

ABSTRACT
In the discovery of secondary metabolites, analysis of sequence data is a promising exploration path that remains largely underutilized due to the lack of computational platforms that enable such a systematic approach on a large scale. In this work, we present IMG-ABC (https://img.jgi.doe.gov/abc), an atlas of biosynthetic gene clusters within the Integrated Microbial Genomes (IMG) system, which is aimed at harnessing the power of “big” genomic data for discovering small molecules. IMG-ABC relies on IMG’s comprehensive integrated structural and functional genomic data for the analysis of biosynthetic gene clusters (BCs) and associated secondary metabolites (SMs). SMs and BCs serve as the two main classes of objects in IMG-ABC, each with a rich collection of attributes. A unique feature of IMG-ABC is the incorporation of both experimentally validated and computationally predicted BCs in genomes as well as metagenomes, thus identifying BCs in uncultured populations and rare taxa. We demonstrate the strength of IMG-ABC’s focused integrated analysis tools in enabling the exploration of microbial secondary metabolism on a global scale, through the discovery of phenazine-producing clusters for the first time in Alphaproteobacteria. IMG-ABC strives to fill the long-existent void of resources for computational exploration of the secondary metabolism universe; its underlying scalable framework enables traversal of uncovered phylogenetic and chemical structure space, serving as a doorway to a new era in the discovery of novel molecules.
IMPORTANCE
IMG-ABC is the largest publicly available database of predicted and experimental biosynthetic gene clusters and the secondary metabolites they produce. The system also includes powerful search and analysis tools that are integrated with IMG’s extensive genomic/metagenomic data and analysis tool kits. As new research on biosynthetic gene clusters and secondary metabolites is published and more genomes are sequenced, IMG-ABC will continue to expand, with the goal of becoming an essential component of any bioinformatic exploration of the secondary metabolism world.
INTRODUCTION
Secondary metabolites (SMs) are small organic compounds that are not essential to the life of an organism but have, nevertheless, a broad biological activity spectrum. SMs derived from plants have been used for thousands of years in the form of natural extracts due to their pharmacological properties (1), while in the modern era, SMs from plants and microorganisms have been a rich source of therapeutics (2). Certain classes of SMs, such as terpenes, are good candidates for biofuel production (3). SMs serve as important chemical agents of communication between bacteria in complex communities or in symbiotic relationships, such as in plant-microbe interactions (4). In this context, some SMs provide biological control of plant pathogens and thus may have important agricultural applications (5). Discovery of new SMs, therefore, will benefit the development of novel biotechnological applications and provide better understanding of the interactions within complex communities.
Traditionally, microbial SMs have been isolated by screening cultured microbes for the desired pharmacological and/or biological activity (6). This approach has its limitations, since many microorganisms are difficult or impossible to obtain in pure culture. Additionally, the bioactive chemicals may not be produced due to the absence of specific environmental stimuli or may remain undetectable through conventional screening methods (7–9). The rapid growth of genomic data from both isolate organisms and microbial communities (metagenomes) (10), in conjunction with the development of tools for computational identification and classification of biosynthetic gene clusters (BCs) (11, 12), present a new opportunity for the discovery of SMs with novel chemical structures (13). Computationally identified BCs can be cloned or synthetically reconstructed and expressed in heterologous systems, which can then be monitored for the production of potentially novel metabolites. Further, the application of this approach to uncultured strains in single cells and metagenomes enables the detection of SM pathways in microbial dark matter (14).
In response to an increasing interest in the identification of novel microbial SMs, a number of databases have been developed in recent years with information on BCs and SMs. StreptomeDB is a database focused on bioactive molecules produced by members of the Streptomyces genus (15). DoBISCUIT provides carefully curated annotations of a limited number of BCs (16), while Clustermine360 focuses on polyketide synthase (PKS) and nonribosomal peptide synthetase (NRPS) type clusters and allows for user submissions of clusters (17). ClustScanDB provides detailed information about thiotemplate modular systems (18). Although some of these efforts provide useful high-quality manually curated annotations, they come at the cost of narrow specificity and often have no or limited maintenance. Most of these systems are limited either by specific classes of organisms (e.g., Streptomyces) or biosynthetic cluster types (e.g., NRPS/PKS). Additionally, some have relatively few records and do not provide the tools for in-depth sequence analyses, while others are no longer updated.
