF2α by means of a commercial kit for competitive enzyme-linked immunoassay (Cayman Chemicals, Ann Arbor, MI, USA). The results were expressed as pg of 8-iso-PGF2α per mL.
Statistical analysis
The results are given as mean±SE. The statistical significance of data was evaluated by one-way analysis of variance (ANOVA) followed by post hoc analysis by Student-Newman-Keuls test, and P values lower than 0.05 were considered significant; “n” indicates the number of experiments. All statistical procedures were performed by personal computer programs. Drugs The following drugs and reagents were used: indomethacin, ketoprofen, diclofenac, piroxicam, human native low density lipoproteins, butyldihy-droxytoluene, HEPES, EDTA, phenylmethylsulphonyl fluoride, leupeptin, aprotinin, sodium orthovanadate, glycerol, Triton-X, Tween-20, sodium dodecylsulfate, hexadecyltrimethylammonium bromide (Sigma Chemicals Co., St. Louis, MO, USA); lansoprazole (kindly provided by Takeda Italia Farmaceutici, Rome, Italy); diethyl ether (Mallinckrodt Baker BV, Deventer, The Netherlands); polyacrylamide (Bio-Rad, Hercules, CA, USA); goat anti-rat COX-1 and COX-2 antibodies, peroxidase-conjugated rabbit anti-goat antibody (Santa Cruz Biotechnology Inc., Santa Cruz, CA, USA). Other reagents were of analytical grade. Lansoprazole and test NSAIDs were suspended in 1% methocel and administered in a volume of 0.5 mL.
RESULTS
Animals subjected to NSAID-induced gastric mucosal damage
Effects of lansoprazole on gastric mucosal damage induced by NSAIDs Intragastric administration of test NSAIDs caused various degrees of macroscopically evident mucosal damage (not shown). Histological examination of gastric sections showed that treatment with test NSAIDs elicited necrotic mucosal lesions associated with widespread areas of infiltration by polymorphonuclear cells in the upper part of the submucosal layer and the lower part of mucosa (Figure 1). Histomorphometric analysis of gastric tissue revealed negligible lesions of surface epithelium in control animals, as well as those receiving lansoprazole alone, with a total damage accounting for (0.27 ± 0.12)% (control), (0.18 ± 0.11)% (lansoprazole 18 µmol/kg) and (0.09 ± 0.03)% (lansoprazole 90 µmol/kg). By contrast, in animals treated with indomethacin, diclofenac, piroxicam or ketoprofen, histomorphometry displayed a necrotic damage affecting (6.38 ± 0.32)%, (4.48 ± 0.26)%, (6.43 ± 0.54)%, and (5.62 ± 0.39)% of gastric mucosa, respectively (Figure 2). Under these conditions, pretreatment with lansoprazole (90 µmol/kg) caused marked reductions of NSAID-induced gastric mucosal injury (Figure 2), which was associated with a concomitant decrease in the degree of infiltration by polymorphonuclear cells (Figure 1). When assayed at the dose of 18 µmol/kg, lansoprazole exerted a slight influence on NSAID-induced mucosal damage, achieving a level of statistical significance only in animals exposed to diclofenac (Figure 2).
Figure 1.
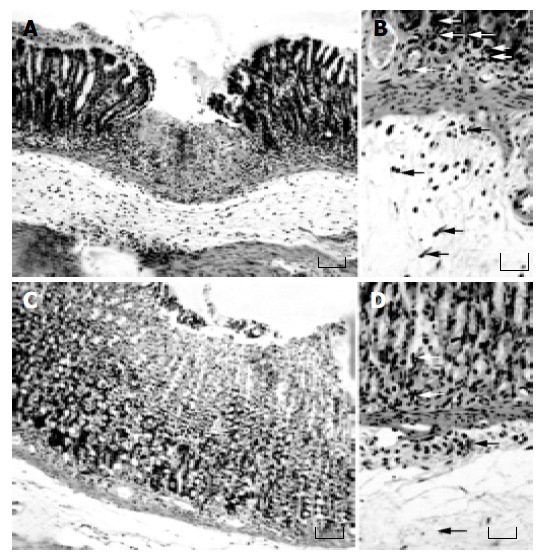
H&E-stained sections of rat gastric mucosa. A, treatment with indomethacin 100 µmol/kg, A: severe lesion with necrosis of gastric mucosa; B: several polymorphonuclear cell scan be detected (arrows); C and D: Rats treated with lansoprazole 90 mmol/kg plus indomethacin 100 µmol/kg, C: less severe lesion with lysis of mucosal cells; D: few polymorphonuclear cells can be evidenced (arrows). A and C: original magnification 100; bar = 150 µm. B and D: original magnification 170 x; bar = 60 µm.
