

Abstract
Background:
Lung administration of antibiotics by nebulization is promising for improved treatment efficiency for pulmonary infections, as it increases drug concentration at sites of infection while minimizing systemic side effects. For poorly soluble molecules like rifampicin, lipid particulate system may improve lung delivery.
Materials and Methods:
We investigated rifampicin-loaded freeze-dried liposomes. Various formulations were prepared with different drug lipid ratios and one formulation was optimized. Optimized colloidal liposome formulation was freeze-dried and subsequently subjected for various evaluation and characterization parameters such as in-vitro dissolution, in-vitro antitubercular activity, aerodynamic characters, surface morphology, and thermal behavior. The optimized formulation of rifampicin-loaded freeze-dried liposome and free rifampicin was subjected for the in-vivo drug disposition study in Wister rat model by intra-tracheal instillation in comparison with an oral route of administration.
Results:
The results of pharmacokinetic study for both free drug and the formulation suggested that liposomes released the drug in a controlled manner for a longer period of time. The enhanced efficiency of drug incorporated into liposomes suggested that the delivery of encapsulated drugs to macrophages was more rapid than that of free drug.
Conclusion:
Therefore, the pharmacokinetic and drug disposition studies provided a sound basis for predicting the successful treatment for tuberculosis.
KEY WORDS: Freeze-dried liposomes, intra-tracheal instillation, nebulization, pulmonary delivery, rifampicin, tuberculosis
INTRODUCTION
Lung the lung has the capability capability to absorb active pharmaceutical compounds delivered either for systemic or local treatment. Pulmonary delivery of such active drugs has become an alternative target and of tremendous scientific and biomedical interest in the health care research area.[1] Specially designed drug delivery systems containing inhaled drugs can be efficiently delivered to the respiratory tract to achieve sustained release pattern. The advantages for sustained release drug delivery to the respiratory tract are numerous, and include extended duration of action, reduction in drug use, improved management of therapy, improved compliance, reduction in side effects,[2] together with potential cost savings that exist for sustained release therapy. For the administration of drugs that are intended to act locally in the lungs, direct deposition to the lungs by some type of aerosol delivery technology,[3] as nebulization of liquid solutions, offers the advantage of increased drug localization at the site of action and decreased possibility of side effects compared to systemic administration. If this administration route is combined with the sustained release and/or targeting potential of novel drug delivery systems, the therapeutic advantages are even more pronounced. Liposomal aerosols in pulmonary therapy offer the additional advantage of uniform deposition of locally active drugs. Between the various available aerosol delivery technologies, nebulizers offer the additional advantage of delivering liposomal drugs without further processing.[4]
Tuberculosis (TB) being the second most deadly infectious disease is still a leading killer of young adults worldwide. Despite potentially curative pharmacotherapies being available for over 50 years, the duration of the treatment and the pill burden can produce patient non-compliances. Thus, low compliance and adherence to administration schedules remain the main reasons for therapeutic failure and contribute in the development of multi-drug-resistant (MDR) strains. An important consideration in the treatment of pulmonary infections is the fact that the multi-drug resistant TB has the capability of surviving intracellulary in the host macrophages for long period of time.[5] Therefore, the ability of the antibacterial agent to eradicate the microorganisms within the macrophage is of key importance. However, most of the anti-mycobacterial drugs presently in use fail to penetrate macrophages. For this reason, many researchers are considering the use of appropriately engineered delivery systems for these drugs, in order to make them therapeutically effective.
The current treatment of TB involves systemic therapy with multiple drugs. However, pulmonary infections do not always respond well to such systemic therapy, due to insufficient drug diffusion into pulmonary tissue and lumen.[6] Bacteria that have persisted over the treatment may develop resistance, but higher drug doses to compensate for poor diffusion may lead to systemic toxicity. Additionally, most patients find it difficult in adhering to treatments that necessitate to several administrations a day to maintain efficient antibiotics concentration. Therefore, it is imperative to develop new ways to deliver and use antibiotics to avoid the spread of resistance and to improve patient's compliance by decreasing dosing frequency. Antibiotic delivery systems administered through pulmonary route aim to deliver high-drug concentrations directly at the site of infection while minimizing systemic bio-distribution and toxicity. Rifampicin (RIF) is a so-called concentration-dependent antibiotic.[7] The rate and extent of bacterial eradication is related to the attainment of sufficiently high and maximum concentration. The bacterial activity is directly proportional to concentration at the target site. In recent years, one of the best ways to achieve higher drug levels in the lungs has been the development of new particulate sustained release formulations that are directly delivered to the lungs via pulmonary route. One of the most non-invasive approaches to the drug delivery is via inhalation. The high frequency of pulmonary TB demands the development of novel drug delivery approaches that enhances bioavailability of the drug at the level of lungs.[8] Preparation of drug-loaded freeze-dried liposomes, designed for delivery to lungs after rehydration/nebulization, are reported to be effective in targeting antibacterial agents to macrophages of the lung.[9,10] The present study was carried out with an aim to develop the particulate-based pulmonary drug delivery systems for encapsulation and release of anti-TB drug in freeze-dried liposomal form in order to make a more effective, compliant and affordable TB pharmacotherapy. Rifampicin (RIF) was used in the present study because it is a frontline drug for tuberculosis and resistance to RIF can develop rapidly. Thereby, there is a therapeutic rationale for delivering RIF-loaded liposomes, in order to deliver it to alveolar macrophages where a large number of tubercule bacilli are present.
