

Abstract
Pyrazinamide, a highly specific agent against Mycobacterium tuberculosis is used as first-line drug to treat tuberculosis. The current work aims to formulate polymeric nanoparticles based drug delivery system to sustain the release profile and reduce the dosing frequency of pyrazinamide. Further aim was to target the macrophages within body fluid. These polymeric nanoparticles were prepared by simultaneous double-emulsion (W/O/W) solvent evaporation/diffusion technique. The prepared dispersions were characterized for various biopharmaceutical parameters such as particle size, zeta potential, polydispersity index, drug loading capacity, entrapment efficiency and targeting to alveolar macrophages. The formulated polymeric nanoparticles were in the particle size range of 45.51 to 300.4 nm with a maximum drug entrapment efficiency of 80.9%. The stability study of optimized batch conducted at 40±2°/75±5% relative humidity showed no significant changes up to 90 days. X-Ray Diffraction spectrum exhibits the transformation of crystalline form of drug to amorphous in the formulation. Scanning Electron Microscope image showed nanoparticles spherical in shape with smooth surface. In vitro release profiles were biphasic in nature with burst release followed by controlled release over a period of 24 h obeying diffusion mechanism. In vivo and ex vivo studies results of the study show significant uptake of the nanoparticles by alveolar macrophages through fluorescent micrograph. Polymeric nanoparticles formulation of pyrazinamide could encompass significant uptake by alveolar macrophages, the high first-pass metabolism, sustain the release of drug leading to reduction in dose, toxicity and improvement of patient compliance.
Keywords: Double-emulsion, polymeric nanoparticles, pulmonary tuberculosis, alveolar macrophages
Tuberculosis (TB), a highly contagious persistent infection is caused by Mycobacterium tuberculosis and Mycobacterium bovis. Tuberculosis is the world's second most common cause of death after HIV/AIDS. In 1993, World Health Organization (WHO) declared TB as a global emergency. Presence of immunosuppressive condition like diabetes, alcoholism, malnutrition, chronic lung disease and HIV/AIDS may increase the chances of TB infection[1]. The disease typically attacks the lungs but can also develop as extra pulmonary TB in the central nervous or circulatory systems or elsewhere in the body. Untreated active TB has approximately a 50% of mortality rate[2]. Despite potential curative pharmacotherapies being available for over 50 years, the length of treatment and pill burden can hamper patient lifestyle. Thus, low compliance and adherence to administration schedules remain the major reasons for therapeutic failure and contribute to the development of multi-drug-resistant (MDR) strains.
Pyrazinamide, a first-line antitubercular agent, mainly bacteriostatic, but can also be bactericidal on actively replicating tuberculosis bacteria[3]. It is used for first two months of treatment to reduce the duration to six months but regimen without pyrazinamide must be taken for nine months or more. Under slightly acidic pH, pyrazinamide converts to its active moiety pyrazinoic acid (POA) in bacilli, which further interfere with fatty acid syntheses I (FAS) required for growth and replication[4]. The most severe side effect of pyrazinamide is hepatotoxicity, which is dose-dependent. The old dose for pyrazinamide was 40-70 mg/kg and the incidence of drug-induced hepatitis has fallen significantly since the recommended dose has been reduced[5].
Polymeric nanoparticle-based drug delivery systems form the crux of nano medicine. They are suitable for targeting chronic diseases such as tuberculosis as these cross the biological barriers and target cellular reservoirs of Mycobacterium tuberculosis. Experimental data supports the possibility of intermittent chemotherapy with key first-line as well as second-line antitubercular drugs by employing synthetic or natural carriers, chiefly polymers. Besides sustained release of drugs in plasma and organs, other potential advantages of this system include the possibility of selecting various routes of chemotherapy, reduction in drug dosage, adverse effects, drug interactions and targeting both drug-resistant and latent bacteria. In recent years, biodegradable polymeric nanoparticles, particularly those coated with hydrophilic polymer such as polyethylene glycol, have been used because of their ability to circulate for a prolonged period time, targeting to a particular organ, as DNA carriers in gene therapy, and to deliver proteins, peptides and genes[6]. The major goals in designing nanoparticles as a delivery system are to control particle size, surface properties and release of pharmacologically active agents in order to achieve site-specific action of the drug at therapeutically optimal rate and dose regimen. Though liposomes have been used as potential carriers with unique advantages including protecting the drug from degradation, targeting to site of action and reducing toxicity/side effects, their applications are limited due to inherent problems such as low encapsulation efficiency, rapid leakage of water-soluble drug in the presence of blood components and poor storage stability. On the other hand, polymeric nanoparticles offer some specific advantages over liposomes. For instance, they help to increase the stability of drugs/proteins and possess useful controlled release properties. Nanocarriers with optimized physicochemical and biological properties are taken up by cells more easily than larger molecules, so they can be successfully used as delivery tools for currently available bioactive compounds[7].
The current work encompasses the design of a novel nanoscopic oral drug delivery portal, polymeric nanoparticles bearing pyrazinamide. Prepared nanoparticles were characterized for their vesicular size, stability, surface morphology, entrapment efficiency and in vitro drug release profile for monitoring the efficient release of pyrazinamide.