In order to alleviate some of the above limitations and to pave the way from big data to small molecules, we have developed IMG-ABC, an atlas of biosynthetic gene clusters within the Integrated Microbial Genomes (IMG) system. IMG-ABC is a database of biosynthetic gene clusters and the chemicals they are known to produce and, through its integration with the IMG data management system (10, 19), it provides the following capabilities: (i) access to an exhaustive collection of both predicted and published BCs in over 23,000 public isolate microbial genomes and more than 2,200 metagenomes; (ii) class-agnostic inclusion of all classes and types of BCs and SMs; (iii) integration with structural and functional annotations and metadata from IMG and the Genomes Online Database (GOLD) (20); (iv) enhanced search and analysis capabilities; (v) expert user curation capability; (vi) a track record of continuous maintenance and user support.
As newly sequenced data sets are integrated into IMG, they are automatically processed through IMG’s BC prediction pipeline, constantly feeding IMG-ABC with new putative BCs. We expect IMG-ABC to become the primary starting point for the sequence analysis of BCs and to provide the knowledge base needed for the heterologous expression of BCs that produce novel and potentially useful chemical compounds.
RESULTS
Database structure and content.
A schematic overview of the IMG-ABC object structure with associated attributes for BCs and SMs is illustrated in Fig. 1. Experimentally described BCs are linked with the SMs they produce, when known. The result of this computationally intensive effort was the creation of a large database of predicted and experimentally verified BCs. IMG-ABC contains more than 750,000 BCs in publicly available records, most of them from bacterial isolate genomes (Table 1) from 57 phyla (Fig. 2A), reflecting the diversity of available genomic sequences in the IMG database. A subset of BCs from isolate genomes can be assigned to one or more BC enzymatic types based on the presence of signature core enzymes (Fig. 2B). The most common enzymatic activity found in the predicted clusters is that of nonribosomal peptide synthetase (21), followed by type I PKS (22). Integration with IMG allows access to metadata annotations from GOLD (20), enabling the connection of information on secondary metabolism and habitat (Fig. 2C). Since Streptomyces are an excellent source of natural products and have been studied in this respect more than other genera, more than 60% of SMs with a known chemical structure in IMG-ABC have been isolated from Actinobacteria (Fig. 2D).
FIG 1 .
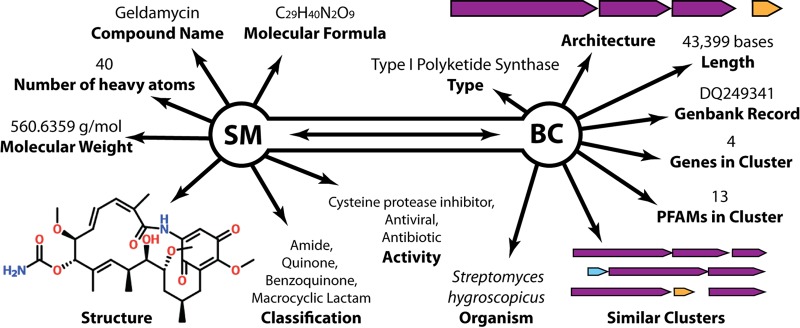
IMG-ABC object structure overview. IMG-ABC contains two main classes of objects with a variety of attributes describing predicted and experimentally studied biosynthetic gene clusters and the secondary metabolites associated with the latter.
TABLE 1 .
IMG-ABC content (BCs) within various domains of life and DNA fragmentsa
| Domain or DNA fragment (no. of public samples with BCs) | No. of BCs |
|---|---|
| Bacteria (23,423) | 441,881 |
| Metagenomes (2,230) | 260,462 |
| Eukaryota (186) | 42,072 |
| Archaea (498) | 4196 |
| Viruses (920) | 1223 |
| Bacterial genome fragments (1,116) | 1391 |
| Bacterial plasmids (197) | 271 |
| Eukaryotic genome fragments (139) | 164 |
| Eukaryotic plasmids (7) | 7 |
| Other plasmids (1) | 1 |
| Archaeal genome fragments (2) | 1 |
| Total | 758,469 |
Distribution of BCs within various domains of life, genome fragments, and plasmids in IMG and IMG/M (as of 27 January 2015).
FIG 2 .

Data content of IMG-ABC. (A) Biosynthetic clusters in isolate bacteria come from a variety of phyla, the most predominant being Gammaproteobacteria. (B) Distribution of the top classes of enzymatic activities (BC types) found in bacteria and genome fragments available in IMG-ABC. Abbreviations: nrps, nonribosomal peptide synthetase; t1pks, type I PKS; t3pks, type III PKS; t2pks, type II PKS; hserlactone, homoserine lactone; transatpks, trans-AT PKS. (C) Ecosystem sources for the 10 most common BC enzymatic types. (D) Sources of experimentally verified bacterial secondary metabolites, summarized at the phylum level.