MATERIALS AND METHODS
RIF IP was obtained as a gift sample from Micro Labs Ltd Hosur, Tamil Nadu, India. Soya lecithin, cholesterol and dialysis membrane were purchased from, Hi Media Laboratories, Mumbai, India. Ultra-pure water was obtained from a Milli-Q Plus PF water purification system and used throughout the study. All other chemicals were of analytical grade and used without further purification. The institutional animal ethical committee permission was obtained prior to carry out the experiments.
Preparation of rifampicin-loaded liposomes
In the present study, neutral liposomal vesicles were prepared[11] using soya lecithin which is of natural origin. The drug-loaded liposomes were formulated keeping a constant drug: Lipid ratio and varying the ratio of soya lecithin: cholesterol. The details of formulation composition are presented in Table 1. Liposomes were prepared by the thin film hydration method using rotary flash evaporator (Remi Laboratories and Instruments Ltd, Mumbai). Accurately weighed quantities of drug and soya lipids were dissolved in a mixture of chloroform and ethanol in 2:1 ratio in a 100 mL round bottom flask with a round glass neck. The flask was attached to a rotary evaporator immersed in a 40°C water bath, and rotated under vacuum at a speed of 180 rpm until all the organic solvents were evaporated and a dry thin film was deposited on the walls of the flask. The flask was left in a vacuum desiccator overnight to ensure complete removal of residual solvent. Hydration of the thin film with pH 7.4 phosphate buffer saline (PBS) was done for 30–40 min and vortexed till all the lipid film comes out. Liposome formation was identified by the conversion of the buffer into a colloidal milky suspension. The flask was removed and liposomes were transferred into a container and stored under refrigerated conditions. Based on the results of encapsulation efficiency and the in-vitro drug release study FL4 formulation was selected for lyophilization and further evaluation.
Table 1.
Formulation details of liposomes
Lyophilization of liposomal suspension
One of the factors which limit the development of liposome as a delivery system is leakage of the entrapped drug across the lipid membrane. This effect may be retarded by storing the liposomal suspension at low temperature. The freezing and thawing of the liposomal suspension may also be responsible for damage of the vesicle membrane. This effect, in turn, may be minimized by the use of cryoprotectant which acts rapidly by restricting osmotic dehydration and the consequent ionic damage which occurs due to ice-crystal formation outside, but not inside the vesicles of liposome. Liposomal dispersions were prepared by hydrating a dry liquid film with aqueous phase containing sucrose in 1:1 ratio with lipid. Non-encapsulated ripampicin was removed by dialysis for 30 min. Thus prepared liposome was lyophilized under optimum condition and temperatures for 24 h. The dispersion was frozen to − 40°C and freeze drying was performed in freeze drier (OPR 6 DB-5503, Operon, Korea) till a dry powder was obtained.[12,13] The whole drying process took 24 h with primary drying at −40°C for 16 h and secondary drying at −20°C for 8 h. The lyophilized product was subjected for further characterization and evaluation.
Evaluation of liposomes
Vesicle shape
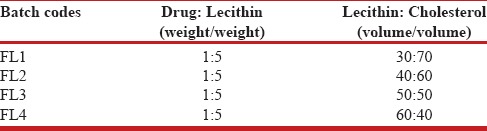
Formation of spherical vesicles was confirmed by examining the suspension under an optical microscope. The vesicle shape was determined by an optical invert microscope under suitable magnification. The samples were placed on the clean glass slide and covered with cover slip and observed. Photographs of the vesicles were taken using a suitable camera. The vesicle shape is shown in Figure 1.
Figure 1.
Vesicle shape of liposomes in colloidal suspension. Liposomes Vesicles
Percent drug encapsulated
Since the in-vitro analysis of drug and drug content assay in formulations need to be carried out in pH 7.4 phosphate buffer solution, a standard graph using this as a medium was thought to be appropriate. The different concentrations of drug solution were prepared in pH 7.4 phosphate buffer solutions. These solutions were scanned at a wavelength range of 400–500 nm in order to determine the λ max value. The drug exhibited a λ max value of 475 nm in pH 7.4 phosphate buffer. With the buffer solutions linearity was found in the concentration range of 2–20 mcg/ml. The standard deviation of six determinations was calculated. A standard graph of absorbance vs. concentration was plotted. The regression co-efficient for standard graph of RIF with the buffer pH 7.4 was found to be 0.9985.