MATERIALS AND METHODS
Pyrazinamide was a gift sample obtained from Lupin Ltd. (Hyderabad). Eudragit RS-100 was gift sample obtained from Evonik Industries (UK). Span 80, poloxamer and polyvinyl alcohol were purchased from S. D. Fine Chem. Ltd. (Mumbai). All other reagents used were of analytical grade.
Preparation of polymeric nanoparticles:
Pyrazinamide (PYZ)-loaded polymeric nanoparticles (PNPs) were prepared using double-emulsion solvent evaporation/diffusion technique[8,9]. Various formulations were prepared to know the effect of polymer and surfactant concentration and were assigned formulation code F1 to F16 as shown in Table 1. Firstly, polymeric solution was prepared by dissolving specific amount of Eudragit RS-100 dissolved in 20 ml organic mixture consisting of span 80 (2% v/v) dissolved in dichloromethane. Then, followed by emulsification of aqueous drug solution (2% w/v) in polymeric organic solution under magnetic stirring (Remi Magnetic Stirrer) at 1000-1200 rpm for 15 min to get primary W/O emulsion. Further this was added to 25 ml distilled water containing either poloxamer or polyvinyl alcohol (PVA) under stirring for 10 min, to achieve a stable W/O/W double emulsion[10,11]. The obtained polymeric nanoparticles were separated by ultracentrifugation (Remi cooling centrifuge) at 11 000 rpm for 40 min and washed with distilled water[12,13]. Finally, the products were freeze dried by using lyophilizer and stored at -4°. Schematic representation of polymeric nanoparticle preparation is shown in fig. 1.
TABLE 1.
COMPOSITION OF FORMULATIONS AND EVALUATION RESULTS
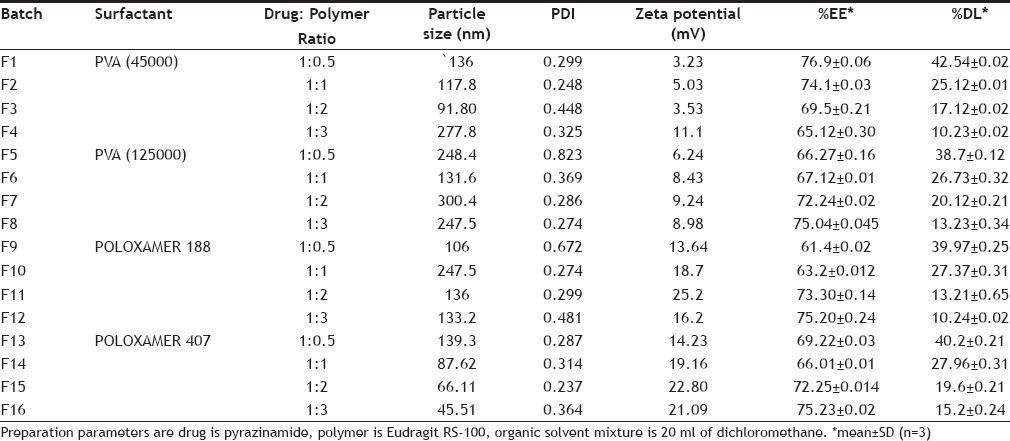
Fig. 1.
Schematic representation of nanoparticles preparation. O is oil phase containing Eudragit RS 100, dichloromethane and surfactant, W is aqueous solution of pyrazinamide, W/O is the primary emulsion and W2 is aqueous phase comprising of different concentrations of polyvinyl alcohol and Polaxamers.
Particle size and polydispersity index:
Particle size and polydispersity of nanoparticles were determined by Photon Correlation Spectroscopy (PCS) using Delsa Nano C (Beckman Coulter Counter, USA). Samples suitably diluted were transferred to a polystyrene cuvette and placed inside a temperature regulated cell-scattering enclosure. Analysis was carried out for 60 s at 165° scattering angle of detection. Polydispersity index (PDI) was determined as a measurement of particle size homogeneity of the prepared nanoparticles[14]. A small value of PDI (<0.3) indicates homogeneous vesicle population[15]. The measurements of the correlation function were analyzed and the diffusion coefficient was obtained.
Zeta potential determination:
Zeta potential (ζ) is the charge on particle surface and it is the inherent property of a particle. The value of ζ can be correlated to the stability of colloidal dispersions. ζ indicates the degree of repulsion between adjacent, similarly charged particles in dispersion[16]. For molecules and particles that are small enough, a high ζ will confer stability, i.e,. the solution or dispersion will resist aggregation. When the potential is low, attraction exceeds repulsion and the dispersion breaks and flocculate. So, colloids with high ζ (negative or positive) are electrically stabilized while colloids with low ζ tend to coagulate or flocculate.
Entrapment efficiency and percent drug loading:
Entrapment efficiency (EE) and percent drug loading (% DL) were determined using ultracentrifugation method (indirect method) i.e. by measuring the concentration of free drug in aqueous phase[17]. Nanoparticles containing emulsion were centrifuged at 11 000 rpm for 40 min. From the supernatant, 0.1 ml was taken into 10 ml graduated test tube and volume was made up to 10 ml with distilled water, finally filtered using Whatman filter paper. The amount of PYZ was analyzed by UV spectroscopy at a wavelength of 268 nm. The entrapment efficiency and percent drug loaded was calculated using the Eqns. 1 and 2 which are, EE=(Wt-Wf)/Wt×100…1 and % DL=(Wt-Wf)/Wn×100….2, where Wt represents the amount of total PYZ, Wf is the amount of free PYZ in the supernatant, and Wn is the weight of nanoparticles after freeze-drying. All experiments were performed in triplicate and expressed as mean±SD values.