Figure 2.
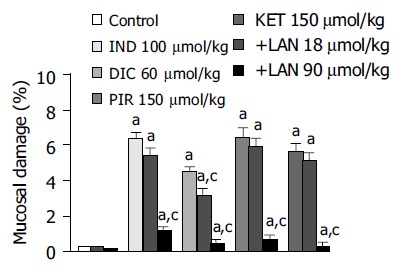
Histomorphometric evaluation of gastric mucosal damage in rats subjected to intragastric administration of indomethacin (IND, 100 µmol/kg), diclofenac (DIC, 60 µmol/kg), piroxicam (PIR, 150µmol/kg) or ketoprofen (KET, 150 µmol/kg) either alone or in the presence of lansoprazole (LAN, 18 and 90 µmol/kg). Each column indicates the mean value obtained from 6-8 animals ± SE (vertical lines). aP < 0.05 vs control values; cP < 0.05 between the respective values obtained in animals treated with a test NSAID alone.
Assay of MPO, MDA, and GSH in gastric mucosa
MPO levels in the gastric mucosa of control animals were 3.57 ± 0.78 ng/100 mg, and this value was not significantly affected by treatment with lansoprazole alone (90 mmol/kg). In this set of experiments, indomethacin, and piroxicam induced marked increments of mucosal MPO concentrations (+116.8% and +131.1%, respectively), which no longer occurred after pretreatment of animals with lansoprazole (Figure 3A).
Figure 3.
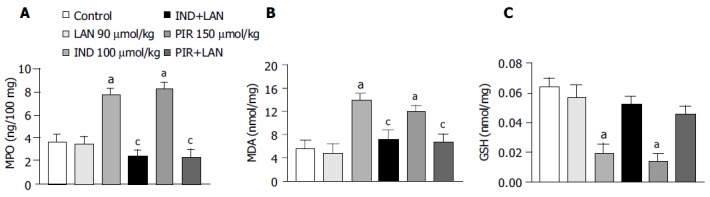
MPO (A), MDA (B) and GSH (C) levels in the gastric mucosa of rats treated with indomethacin (IND, 100 µmol/kg) or piroxicam (PIR, 150 µmol/kg), either alone or in the presence of lansoprazole (LAN, 90 µmol/kg). Each column represents the mean value obtained from 7 to 10 animals ± SE. aP < 0.05 vs control values; cP < 0.05 between the respective values obtained in animals treated with indomethacin or piroxicam alone.
MDA levels in the gastric mucosa of control rats were 5.6 ± 1.4 nmol/mg tissue. Lansoprazole alone (90 µmol/kg) did not influence basal MDA concentrations. Treatment with indomethacin or piroxicam caused a significant increase in mucosal MDA concentrations (+148.2% and +112.5%, respectively), and pretreatment with lansoprazole significantly prevented the tissue accumulation of MDA promoted by test NSAIDs (Figure 3B).
In control animals, mucosal GSH levels reached 0.064 ± 0.006 nmol/mg. Under these conditions, lansoprazole alone (90 µmol/kg) did not significantly affect the mucosal concentration of endogenous sulfhydryl compounds. Treatment with indomethacin or piroxicam elicited a significant reduction of mucosal GSH levels (-70.3% and -79.7%, respectively). However, the decrease in gastric GSH concentration caused by test NSAIDs no longer occurred upon pretreatment of animals with lansoprazole (Figure 3C).
Assay of PGE2 in gastric mucosa
Basal PGE2 levels in the gastric mucosa of control animals accounted for 151.9 ± 13.3 ng/g, this value being not significantly affected by administration of lansoprazole alone (90 µmol/kg). All test NSAIDs caused a significant reduction in mucosal PGE2 levels. Marked inhibitory effects were observed in animals treated with indomethacin, piroxicam and ketoprofen (-74.8%, -79% and -86.6%, respectively), whereas a moderate inhibitory action could be detected in diclofenac-treated rats (-48.4%). Pretreatment with lansoprazole did not significantly interfere with the inhibitory actions of test NSAIDs on mucosal PGE2 production (Figure 4).
Figure 4.