The percent drug encapsulated into the liposomes was determined using the dialysis method. The dialysis membrane-150 (Himedia, India), having cut-off size 12,000 MW and pore size 2.4 nm was selected for dialysis. The dialysis membrane was activated by soaking in Millipore water for 2 hours. One end of the membrane was tied with the thread and then the liposomal solution equivalent to definite amount of drug was poured and tied with a thread. The bag is suspended in a beaker and placed on the magnetic stirrer. During this, unencapsulated drug (free drug) will escape from the colloidal system. Samples were withdrawn at particular time intervals till the readings become constant, and medium was replaced with the same amount of fresh medium. The percent drug encapsulation in all the formulations was calculated by subtracting the amount of free drug (drug which is not encapsulated into vesicles) from the total drug content. The percent drug encapsulated was calculated using the following expression: % Drug Encapsulated = [Total Drug Content − Free drug/Total Drug Content] ×100. The results are shown in Table 2.
Table 2.
Data showing drug entrapment efficiency

In-vitro release study for liposomal suspension
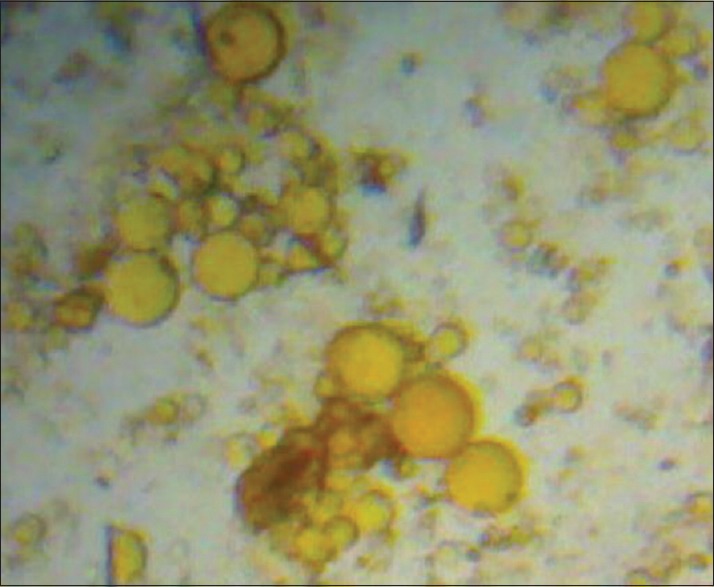
The in-vitro drug release from the liposomal formulations was determined by the dialysis method.[14,15] The liposomal dispersion was transferred to the activated dialysis bags. The bags were tied to the blade of mechanical stirrer and suspended in 250 mL beaker containing 100 mL PBS of pH 7.4. The beakers were placed on magnetic stirrer (Remi Laboratory and Instruments, Mumbai, India) rotating at 50 rpm. The temperature was maintained at 37±1°C throughout the experiment and samples were withdrawn periodically up to 10 hours. The absorbance was measured at 475 nm using the UV–visible spectrophotometer (Simadzu Pharmaspec 1700, Japan). The amount of drug release was determined by using the calibration curve. The results of drug release profile are presented in Figure 2. The best colloidal liposomal solution based on the results of in-vitro drug release in PBS pH 7.4 was lyophilized and subjected for further evaluation and characterization.
Figure 2.
In-vitro drug release profile of rifampicin-loaded colloidal liposomes in pH 7.4 buffer. Colloidal liposome formulations from F1 to F4
Drug content for lyophilized liposomal powder
The drug content in the lyophilized liposome formulation (FDFL4) was estimated using the UV–spectrophotometric method based on the measurement of absorbance at 475 nm in phosphate buffer pH 7.4. A known amount of formulation was added to 10 mL of methanol and kept it for 2 hours. The content was vortexed followed by centrifugation. The supernatant solution was passed through nylon filter and diluted suitably with phosphate buffer pH 7.4. Then the amount of drug present in the solution was estimated using UV–visible spectrophotometer. Then the percentage of drug present was calculated using the standard graph. The results are shown in Table 3.
Table 3.
Data obtained from evaluation of freeze dried liposomal formulation

Determination of residual moisture
The percentage of residual moisture in the FDFL4 was determined using a Karl Fischer coulometer. Anhydrous methanol was used to extract water from the lyophilized product. The moisture present was determined by using the expression: Residual moisture=[Volume consumed × KF factor]/Weight taken. The results are shown in Table 3.
pH of the reconstituted solution
The FDFL4 formulation was reconstituted with pH 7.4 PBS and pH of the solution was checked through pH meter. The results are shown in Table 3.
Differential scanning calorimetry
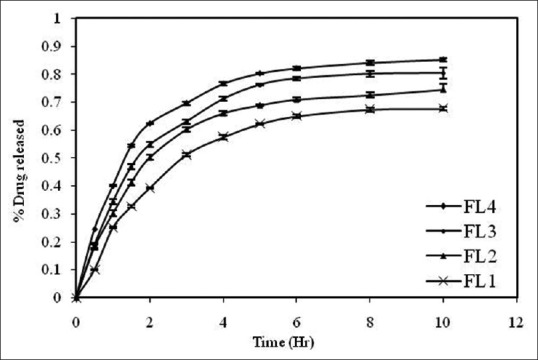
DSC is a thermo analytical technique in which the difference in the amount of heat required to increase the temperature of a sample and reference are measured as a function of temperature. DSC (METTLER TOLEDO, 823e, JAPAN) experiments were carried out[16,17] in order to characterize the physical state of the drugs. Sample of FDFL4 was placed in aluminum pans and thermatically sealed. The heating rate was 10°C per minute using nitrogen as the purge gas. The results of thermal analysis are given in Figure 3.