X-ray diffraction study:
X-Ray diffraction patterns of PYZ, Eudragit RS-100 and PYZ-loaded polymeric nanoparticles were collected in transmission using a Miniflex II Desktop X-ray diffractometer (Rigaku, Japan) with monochromatic CuKα1 radiation (Radiation; λ=1.5406 A°) generated at 30 kV. Powder diffractometer operated on Bragg-Brentano geometry was fitted with a curved crystal graphite monochromator in the diffraction beam from range 11-45° (2θ) and with a step size of 0.020. The powder was packed into the rotating sample holder. The scanning rate was set at 10°/min.
Morphology:
Morphological characteristics were observed by SEM (Scanning Electron Microscope-Quanta 200). The formulation was spread on circular aluminum stub pre-coated with silver glue (for enhancing conductivity to electrons) and dried in vacuum oven to form a dry film specimen. The sample was then examined and photographed with SEM at an accelerating voltage of 20 kV in varying magnifications[18].
In vitro drug release studies:
In vitro drug release of PYZ from the polymeric nanoparticles was performed using dialysis bag diffusion method[19]. Dialysis membrane (Himedia, thickness 0.025 mm, with dimensions 6×2.5 cm) previously soaked in distilled water for 24 h was used in the study. The drug release studies of pure PYZ solution and PYZ-loaded Eudragit RS-100 nanoparticles were carried out in 250 ml of phosphate buffer (pH 6.8) on a magnetic stirrer at 50 rpm with temperature maintained at 37±2° by constant heating equipment (IKA Auto Temp Regulator, Germany). Formulation sample of volume 5 ml was filled in the dialysis pouch and the two ends were sealed. Aliquot samples of 5 mL were withdrawn at regular time intervals maintaining sink condition. The amount of PYZ released was analyzed using a UV spectrophotometer (Elico, Hyderabad) at the λmax value of 268 nm. All experiments were performed in triplicate.
Drug release kinetics:
In order to understand the kinetics and mechanism of drug release, results of in vitro drug release study of the prepared PNPs were fitted into various kinetic equations like zero order (cumulative % remaining vs. time), first order (log % drug remaining vs. time), Higuchi's model (cumulative % drug release vs. square root of time), Peppas (log % drug release vs. log time)[20,21,22,23]. Rate constant (K) and regression coefficient (R2) values were calculated for the linear curve obtained by regression analysis of the above plots.
Stability studies:
Stability study of optimized batch of nanoparticles suspension was performed under accelerated stability conditions (40±2°/75±5% RH) kept in stability testing chamber for three months according to ICH guidelines[24]. The samples were withdrawn at different interval (0, 1 and 3 months) and evaluated for particle size, entrapment efficiency and in vitro drug release studies.
Preparation of fluorescein labelled polymeric nanoparticles:
Ten milligrams of PYZ was dissolved along with 3 mg of fluorescein sodium and the preparation was continued as mentioned for polymeric nanoparticles. The prepared fluorescein labeled nanoparticles were suspended in distilled water containing 2% mannitol (cryoprotectant) and then lyophilized.
Comparative ex vivo/in vivo uptake studies with drug solution and nanoparticles on Broncho-alveolar macrophages:
Charles Foster strain rats of either sex (approx. weight 210 g or 6-8 week old) were selected for the experiment and allowed to free access of tap water and standard pellet food in cages and maintained on a 12 h light/dark cycle at room temperature (21-25°) and relative humidity 45-55%. Animals are to be acclimatized to study area conditions for at least 5 days before the experiment. General and environmental conditions were strictly monitored[25,26]. All experiments were performed in according to the study protocol approved by the Institutional Animal Ethics Committee (Reg. No, 516/01/A/CPCSEA) constituted under Committee for the Purpose of Control and Supervision on Experimental Animals, India. Rats were anaesthetized by exposing it to small amount of diethyl ether added at the bottom of anaesthetizing chamber. Breathing of the rat was watched carefully and taken out when slowed down. Then the rat was sterilized by spraying 70% ethanol and kept in a tissue culture hood. An incision was made at upper thoracic part and a polyethylene tube was inserted into the trachea and tied along with it. Through the use of 1 ml syringe inserted in the tube, lung was lavaged 10 times with 2 ml aliquots of Ca2+ and Mg2+ free Hank's balanced salt solution. Then the lavaged suspension was centrifuged at 4° and the pellet was resuspended in PBS buffer. The cell viability was checked with trypan blue exclusion test (the cells were treated with 100 μl of 0.4% dye solution and counted with Neubauer Haemocytometer). The test was based on principle that live cells possess intact cell membranes that exclude the permeation of this dye and therefore, their cytoplasm remains clear and colourless. On the other hand, nonviable cells taken up the dye and have a blue cytoplasm. Cells with seeding density of 1×106 cells/ml were plated in 6-well culture plates in incomplete medium Eagle's Minimal Essential Medium (EMEM) without 10% fetal calf serum (FCS). After 2 h of incubation at 37° and 5% CO2, the medium was replaced by fresh medium of EMEM with 10% FCS. Then the culture plates were kept overnight for incubation at 37° and 5% CO2.
