

ABSTRACT
Bacterial infection can trigger cellular stress programs, such as the unfolded protein response (UPR), which occurs when misfolded proteins accumulate within the endoplasmic reticulum (ER). Here, we used the human pathogen methicillin-resistant Staphylococcus aureus (MRSA) as an infection model to probe how ER stress promotes antimicrobial function. MRSA infection activated the most highly conserved unfolded protein response sensor, inositol-requiring enzyme 1α (IRE1α), which was necessary for robust bacterial killing in vitro and in vivo. The macrophage IRE1-dependent bactericidal activity required reactive oxygen species (ROS). Viable MRSA cells excluded ROS from the nascent phagosome and strongly triggered IRE1 activation, leading to sustained generation of ROS that were largely Nox2 independent. In contrast, dead MRSA showed early colocalization with ROS but was a poor activator of IRE1 and did not trigger sustained ROS generation. The global ROS stimulated by IRE1 signaling was necessary, but not sufficient, for MRSA killing, which also required the ER resident SNARE Sec22B for accumulation of ROS in the phagosomal compartment. Taken together, these results suggest that IRE1-mediated persistent ROS generation might act as a fail-safe mechanism to kill bacterial pathogens that evade the initial macrophage oxidative burst.
IMPORTANCE
Cellular stress programs have been implicated as important components of the innate immune response to infection. The role of the IRE1 pathway of the ER stress response in immune secretory functions, such as antibody production, is well established, but its contribution to innate immunity is less well defined. Here, we show that infection of macrophages with viable MRSA induces IRE1 activation, leading to bacterial killing. IRE1-dependent bactericidal activity required generation of reactive oxygen species in a sustained manner over hours of infection. The SNARE protein Sec22B, which was previously demonstrated to control ER-phagosome trafficking, was dispensable for IRE1-driven global ROS production but necessary for late ROS accumulation in bacteria-containing phagosomes. Our study highlights a key role for IRE1 in promoting macrophage bactericidal capacity and reveals a fail-safe mechanism that leads to the concentration of antimicrobial effector molecules in the macrophage phagosome.
INTRODUCTION
The endoplasmic reticulum (ER) exhibits stress in response to many environmental insults, including infection by microbial pathogens. Recent evidence supports a functional role for ER stress in immunity, but the host defense mechanisms controlled by this stress response are not fully understood. During ER stress, cells induce an extensive adaptive program, termed the unfolded protein response (UPR), to alleviate the stress. In mammalian cells, the UPR is controlled by three resident ER sensors, of which inositol-requiring enzyme 1α (IRE1α) is the most evolutionarily conserved (1). Oligomerization of IRE1 triggers autophosphorylation and activation of the cytoplasmic endonuclease domain (2). The endonuclease splices a specific target mRNA, removing a 26-nucleotide intron, to produce an active transcription factor, X-box binding protein-1 (XBP1) (3, 4). XBP1 is required for normal differentiation and functions of many immune cell types, including plasma cells, dendritic cells, and CD8+ T cells (5–7). Mice deficient in the IRE1 arm of the UPR fail to control infection by polyomavirus or the bacterial pathogen Francisella tularensis (6, 8), demonstrating the importance of this pathway in host defense.
More recently, IRE1 has been implicated in multiple signaling pathways that lead to immune activation and inflammation. Bacterial ligands induce IRE1 activation in macrophages in a Toll-like receptor (TLR)-dependent manner (8). In this case, IRE1 augmented inflammatory cytokine output through splicing of Xbp1, which led to XBP1-mediated increases in cytokine gene expression. Moreover, exogenous cholera toxin traffics to the ER and activates IRE1 by directly binding to the sensor, stimulating decay of endogenous mRNA (9). This process, termed regulated IRE1-dependent decay (RIDD), engages cytosolic sensing machinery to activate type I interferon and NF-κB-dependent cytokine transcription. Last, IRE1 activation by metabolic stress led to interleukin-1β (IL-1β) production by the inflammasome (10). Thus, the evidence for involvement of IRE1 in modulating inflammatory cytokine production is compelling. However, the ER, as the largest intracellular organelle, also profoundly influences other cellular functions relevant to infection, such as vesicle trafficking and antigen presentation, although the role of IRE1 in these processes has not been well studied.
Interaction of the ER with the phagosome is a critical aspect of phagocyte function. The ER enables phagocytosis by fusing with the nascent phagosome to provide a source of membrane (11). In addition, ER resident proteins are delivered to phagosomes through the activity of the SNARE Sec22B to promote antigen cross-presentation (12). As the compartment for production and sorting of vesicular proteins, the ER also plays a fundamental role in populating phagolysosomes with enzymes that contribute to antimicrobial function (13). Although the ER shapes phagosomal function, the role of the IRE1 ER stress sensor in controlling bacterial killing has not been explored. Here, we show that macrophage IRE1 is strongly activated by live MRSA infection, inducing sustained production of antimicrobial reactive oxygen species within the bacteria-containing phagosome.
RESULTS
IRE1-dependent innate immune signaling augments bacterial killing and resistance to MRSA subcutaneous infection.
To elucidate mechanisms of IRE1-driven antimicrobial immunity, we used community-acquired methicillin-resistant Staphylococcus aureus strain USA300, an important human pathogen that can be taken up by macrophages but does not replicate robustly within the cells (14). We first determined whether MRSA induces IRE1 activation in a TLR-dependent manner. Wild-type bone marrow-derived macrophages (BMDM) or Tlr2/4/9−/− or Myd88−/− BMDM were infected with MRSA, and IRE1 activation was assessed by measuring the extent of Xbp1 splicing (Fig. 1A). MRSA infection increased Xbp1 splicing in wild-type macrophages, but not in macrophages lacking TLR2/4/9 or Myd88. However, treatment by thapsigargin, a potent ER Ca2+ ATPase inhibitor, induced strong activation of IRE1 in both wild-type and knockout macrophages, indicating that the TLR pathway is not required for the general ER stress response. To investigate the functional consequences of TLR-induced IRE1 activation in innate immune effector function, we assessed whether IRE1 contributes to macrophage killing of MRSA. Since IRE1-deficient mice exhibit embryonic lethality (15), we knocked down IRE1 in the RAW 264.7 peritoneal macrophage cell line by stably expressing shRNA specific for IRE1 or a nontargeted (NT) control shRNA (Fig. 1B). These cells were infected with MRSA, CFU were enumerated, and the percent intracellular bacterial killing was calculated as follows: [(CFU2h pi − CFU4h pi)/CFU2h pi]. Bacterial killing was significantly decreased at 4 and 8 h postinfection (h pi) in IRE1 knockdown (KD) cells compared to NT controls (Fig. 1C). Since Xbp1 splicing is the most-well-characterized function of activated IRE1, we asked whether XBP1 contributed to the macrophage antimicrobial phenotype we observed during MRSA infection. Knockdown of XBP1 decreased the ability of macrophages to kill MRSA (see Fig. S1A and B in the supplemental material). Together, these data showed the requirement of TLR signaling in activating IRE1 during MRSA infection and established a role of this ER stress sensor in promoting macrophage bactericidal function.
FIG 1 .
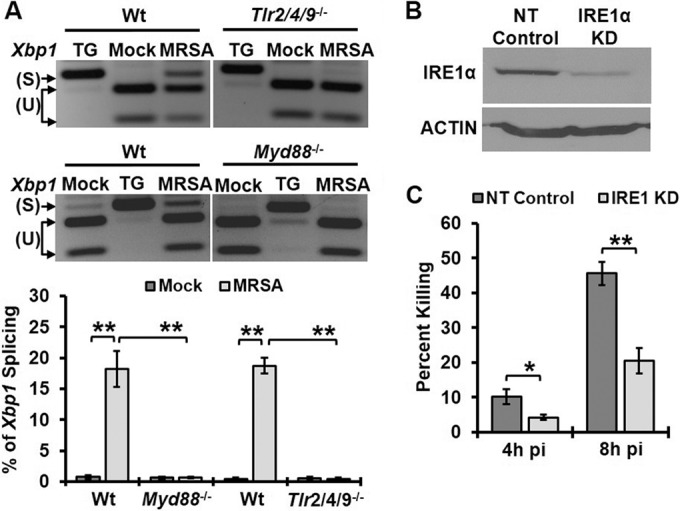
IRE1 activated through TLR signaling enhances macrophage bactericidal activity. (A) RT-PCR analysis of Xbp1 mRNA splicing in BMDM when left untreated (mock), treated with 5 µM thapsigargin (TG), or infected with MRSA for 8 h at an MOI of 20. PCR products were digested with PstI endonuclease. Because unspliced mRNA contains a PstI site within the 26 spliced region, the digested PCR products yield two smaller fragments representing unspliced (U) Xbp1 and one larger fragment representing spliced (S) Xbp1. RT-PCR images are representative of ≥3 independent experiments, and the percent spliced Xbp1 was calculated based on band densitometry as follows: [Xbp1s/(Xbp1s + Xbp1u)]. (B) Immunoblots of cell lysate from RAW 264.7 macrophages stably transduced with lentivirus-encoded shRNA for nontarget (NT-control) or IRE1 KD, probed with an anti-IRE1 antibody or anti-actin antibody as a loading control. (C) NT-control and IRE1 KD macrophages were infected with MRSA (MOI, 20). The percent killing was quantified by the percent difference in CFU at the indicated time points relative to results at 2h pi: [(CFU2h pi − CFU4h pi)/CFU2h pi]. Results presented are averages of ≥3 independent experiments ± standard deviations. *, P < 0.05; **, P < 0.01.