Figure 3.
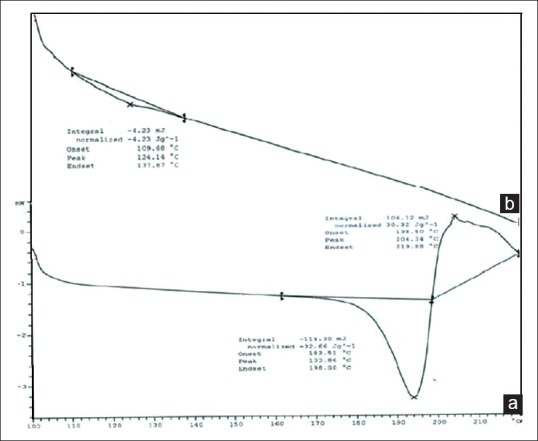
Differential Scanning Calorimetry thermograms. (a) Pure Rifampacin. (b) Formulation FDLH4
Scanning electron microscopy
The surface morphology of the particles was evaluated by scanning electron microscopy (Joel 840 A, Japan). The FDFL4 was scattered individually onto a thin film of epoxy resin and coated with a platinum layer and mounted onto the hub of SEM instrument and the particles were focused at different magnifications at room temperature and photographed with direct data capture of the images onto a personal computer. The results are shown in Figure 4.
Figure 4.

Scanning Electron Microphotographs. (a) Pure Rifampicin. (b) Formulation FDFL4
Particle size analysis
The particle size analysis of FDFL4 was done using Mastersizer-2000 Ver 2.0 (Malvern Instruments, Malvern U.K) with a range of detection of 0.02–2000 μ. The data analysis was done with Mastersizer 2000 Ver 5.22. The mean particle size was then calculated. The results are shown in Table 3.
In-vitro anti-tubercular activity
The FDFL4 formulation and pure RIF were evaluated for in-vitro anti-tubercular activity. The study was performed and interpreted for Mycobacterium tuberculosis H37 RV according to the approved procedure of a macro dilution anti-microbial susceptibility testing method.[18] Samples were taken at concentrations of 100 μg/mL, 50 μg/mL and 25 μg/mL in dimethyl formamide after passing through a membrane filter and added to the media. Ciprofloxacin (10 μg/mL) and streptomycin (7.5 μg/mL) were taken as reference standards. Mycobacterium tuberculosis was inoculated with standard as well as test samples and incubated at 37°C for 4 weeks. The bottles were inspected for growth of the mycobacterium twice a week for a period of 3 weeks. Readings were taken at the end of fourth week. The growth of the bacilli results in turbidity which indicates resistance to the samples. The results are presented in Table 4 and Figure 5.
Table 4.
In-vitro anti-tubercular activity of FDFL4 formulation

Figure 5.
Photographs showing in vitro anti-tubercular activity. (a) Pure Rifampicin. (b) Formulation FDFL4
Aerodynamic characterization (in-vitro lung deposition study)
Aerodynamic characterization of FDFL4 was carried out on an 8- stage Anderson Cascade Impactor with a preseparater (Graseby-Andersen, Atlanta, GA, USA) operating at an air flow rate at 60 l/min which stimulate a typical human respiratory flow rate. The samples were collected in a buffer solution in a three stage glass impinge and analyzed. Mass median aerodynamic diameter (MMAD), geometric standard deviation (GSD) and fine particle fractions (FPF) were calculated according to USP 27 NF 22. The results are presented in Table 5.
Table 5.
Aerodynamic characterization of spray-dried microspheres (in-vitro lung deposition data)

In-vitro drug release study
Drug release from FDFL4 formulation and pure drug was carried out using modified dissolution method.[19,20] The media was simulative intestinal fluid pH 7.4 and simulated lung fluid pH 5.2 corresponding to the endosomal pH of alveolar macrophages and 200 μg/ml ascorbic acid added as an antioxidant to prevent oxidative degradation. A known mass of powdered liposomes sample was suspended in tubes containing the buffer. The tubes were placed in a shaker bath (Remi RS-B12), operated at 90 cycles/min at 37±1°C running for 72 hours. At predetermined intervals of time, 5 mL of the sample was withdrawn and analyzed for drug content at 475 nm spectrophotometrically following suitable dilutions. The study was performed in triplicate. The in-vitro drug release profile is presented in Figure 6.
Figure 6.