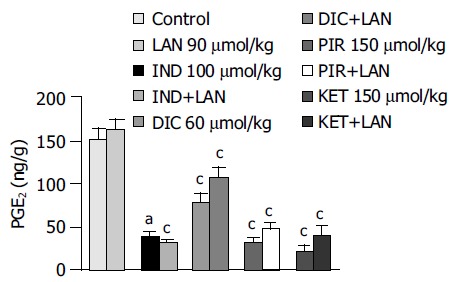
PGE2 levels in the gastric mucosa of rats treated with indomethacin (IND, 100 µmol/kg), diclofenac (DIC, 60 µmol/kg), piroxicam (PIR, 150 µmol/kg) or ketoprofen (KET, 150 µmol/kg), either alone or in the presence of lansoprazole (LAN, 90 µmol/kg). Each column represents the mean value obtained from 6 to 8 animals ± SE (vertical lines). aP < 0.05 vs control values; cP < 0.05 between the respective values obtained in animals treated with a test NSAID alone.
Western blot assay of COX-1 and COX-2 in gastric mucosa
The protein expression of COX isoforms was evaluated by Western blot analysis of gastric mucosal samples obtained from animals subjected to vehicle, indomethacin, lansoprazole or lansoprazole plus indomethacin administration. Western blot assay revealed the expression of both COX-1 and COX-2 in the gastric mucosa of control animals as well as in rats treated with lansoprazole alone (90 µmol/kg) (Figure 5). The densitometric analysis of immunoreactive bands showed that the relative expression of COX-1 or COX-2, in the presence of lansoprazole, did not differ significantly from that estimated in control animals. Treatment with indomethacin was followed by a marked enhancement of COX-2, but not COX-1, expression in the gastric mucosa. A similar effect was exerted by indomethacin upon pre-treatment of animals with lansoprazole (Figure 5).
Figure 5.
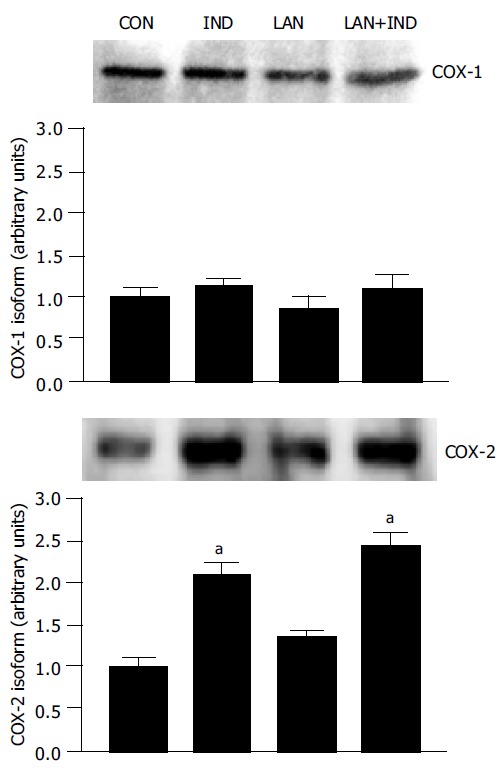
Western blot analysis of COX-1 and COX-2protein expression in gastric mucosal lysates obtained from animals treated with indomethacin (IND), lansoprazole (LAN, 90 µmol/kg) or lansoprazole plus indomethacin (LAN+IND). aP < 0.05 vs the control values (CON).
Animals subjected to pylorus ligation
In control animals subjected to pylorus ligation for 2 h and pretreated with lansoprazole vehicle the gastric acid output accounted for 153.7 ± 5.8 µEqH+/2 h (n = 6). The administration of lansoprazole (18 or 90 µmol/kg) at the time of pylorus ligation caused a significant decrease in acidity (-81.4% and -93.1%, respectively; Figure 6).
Figure 6.
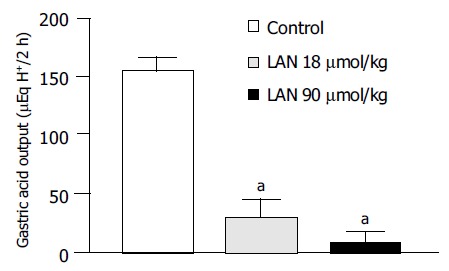
Rats subjected to pylorus ligation. Effect of lansoprazole (LAN, 18 and 90 µmol/kg) on gastric acid secretion. Each column represents the mean value obtained from six experiments ±SE(vertical lines). aP < 0.05 vs the control values.