For ex vivo studies, nanoparticles labelled with fluorescein have been added to the culture plates containing the previously harvested alveolar macrophages (AMs). And for in vivo studies 5 ml of fluorescein labeled formulation/fluorescein labeled drug solution was administered orally 2 h before the isolation of broncho alveolar macrophages. Incubation was carried out for 4 h at 37° and 5% CO2. The cells were removed from the wells and subjected to two cycles of washing/centrifugation (5000 rpm/3 min) with ice cold phosphate buffer saline (PBS). Finally cells were fixed with an aqueous solution of 8% v/v formaldehyde and examined under confocal laser scanning microscope (Nikon Eclipse E800, filter 520-560 nm, Excitation wavelength 488 nm)[27].
RESULTS AND DISCUSSION
The average particle size of prepared nanoparticles was found to be in the range of 45.51 nm to 300.4 nm as shown in Table 1. Both the polymer (Eudragit RS-100) and stabilizing agent (poloxamer or PVA) exhibit significant effect on particle size. Increase in polymer concentration has led to increase in viscosity of organic phase, reducing net shear stress and promoting formation of larger droplet. Nanoparticles <10 nm can be rapidly cleared by kidneys in contrast to larger nanoparticles which are cleared by cells of mononuclear phagocyte system. It was observed that nanoparticles <100 nm have a higher potential to circulate in blood for long periods of time and experience reduced hepatic filtration[28]. With the use of surfactants (PVA or poloxamer), a significant reduction in particle size was observed with varying molecular weights. With the use of poloxamer of low molecular weight, an optimum particle size of nanoparticle could be obtained when compared to PVA. PDI of PZY-PNs was observed in the range of 0.237 to 0.672 with a low coefficient of variation value of 0.11. Generally the value of PDI ranges from Zero to One. From these results, it was found that presence of surfactant has led to smaller particles, with a satisfactory PDI and this may be attributed to the fact that surfactant ensures a good emulsification process and, therefore, leading to formation of smaller particles with uniform size distribution.
Nanoparticles with a ζ value between -10 and +10 mV are considered approximately neutral, while nanoparticles with zeta potentials of greater than +25 mV or less than -25 mV are considered strongly cationic and strongly anionic, respectively. The mucin molecules, are negatively charged at physiological pH because of sialic acid residues in the oligosaccharide units, interact with the positively charged nanoparticles, leading to the enhanced mucoadhesive properties of the carrier system[29]. Of all formulations, F11 exhibits highest ζ value of +25.2 mV.
The percentage entrapment efficiency was found to be high in respect of hydrophilic nature of drug, ranging between 61.4 to 80.9 % and drug loading (% w/w) in the range of 13.21 to 42.7%, represented in Table 1. From these results, it is clear that EE increases with increase in polymer concentration (Eudragit RS-100), whereas %DL decreases with increase in polymer concentration. This may be due to increase in viscosity of organic phase, which increases the drug resistance and diffusion into aqueous phase, thereby enhancing the entrapment of drug. With the use of poloxamer, a significant reduction in particle size was observed which led to low entrapment of drug when compared to PVA.
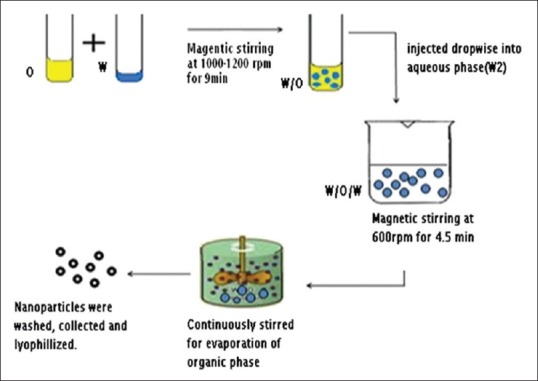
The diffraction pattern of pure PYZ exhibits sharp peaks at diffraction angles of 2θ at 17.6° indicating the presence of drug in crystalline nature. The XRD pattern of PYZ loaded polymeric nanoparticles had shown the characteristic pure drug peak at 17.6° (2θ) without any shift/change, indicating that there is no significant incompatibility between drug and other excipients within polymeric nanoparticles. The XRD spectra of PYZ, Eudragit RS-100, PYZ-Eudragit RS-100 physical mixture and PYZ-loaded polymeric nanoparticles are shown in fig. 2.
Fig. 2.
X-ray difractograms of the formulation, physical mixture, Eudragit and pyrazinamide.
X-ray difractograms of (a) formulation, (b) pyrazinamide-Eudragit RS-100 physical mixture, (c) Eudragit RS-100 and d) pyrazinamide.
The external morphology study using ESEM revealed that all nanoparticles are spherical in shape with smooth texture, but exhibits substantial agglomeration. The degree of nanoparticles fusion is notable in fig. 3. This may be due to removal of solvent during lyophilization which affected the particle equilibrium resulting in coalescence and agglomeration.
Fig. 3.

Scanning electron micrograph of lyophilized optimized batch F11.