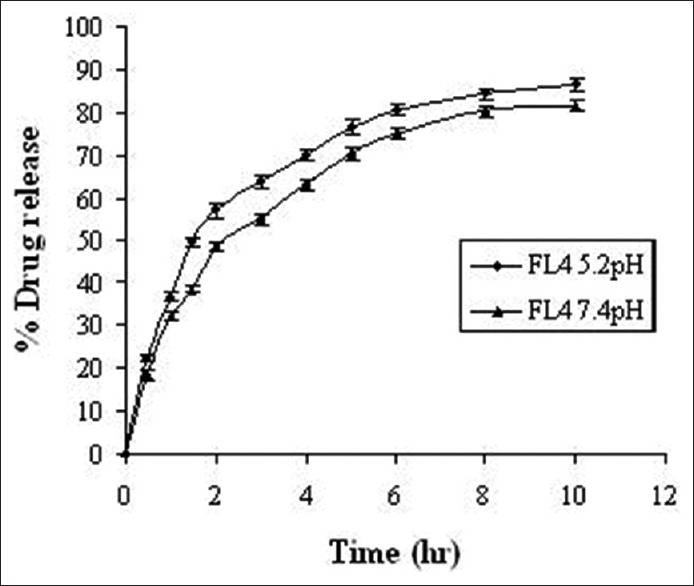
In-vitro drug release profile of FL4 in pH 7.4 and pH 5.2 buffers. FL4: Rifampicin-loaded lyophilized liposomes
In-vivo drug disposition study
All experimental procedures were in accordance with guidelines for animal welfare set out by the Medical Research Council of India. Animal experiments were approved by institutional animal ethical committee, Al-Ameen College of Pharmacy, Bangalore (AACP/IAEC/P-34/2006. Dt. 26.05.2006). Adult Wister rats of either sex weighing 100-150 ± 10 gm were housed in polypropylene cages with free access to pelletized chow and tap water. The animals were exposed to alternate cycles of 12 hours light and darkness. The temperature was maintained at approximately 26°C to 28°C.
For single dose drug deposition studies, the rats were randomized into four groups (n = 6). Group 1, free drug instilled: Group 2, drug-loaded liposomes instilled; Group 3, oral free drug; Group 4, oral drug-loaded liposomes. The formulations equivalent to the drug was used at therapeutic dosage of 12 mg/kg body weight. The surgery was performed after anesthetizing animals with an intraperitoneal injection of urethane.[21,22,23,24] Anesthetized animals were placed in supine position on a 45° slanted support, and a small middle incision was made over the trachea. The trachea was exposed by blunt dissection of the sternohyoideus muscle. A small hole was made in the trachea between the fifth and sixth tracheal rings using a 20-gauge needle. A short about 10-20 cm length of PE50 tubing was inserted into the hole and advanced to the bifurcation of the trachea. Formulation of 0.5 mL (lyophilized drug-loaded liposomal powder, and free drug separately suspended in a sterile phosphate buffer pH 5.2) was slowly instilled over a 1 min period using a 1 mL syringe attached to the PE50 tubing. Following instillation, the tubing was withdrawn and a small drop of cyanoacrylate adhesive was placed over the hole to seal the opening. The skin was clothed with 3-0 Dexon sutures. The animals were removed from anesthesia and allowed to recover under a heating lamp. After recovery, animals were housed in individual plastic cages with access to food and water for remainder of the study. The free drug and encapsulated formulations were also administered intragastrically to oral groups.
Blood was sampled from the tail vein at several time points and plasma was separated. To 2 mL of plasma in a 15 mL test tube, 0.4 mL of 10% v/v aqueous acetic acid was added to adjust the pH to 4.2. Drug was extracted[25] by shaking with 7 mL of diethyl ether: dichloromethane (2:1v/v). The sample was vortexed for 10 min followed by centrifugation for 10 min at 10,000 rpm and the content was transferred to a tapered test tube and evaporated to dryness under a stream of liquid nitrogen. The resulting residue was reconstituted in 5 μL mobile phase and injected into LCMS system. The chromatograms were acquired using the computer-based software supplied by Aligent technologies. The data was processed by peak area ratio. The concentration of the RIF in plasma at different time point was calculated from the following equation using regression analysis of spiked calibration standard with the reciprocal of the square of the drug concentration as weighing factor (1/concentration × concentration). Y = mx + b, where, x = concentration of analyte, m = slope of the concentration curve, Y = peak area of analyte to internal standard and b = y-axis intercept of the calibration curve. The results are shown in Figure 7.
Figure 7.
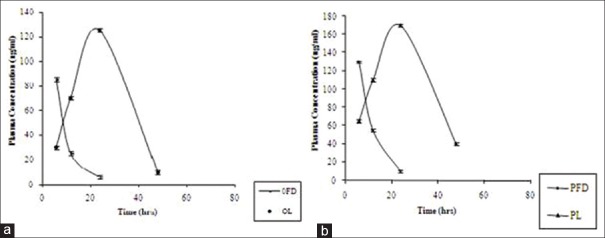
In vivo Plasma drug profile of free Rifampicin and its lyophilized liposome formulation. (a) Oral administration. (b) Pulmonary administration
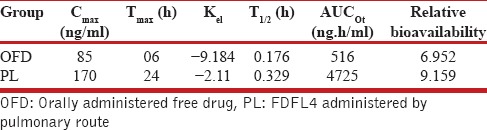
Serum RIF's levels were measured to evaluate pharmacokinetics following administration of drug -oaded liposomes and free drug. The serum RIF's concentrations at each sampling time point were plotted against time in hours. Maximum serum concentration (Cmax), time in hours to achieve Cmax (Tmax), area under the plasma concentration curve (AUC), serum elimination constant (Kel) and RIF serum half-life (t1/2) were determined from RIF serum concentration-time curve. The area under the curve was calculated by the trapezoidal rule and further used to compute relative bioavailability using the following equation: Relative bioavailability = (ACU0-t of oral encapsulated drug/ACU0-t of oral free drug) × (dose of oral free drug/dose of oral encapsulated drug). The results are presented in Table 6.