In vitro assay of antioxidant activity of lansoprazole
In control experiments, 8-iso-PGF2α generation promoted by CuSO4-induced oxidation of LDLs accounted for 1 261.3 ± 113.7 pg/mL. When the oxidative reaction was carried out in the presence of increasing concentrations of lansoprazole (1-300 µmol/L), the production of 8-iso-PGF2α underwent a progressive decrease. Under these conditions, the antioxidant activity of lansoprazole achieved a significant level at 3 µmol/L (-17%), and a maximal effect could be detected at the concentration of 100 µmol/L (-85.9%, Figure 7).
Figure 7.
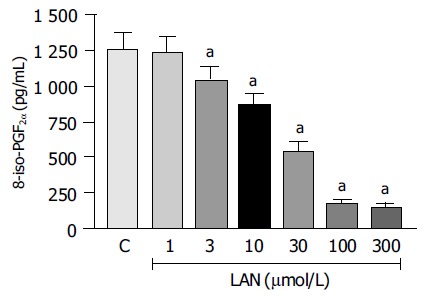
8-Iso-PGF2α levels generated in vitro upon oxidation of native LDLs by CuSO4, either in the absence (C, control) or in the presence of increasing concentrations of lansoprazole (LAN). Each column represents the mean value obtained from 5-6 experiments±SE (vertical lines). aP < 0.05 vs the control values.
DISCUSSION
It is currently acknowledged that the ability of PPIs to prevent NSAID-associated gastric ulcers in clinical settings results from their inhibition of acid secretion[1]. However, several lines of evidence indicate that acid-independent mechanisms may contribute to the antiulcer actions of PPIs[9,10]. In the present study, the protective properties of lansoprazole were examined in a rat model of mucosal injury evoked by NSAIDs associated with high risk of adverse digestive events in clinical practice[4]. The histomorphometric analysis of gastric sections showed that the damaging effects of test NSAIDs were prevented by lansoprazole, and evidence was also obtained that the ability to interfere with the oxidative and inflammatory injuries evoked by NSAID treatment plays a relevant role in the gastroprotective action of this benzimidazole derivative.
NSAIDs assayed in the present investigation elicited areas of mucosal necrosis which were associated with destruction of glandular architecture, submucosal edema, and extensive infiltration by polymorphonuclear cells. This pattern of gastric damage was also characterized by a marked increase in mucosal MPO levels, regarded as a reliable marker of tissue neutrophil infiltration, which occurred together with an increment of mucosal MDA content and a decrease in GSH concentration. MDA is an end product resulting from peroxidation of polyunsaturated fatty acids and related esters within cell membranes, and the measurement of this substance represents a suitable index of oxidative tissue injury[21]. On the other hand, sulfhydryl compounds are involved in the maintenance of gastric integrity, particularly when reactive oxygen species are implicated in the pathophysiology of tissue damage[22,23]. Indeed, GSH participates in many aspects of oxidative metabolism, including the neutralization of hydroperoxides and the maintenance of the physiological sulfhydryl status of proteins[23,24]. Overall, the present findings are consistent with previous evidence indicating that NSAIDs, acting through both local and systemic mechanisms, promote ischemic and inflammatory alterations which result in gastric neutrophil infiltration, release of oxygen metabolites, and cell membrane peroxidation[1,3]. In particular, focal ischemia, characterized by neutrophil adherence to vascular endothelium, capillary congestion and intravascular fibrin deposition, has been recognized as an early event in the pathogenesis of NSAID-induced gastric oxidative injury, and these alterations are further amplified by subsequent tissue infiltration with polymorphonuclear cells, the activation of which causes a massive release of reactive oxygen radicals and other inflammatory mediators responsible for epithelial damage[1,25]. In these respects, rat models, as that adopted in the present study, can be regarded as predictive of NSAID-induced gastric alterations occurring in clinical settings, since gastric erosions elicited by aspirin in healthy volunteers were accompanied by marked increments of mucosal MPO activity and lipid peroxidation[5].
It is recognized that acid secretion can facilitate or exacerbate the damage to gastric mucosa evoked by exogenous agents, including NSAIDs, and acid suppression is then regarded as the main target of most pharmacological treatments designed to prevent or heal gastric ulcers[26]. However, NSAID-induced ulcers result mainly from alterations of protective mechanisms, and some arguments suggest that acid secretion may not represent a pivotal step in the pathophysiology of NSAID gastropathy[1,3]. For instance, achlorhydria does not protect patients against upper digestive ulcers during NSAID use[27], and H2 receptor antagonists appear to be scarcely effective in preventing NSAID-associated gastric ulcers under both experimental and clinical conditions[28]. In the present study, NSAID-induced gastric lesions were extensively prevented by lansoprazole 90 µmol/kg, whereas rather weak effects were detected with 18 µmol/kg, a dose shown to ensure a marked blockade of acid secretion in pylorus-ligated rats. On these bases, it can be proposed that acid-independent mechanisms concur with inhibition of acid secretion for gastric protection afforded by lansoprazole against NSAIDs. In particular, since in our experiments lansoprazole prevented mucosal MDA generation elicited by test NSAIDs, it is conceivable that this drug is endowed with antioxidant properties accounting for its gastroprotective actions.