IRE1 deficiency decreased bacterial killing by macrophages in vitro, but it might not have the same effect on innate immune function in vivo. In order to investigate the role of IRE1 in innate immunity to MRSA in vivo, we first tested the in vitro efficacy of two commercially available IRE1 endonuclease inhibitors, 4µ8C and STF-083010, in macrophages (16, 17). When RAW 264.7 cells were treated with either of the IRE1 inhibitors, we observed no Xbp1 splicing (Fig. 2A), and the inhibitor-treated cells were less capable of killing MRSA than were dimethyl sulfoxide (DMSO)-treated cells (Fig. 2B). Similar results were observed in IRE1 inhibitor-treated human THP-1 monocytes (see Fig. S2 in the supplemental material). If IRE1 contributes to antimicrobial function in vivo, we predict that IRE1 inhibitors would exacerbate MRSA infection. We used a skin abscess model of MRSA infection in which innate immune function plays a major role in early host defense and bacterial clearance (18, 19). The IRE1 inhibitors 4µ8C and STF-083010 were administered intraperitoneally prior to and during infection with MRSA, which was inoculated subcutaneously into the shaved flanks of animals. Inhibitor-treated mice developed larger lesions and had higher bacterial burdens than mice treated with vehicle controls (Fig. 2C and D). Taken together, these data indicate that activation of the IRE1 ER stress sensor enhances bacterial killing in vitro and contributes to the innate immune defense against bacterial infection in vivo.
FIG 2 .
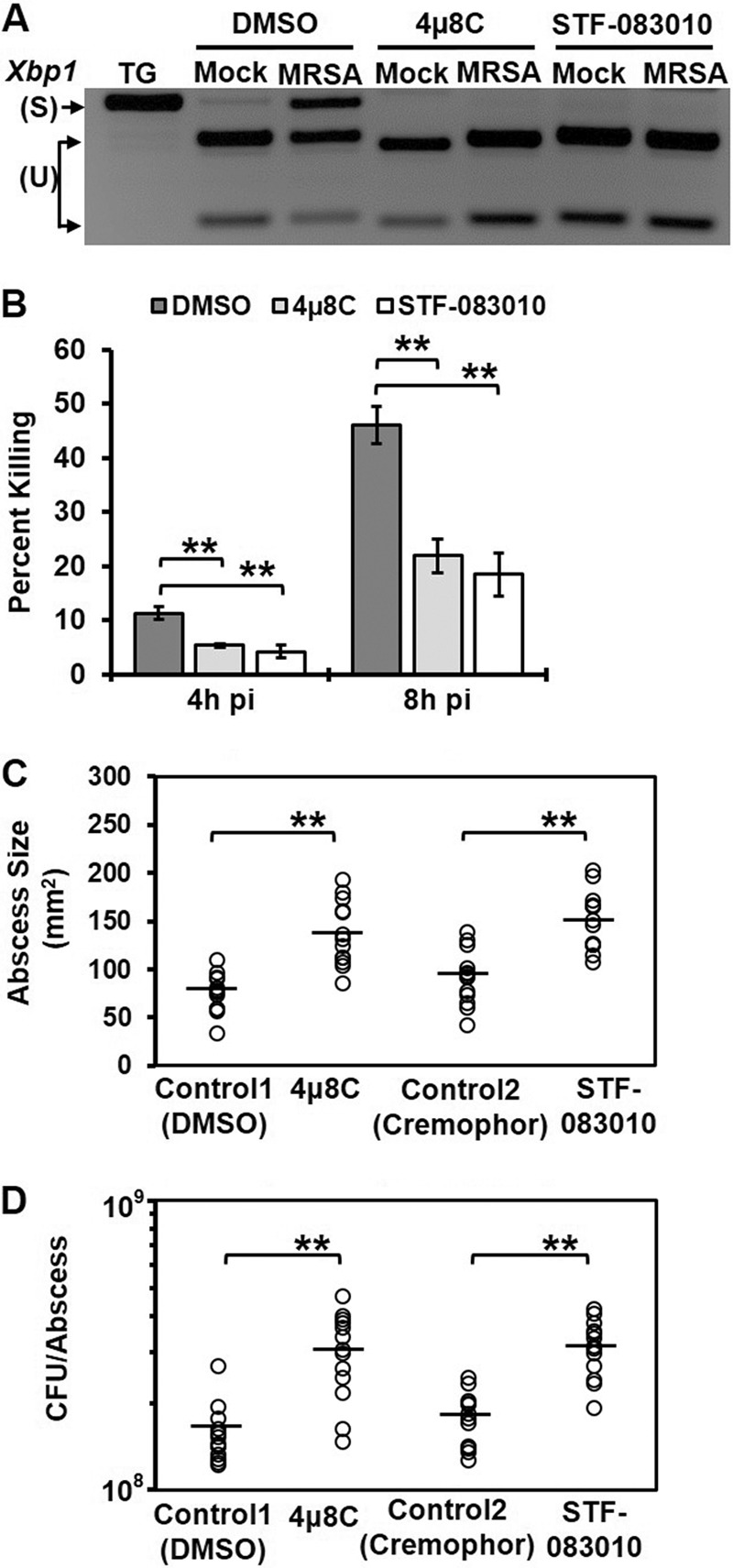
IRE1 endonuclease is required for macrophage bactericidal activity and resistance to MRSA infection. (A) RT-PCR analysis results of Xbp1 mRNA splicing in mock- or MRSA-infected RAW 264.7 cells treated with DMSO control or small-molecule IRE1 inhibitors 4µ8C (25 µM) or STF-083010 (60 µM). (B) The percent killing was quantified as described for Fig. 1C and is presented as the average of ≥3 independent experiments ± the standard deviation. RAW 264.7 macrophages were infected with MRSA (MOI, 20) in the presence of the DMSO control or IRE1 inhibitor 4µ8C (25 µM) or STF-083010 (60 µM). (C) Skin abscess size (in square millimeters) from C57BL/6 mice infected subcutaneously with 108 CFU MRSA at 3 days pi. Mice were injected intraperitoneally with DMSO control (5%), 4µ8C (10 mg/kg), cremophor EL control (15%), or STF-083010 (30 mg/kg) 1 day prior to and each day during infection. (D) Bacterial burden in skin abscesses from mice infected as described for panel C. Abscess size and bacterial load are shown for mice infected in 2 independent experiments; horizontal lines represent the means. *, P < 0.05; **, P < 0.01.
ROS-mediated intracellular killing of MRSA in macrophages requires IRE1.
Reactive oxygen species (ROS) can be a potent chemical weapon against S. aureus (20). To determine if ER stress-driven ROS production aids bacterial killing, we investigated whether IRE1 and ROS function in the same pathway that leads to MRSA killing. Macrophages treated with diphenyleneiodonium (DPI), which inhibits ROS production, or the ROS scavenger N-acetyl cysteine (NAC) were significantly impaired in their ability to kill MRSA (Fig. 3A and B). Addition of the IRE1 inhibitor 4µ8C to DPI- or NAC-treated macrophages did not further decrease MRSA killing. DPI or NAC might reduce IRE1-dependent bactericidal activity by preventing IRE1 activation or by interfering directly in ROS-mediated killing. We therefore tested Xbp1 splicing in the presence of DPI or NAC (Fig. 3C). DPI treatment eliminated infection-induced IRE1 activation, suggesting that in this case DPI prevented MRSA killing by interfering with induction of IRE1. In contrast, NAC, a ROS scavenger, did not prevent IRE1 activation but still decreased bactericidal activity. Therefore, these data point to IRE1-induced ROS generation as a possible mechanism for bacterial killing. We then investigated whether IRE1-driven bactericidal activity was mediated via Nox2, the major phagocyte NADPH oxidase responsible for the oxidative burst (21, 22). Macrophages isolated from Nox2-deficient mice (Cybb−/y) induced IRE1 activation similarly to wild-type macrophages upon MRSA infection (Fig. 3D). These results are in contrast to those from a previous study that showed that IRE1 was not activated in Nox2-deficient macrophages upon addition of TLR ligand (8). However, MRSA was not used in that study, so it may be that MRSA infection stimulates additional signaling pathways that impact the ER stress response. Nox2-deficient macrophages showed a modest reduction in MRSA killing compared to wild-type macrophages (Fig. 3E). Treatment of these macrophages with DPI or 4µ8C further suppressed their ability to kill MRSA. These data suggest that Nox2 does not comprise the major ROS-generating machinery required for IRE1-dependent antimicrobial function in macrophages, but ROS production is essential for this process.