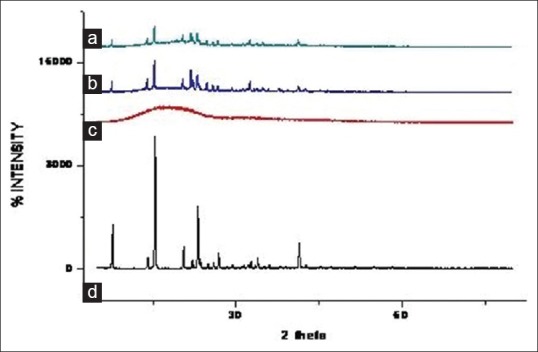
The in vitro release study was performed in phosphate buffer (pH 6.8) up to 24 h and results allow for the following observations and inferences. When PYZ pure drug was studied for its in vitro dissolution profile, a high percent of PYZ was dissolved within one hour. Almost all the drug (90%) was dissolved within 6 h and thereafter no further release was observed, as shown in fig. 4.
Fig. 4.
In vitro dissolution profiles of PYZ polymeric nanoparticles. Dissolution profile of pyrazinamide from nanoparticles of F1 ( ), F2 (
), F2 ( ), F3 (
), F3 ( ), F4 (
), F4 ( ), F5 (
), F5 ( ), F6 (
), F6 ( ), F7 (
), F7 ( ), F8 (
), F8 ( ), F9 (
), F9 ( ), F10 (
), F10 ( ), F11 (
), F11 ( ), F12 (
), F12 ( ), F13 (
), F13 ( ), F14 (
), F14 ( ), F15 (
), F15 ( ), F16 (
), F16 ( ).
).
Formulations F1 to F4 prepared using PVA (45 000), F5 to F8 prepared using PVA (125 000), F9 to F12 prepared using poloxamer 188, F13 to F16 prepared using poloxamer 407 have sustained the drug release as time prolonged. These formulations showed a biphasic release profile, an initial rapid release phase up to 11 h, followed by a slower release phase controlled over 24 h, as shown in fig. 5. The initial rapid release may be due to presence of drug on the surface of particles, free drug in the solution. It was observed that, with increase in concentration of polymer, the release rate was retarded. On an average, around 79, 80, 77 and 78% of PYZ was released from formulations prepared using PVA (45 000), PVA (125 000), poloxamer 188 and poloxamer 407, respectively. Sustained release pattern of the drug is more pronounced in presence of poloxamer as stabilizing agent when compared to polyvinyl alcohol. This may be attributed to the alteration in stability of polymeric matrix system in presence of poloxamer consisting of both hydrophobic chains (polyoxy propylene) and hydrophilic chains (polyoxy ethylene), in contrast to polyvinyl alcohol, which is hydrophilic (hydroxyl groups) in nature.
Fig. 5.
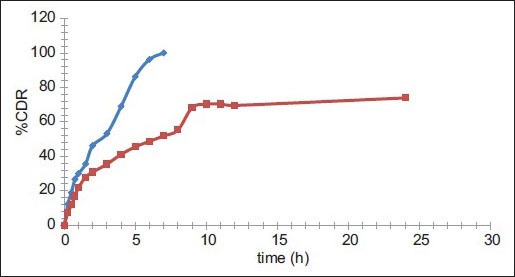
Comparative in vitro dissolution profile of pure PYZ and optimized batch F11.
Comparative in vitro dissolution profile of pyrazinamide (PYZ, ) and optimized batch (F11,
) and optimized batch (F11, ).
).
Different release characterization models were applied to the drug release profiles, and R2 and K values are reported in Table 2. The regression coefficient (R2) was used as an indicator of best fitting for each models considered. Majority of the formulations, when tested for goodness of fit for PYZ showed higher R2 values in zero order release compared to first order release kinetics. Applicability of the release curves to zero order model throughout the whole series of formulations investigated indicated that the drug release is independent of its concentration i.e., the same amount of drug is released in every interval of time.
TABLE 2.
DRUG RELEASE KINETICS FOR FORMULATIONS OF PYZ
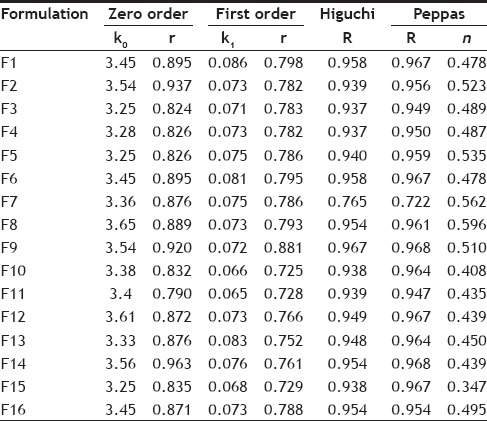
The release data was subjected to a goodness of fit testing to Higuchi model, showing a higher R2 values than the other models, indicating that the drug release is principally controlled by diffusion. Eudragit RS-100 being completely insoluble in water, there exist a less chance of drug release due to surface erosion. Moreover, the values of release exponent (n) was less than 0.45, when the data was fit to Peppa's model determined for the formulations F9 to F16 (using poloxamer). This further confirms that the drug release from these carrier systems is directly proportional to square root of time and is based on the Fickian diffusion (Case I) mechanism of release. The value of release exponent (n) in the Peppas model was higher than 0.45, in case of the formulations prepared using PVA (F1 to F8). This indicates that the formulations followed non-Fickian diffusion kinetics (anomalous transport), i.e. the release is ruled by both diffusion of the drug and dissolution of the polymer.