At least two mechanisms can be advocated to explain the defence exerted by lansoprazole against gastric oxidative damage associated with NSAID treatment, as this drug might directly scavenge reactive oxygen species or interfere with the oxidative metabolism arising from the activation of polymorphonuclear cells. To address this issue, we carried out a series of experiments on an in vitro model, and evidence was obtained that lansoprazole directly protects native LDLs from copper-induced oxidation. These results agree with the experiments performed by Lapenna et al[29] where omeprazole was shown to significantly scavenge hypochlorous acid as well as to inhibit iron- and copper-driven oxidative reactions in appropriate in vitro systems. When taking into account indirect antioxidant mechanisms, it is important to consider that the increased output of free oxygen radicals, arising from activated polymorphonuclear cells, requires the acidification of phagolysosomes, and that such a process is accomplished by a proton pump which is fully susceptible to blockade by benzimidazole derivatives[30,31]. Indeed, omeprazole blocked the oxidative burst of isolated polymorphonuclear cells[30,32]. In addition, lansoprazole inhibited the output of free oxygen radicals from neutrophils activated by Helicobacter pylori[33], and counteracted the increase in plasma levels of peroxidated lipids in patients with duodenal ulcer[34]. Overall, it can be proposed that lansoprazole, acting via both direct and indirect antioxidant mechanisms, protects the gastric mucosa against oxidative injury caused by NSAID-induced focal ischemia and neutrophil activation.
Sulfhydryl radicals take a significant part in mechanisms deputed to the defence of tissues against oxidative injury, and there is evidence to suggest that sulfhydryl donor drugs can afford protection against gastric mucosal injuries elicited by various necrotizing agents, stress or ischemia[22]. Adequate levels of sulfhydryl compounds appear also to be an important requirement for prevention of NSAID-induced gastropathy, since previous reports showed that a mucosal depletion of endogenous sulfhydryl radicals contributed to the pathogenesis of gastric lesions evoked by different NSAIDs[14,15], and GSH concentrations were significantly decreased in mucosal bioptic specimens obtained from patients with NSAID-induced gastric bleeding[35]. Consistent with these observations, in the present study animals treated with indomethacin or piroxicam displayed a marked reduction in mucosal GSH levels, and in both cases the decreasing action could be reversed by pretreatment with lansoprazole. The latter finding can be interpreted in light of the antioxidant properties of lansoprazole, through which this drug is expected to preserve mucosal sulfhydryl compounds from the excess of gastric scavenging activity required to counteract NSAID-induced oxidation, and therefore it is likely that an increased bioavailability of endogenous sulfhydryls plays a significant role in the prevention afforded by lansoprazole against gastropathy associated with NSAID therapy. In keeping with this view, lansoprazole was previously shown to interfere with the decrease in mucosal GSH concentrations in a rat model of gastric necrosis and oxidative injury caused by intraluminal application of acidified ethanol[36].
The implication of prostaglandins in the antiulcer effects of PPIs is currently debated. In previous studies in rat models, single-dose administration of lansoprazole did not influence gastric prostaglandin production[36,37]. Furthermore, omeprazole failed to affect the release of prostaglandins from cultured gastric mucosal cells[38]. More recently, Tsuji et al[10] observed that, after a treatment course of 14 d, lansoprazole promoted a gastrin-dependent increment of both gastric COX-2 expression and PGE2 concentration in rats. In the present study, a series of experiments was designed to evaluate PGE2 levels and the expression of COX isoforms in the gastric mucosa. PGE2 mucosal concentrations were consistently decreased after administration of test-NSAID, as expected, but such inhibitory effects were not influenced by pretreatment with lansoprazole. Moreover, indomethacin enhanced the mucosal expression of COX-2, but not COX-1, in line with previous findings[39], whereas the expression of both isoforms was not modulated by lansoprazole neither in the absence nor in the presence of indomethacin. Therefore, lansoprazole does not appear to exert positive influences on gastric PGE2 levels, at least upon blockade of COX isoforms by NSAID treatment. In agreement with these observations, omeprazole prevented indomethacin-induced gastric ulcers in rabbits, without interfering with the concomitant decrease in mucosal PGE2 formation[40].