FIG 3 .

IRE1-dependent killing of MRSA occurs through ROS production. (A) Percent MRSA killing by RAW 264.7 macrophages treated with DMSO, 5 µM DPI, or 5 µM DPI plus 25 µM 4µ8C. (B) The percent MRSA killing by RAW 264.7 macrophages treated with DMSO, 5 mM NAC, or 5 mM NAC plus 25 µM 4µ8C. (C) RT-PCR analysis results for Xbp1 mRNA splicing in mock- or MRSA-infected RAW 264.7 macrophages treated with DMSO control, 5 µM DPI, or 5 mM NAC. TG was used as a positive control for IRE1 activation. (D) BMDM isolated from wild-type and Nox2-deficient mice (Cybb−/y) were infected with MRSA (MOI, 20), and spliced Xbp1 was assessed at 8h pi by RT-PCR. TG was used as a positive control for IRE1 activation. (E) Percent MRSA killing by wild-type or Nox2-deficient (Cybb−/y) macrophages in the presence of DMSO, 5 µM DPI, or 25 µM 4µ8C. Graphs illustrate mean results from ≥3 independent experiments with standard deviations. *, P < 0.05; **, P < 0.01.
IRE1 promotes sustained macrophage ROS production.
Immune cells can generate ROS in a rapid intensive burst or in a sustained manner over many hours (23, 24). To elucidate the role of IRE1 in enhancing macrophage ROS production, we measured ROS at different times during MRSA infection by incubating RAW 264.7 cells with the ROS fluorescent indicator chloromethyl 2′,7′-dichlorodihydrofluorescein diacetate (CM-H2DCFDA), followed by flow cytometry analysis (Fig. 4A). At 1h pi, ROS was produced by most macrophages. This early oxidative burst was susceptible to DPI inhibition, but inhibition of IRE1 had little effect on either the number of ROS+ macrophages or the mean fluorescence intensity at 1h pi. ROS production was sustained through 8h pi, and late ROS generation was strongly susceptible to IRE1 inhibition. Silencing of IRE1 showed a similar defect in late ROS generation inhibition (see Fig. S3A in the supplemental material). By immunofluorescence analysis, approximately 40% of infected macrophages at 8h pi were observed to have at least one ROS+ MRSA-containing phagosome. We chose this method of analysis because it afforded snapshot measurements of cells with ROS-positive phagosomes at the indicated times during infection. Thus, we could not discriminate between ROS-negative phagosomes and ROS-detoxified phagosomes. IRE1 inhibition or silencing not only reduced total ROS production but also accumulation in MRSA-containing phagosomes (Fig. 4B; see also Fig. S3B). Knockdown of XBP1 similarly decreased total ROS in MRSA-infected cells (see Fig. S3C and D). These results point to a role for IRE1 in stimulating sustained global ROS production in association with elevated ROS in bacteria-containing phagosomes.
FIG 4 .

IRE1 contributes to sustained ROS production by macrophages. (A) Global ROS production as assessed by flow cytometry using CM-H2DCFDA dye. RAW 264.7 macrophages were infected with MRSA (MOI, 20) in the presence of DMSO, 5 µM DPI, or 25 µM 4µ8C. Flow cytometry was performed on live cells treated with CM-H2DCFDA for 45 min prior to analysis. (Left) The percentage of ROS+ cells was determined by gating against stained mock-infected control cells. Representative plots are shown with the mean percentages of ROS+ cells from ≥3 independent experiments ± the standard deviations. (Right) Quantification of mean fluorescence intensity under the indicated conditions was calculated as the geometric mean, using FlowJo software for results from ≥3 independent experiments ± the standard deviation. (B) Cells with ROS+ phagosomes were imaged with an Olympus IX70 inverted live-cell fluorescence microscope. RAW 264.7 macrophages were infected with MRSA-mCherry (MOI, 20) in the presence of DMSO or 25 µM 4µ8C. CM-H2DCFDA was added at 7h pi for 30 min, and cells were imaged at 8h pi. Cells with ROS+ phagosomes were measured by counting cells with at least one ROS-enriched area colocalized with MRSA from at least 100 ROS+ infected cells. Representative images are shown with the mean percentage of cells with ROS+ phagosomes from ≥3 independent experiments ± the standard deviations. Cells are outlined with a white line in the low-magnification merged images for clarification. *, P < 0.05; **, P < 0.01.
The Sec22B SNARE enables IRE1-dependent MRSA killing.
Our observations revealed that IRE1, an ER-resident protein, controls killing of MRSA within spatially distinct bacterial phagosomes. We therefore hypothesized that ER-to-phagosome trafficking of proteins that facilitate antimicrobial function might be one possible mechanism by which IRE1 indirectly promotes MRSA killing. Previous studies identified the SNARE protein Sec22B as a key player in antigen cross-presentation that mediates trafficking of ER-resident proteins to phagosomes (12). To test this hypothesis, we assessed the requirement for Sec22B in IRE1-mediated bactericidal activity in macrophages. We stably knocked down Sec22B protein in RAW 264.7 macrophages (Sec22B KD), which was confirmed by immunoblot analysis (Fig. 5A). We first tested whether NT-control and Sec22B KD cells could still trigger IRE1 activation upon infection (Fig. 5B). Sec22B KD cells exhibited similar levels of spliced Xbp1 as NT-control cells. Next, these cells were infected with MRSA, and the percentage of MRSA killing was quantified. Silencing Sec22B impaired macrophage killing of MRSA compared to NT-control cells (Fig. 5C). Inhibition of IRE1 or ROS production in Sec22B KD cells did not further decrease bacterial killing, in contrast to NT-control macrophages (Fig. 5D). These results suggest that IRE1, Sec22B, and ROS may act through a common mechanism to enhance MRSA killing, either in the same pathway or in parallel pathways that converge at the phagosome. We would predict that Sec22B acts on the MRSA-containing phagosome through a trafficking mechanism. To test this possibility, we transiently knocked down putative Sec22B partner t-SNAREs syntaxin 4A (STX4A) and syntaxin 5A (STX5A) (12, 25) in RAW 264.7 macrophages and tested the ability of these macrophages to kill MRSA (Fig. 5E and F). Reducing expression of either STX4A or STX5A compromised macrophage antimicrobial function. Taken together, our data implicate Sec22B-dependent trafficking in IRE1-driven macrophage killing of bacterial pathogens.
FIG 5 .

Sec22B enhances MRSA killing by macrophages via an IRE1- and ROS-mediated mechanism. (A) Sec22B and actin immunoblots of cell lysates from NT-control and Sec22B KD macrophages. (B) RT-PCR results from an Xbp1 splicing assay of mock-infected and infected NT-control or Sec22B KD macrophages at 8h pi. (C) NT-control and Sec22B macrophages were infected with MRSA (MOI, 20), and the percent killing was quantified. (D) Percentage of MRSA killing by NT-control and Sec22B KD macrophages in the presence of DMSO, 5 µM DPI, or 25 µM 4µ8C. (E) RAW 264.7 cells were transiently transfected with NT-control or Stx4a siRNA. STX4A, and actin immunoblot assays were performed on cell lysates from a representative experiment (top panel).The percent MRSA killing by these transfected cells averaged over 3 independent experiments is shown in the bottom panel. (F) STX5A and actin immunoblotting results with cell lysates (top) and the percent MRSA killing (bottom) from transiently transfected RAW 264.7 cells with NT-control or Stx5a siRNA. The graphs represent mean results from ≥3 independent experiments ± standard deviuations. *, P < 0.05; **, P < 0.01.
Sec22B is required for local accumulation of ROS in the bacterial phagosome.
We envisioned at least two possibilities to explain how Sec22B might promote bacterial killing. First, Sec22B could be required for global induction of ROS, perhaps via delivery of bacterial ligands from the phagosome to the ER (26). Second, Sec22B could promote local production or accumulation of ROS in the phagosome, perhaps by delivering the machinery of ROS production from the ER to the phagosome or by altering phagolysosome fusion (12). To test the first possibility, we assessed whether Sec22B contributes to global ROS production. NT-control and Sec22B KD cells were infected with MRSA, and total ROS was measured by flow cytometry at 1 and 8h pi. There was no decrease in the amount of global ROS produced upon MRSA infection between NT-control and Sec22B KD macrophages (Fig. 6A). We therefore tested the second possibility by determining whether Sec22B was required for IRE1-dependent accumulation of phagosomal ROS, which we previously observed (Fig. 6B). Sec22B KD and NT-control cells were infected with MRSA, and the percentage of cells with ROS+ MRSA-containing phagosomes was measured. Similar to wild-type RAW 264.7 cells, approximately 40% of NT-control macrophages contained at least one ROS+ MRSA-containing phagosome at 8h pi (Fig. 6C). Silencing Sec22B reduced the number of cells with one or more ROS+ phagosome to approximately 10%, even though total ROS production was not affected. Transient silencing of STX4A or STX5A also decreased the number of macrophages with at least one ROS+ MRSA-containing phagosome at 8h pi (Fig. 6D). Our data suggest the possibility that Sec22B, STX4A, and STX5A contribute to IRE1-dependent killing by directly or indirectly enabling ROS accumulation in the bacterial phagosome.