Table 6.
Pharmacokinetics of drug-loaded formulations compared with oral free drug
Statistical analysis of data
The pharmacokinetic data, including bioavailability, were analyzed by analysis of student's unpaired t-test and differences at P < 0.05 were considered significant.
RESULTS
Liposomal formulations were subjected for various evaluation and characterization studies. The vesicle shape of the liposomes was determined by optical invert microscope under suitable magnification and it is evident from Figure 1 that the shape of the liposomes was almost spherical. The data of percent drug encapsulated in the liposomes was presented in Table 2 and found to be in the range of 66.52-79.25%. The freeze-dried liposome formulation (FDFL4) was subjected for further characterization and evaluation. The percentage drug content for the FDFL4 was found to be highest and satisfactory than other formulations. Percentage residual moisture content in the FDFL4 was found to be 1.87, which is in the acceptable limit. The pH of the reconstituted solution of FDFL4 in phosphate buffer pH 7.4 solution was found to be almost neutral. The particle size of FDFL4 was found to be 6.373 μm. The results of above four parameters are depicted in Table 3. The in-vitro drug release from FDFL4 was carried out both in simulated lung fluid (pH 5.2 buffer) and simulated intestinal fluid (pH 7.4 buffer without enzymes up to 10 hours. The pattern of in-vitro drug release profile from FDFL4 was examined and is presented in Figure 2. The release data indicated that, the formulation FDFL4 has shown maximum 86.62% drug release in simulated lung fluid and 82.124% in simulated intestinal fluid at the end tenth hour. The drug release pattern of FDFL4 had shown 59% and 63% of drug release within 3 hours in simulated intestinal and simulated lung fluid, respectively.
The FTIR spectra of pure RIF and FDFL4 were determined and all the functional groups of drug were found to be intact in the formulation and it was further confirmed by DSC. The DSC curve of pure RIF showed a single melting peak at 193°C and started to degrade as it melted. The endothermic peak of RIF was disappeared in the thermogram of FDFL4. The surface morphology of pure drug and FDFL4 was studied by SEM. The characteristic needle-shaped crystals were observed in photomicrographs of pure RIF. A prominent change in the surface morphology was observed with photomicrographs of RIF-encapsulated powdered liposomes. The morphology was found to be porous, continuous, amorphous, and uniform texture with clustered having sharp edges.
In-vitro anti-tubercular activity of RIF and FDFL4 was investigated. The formulation FDFL4 had shown better in-vitro anti-tubercular activity against M. tuberculosis H37 RV strain than the pure RIF alone. The study demonstrated that the RIF-encapsulated freeze-dried liposome formulation was showing no growth of micro-organisms at 50 μg/mL and 100 μg/mL concentrations, thus, exhibiting maximum effectiveness when compared to pure RIF alone.
The FDFL4 formulation had an average particle size of 6.373 μ. Aerodynamic characterization of FDFL4 was carried out using an 8-stage Anderson Cascade Impactor, then the mass median aerodynamic diameter (MMAD), geometric standard deviation (GSD) and fine particle fractions (FPF) were calculated. Aerodynamic characterization reveled that nearly 75% of FDFL4 particles were in the respirable size range, with a MMAD of 5.546 μ and GSD of 1.832 μ. The FPF of FDFL4 was 25.52%.
A single dose drug disposition study was carried out for formulation FDFL4 in rats. When FDFL4 was administered through both intratracheal instillation and oral route, RIF was detected in plasma from 4 hours onwards and observed up to 48 hours. In contrast, free drug administered by both the routes was cleared from the circulation within 24 hours. All the pharmacokinetic parameters calculated were found to be better for encapsulated liposome upon administered through pulmonary route, resulting in increased relative bioavailability compared with free drug. The prolonged plasma retention of RIF encapsulated in liposome was achieved compared with free drug given by both oral and pulmonary route. In the encapsulated form, RIF stayed in the plasma for up to 48 hours.
DISCUSSION
The formulation FL4 showed better percent drug encapsulation. As the cholesterol concentration increased the drug encapsulation decreased, this may be due to the increased lipophilic properties of lipid bilayer. The in-vitro drug release from the liposomes was shown to be dependent on concentration of cholesterol used. As the concentration of cholesterol increased the drug release decreased. Therefore, the formulation FL4 showed better drug release and was chosen for lyophilization.