In conclusion, the present results support the view that, besides the inhibition of acid secretion, the protective effects exerted by lansoprazole against NSAID-induced gastric damage can be ascribed to a reduction of gastric oxidative injury, which is also responsible for an increased bioavailability of mucosal sulfhydryl compounds. It is also suggested that lansoprazole does not exert any modulator influence on the down-regulation of gastric prostaglandin formation associated with NSAID treatment.
Footnotes
Science Editor Zhu LH Language Editor Elsevier HK
References
- 1.Wallace JL. Pathogenesis of NSAID-induced gastroduodenal mucosal injury. Best Pract Res Clin Gastroenterol. 2001;15:691–703. doi: 10.1053/bega.2001.0229. [DOI] [PubMed] [Google Scholar]
- 2.Vane JR, Botting RM. Mechanism of action of antiinflammatory drugs. Int J Tissue React. 1998;20:3–15. [PubMed] [Google Scholar]
- 3.Whittle BJ. Gastrointestinal effects of nonsteroidal anti-inflammatory drugs. Fundam Clin Pharmacol. 2003;17:301–313. doi: 10.1046/j.1472-8206.2003.00135.x. [DOI] [PubMed] [Google Scholar]
- 4.Hawkey CJ. Nonsteroidal anti-inflammatory drug gastropathy. Gastroenterology. 2000;119:521–535. doi: 10.1053/gast.2000.9561. [DOI] [PubMed] [Google Scholar]
- 5.Pohle T, Brzozowski T, Becker JC, Van der Voort IR, Markmann A, Konturek SJ, Moniczewski A, Domschke W, Konturek JW. Role of reactive oxygen metabolites in aspirin-induced gastric damage in humans: gastroprotection by vitamin C. Aliment Pharmacol Ther. 2001;15:677–687. doi: 10.1046/j.1365-2036.2001.00975.x. [DOI] [PubMed] [Google Scholar]
- 6.McCarthy DM. Prevention and treatment of gastrointestinal symptoms and complications due to NSAIDs. Best Pract Res Clin Gastroenterol. 2001;15:755–773. doi: 10.1053/bega.2001.0233. [DOI] [PubMed] [Google Scholar]
- 7.Matheson AJ, Jarvis B. Lansoprazole: an update of its place in the management of acid-related disorders. Drugs. 2001;61:1801–1833. doi: 10.2165/00003495-200161120-00011. [DOI] [PubMed] [Google Scholar]
- 8.Welage LS. Pharmacologic properties of proton pump inhibitors. Pharmacotherapy. 2003;23:74S–80S. doi: 10.1592/phco.23.13.74s.31929. [DOI] [PubMed] [Google Scholar]
- 9.Blandizzi C, Natale G, Gherardi G, Lazzeri G, Marveggio C, Colucci R, Carignani D, Del Tacca M. Acid-independent gastroprotective effects of lansoprazole in experimental mucosal injury. Dig Dis Sci. 1999;44:2039–2050. doi: 10.1023/a:1026626519534. [DOI] [PubMed] [Google Scholar]
- 10.Tsuji S, Sun WH, Tsujii M, Kawai N, Kimura A, Kakiuchi Y, Yasumaru S, Komori M, Murata H, Sasaki Y, et al. Lansoprazole induces mucosal protection through gastrin receptor-dependent up-regulation of cyclooxygenase-2 in rats. J Pharmacol Exp Ther. 2002;303:1301–1308. doi: 10.1124/jpet.102.035204. [DOI] [PubMed] [Google Scholar]
- 11.Wallace JL, Granger DN. The cellular and molecular basis of gastric mucosal defense. FASEB J. 1996;10:731–740. doi: 10.1096/fasebj.10.7.8635690. [DOI] [PubMed] [Google Scholar]
- 12.Takeuchi K, Mizoguchi H, Araki H, Komoike Y, Suzuki K. Lack of gastric toxicity of nitric oxide-releasing indomethacin, NCX-530, in experimental animals. Dig Dis Sci. 2001;46:1805–1818. doi: 10.1023/a:1010638528675. [DOI] [PubMed] [Google Scholar]
- 13.Wallace JL, Reuter B, Cicala C, McKnight W, Grisham M, Cirino G. A diclofenac derivative without ulcerogenic properties. Eur J Pharmacol. 1994;257:249–255. doi: 10.1016/0014-2999(94)90136-8. [DOI] [PubMed] [Google Scholar]