FIG 6 .

Sec 22B controls sustained ROS accumulation in phagosomes. (A) Flow cytometry analysis results for global ROS production in NT-control- and Sec22B KD-infected macrophages. (Left) Representative histogram plots are shown, with the percentage of ROS+ cells. (Right) Geometric mean fluorescence intensity of ROS production. (B) Live cell fluorescent images of NT-control and Sec22B KD macrophages infected with MRSA-mCherry (MOI, 20) and stained with a ROS fluorescence indicator at 8h pi. (C) The percentage of cells with ROS+ phagosomes was quantified from NT-control and Sec22B KD macrophages. The percentage of cells was determined from the number of cells with at least one enriched area of ROS colocalized with MRSA from at least 100 ROS+ infected cells. (D) The percentage of cells with ROS+ phagosomes was quantified from NT-control, STX4A KD, or STX5A KD macrophages as described for panel C. Graphs represent mean results from ≥3 independent experiments ± standard deviations. *, P < 0.05; **, P < 0.01.
Robust IRE1 activation driven by viable bacteria sustains ROS production.
MRSA stimulated a notable IRE1-independent oxidative burst by 1h pi (Fig. 4A), and so we questioned why the sustained ROS production driven by IRE1 would be necessary for bacterial killing. We hypothesized that live MRSA can evade the initial oxidative burst, and therefore sustained ROS generation might be important for killing pathogens that overcome this initial barrier. We first tested whether live and dead MRSA could stimulate IRE1 activation to the same degree by infecting RAW 264.7 macrophages and measuring Xbp1 splicing (Fig. 7A). Dead MRSA, inactivated either by paraformaldehyde (PFA) or heat treatment, induced significantly less Xbp1 splicing than live MRSA but more than polystyrene latex beads. Consistent with these data, dead MRSA did not stimulate sustained ROS production at 8h pi, even though the early oxidative burst at 1h pi was comparable between macrophages infected with live or dead MRSA (Fig. 7B). To determine whether live MRSA could evade the early oxidative burst, we infected macrophages with live or dead MRSA and evaluated localized ROS production in the phagosome at 1h pi (Fig. 7C). Macrophages infected with dead MRSA showed a robust association of ROS with phagosomes, while live MRSA-infected macrophages showed almost no ROS localized to the phagosomes at 1h pi, despite similar levels of global ROS. These results support the idea that live MRSA evades the initial oxidative burst, perhaps through the expression of ROS-detoxifying enzymes (27). Thus, the persistent ROS production mediated by IRE1 makes a key contribution to killing MRSA later in infection (Fig. 4). Taken together, our data support a model where IRE1-dependent sustained ROS production acts as a fail-safe mechanism to kill vacuolar pathogens that escape the initial oxidative burst.
FIG 7 .

Viable MRSA induces stronger IRE1 activation and is required for sustained ROS production. (A) Xbp1 splicing assay of RNA isolated from RAW 264.7 macrophages that were untreated (mock) or treated with TG and infected with live MRSA, stimulated with PFA-fixed MRSA, stimulated with heat-killed MRSA, or stimulated with latex beads. A representative RT-PCR image (top) and the percentage of Xbp1 splicing (bottom) are shown. The graph represents mean results from ≥3 independent experiments ± standard deviations. (B) Flow cytometry histograms of global ROS production by RAW 264.7 macrophages infected with live MRSA or stimulated with dead MRSA. The percentage of ROS+ cells and the mean fluorescent intensity shown represent means of ≥3 independent experiments ± standard deviations. The mean fluorescent intensity is reported as the geometric mean. (C) ROS localization in RAW 264.7 macrophages infected with live MRSA or stimulated with dead MRSA for 1 h. Representative fluorescence images and the percentages of cells with at least one ROS+ phagosome are shown from ≥3 independent experiments ± standard deviations. *, P < 0.05; **, P < 0.01.
DISCUSSION
Hallmarks of ER stress are found in many human diseases, especially those associated with cellular damage and inflammation (28). IRE1, a gatekeeper of the ER stress program, has recently emerged as a key regulator of inflammation and immunity. Compelling evidence has revealed an important role for IRE1 in controlling inflammatory cytokine output (8–10), but given the profound effects of ER stress on cellular function, we hypothesized that IRE1 also controlled other mechanisms of immunity. Here, we demonstrated that IRE1 activation by MRSA infection enhances macrophage bacterial killing and promotes host immunity in vivo. IRE1-dependent microbicidal activity required XBP1 and ROS but did not impact the initial phagosomal oxidative burst. Instead, IRE1 stimulated sustained production of ROS that could be observed in the phagosome many hours postinfection. Sec22B, a regulator of ER-phagosome trafficking, along with its t-SNARE partners STX4A and STX5A, enabled ROS accumulation in bacteria-containing phagosomes. Notably, robust IRE1 activation and subsequent ROS production only occurred upon infection with live MRSA that evaded the early oxidative burst. These results led us to propose a model where IRE1 augments the host defense by enhancing sustained ROS production to license bacterial killing in the phagosome.
ROS act as an essential component of host defense against bacterial infection by exerting antimicrobial effects or mediating immune signaling. While Nox2 is a well-established NADPH oxidase that can generate ROS in innate immune cells, ROS can also be produced by other enzymatic machinery, such as mitochondria, Nox1, and Nox4 (29–31). In fact, many pathogens, including Salmonella enterica serovar Typhimurium, Coxiella burnetti, and Francisella tularensis, can evade Nox2-dependent oxidative killing by preventing enzyme recruitment, inhibiting ROS production, or promoting its detoxification (32–34). Therefore, immune cells must utilize additional antimicrobial effector mechanisms to control these infections. Our data suggest that one such mechanism in macrophages is IRE1-dependent sustained ROS production. Interestingly, a recent study showed that bacteria encounter different levels of ROS in vivo, depending on the cell type (35). Salmonella encountered high levels of ROS in neutrophils and inflammatory monocytes, where Nox2 is required for killing. However, Salmonella found in splenic macrophages encountered lower levels of ROS and Nox2-independent killing. Consistent with these observations, we found that macrophages could kill MRSA by a Nox2-independent mechanism that requires IRE1. IRE1 was only strongly engaged by live MRSA, which evaded the initial oxidative burst, suggesting that signals from surviving MRSA in the phagosome trigger the fail-safe mechanism. Since TLR2, -4, or -9 signaling is required for IRE1 activation in infected cells, we speculate that live MRSA may present higher levels of certain TLR ligands, such as secreted bacterial lipopeptides, that are not provided by killed MRSA. ROS eventually accumulated in MRSA-containing phagosomes and was important for killing. Our data favor a model in which ROS control MRSA killing, but whether they act directly to kill bacteria in phagosomes or facilitate other antimicrobial effector mechanisms remains to be elucidated.
Nascent phagosomes acquire their antimicrobial capacity by fusion with lysosomes, a degradative compartment loaded with cargo produced and sorted through the ER/Golgi apparatus network (13). We showed that IRE1, a resident of the ER, enhances bacterial killing in the phagosome, even though these compartments are spatially distinct. These results suggest that trafficking between these compartments may be important for IRE1-dependent bactericidal function. A recent study demonstrated that the SNARE protein Sec22B, which resides in the ER and the ER-Golgi appparatus intermediate compartment (ERGIC), controls antigen cross-presentation by promoting protein trafficking between the ER and the phagosome (12). Consistent with these observations, our data showed that Sec22B is required for IRE1-mediated antimicrobial function. Notably, Sec22B was required for ROS accumulation in the phagosome. We envision several mechanisms by which Sec22B could facilitate phagosomal ROS accumulation and IRE1-dependent killing. Sec22B could deliver cargo needed for ROS generation, such as Nox4 or protein disulfide isomerase (PDI), to phagosomes (36). Alternatively, Sec22B could delay phagolysosome fusion, enhancing ROS generation in the phagosome by preventing degradation of ROS-producing machinery. Indeed, Sec22B is known to delay phagolysosome fusion in models of antigen presentation (12). Lastly, Sec22B-mediated trafficking may simply deliver additional membrane to the phagosome, protecting it from MRSA escape (37), thereby facilitating ROS accumulation in this limiting compartment. Collectively, these data emphasize the importance of Sec22B in macrophage effector function and reveal UPR-mediated control of a novel mechanism that enables ROS to be concentrated within the bacteria-containing phagosome.