The freeze-dried liposome formulation (FDFL4) was subjected for further characterization and evaluation. Percentage residual moisture content in the FDFL4 was found to be in the acceptable limit. The pH of the reconstituted solution of FDFL4 in phosphate buffer pH 7.4 solution was found to be almost neutral. The results of in-vitro drug release pattern of FDFL4 indicated that the drug release pattern of FDFL4 was better in simulated lung fluid than in physiological fluid. The liposome formulation had shown slow and sustained release of drug.
The FTIR spectra of pure RIF and FDFL4 were determined and there was no indication of drug polymer interactions which indicated that the drug found to be intact in the formulation and it was further confirmed by DSC.
The DSC curve of pure RIF showed a single melting peak at 193°C indicating degradation of drug as it melted. The endothermic peak of RIF was disappeared in the thermogram of FDFL4 which might be due to the amorphous state of the encapsulated drug in the liposomes. Since, there was no shift in lipid peak, it can be concluded that there is no significant interaction occurring between the RIF and lipid used.
The surface morphology of pure drug and FDFL4 was studied by SEM. The characteristic needle-shaped crystals of pure RIF indicated that drug is in crystalline form. A prominent change in the surface morphology was observed with photomicrographs of RIF-encapsulated powdered liposomes. Powder surface was found to be porous, continuous, and uniform texture with clustered having sharp edges suggested that the drug is converted in to amorphous form.
The results of in-vitro anti-tubercular activity of pure RIF and FDFL4 suggested that the components of liposomal vesicles helped to convert the crystalline drug into amorphous form which ultimately leads to enhance the solubility of drug as well as its anti-tubercular activity when compared to pure drug alone.
The results of aerodynamic characteristics suggested that the particles formed were within the respirable size range and nebulization efficiency of the encapsulated liposomes depends on the percentage of aerosolized drug that remains encapsulated after nebulization. These studies are of particular importance to have information of the capability of particles to be aerosolized and also to retain the entrapped drug during the nebulization process.
The results single dose drug disposition study in rats supported the role of liposomes in enhancement of drug permeation through alveolar epithelium by altering physico-chemical properties of the drug. Liposome encapsulation of RIF also acted as a bio-degradable reservoir, which prolonged pulmonary residence time of drug.
CONCLUSION
To overcome the daily dosing problem of TB therapy, the application of drug delivery systems is a novel strategy. Present study clearly indicates that rifampicin-encapsulated liposomes have a tremendous potential as direct drug delivery systems to lungs. Main advantages of these drug delivery systems include the sustained release of drug over longer durations and reduction in systemic drug toxicity. The developed liposome formulation demonstrated improved bio-availability of rifampicin when administered by pulmonary route in comparison with oral route. The components of liposomal vesicles may be suitably changed to achieve higher bio-availability. Hence, TB can be treated effectively with a significant reduction in the severity of disease compared with free drug. Therefore, above-developed formulations offer promising potential as an alternative chemotherapy for treatment of TB in improving patient compliance and lowering cost, dosage and toxic effects of the therapy. The drug delivery system developed in the present study was found to be suitable for administration to the patients in dry powder inhaler form which is evidenced by the aerodynamic characterization data. The developed formulations can be nebulized with conventional nebulizers, efficiently and conveniently, resulting in particle size ranges compatible with pulmonary deposition. This novel approach opens the door for future in-vivo testing in a suitable infected animal model and further clinical trials.
Footnotes
Source of Support: Nil
Conflict of Interest: None declared.
REFERENCES
- 1.Patil JS, Sarasija S. Pulmonary drug delivery strategies: A concise, systematic review. Lung India. 2012;29:44–9. doi: 10.4103/0970-2113.92361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hardy JG, Chadwick TS. Sustained release drug delivery to the lungs: An option for the future. Clin Pharmacokinet. 2000;39:1–4. doi: 10.2165/00003088-200039010-00001. [DOI] [PubMed] [Google Scholar]
- 3.Suarez S, Hickey AJ. Drug properties affecting aerosol behavior. Respir Care. 2000;45:652–66. [PubMed] [Google Scholar]
- 4.Lange CF, Hancock RE, Samuel J, Finlay WH. In vitro aerosol delivery and regional airway surface liquid concentration of a liposomal cationic peptide. J Pharm Sci. 2001;90:1647–57. doi: 10.1002/jps.1115. [DOI] [PubMed] [Google Scholar]