- 14.Avila JR, de la Lastra CA, Martin MJ, Motilva V, Luque I, Delgado D, Esteban J, Herrerias J. Role of endogenous sulphydryls and neutrophil infiltration in the pathogenesis of gastric mucosal injury induced by piroxicam in rats. Inflamm Res. 1996;45:83–88. doi: 10.1007/BF02265120. [DOI] [PubMed] [Google Scholar]
- 15.Alarcón de la Lastra C, Nieto A, Martín MJ, Cabré F, Herrerías JM, Motilva V. Gastric toxicity of racemic ketoprofen and its enantiomers in rat: oxygen radical generation and COX-expression. Inflamm Res. 2002;51:51–57. doi: 10.1007/BF02683999. [DOI] [PubMed] [Google Scholar]
- 16.Natale G, Lazzeri G, Blandizzi C, Gherardi G, Lenzi P, Pellegrini A, Del Tacca M. Seriate histomorphometry of whole rat stomach: an accurate and reliable method for quantitative analysis of mucosal damage. Toxicol Appl Pharmacol. 2001;174:17–26. doi: 10.1006/taap.2001.9193. [DOI] [PubMed] [Google Scholar]
- 17.Pacheco I, Otaka M, Jin M, Sasahara H, Iwabuchi A, Odashima M, Konishi N, Wada I, Masamune O, Watanabe S. Corticosteroid pretreatment prevents small intestinal mucosal lesion induced by acetic acid-perfusion model in rats. Dig Dis Sci. 2000;45:2337–2346. doi: 10.1023/a:1005519304829. [DOI] [PubMed] [Google Scholar]
- 18.Tashima K, Fujita A, Takeuchi K. Aggravation of ischemia/reperfusion-induced gastric lesions in streptozotocin-diabetic rats. Life Sci. 2000;67:1707–1718. doi: 10.1016/s0024-3205(00)00754-2. [DOI] [PubMed] [Google Scholar]
- 19.Blandizzi C, Gherardi G, Marveggio C, Natale G, Carignani D, Del Tacca M. Mechanisms of protection by omeprazole against experimental gastric mucosal damage in rats. Digestion. 1995;56:220–229. doi: 10.1159/000201247. [DOI] [PubMed] [Google Scholar]
- 20.Lubrano V, Vassalle C, Blandizzi C, Del Tacca M, Palombo C, L'Abbate A, Baldi S, Natali A. The effect of lipoproteins on endothelial nitric oxide synthase is modulated by lipoperoxides. Eur J Clin Invest. 2003;33:117–125. doi: 10.1046/j.1365-2362.2003.01083.x. [DOI] [PubMed] [Google Scholar]
- 21.Kwiecień S, Brzozowski T, Konturek SJ. Effects of reactive oxygen species action on gastric mucosa in various models of mucosal injury. J Physiol Pharmacol. 2002;53:39–50. [PubMed] [Google Scholar]
- 22.Szabo S. Mechanisms of gastric mucosal injury and protection. J Clin Gastroenterol. 1991;13 Suppl 2:S21–S34. [PubMed] [Google Scholar]
- 23.Loguercio C, Di Pierro M. The role of glutathione in the gastrointestinal tract: a review. Ital J Gastroenterol Hepatol. 1999;31:401–407. [PubMed] [Google Scholar]
- 24.Hayes JD, McLellan LI. Glutathione and glutathione-dependent enzymes represent a co-ordinately regulated defence against oxidative stress. Free Radic Res. 1999;31:273–300. doi: 10.1080/10715769900300851. [DOI] [PubMed] [Google Scholar]
- 25.Anthony A, Sim R, Dhillon AP, Pounder RE, Wakefield AJ. Gastric mucosal contraction and vascular injury induced by indomethacin precede neutrophil infiltration in the rat. Gut. 1996;39:363–368. doi: 10.1136/gut.39.3.363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lehmann F, Hildebrand P, Beglinger C. New molecular targets for treatment of peptic ulcer disease. Drugs. 2003;63:1785–1797. doi: 10.2165/00003495-200363170-00002. [DOI] [PubMed] [Google Scholar]
- 27.Janssen M, Dijkmans BA, Vandenbroucke JP, Biemond I, Lamers CB. Achlorhydria does not protect against benign upper gastrointestinal ulcers during NSAID use. Dig Dis Sci. 1994;39:362–365. doi: 10.1007/BF02090209. [DOI] [PubMed] [Google Scholar]