ROS production during ER stress is modulated by several mechanisms (38). For example, when ER function is perturbed by exposure to tunicamycin, ROS levels increase through activity of ER oxidoreductin 1 (ERO1), a key enzyme in the oxidative protein-folding pathway (39). Silencing Ero1 by RNA interference (RNAi) induced ER stress but inhibited ROS production, suggesting that ROS production occurs subsequent to ER stress. ER stress triggered by oxidized low-density lipoprotein also induced ROS production in a Nox4-dependent manner (29, 40). Notably, expression of ERO1 and NOX4 is induced by IRE1, and both of these enzymes are localized primarily in the ER (29, 41–43). Activation of a second ER stress sensor, PERK, by external oxidants opposed cellular ROS production by increasing glutathione biosynthesis (39). Thus, the selective activation of IRE1 by innate immune signaling pathways could increase the generation of ROS while preventing PERK-dependent antioxidant synthesis (8). IRE1 can also increase expression of thioredoxin-interacting protein (TXNIP), a factor that binds to the catalytic center of thioredoxin and inhibits its antioxidant activity (44). Thus, previous studies indirectly suggested that IRE1 could influence ROS, but a direct demonstration of ROS induction in response to IRE1 activation had not been reported. Our data conclusively show that macrophage IRE1 induces sustained ROS production during bacterial infection.
Mechanisms to balance ROS production by the immune system are critical for long-term health. ROS generation can be a potent antimicrobial mechanism, as patients with defects in the Nox2 NADPH oxidase are susceptible to recurrent and severe bacterial infections (45). ROS can also promote wound healing by serving as a chemoattractant to recruit leukocytes to the site of injury (46). However, excess ROS production can cause tissue injury by either direct oxidant damage or by increasing recruitment of inflammatory cells to the damaged area (47). As a consequence, ROS production paradoxically promotes both health and disease in different contexts. In our MRSA infection model, IRE1 inhibition was deleterious early in infection, exacerbating abscess size and bacterial burden. In contrast, a recent study revealed that IRE1 inhibition protected mice from TLR-induced inflammatory arthritis by suppressing cytokine production (48). Since our data demonstrate a clear role for IRE1 in modulating ROS production, defining the contribution of ER stress-induced ROS to disease pathology in different infectious or inflammatory contexts is warranted and may provide opportunities for therapeutic synergies with other treatments for infection and inflammation.
MATERIALS AND METHODS
Mouse strains and cell cultures.
Wild-type male C57BL/6 and Nox2-deficient (Cybb−/y) mice were purchased from Jackson Laboratories. MYD88-deficient and control macrophages were a gift from N. Lukacs (University of Michigan Medical School) (49). The TLR2/4/9 knockout femurs were a gift from T. Merkel (FDA), who bred the strain using individual TLR knockout mouse strains on a C57BL/6 background (50, 51). BMDMs were prepared by flushing mouse femurs in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 100 units/ml of penicillin-streptomycin (Pen/Strep; Invitrogen). Cells were differentiated by incubation in BMDM medium (50% DMEM, 2 mM l-glutamine, 1 mM sodium pyruvate, 30% L929-conditioned medium, 20% heat-inactivated fetal bovine serum [FBS; Invitrogen], 55 µM 2-mercaptoethanol, and Pen/Strep). RAW 264.7 and THP-1 cells were cultured in RPMI 1640 containing 2 mM l-glutamine and 10% heat-inactivated FBS. THP-1 cells were differentiated by stimulation with 100 nM of phorbol 12-myristate 13-acetate (Sigma-Aldrich) for 24 h. Cells were incubated at 37°C in 5% CO2.
Macrophage infections.
For the MRSA killing assay, macrophages were seeded in a 24-well plate at a density of 1.5 × 105 cell/well. For the Xbp1 splicing assay, cells were seeded in 60-mm tissue culture treated dishes at 1 × 106 cell/dish. Strain USA300 LAC, a community-associated MRSA strain, was cultured on tryptic soy agar (Becton, Dickinson), and selected colonies were grown overnight at 37°C with shaking (200 rpm) in liquid trypic soy broth (TSB) medium. For infections, bacteria were pelleted, washed, and resuspended in phosphate-buffered saline (PBS). The bacterial inoculum was estimated based on the optical density at 600 nm and verified by plating serial dilutions on TSB plates to determine CFU. Macrophages were preincubated with the indicated inhibitors for 30 min, and all inhibitors were maintained throughout the experiment at the following concentrations: 25 µM 4µ8C, 60 µM STF-083010, 5 µM DPI, and 5 mM NAC. Macrophages were infected at a multiplicity of infection (MOI) of 20 in culture medium without antibiotic for 1 h. Infected macrophages were washed three times with PBS and incubated in medium containing 100 µg/ml of gentamicin to kill extracellular bacteria. The number of intracellular bacteria was determined by washing infected macrophages once with PBS and lysing with 0.1% NP-40, and bacterial CFU were enumerated. The percentage of killed MRSA was calculated using the following formula [(CFU2h pi − CFUindicated time point)/CFU2h pi], which represents the percent difference between CFU counts at the indicated time points relative to results at 2h pi.
ROS measurements.
Macrophages were plated in 60-mm nontreated dishes for flow cytometry and 35-mm glass-bottom dishes (MatTek) for immunofluorescence microscopy. Macrophages were treated for 30 min with the indicated inhibitors or control solvent prior to infection with fluorescent-tagged MRSA harboring pSarA-mCherry at an MOI of 20 (52). Infection was synchronized by centrifugation for 5 min at 1,500 rpm. Infected macrophages were washed and incubated in medium containing 100 µg/ml gentamicin. Culture medium was removed, cells were washed with PBS, and then incubated with CM-H2DCFDA (Invitrogen) at a final concentration of 2.5 µM in Ringer buffer (155 mM NaCl, 5 mM KCl, 1 mM MgCl2 ⋅ 6H2O, 2 mM NaH2PO4 ⋅ H2O, 10 mM HEPES, and 10 mM glucose). Cells were incubated for 30 min at 37°C, washed three times with cold medium, and incubated for an additional 15 min at 37°C in warm medium. Cells were washed with PBS and either imaged with an Olympus IX70 inverted live-cell fluorescence microscope or removed from plates with cold PBS and subjected to flow cytometry for total ROS analysis. Fluorescence data were further analyzed by using MetaMorph (microscopy) or FlowJo (flow cytometry) software. For immunofluorescence analysis, cells with at least one ROS+ MRSA-containing phagosome were counted as positive. MRSA-containing phagosomes were considered ROS+ when the fluorescence intensity of the phagosome was ≥1.5 relative to an area of the cell adjacent to the phagosome. For flow cytometry analysis, the frequencies of ROS+ cells were determined by gating against stained mock-infected cells. The mean fluorescence intensity for each condition was determined as the geometric mean.
Mouse infection.
Subcutaneous MRSA infection was performed as previously described (19). Male C57BL/6 mice were shaved on the right flank. Four groups were used, with each group injected intraperitoneally with 10 mg/kg of body weight 4µ8C, 30 mg/kg STF-083010, control solvent 1 (5% DMSO), or control solvent 2 (15% Cremophor EL). Inhibitors and control solvents were administered 1 day prior to infection and each day throughout the experiment. On the second day, mice were inoculated with 1 × 108 bacteria in 100 µl of PBS subcutaneously on the shaved area of the skin via a 27-gauge needle. Mice were sacrificed on day 3 postinfection, and skin lesions were measured using a digital caliper (Fisher Scientific). The skin lesion was excised and homogenized in PBS, and total CFU per mouse lesion were enumerated by serial dilution and plating on TSB agar.
RNAi knockdown and immunoblotting.
XBP1, STX4A, and STX5A were transiently knocked down by transfecting RAW 264.7 cells with specific siGENOME SMART pool small interfering RNA (siRNA) or nontarget siRNA by using DharmaFECT 4 transfection reagent (Dharmacon) according to the manufacturer’s instructions. The generation of lentivirus for shRNA knockdown was done by using HEK293T packaging cells, which were grown in DMEM with 10% FBS. The virus particles were produced by transfecting the cells with the TRC shRNA-harboring plasmid pLKO.1, along with the packaging plasmids (pHCMV-G and pHCMV-HIV-1) (53), by using FuGene-HD transfection reagent (Promega). Medium was changed after 24 h, and virus particles were collected after 72 h posttransfection. A total of 2 ml of medium containing virus was concentrated 10-fold by ultracentrifugation at 24,000 rpm for 2 h at 4°C and used to transduce RAW 264.7 cells. Transduced cells were selected with puromycin (4 µg/ml). The mouse Ire1-α- and Sec22b-specific shRNA plasmids with the corresponding antisense sequences (TTTCTCTATCAATTCACGAGC and AAACTCGATGAAGGAATAGGG, respectively) were purchased from Open Biosystems. The nontarget control shRNA plasmid was purchased from Sigma-Aldrich. The efficiency of knockdown was monitored by immunoblot analysis using anti-IRE1 antibody (clone 14C10; Cell Signaling), anti-Sec22B (clone 29-F7; Santa Cruz Biotechnology), anti-XBP1 antibody (clone ab37152; Abcam), anti-STX4A antibody (clone H-16; Santa Cruz Biotechnology), or anti-STX5A antibody (clone FL-301; Santa Cruz Biotechnology), and anti-actin antibody was used as a loading control (Fisher Scientific).