The optimization was aimed at maximizing entrapment efficiency and percent loading of PYZ in the formulation with optimum particle size, effective PDI, positive ζ value and controlled in vitro drug release profile. Among the entire formulations, F11 prepared using Eudragit RS-100, poloxamer 188 (1% w/v), was optimized which showed best results in terms of desired particle size of 136 nm, smaller PDI value of 0.29, high ζ value of +25 mv, good EE of 73.3% and % DL of 13.21% with a controlled release profile for more than 24 h. Hence, it was selected for further studies.
The results of stability studies are given in Table 3. It was found that the optimized formulation was stable for two months as no significant change in particle size, zeta potential and entrapment efficiency was observed. However, the particle size increased from 133 nm to 213 nm at the end of 3 mo. Here the contributing factor of stability of system was the protective colloidal nature of non-ionic polymer (surfactant). Poloxamer coated the particle surface uniformly and extended the polymer chain to surrounding medium which provided steric hindrance between particles, thus preventing aggregation of particles. There was no significant change in EE of PNPs for 3 months. At the end of 3rd month, it was decreased from 69 to 50 % which clearly indicate drug leakage. The drug release characteristics of PNPs tested remains unaltered during storage at 40±2°/75±5% RH, as shown in fig. 6. Results indicate that the optimized formulation was quite stable under accelerated conditions of temperature and humidity as the controlled release characteristics of the optimized PNPs remained unaltered.
TABLE 3.
EFFECT OF STORAGE CONDITION ON PNPS
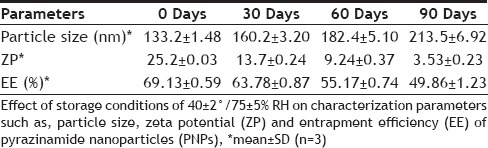
Fig. 6.
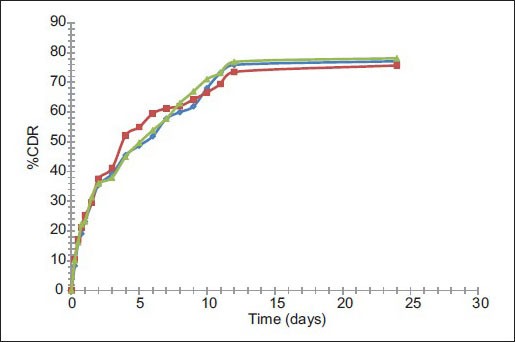
Release profile of PYZ from F11 before and after storage for 3 months & 6 months.
Release profile of pyrazinamide from optimized formulation (F11, ) after 3 months (FF3, ) and after 6 months (FF6, ) at 40±2°/75±5% RH.
Confocal laser scanning microscopy (CLSM) micrographs, visualized nanoparticles present in intracellular and within the nucleus. After 2 h of administration of formulation, strong intracellular fluorescence was observed in the cytosol of alveolar macrophages, which indicates endocytosis in macrophages (fig. 7a). Translating this prompt internalization and distribution behavior of the formulation in terms of its therapeutic effectiveness, the carrier system (PNs) could be proposed to be more interesting since more amount of drug could be internalized within the alveolar macrophages for both in vivo and ex vivo uptake studies, which constitute the desired site of action for antitubercular drugs[30,31,32]. Such treatment modality is expected to avoid the chances of microbial persistence and recurrent infections. After performing these studies it was found that uptake into the macrophages was more efficient in case of fluorescein labelled nanoparticles (fig. 7b) whereas the drug was uptake in case of fluorescein labelled normal drug solution (fig. 7c) was very minimal due to less interaction with macrophages and the physicochemical characteristics of the drug (hydrophilic in nature) alone. The PZY-loaded nanoparticles have been efficiently up taken by alveolar macrophages due to hydrophobic (lipophilic) nature of the polymer.
Fig. 7.
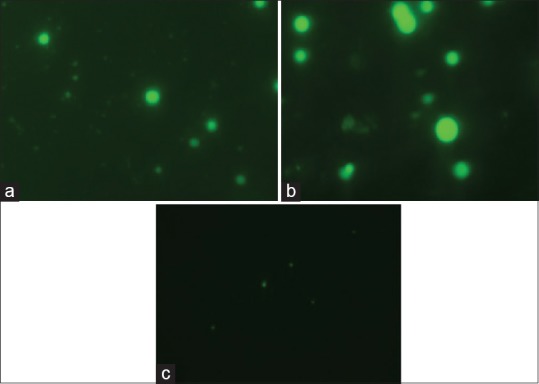
CLSM images of alveolar macrophages exposed to fluorescein labeled nanoparticles ex vivo and in vivo and fluorescein labeled drug solution in vivo.
Confocal laser scanning microscopy (CLSM) images of alveolar macrophages exposed to (a) fluorescein labeled nanoparticles ex vivo, (b) fluorescein labeled nanoparticles in vivo and (c) fluorescein labeled drug solution in vivo.