- 5.Quenelle DC, Winchester GA, Staas JK, Barrow EL, Barrow WW. Treatment of tuberculosis using a combination of sustained-release rifampin-loaded microspheres and oral dosing with isoniazid. Antimicrob Agents Chemother. 2001;45:1637–44. doi: 10.1128/AAC.45.6.1637-1644.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Li J, Rayner CR, Nation RL, Owen RJ, Spelman D, Tan KE, et al. Heteroresistance to colistin in multidrug-resistant Acinitobacter baumannii. Antimicrob Agents Chemother. 2006;50:2946–50. doi: 10.1128/AAC.00103-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gumbo T, Louie A, Deziel MR, Liu W, Parsons LM, Salfinger M, et al. Concentration-dependent Mycobacterium tuberculosis killing and prevention of resistance by rifampin. Antimicrob Agents Chemother. 2007;51:3781–8. doi: 10.1128/AAC.01533-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Patil JS, Suresh S. Physicochemical characterization, in vitro release and permeation studies of resipirable rifampicin-cyclodextrin inclusion complexes. Indian J Pharm Sci. 2009;71:638–43. doi: 10.4103/0250-474X.59545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zaru M, Sinico C, De Logu A, Caddeo C, Lai F, Manca ML, et al. Rifampicin-loaded liposomes for the passive targeting to alveolar macrophages: In vitro and in vivo evaluation. J Liposome Res. 2009;19:68–76. doi: 10.1080/08982100802610835. [DOI] [PubMed] [Google Scholar]
- 10.Zaru M, Mourtas S, Klepetsanis P, Fadda AM, Antimisiaris SG. Liposomes for drug delivery to the lungs by nebulization. Eur J Pharm Biopharm. 2007;67:655–66. doi: 10.1016/j.ejpb.2007.04.005. [DOI] [PubMed] [Google Scholar]
- 11.Voss A, Finlay WH. Deagglomeration of dry powder pharmaceutical aerosols. Int J Pharm. 2003;248:39–50. doi: 10.1016/s0378-5173(02)00319-8. [DOI] [PubMed] [Google Scholar]
- 12.Coates MS, Fletcher DF, Chan HK, Raper JA. Effects of design on the performance of a dry powder inhaler using computational fluid dynamics. Part 1: Grid structure and mouthpiece length. J Pharm Sci. 2004;11:2863–76. doi: 10.1002/jps.20201. [DOI] [PubMed] [Google Scholar]
- 13.Cipolla D, Farr SJ, Gonda I, Otulana B. Delivery of biologics to the lung. In: Hansel TT, Barnes PJ, Bolliger CT, editors. New Drugs for Asthma, Allergy and COPD: Progress in Respiratory Research. Vol. 31. Switzerland: Karger Publisher; 2001. pp. 20–33. [Google Scholar]
- 14.Nagarsenker MS, Londhe VY. Preparation and evaluation of a liposomal formulation of sodium cromoglicate. Int J Pharm. 2003;251:49–56. doi: 10.1016/s0378-5173(02)00583-5. [DOI] [PubMed] [Google Scholar]
- 15.Glavas-Dodov M, Fredro-Kumbaradzi E, Goracinova K, Simonoska M, Calis S, Trajkovic-Jolevska S, et al. The effect of lyophilization on the stability of liposomes containing 5-FU. Int J Pharm. 2005;291:79–86. doi: 10.1016/j.ijpharm.2004.07.045. [DOI] [PubMed] [Google Scholar]
- 16.Jakobsen DF, Frokjaer S, Larsen C, Niemann H, Buur A. Application of isothermal micro-calorimetry in preformulation: Hygrosopicity of drug substances. Int J Pharm. 1997;156:67–77. [Google Scholar]
- 17.Dubernet C. Thermoanalysis of microspheres. Thermo Chim Acta. 1995;248:259–69. [Google Scholar]
- 18.Andrews JM. Determination of minimum inhibitory concentrations. J Antimicrob Chemother. 2001;48:5–16. doi: 10.1093/jac/48.suppl_1.5. [DOI] [PubMed] [Google Scholar]
- 19.O’Hara P, Hickey AJ. Respirable PLGA microspheres containing rifampicin for the treatment of tuberculosis: Manufacture and characterization. Pharm Res. 2000;17:955–61. doi: 10.1023/a:1007527204887. [DOI] [PubMed] [Google Scholar]
- 20.Davies NM, Feddah MR. A novel method for assessing dissolution of aerosol inhaler products. Int J Pharm. 2003;255:175–87. doi: 10.1016/s0378-5173(03)00091-7. [DOI] [PubMed] [Google Scholar]
- 21.Sanna V, Kirschvink N, Gustin P, Gavini E, Roland I, Delattre L, et al. Preparation and in vivo toxicity study of solid lipid microparticles as carrier for pulmonary administration. AAPS PharmSciTech. 2003;5:e27. doi: 10.1208/pt050227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Garcia-Contreras L, Morçöl T, Bell SJ, Hickey AJ. Evaluation of novel particles as pulmonary delivery systems for insulin in rats. APPS Pharm Sci. 2003;5:E9. doi: 10.1208/ps050209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shahiwala A, Misra A. A preliminary pharmacokinetic study of liposomal leuprolide dry powder inhaler: A technical note. AAPS Pharm Sci Tech. 2005;6:E482–6. doi: 10.1208/pt060360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Enna SJ, Schanker LS. Absorption of drugs from the rat lung. Am J Physiol. 1972;223:1227–31. doi: 10.1152/ajplegacy.1972.223.5.1227. [DOI] [PubMed] [Google Scholar]
- 25.Hemanth Kumar AK, Chandra I, Geetha R, Chelvi KS, Lalitha V, Prema G. A validated high-performance liquid chromatography method for the determination of rifampicin and desacetyl rifampicin in plasma and urine. Indian J Pharmacol. 2004;36:231–3. [Google Scholar]