Xbp1 splicing.
Total RNA was prepared from cultured cells using the RNeasy kit (Qiagen) and was treated with RNase-free DNase (Qiagen). cDNA synthesis was performed using 2 µg of total RNA, murine leukemia virus reverse transcriptase (RT; Invitrogen), and random hexamers (Applied Biosystems). Spliced and unspliced Xbp1 transcripts were amplified by semiquantitative RT-PCR using the pair primers forward, 5′-GAACCAGGAGTTAAGAACACG-3′, and reverse, 5′-AGGCAACAGTGTCAGAGTCC-3′. PCR conditions were as follows: 95°C for 5 min, and 30 amplification cycles of 95°C for 30 s, 50°C for 45 s, and 72°C for 1 min. The PCR product was purified and digested with PstI restriction endonuclease to discriminate between unspliced and spliced Xbp1 (54). Spliced and unspliced DNA fragments were resolved by electrophoresis on a 2.5% agarose gel. Band intensities were measured using ImageJ software, and the percent spliced was calculated using the following formula: [Xbp1s/(Xbp1s + Xbp1u)], i.e., the Xbp1 spliced band density (Xbp1s) relative to total spliced and unspliced (Xbp1u) band densities.
Statistical analysis.
Student’s paired two-tailed t test was used to analyze the data. The means of at least three independent experiments are reported, with error bars showing standard deviations (SD). P values of less than 0.05 were considered significant (*, P < 0.05; **, P < 0.01). All statistically significant comparisons within experimental groups are indicated in the figures. Statistically significant differences between experimental groups are only marked in specific graphs as indicated in the figure legends.
SUPPLEMENTAL MATERIAL
XBP1 enhances MRSA killing by macrophages. (A) XBP1 and actin immunoblotting results for cell lysates from transiently transfected RAW 264.7 cells with NT-control or Xbp1 siRNA which were then infected with MRSA for 8 h. (B) The percentage of MRSA killing by RAW 264.7 macrophages transfected with NT-control or Xbp1 siRNA. The percent killing is presented as the mean of ≥3 independent experiments ± standard deviations. *, P < 0.05; **, P < 0.01. Download
IRE1 is activated by human macrophages and enhances MRSA killing. (A) RT-PCR analysis of Xbp1 mRNA splicing in differentiated THP-1 cells left untreated (mock), treated with 5 µM thapsigargin (TG), treated with 25 µM 4µ8C, or infected with MRSA for 8 h (MOI, 20) in the presence of DMSO or 4µ8C. RT-PCR results (top) are representative of ≥3 independent experiments, and the percent spliced Xbp1 (bottom) was calculated based on band densitometry and the following formula: [Xbp1s/(Xbp1s + Xbp1u)]. (B) The percentage of MRSA killing by differentiated THP-1 cells in the presence of DMSO or 4µ8C. The graph shows mean results from ≥3 independent experiments ± standard devaitions. *, P < 0.05. Download
IRE1 KD prevents sustained ROS production by macrophages. (A) Total cellular ROS production in live cells, assessed by flow cytometry using CM-H2DCFDA. The percentages of ROS+ cells were determined by gating against stained mock-infected cells. Representative histograms are shown (left) with the mean percentages of ROS+ cells from ≥3 independent experiments ± standard deviations. Mean fluorescent intensity (right) for the indicated conditions was calculated as the geometric mean from ≥3 independent experiments ± the standard deviation. (B) Live-cell fluorescence images of ROS association with MRSA-containing phagosomes. RAW 264.7 macrophage cells expressing NT-control or IRE1 shRNA were infected with MRSA-mCherry (MOI, 20) and stained with the ROS fluorescence indicator CM-H2DCFDA (green) at 8h pi. The percentages of cells with ROS+ phagosomes were determined by counting cells with at least one enriched ROS area localized with MRSA from at least 100 ROS+-infected cells. Representative images are shown with mean percentages of cells with ROS+ phagosome from ≥3 independent experiments ± standard deviations. (C) The percentage of ROS+ cells was quantified by flow cytometry from NT-control or XBP1 KD macrophages at 8h pi. The percentage of ROS+ cells were measured by gating against stained mock-infected cells. (D) Mean fluorescent intensity (MFI) was measured from NT-control or XBP1 KD macrophages. The cells were either left untreated (mock) or infected with MRSA for 8 h. MFI was analyzed as the geometric mean by using FlowJo software. (C and D) Graphs presenting means of ≥3 independent experiments ± standard deviations. *, P < 0.05. Download
ACKNOWLEDGMENTS
This work was supported by NIH awards R01 AI099065 (B.R.B.) and R21 AI101777 (M.X.O). K.B. was supported by NIH T32 AI008413. B.H.A. was supported by NIH T32 AI007538, T32 HLL007517, and an American Heart Association postdoctoral fellowship.
We thank O’Riordan lab members for many helpful discussions. We gratefully acknowledge Samuel Straight and the Center for Live Cell Imaging at the University of Michigan Medical School for assistance with fluorescence microscopy analysis. We thank N. Lukacs (University of Michigan Medical School) for Myd88−/− femurs and T. Merkel (FDA) for Tlr2/4/9−/− femurs.
Footnotes
Citation Abuaita BH, Burkholder KM, Boles BR, O’Riordan MX. 2015. The endoplasmic reticulum stress sensor inositol-requiring enzyme 1α augments bacterial killing through sustained oxidant production. mBio 6(4):e00705-15. doi:10.1128/mBio.00705-15.
Contributor Information
Wayne Lencer, Children's Hospital Boston.
Christine A. Biron, Brown University.
REFERENCES
- 1.Ron D, Walter P. 2007. Signal integration in the endoplasmic reticulum unfolded protein response. Nat Rev Mol Cell Biol 8:519–529. doi: 10.1038/nrm2199. [DOI] [PubMed] [Google Scholar]
- 2.Korennykh AV, Egea PF, Korostelev AA, Finer-Moore J, Zhang C, Shokat KM, Stroud RM, Walter P. 2009. The unfolded protein response signals through high-order assembly of Ire1. Nature 457:687–693. doi: 10.1038/nature07661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Calfon M, Zeng H, Urano F, Till JH, Hubbard SR, Harding HP, Clark SG, Ron D. 2002. IRE1 couples endoplasmic reticulum load to secretory capacity by processing the XBP-1 mRNA. Nature 415:92–96. doi: 10.1038/415092a. [DOI] [PubMed] [Google Scholar]
- 4.Lee K, Tirasophon W, Shen X, Michalak M, Prywes R, Okada T, Yoshida H, Mori K, Kaufman RJ. 2002. IRE1-mediated unconventional mRNA splicing and S2P-mediated ATF6 cleavage merge to regulate XBP1 in signaling the unfolded protein response. Genes Dev 16:452–466. doi: 10.1101/gad.964702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Reimold AM, Iwakoshi NN, Manis J, Vallabhajosyula P, Szomolanyi-Tsuda E, Gravallese EM, Friend D, Grusby MJ, Alt F, Glimcher LH. 2001. Plasma cell differentiation requires the transcription factor XBP-1. Nature 412:300–307. doi: 10.1038/35085509. [DOI] [PubMed] [Google Scholar]
- 6.Iwakoshi NN, Pypaert M, Glimcher LH. 2007. The transcription factor XBP-1 is essential for the development and survival of dendritic cells. J Exp Med 204:2267–2275. doi: 10.1084/jem.20070525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kamimura D, Bevan MJ. 2008. Endoplasmic reticulum stress regulator XBP-1 contributes to effector CD8+ T cell differentiation during acute infection. J Immunol 181:5433–5441. doi: 10.4049/jimmunol.181.8.5433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Martinon F, Chen X, Lee AH, Glimcher LH. 2010. TLR activation of the transcription factor XBP1 regulates innate immune responses in macrophages. Nat Immunol 11:411–418. doi: 10.1038/ni.1857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cho JA, Lee AH, Platzer B, Cross BC, Gardner BM, De Luca H, Luong P, Harding HP, Glimcher LH, Walter P, Fiebiger E, Ron D, Kagan JC, Lencer WI. 2013. The unfolded protein response element IRE1α senses bacterial proteins invading the ER to activate RIG-I and innate immune signaling. Cell Host Microbe 13:558–569. doi: 10.1016/j.chom.2013.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 10.Oslowski CM, Hara T, O’Sullivan-Murphy B, Kanekura K, Lu S, Hara M, Ishigaki S, Zhu LJ, Hayashi E, Hui ST, Greiner D, Kaufman RJ, Bortell R, Urano F. 2012. Thioredoxin-interacting protein mediates ER stress-induced beta cell death through initiation of the inflammasome. Cell Metab 16:265–273. doi: 10.1016/j.cmet.2012.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gagnon E, Duclos S, Rondeau C, Chevet E, Cameron PH, Steele-Mortimer O, Paiement J, Bergeron JJ, Desjardins M. 2002. Endoplasmic reticulum-mediated phagocytosis is a mechanism of entry into macrophages. Cell 110:119–131. doi: 10.1016/S0092-8674(02)00797-3. [DOI] [PubMed] [Google Scholar]