The present study revealed that PYZ polymeric nanoparticles formulated by simultaneous double-emulsion (W/O/W) solvent evaporation/diffusion technique were effective in providing zero order release profile. With increase in concentration of polymer (Eudragit RS-100), the release rate was retarded higher and a suitable ratio of drug:polymer (1:2 used in F11) was able to best control drug release better for more than 24 h. On the basis of result of these sixteen formulations F11 prepared using PYZ (100 mg), Eudragit RS-100 (200 mg), poloxamer (1% w/v) showed best results in terms of optimum particle size, high PDI, positive ζ value, good entrapment efficiency and % drug loading was selected as the optimized formulation. The release kinetics followed Higuchi model obeying Fickian diffusion of mechanism of drug release from the nanoparticle system. The stability study of optimized batch showed insignificant changes in particle size, zeta potential, entrapment efficiency and in vitro dissolution characteristics up to 90 days. In vivo and ex vivo studies of NPs on alveolar macrophages were done by labeling the NPs with fluorescein dye, CLSM and fluorescent micrograph. The results of the study show significant uptake of the NPs by alveolar macrophages. Thus the objectives of this research project undertaken were achieved by the development of polymeric nanoparticle based controlled drug delivery system of PYZ with optimum particle size and good entrapment efficiency for uptake within macrophages.
Financial support and sponsorship:
Nil.
Conflict of interest:
There are no conflicts of interest.
Acknowledgements:
The authors are grateful to AICTE, New Delhi, India for granting GPAT Fellowship during 2011-2013, to carry out this work. Also authors acknowledge Lupin Pharmaceuticals, Hyderabad for providing PYZ gift sample.
Footnotes
Varma, et al.: Pyrazinamide Polymeric Nanoparticles for Alveolar Macrophage Targeting
REFERENCES
- 1.Gebrekristos HT, Lurie MN, Mthethwa N, Karim QA. Disclosure of HIV status: Experiences of Patients Enrolled in an Integrated TB and HAART Pilot Programme in South Africa. Afr J AIDS Res. 2009;8:1–6. doi: 10.2989/AJAR.2009.8.1.1.714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Golden MP, Vikram HR. Extra pulmonary tuberculosis: An overview. Amer Fam Phy. 2005;72:1761–8. [PubMed] [Google Scholar]
- 3.American Thoracic Society/Centres for Disease Control/Infectious Diseases Society of America: Treatment of Tuberculosis. Am J Respir Crit Care Med. 2003;167:602–2. doi: 10.1164/rccm.167.4.603. [DOI] [PubMed] [Google Scholar]
- 4.Zimhony O, Cox JS, Welch JT, Vilchèze C, Jacobs WR. Pyrazinamide inhibits the eukaryotic-like fatty acid synthetase I (FAS) of Mycobacterium Tuberculosis. Nat Med. 2000;6:1043–7. doi: 10.1038/79558. [DOI] [PubMed] [Google Scholar]
- 5.Controlled clinical trial of 4 short- course regimens of chemotherapy (three 6-month and one 8-month) for pulmonary tuberculosis. East and Central African/British Medical Research Council Fifth Collaborative Study. Tubercule. 1986;67:5–15. doi: 10.1016/0041-3879(86)90027-9. [DOI] [PubMed] [Google Scholar]
- 6.Kumar R, Majetri NV. Nano and microparticles as controlled drug delivery devices. J Pharm Pham Sci. 2000;3:234–58. [PubMed] [Google Scholar]
- 7.Suri SS, Fenniri H, Singh B. Nanotechnology-based drug delivery systems. J Occup Med Toxicol. 2007;2:16. doi: 10.1186/1745-6673-2-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rao JP, Geckeler KE. Polymer nanoparticles: Preparation techniques and size-control parameters. Prog Poly Sci. 2011;36:887–913. [Google Scholar]
- 9.Singh B, Fenniri H, Suri SS. Nanotechnology-based drug delivery systems. J Occup Med Toxicol. 2007;2:16. doi: 10.1186/1745-6673-2-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kumari A, Yadav SK, Yadav C. Biodegradable polymeric nanoparticles based drug delivery systems. Coll Surf B Biointerfaces. 2010;75:1–18. doi: 10.1016/j.colsurfb.2009.09.001. [DOI] [PubMed] [Google Scholar]
- 11.Ahmad Z, Pandey R, Sharma S, Khuller GK. Pharmacokinetic and pharmacodynamic behaviour of antitubercular drugs encapsulated in alginate nanoparticles at two doses. Int J Antimicrob Agents. 2006;27:409–16. doi: 10.1016/j.ijantimicag.2005.12.009. [DOI] [PubMed] [Google Scholar]
- 12.Yang YY, Chung TS, Ng NP. Morphology, drug distribution and in vitro release profiles of biodegradable polymeric microspheres containing protein fabricated by double-emulsion solvent extraction/evaporation method. Biomaterials. 2001;22:231–41. doi: 10.1016/s0142-9612(00)00178-2. [DOI] [PubMed] [Google Scholar]