- 28.Scarpignato C, Pelosini I. Prevention and treatment of non-steroidal anti-inflammatory drug-induced gastro-duodenal damage: rationale for the use of antisecretory compounds. Ital J Gastroenterol Hepatol. 1999;31 Suppl 1:S63–S72. [PubMed] [Google Scholar]
- 29.Lapenna D, de Gioia S, Ciofani G, Festi D, Cuccurullo F. Antioxidant properties of omeprazole. FEBS Lett. 1996;382:189–192. doi: 10.1016/0014-5793(96)00155-x. [DOI] [PubMed] [Google Scholar]
- 30.Suzuki M, Mori M, Miura S, Suematsu M, Fukumura D, Kimura H, Ishii H. Omeprazole attenuates oxygen-derived free radical production from human neutrophils. Free Radic Biol Med. 1996;21:727–731. doi: 10.1016/0891-5849(96)00180-3. [DOI] [PubMed] [Google Scholar]
- 31.Agastya G, West BC, Callahan JM. Omeprazole inhibits phagocytosis and acidification of phagolysosomes of normal human neutrophils in vitro. Immunopharmacol Immunotoxicol. 2000;22:357–372. doi: 10.3109/08923970009016425. [DOI] [PubMed] [Google Scholar]
- 32.Wandall JH. Effects of omeprazole on neutrophil chemotaxis, super oxide production, degranulation, and translocation of cytochrome b-245. Gut. 1992;33:617–621. doi: 10.1136/gut.33.5.617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Suzuki M, Nakamura M, Mori M, Miura S, Tsuchiya M, Ishii H. Lansoprazole inhibits oxygen-derived free radical production from neutrophils activated by Helicobacter pylori. J Clin Gastroenterol. 1995;20 Suppl 2:S93–S96. doi: 10.1097/00004836-199506002-00025. [DOI] [PubMed] [Google Scholar]
- 34.Manjari V, Das UN. Oxidant stress, anti-oxidants, nitric oxide and essential fatty acids in peptic ulcer disease. Prostaglandins Leukot Essent Fatty Acids. 1998;59:401–406. doi: 10.1016/s0952-3278(98)90102-8. [DOI] [PubMed] [Google Scholar]
- 35.Savoye G, Miralles-Barrachina O, Déchelotte P, Belmonte-Zalar L, Brung-Lefebvre M, Zalar A, Hochain P, Hervé S, Colin R, Lerebours E, et al. Low levels of gastric mucosal glutathione during upper gastric bleeding associated with the use of nonsteroidal anti-inflammatory drugs. Eur J Gastroenterol Hepatol. 2001;13:1309–1313. doi: 10.1097/00042737-200111000-00008. [DOI] [PubMed] [Google Scholar]
- 36.Natale G, Lazzeri G, Lubrano V, Colucci R, Vassalle C, Fornai M, Blandizzi C, Del Tacca M. Mechanisms of gastroprotection by lansoprazole pretreatment against experimentally induced injury in rats: role of mucosal oxidative damage and sulfhydryl compounds. Toxicol Appl Pharmacol. 2004;195:62–72. doi: 10.1016/j.taap.2003.10.006. [DOI] [PubMed] [Google Scholar]
- 37.Fukuda T, Arakawa T, Shimizu Y, Ohtani K, Higuchi K, Kobayashi K. Effects of lansoprazole on ethanol-induced injury and PG synthetic activity in rat gastric mucosa. J Clin Gastroenterol. 1995;20 Suppl 2:S5–S7. doi: 10.1097/00004836-199506002-00003. [DOI] [PubMed] [Google Scholar]
- 38.Ota S, Takahashi M, Yoshiura K, Hata Y, Kawabe T, Terano A, Omata M. Antiulcer drugs and gastric prostaglandin E2: an in vitro study. J Clin Gastroenterol. 1993;17 Suppl 1:S15–S21. doi: 10.1097/00004836-199312001-00006. [DOI] [PubMed] [Google Scholar]
- 39.Tanaka A, Hase S, Miyazawa T, Takeuchi K. Up-regulation of cyclooxygenase-2 by inhibition of cyclooxygenase-1: a key to nonsteroidal anti-inflammatory drug-induced intestinal damage. J Pharmacol Exp Ther. 2002;300:754–761. doi: 10.1124/jpet.300.3.754. [DOI] [PubMed] [Google Scholar]
- 40.Lee M, Kallal SM, Feldman M. Omeprazole prevents indomethacin-induced gastric ulcers in rabbits. Aliment Pharmacol Ther. 1996;10:571–576. doi: 10.1046/j.1365-2036.1996.34176000.x. [DOI] [PubMed] [Google Scholar]