- 12.Cebrian I, Visentin G, Blanchard N, Jouve M, Bobard A, Moita C, Enninga J, Moita LF, Amigorena S, Savina A. 2011. Sec22b regulates phagosomal maturation and antigen crosspresentation by dendritic cells. Cell 147:1355–1368. doi: 10.1016/j.cell.2011.11.021. [DOI] [PubMed] [Google Scholar]
- 13.Kornfeld S, Mellman I. 1989. The biogenesis of lysosomes. Annu Rev Cell Biol 5:483–525. doi: 10.1146/annurev.cb.05.110189.002411. [DOI] [PubMed] [Google Scholar]
- 14.Kubica M, Guzik K, Koziel J, Zarebski M, Richter W, Gajkowska B, Golda A, Maciag-Gudowska A, Brix K, Shaw L, Foster T, Potempa J. 2008. A potential new pathway for Staphylococcus aureus dissemination: the silent survival of S. aureus phagocytosed by human monocyte-derived macrophages. PLoS One 3:e1409. doi: 10.1371/journal.pone.0001409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhang K, Wong HN, Song B, Miller CN, Scheuner D, Kaufman RJ. 2005. The unfolded protein response sensor IRE1α is required at 2 distinct steps in B cell lymphopoiesis. J Clin Invest 115:268–281. doi: 10.1172/JCI21848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cross BC, Bond PJ, Sadowski PG, Jha BK, Zak J, Goodman JM, Silverman RH, Neubert TA, Baxendale IR, Ron D, Harding HP. 2012. The molecular basis for selective inhibition of unconventional mRNA splicing by an IRE1-binding small molecule. Proc Natl Acad Sci U S A 109:E869–E878. doi: 10.1073/pnas.1115623109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Papandreou I, Denko NC, Olson M, Van Melckebeke H, Lust S, Tam A, Solow-Cordero DE, Bouley DM, Offner F, Niwa M, Koong AC. 2011. Identification of an Ire1α endonuclease specific inhibitor with cytotoxic activity against human multiple myeloma. Blood 117:1311–1314. doi: 10.1182/blood-2010-08-303099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Miller LS, Cho JS. 2011. Immunity against Staphylococcus aureus cutaneous infections. Nat Rev Immunol 11:505–518. doi: 10.1038/nri3010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tseng CW, Sanchez-Martinez M, Arruda A, Liu GY. 2011. Subcutaneous infection of methicillin resistant Staphylococcus aureus (MRSA). J Vis Exp 48:2528. doi: 10.3791/2528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liu GY, Essex A, Buchanan JT, Datta V, Hoffman HM, Bastian JF, Fierer J, Nizet V. 2005. Staphylococcus aureus golden pigment impairs neutrophil killing and promotes virulence through its antioxidant activity. J Exp Med 202:209–215. doi: 10.1084/jem.20050846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Morel F, Doussiere J, Vignais PV. 1991. The superoxide-generating oxidase of phagocytic cells: physiological, molecular and pathological aspects. Eur J Biochem 201:523–546. doi: 10.1111/j.1432-1033.1991.tb16312.x. [DOI] [PubMed] [Google Scholar]
- 22.Robinson JM, Badwey JA. 1995. The NADPH oxidase complex of phagocytic leukocytes: a biochemical and cytochemical view. Histochem Cell Biol 103:163–180. doi: 10.1007/BF01454021. [DOI] [PubMed] [Google Scholar]
- 23.Wheeler ML, Defranco AL. 2012. Prolonged production of reactive oxygen species in response to B cell receptor stimulation promotes B cell activation and proliferation. J Immunol 189:4405–4416. doi: 10.4049/jimmunol.1201433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Maitra U, Singh N, Gan L, Ringwood L, Li L. 2009. IRAK-1 contributes to lipopolysaccharide-induced reactive oxygen species generation in macrophages by inducing NOX-1 transcription and Rac1 activation and suppressing the expression of antioxidative enzymes. J Biol Chem 284:35403–35411. doi: 10.1074/jbc.M109.059501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Xu D, Joglekar AP, Williams AL, Hay JC. 2000. Subunit structure of a mammalian ER/Golgi SNARE complex. J Biol Chem 275:39631–39639. doi: 10.1074/jbc.M007684200. [DOI] [PubMed] [Google Scholar]
- 26.Nilsen NJ, Deininger S, Nonstad U, Skjeldal F, Husebye H, Rodionov D, von Aulock S, Hartung T, Lien E, Bakke O, Espevik T. 2008. Cellular trafficking of lipoteichoic acid and Toll-like receptor 2 in relation to signaling: role of CD14 and CD36. J Leukoc Biol 84:280–291. doi: 10.1189/jlb.0907656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Horsburgh MJ, Clements MO, Crossley H, Ingham E, Foster SJ. 2001. PerR controls oxidative stress resistance and iron storage proteins and is required for virulence in Staphylococcus aureus. Infect Immun 69:3744–3754. doi: 10.1128/IAI.69.6.3744-3754.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rath E, Haller D. 2011. Inflammation and cellular stress: a mechanistic link between immune-mediated and metabolically driven pathologies. Eur J Nutr 50:219–233. doi: 10.1007/s00394-011-0197-0. [DOI] [PubMed] [Google Scholar]
- 29.Lee CF, Qiao M, Schröder K, Zhao Q, Asmis R. 2010. Nox4 is a novel inducible source of reactive oxygen species in monocytes and macrophages and mediates oxidized low density lipoprotein-induced macrophage death. Circ Res 106:1489–1497. doi: 10.1161/CIRCRESAHA.109.215392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.West AP, Brodsky IE, Rahner C, Woo DK, Erdjument-Bromage H, Tempst P, Walsh MC, Choi Y, Shadel GS, Ghosh S. 2011. TLR signalling augments macrophage bactericidal activity through mitochondrial ROS. Nature 472:476–480. doi: 10.1038/nature09973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yeligar SM, Harris FL, Hart CM, Brown LA. 2012. Ethanol induces oxidative stress in alveolar macrophages via upregulation of NADPH oxidases. J Immunol 188:3648–3657. doi: 10.4049/jimmunol.1101278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Vazquez-Torres A, Fang FC. 2001. Salmonella evasion of the NADPH phagocyte oxidase. Microbes Infect 3:1313–1320. doi: 10.1016/S1286-4579(01)01492-7. [DOI] [PubMed] [Google Scholar]
- 33.Mertens K, Samuel JE. 2012. Defense mechanisms against oxidative stress in Coxiella burnetii: adaptation to a unique intracellular niche. Adv Exp Med Biol 984:39–63. doi: 10.1007/978-94-007-4315-1_3. [DOI] [PubMed] [Google Scholar]
- 34.Celli J, Zahrt TC. 2013. Mechanisms of Francisella tularensis intracellular pathogenesis. Cold Spring Harb Perspect Med 3:a010314. doi: 10.1101/cshperspect.a010314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Burton NA, Schürmann N, Casse O, Steeb AK, Claudi B, Zankl J, Schmidt A, Bumann D. 2014. Disparate impact of oxidative host defenses determines the fate of salmonella during systemic infection in mice. Cell Host Microbe 15:72–83. doi: 10.1016/j.chom.2013.12.006. [DOI] [PubMed] [Google Scholar]
- 36.Janiszewski M, Lopes LR, Carmo AO, Pedro MA, Brandes RP, Santos CX, Laurindo FR. 2005. Regulation of NAD(P)H oxidase by associated protein disulfide isomerase in vascular smooth muscle cells. J Biol Chem 280:40813–40819. doi: 10.1074/jbc.M509255200. [DOI] [PubMed] [Google Scholar]