- 13.Astete CE, Sabliov CM. Synthesis and characterization of PLGA nanoparticles. J Biomat Sci Polym Ed. 2006;17:247–89. doi: 10.1163/156856206775997322. [DOI] [PubMed] [Google Scholar]
- 14.Song X, Zhao Y, Hou S, Xu F, Zhao R, He J, et al. Dual agents loaded PLGA nanoparticles: Systematic study of particle size and entrapment efficiency. Eur J Pharm Biopharm. 2008;69:445–53. doi: 10.1016/j.ejpb.2008.01.013. [DOI] [PubMed] [Google Scholar]
- 15.Koppel DE. Analysis of macromolecular polydispersity in intensity correlation spectroscopy: The method of cumulates. J Chem Phys. 1972;57:4814–20. [Google Scholar]
- 16.Kirby BJ. Cambridge, United Kingdom: Cambridge University Press; 2010. Micro and Nanoscale Fluid Mechanics: Transport in Microfluid Devices. ISBN 978-0-521-11903-0. [Google Scholar]
- 17.Tripathi A, Gupta R, Saraf SA. PLGA Nanoparticles of Antitubercular Drug: Drug Loading and Release Studies of a Water-In-Soluble Drug. Int J Pharm Tech Res. 2010;2:2116–23. [Google Scholar]
- 18.Hortola P. SEM Examination of Human Erythrocytes in Uncoated Bloodstains on stone: Use of Conventional as Environmental-like SEM in a Soft Biological Tissue and Hard Inorganic Material. J Microsc. 2005;218:94–103. doi: 10.1111/j.1365-2818.2005.01477.x. [DOI] [PubMed] [Google Scholar]
- 19.Singhvi G, Singh M. Review: In vitro Drug Release Characterization Models. Int J Pharm Studies Res. 2011;2:77–84. [Google Scholar]
- 20.Costa P, Lobo JM. Modeling and comparison of dissolution profiles. Euro J Pharm Bio. 2001;13:123–33. doi: 10.1016/s0928-0987(01)00095-1. [DOI] [PubMed] [Google Scholar]
- 21.Charyulu N, Bera D, Sakhia D, Mandavadaria K, Shetty NG. Formulation and evaluation of bilayer floating tablets of diltiazem hydrochloride for bimodal release. Int J Pharm Res Sci. 2011;3:301–6. [Google Scholar]
- 22.Dash S, Murthy PN, Nath L, Chowdhury P. Kinetic modeling on drug release from controlled drug delivery systems. Acta Pol Pharm. 2010;67:217–23. [PubMed] [Google Scholar]
- 23.Uronnachi EM, Ogbonna JDN, Kenechukwu FC, Attama AA, Okore VC. Formulation and in vitro/in vivo Evaluation of Zidovudine Contained in Solidified Reverse Micellar Delivery System of Immune Compromised Rats. J App Pharm Sci. 2013;3:31–5. [Google Scholar]
- 24.Yoshioka S. Quinine Actinometry as a Method for Calibrating Ultraviolet Radiation Intensity in Light-Stability Testing of Pharmaceuticals. Drug Dev Ind Pharm. 1994;20:2049–62. [Google Scholar]
- 25.Nagayama S, Ogawara K, Fukuoka Y, Higaki K, Kimura T. Time-dependent changes in opsonin amount associated on nanoparticles alter their hepatic uptake characteristics. Int J Pharm. 2007;342:215–21. doi: 10.1016/j.ijpharm.2007.04.036. [DOI] [PubMed] [Google Scholar]
- 26.Zhao F, Zhao Y, Liu Y, Chang X, Chen C, Zhao Y. Cellular uptake, intracellular trafficking, and cytotoxicity of nanomaterials. Small. 2011;7:1322–37. doi: 10.1002/smll.201100001. [DOI] [PubMed] [Google Scholar]
- 27.Li H, Xiao Y, Niu J, Chen X, Ping Q. Preparation of a cationic nanoemulsome for intratumoral drug delivery and its enhancing effect on cellular uptake in vitro. J Nanosci Nanotechnol. 2011;11:8547–55. doi: 10.1166/jnn.2011.4965. [DOI] [PubMed] [Google Scholar]
- 28.Geiser M, Quaile O, Wenk A, Wigge C, Eigeldinger-Berthou S, Hirn S, et al. Cellular uptake and localization of inhaled gold nanoparticles in lungs of mice with chronic obstructive pulmonary disease. Part Fibre Toxicol. 2013;10:19. doi: 10.1186/1743-8977-10-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Carlstedt I, Sheehan JK, Corfield AP, Gallagjer JT. Mucous glycoproteins: A gel of a problem. Essays Biochem. 1985;20:40–76. [PubMed] [Google Scholar]
- 30.Wijagkanalan W, Kawakami S, Takenaga M, Igarashi R, Yamashita F, Hashida M. Efficient targeting to alveolar macrophages by intratracheal administration of mannosylated liposomes in rats. J Control Release. 2008;125:121–30. doi: 10.1016/j.jconrel.2007.10.011. [DOI] [PubMed] [Google Scholar]
- 31.Diab R, Brillault J, Bardy A, Gontijo AVL, Olivier JC. Formulation and in vitro characterization of inhalable polyvinyl alcohol-free rifampicin-loaded PLGA microspheres prepared with sucrose palmitate as stabilizer: Efficiency for ex vivo alveolar macrophage targeting. Int J Pharm. 2012;436:833–9. doi: 10.1016/j.ijpharm.2012.07.036. [DOI] [PubMed] [Google Scholar]
- 32.Schaeffer DA, Riazy M, Parhar KS, Chen JH, Duronio V, Sawamura T, et al. LOX-1 augments oxLDL uptake by lysoPC-stimulated murine macrophages but is not required for oxLDL clearance from plasma. J Lipid Res. 2009;50:1676–84. doi: 10.1194/jlr.M900167-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]