- 37.Sokolovska A, Becker CE, Ip WK, Rathinam VA, Brudner M, Paquette N, Tanne A, Vanaja SK, Moore KJ, Fitzgerald KA, Lacy-Hulbert A, Stuart LM. 2013. Activation of caspase-1 by the NLRP3 inflammasome regulates the NADPH oxidase NOX2 to control phagosome function. Nat Immunol 14:543–553. doi: 10.1038/ni.2595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Santos CX, Nabeebaccus AA, Shah AM, Camargo LL, Filho SV, Lopes LR. 2014. Endoplasmic reticulum stress and Nox-mediated reactive oxygen species signaling in the peripheral vasculature: potential role in hypertension. Antioxid Redox Signal 20:121–134. doi: 10.1089/ars.2013.5262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Harding HP, Zhang Y, Zeng H, Novoa I, Lu PD, Calfon M, Sadri N, Yun C, Popko B, Paules R, Stojdl DF, Bell JC, Hettmann T, Leiden JM, Ron D. 2003. An integrated stress response regulates amino acid metabolism and resistance to oxidative stress. Mol Cell 11:619–633. doi: 10.1016/S1097-2765(03)00105-9. [DOI] [PubMed] [Google Scholar]
- 40.Yao ST, Sang H, Yang NN, Kang L, Tian H, Zhang Y, Song GH, Qin SC. 2010. Oxidized low density lipoprotein induces macrophage endoplasmic reticulum stress via CD36. Sheng Li Xue Bao 62:433–440. (In Chinese.) [PubMed] [Google Scholar]
- 41.Pedruzzi E, Guichard C, Ollivier V, Driss F, Fay M, Prunet C, Marie JC, Pouzet C, Samadi M, Elbim C, O’Dowd Y, Bens M, Vandewalle A, Gougerot-Pocidalo MA, Lizard G, Ogier-Denis E. 2004. NAD(P)H oxidase Nox-4 mediates 7-ketocholesterol-induced endoplasmic reticulum stress and apoptosis in human aortic smooth muscle cells. Mol Cell Biol 24:10703–10717. doi: 10.1128/MCB.24.24.10703-10717.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Frand AR, Kaiser CA. 1998. The ERO1 gene of yeast is required for oxidation of protein dithiols in the endoplasmic reticulum. Mol Cell 1:161–170. doi: 10.1016/S1097-2765(00)80017-9. [DOI] [PubMed] [Google Scholar]
- 43.Kakihana T, Araki K, Vavassori S, Iemura S, Cortini M, Fagioli C, Natsume T, Sitia R, Nagata K. 2013. Dynamic regulation of Ero1alpha and peroxiredoxin 4 localization in the secretory pathway. J Biol Chem 288:29586–29594. doi: 10.1074/jbc.M113.467845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lerner AG, Upton JP, Praveen PV, Ghosh R, Nakagawa Y, Igbaria A, Shen S, Nguyen V, Backes BJ, Heiman M, Heintz N, Greengard P, Hui S, Tang Q, Trusina A, Oakes SA, Papa FR. 2012. IRE1α induces thioredoxin-interacting protein to activate the NLRP3 inflammasome and promote programmed cell death under irremediable ER stress. Cell Metab 16:250–264. doi: 10.1016/j.cmet.2012.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Winkelstein JA, Marino MC, Johnston RB Jr., Boyle J, Curnutte J, Gallin JI, Malech HL, Holland SM, Ochs H, Quie P, Buckley RH, Foster CB, Chanock SJ, Dickler H. 2000. Chronic granulomatous disease. Report on a national registry of 368 patients. Medicine 79:155–169. doi: 10.1097/00005792-200005000-00003. [DOI] [PubMed] [Google Scholar]
- 46.Niethammer P, Grabher C, Look AT, Mitchison TJ. 2009. A tissue-scale gradient of hydrogen peroxide mediates rapid wound detection in zebrafish. Nature 459:996–999. doi: 10.1038/nature08119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lambeth JD, Neish AS. 2014. Nox enzymes and new thinking on reactive oxygen: a double-edged sword revisited. Annu Rev Pathol 9:119–145. doi: 10.1146/annurev-pathol-012513-104651. [DOI] [PubMed] [Google Scholar]
- 48.Qiu Q, Zheng Z, Chang L, Zhao YS, Tan C, Dandekar A, Zhang Z, Lin Z, Gui M, Li X, Zhang T, Kong Q, Li H, Chen S, Chen A, Kaufman RJ, Yang WL, Lin HK, Zhang D, Perlman H, Thorp E, Zhang K, Fang D. 2013. Toll-like receptor-mediated IRE1α activation as a therapeutic target for inflammatory arthritis. EMBO J 32:2477–2490. doi: 10.1038/emboj.2013.183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rudd BD, Schaller MA, Smit JJ, Kunkel SL, Neupane R, Kelley L, Berlin AA, Lukacs NW. 2007. MyD88-mediated instructive signals in dendritic cells regulate pulmonary immune responses during respiratory virus infection. J Immunol 178:5820–5827. doi: 10.4049/jimmunol.178.9.5820. [DOI] [PubMed] [Google Scholar]
- 50.Hemmi H, Takeuchi O, Kawai T, Kaisho T, Sato S, Sanjo H, Matsumoto M, Hoshino K, Wagner H, Takeda K, Akira S. 2000. A Toll-like receptor recognizes bacterial DNA. Nature 408:740–745. doi: 10.1038/35047123. [DOI] [PubMed] [Google Scholar]
- 51.Takeuchi O, Hoshino K, Kawai T, Sanjo H, Takada H, Ogawa T, Takeda K, Akira S. 1999. Differential roles of TLR2 and TLR4 in recognition of gram-negative and gram-positive bacterial cell wall components. Immunity 11:443–451. doi: 10.1016/S1074-7613(00)80119-3. [DOI] [PubMed] [Google Scholar]
- 52.Boles BR, Horswill AR. 2008. Agr-mediated dispersal of Staphylococcus aureus biofilms. PLoS Pathog 4:e1000052. doi: 10.1371/journal.ppat.1000052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kulpa DA, Del Cid N, Peterson KA, Collins KL. 2013. Adaptor protein 1 promotes cross-presentation through the same tyrosine signal in major histocompatibility complex class I as that targeted by HIV-1. J Virol 87:8085–8098. doi: 10.1128/JVI.00701-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yoshida H, Matsui T, Yamamoto A, Okada T, Mori K. 2001. XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell 107:881–891. doi: 10.1016/S0092-8674(01)00611-0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
XBP1 enhances MRSA killing by macrophages. (A) XBP1 and actin immunoblotting results for cell lysates from transiently transfected RAW 264.7 cells with NT-control or Xbp1 siRNA which were then infected with MRSA for 8 h. (B) The percentage of MRSA killing by RAW 264.7 macrophages transfected with NT-control or Xbp1 siRNA. The percent killing is presented as the mean of ≥3 independent experiments ± standard deviations. *, P < 0.05; **, P < 0.01. Download
IRE1 is activated by human macrophages and enhances MRSA killing. (A) RT-PCR analysis of Xbp1 mRNA splicing in differentiated THP-1 cells left untreated (mock), treated with 5 µM thapsigargin (TG), treated with 25 µM 4µ8C, or infected with MRSA for 8 h (MOI, 20) in the presence of DMSO or 4µ8C. RT-PCR results (top) are representative of ≥3 independent experiments, and the percent spliced Xbp1 (bottom) was calculated based on band densitometry and the following formula: [Xbp1s/(Xbp1s + Xbp1u)]. (B) The percentage of MRSA killing by differentiated THP-1 cells in the presence of DMSO or 4µ8C. The graph shows mean results from ≥3 independent experiments ± standard devaitions. *, P < 0.05. Download
IRE1 KD prevents sustained ROS production by macrophages. (A) Total cellular ROS production in live cells, assessed by flow cytometry using CM-H2DCFDA. The percentages of ROS+ cells were determined by gating against stained mock-infected cells. Representative histograms are shown (left) with the mean percentages of ROS+ cells from ≥3 independent experiments ± standard deviations. Mean fluorescent intensity (right) for the indicated conditions was calculated as the geometric mean from ≥3 independent experiments ± the standard deviation. (B) Live-cell fluorescence images of ROS association with MRSA-containing phagosomes. RAW 264.7 macrophage cells expressing NT-control or IRE1 shRNA were infected with MRSA-mCherry (MOI, 20) and stained with the ROS fluorescence indicator CM-H2DCFDA (green) at 8h pi. The percentages of cells with ROS+ phagosomes were determined by counting cells with at least one enriched ROS area localized with MRSA from at least 100 ROS+-infected cells. Representative images are shown with mean percentages of cells with ROS+ phagosome from ≥3 independent experiments ± standard deviations. (C) The percentage of ROS+ cells was quantified by flow cytometry from NT-control or XBP1 KD macrophages at 8h pi. The percentage of ROS+ cells were measured by gating against stained mock-infected cells. (D) Mean fluorescent intensity (MFI) was measured from NT-control or XBP1 KD macrophages. The cells were either left untreated (mock) or infected with MRSA for 8 h. MFI was analyzed as the geometric mean by using FlowJo software. (C and D) Graphs presenting means of ≥3 independent experiments ± standard deviations. *, P < 0.05. Download