Accessing the IMG-ABC system.
IMG-ABC can be accessed directly (https://img.jgi.doe.gov/abc) (Fig. 3) or through its seamless integration with the user interface structure of the Joint Genome Institute’s (JGI) IMG system (https://img.jgi.doe.gov/) via the “ABC” tab. The synergies gained add an extraordinary value to the IMG-ABC database, because the user has immediate access not only to both computationally predicted and experimentally validated BCs and associated SMs but also to a vast array of integrated functionally and phylogenetically annotated genomic/metagenomic data, gene expression data, and functional genomics data. Users also have access to search-based and statistics-based entry points into both BC and SM objects within IMG. Most analysis tools in IMG are accessible to workflows employing an SM or a BC as a starting point. An important added benefit stemming from IMG-ABC’s integration with IMG is that it will help expose secondary metabolism to the 10,000+ registered IMG users, some of whom may have never before considered studying BCs in their favorite organisms, and will empower them to quickly and easily ask questions using a familiar interface.
FIG 3 .

Entry page to the IMG-ABC system. IMG-ABC is accessible either directly at https://img.jgi.doe.gov/abc or through the “ABC” tab in IMG (red arrow). Through these two avenues, the user has access to functions for the analysis and search of SMs and BCs.
Browsing the BC database.
The entry page to the BC statistics section of IMG-ABC displays the summary statistics for the BC data available in the system (Fig. 4A). Additional tabs allow the user to browse BCs by domain, phylum, BC type, SM type, cluster length, gene count, and functional classifications (viz. Enzyme Commission [EC] numbers [23] and Pfams [24]) assigned to genes within them. The summary data for BCs found under each tab are further broken down by experimental versus predicted and also by whether they are found in isolate genomes or in metagenomes. Narrowing results to specific categories is click-enabled for the user to arrive at a limited number of BCs of interest. Browsing BCs by their primary enzymatic mechanism (“BC type”) helps narrow search results to relevant BC types (Fig. 4B). Similarly, since specific Pfams and EC numbers can be directly related to secondary metabolism, browsing BCs by Pfams and EC numbers (Fig. 4C) is a useful feature that enables the analysis of gene clusters, but this feature is not available in any existing application. Once users obtain a listing of BCs in a tabular form by using any of the browsing methods mentioned above, they have the option to add genes of selected BCs to IMG’s “Gene Cart” for further analysis. There is also the capability to directly add the scaffolds where the selected BCs are found to the “Scaffold Cart,” which provides access to IMG’s extensive arsenal of tools for statistical and phylogenetic analyses.
FIG 4 .

BC summary section. (A) The BC statistics section can be navigated via the multiple-tab interface. (B) Listing of the number of BCs associated with specific combinations of BC types. (C) An expandable and interactive hierarchical tree of Enzyme Commission (EC) enzyme classifications.
BC detail interface.
Once users obtain a BC of interest, they can unlock a new set of BC-specific interfaces. The entry tab displays more information pertaining to the BC (Fig. 5A). Additionally, users can export the BC-related sequence in fasta format (for nucleotides or amino acids) or GenBank format. The “Genes in Cluster” tab lists all genes in the BC along with functional annotations and the bidirectional best hit for the gene and genome (Fig. 5C). From this tab, the user can add a subset or all genes to the Gene Cart for further analysis or storage. The “Cluster Neighborhood” tab provides access to an interactive graphical representation of the BC architecture along with its flanking regions. The BC boundary is denoted by a red dashed box, and each gene is colored according to its assigned COG. Clicking on any gene will result in displaying the neighborhoods of the bidirectional best hits for that gene, ordered by descending bit score (Fig. 5D). This function is a powerful way to evaluate potentially related loci and quickly highlight variant cluster architectures or unusual enzymatic activities, thus leading to the discovery of novel secondary metabolites. If the BC viewed is an experimentally validated one, the “Secondary Metabolite” tab will list the SMs that are produced by the BC whenever the chemical structure is available. The last three tabs list the metabolic pathways (KEGG, MetaCyc, IMG pathways) (Fig. 5B) in which the genes participate. KEGG and MetaCyc pathway detail pages show both genes in the selected BC and genes not in this BC but in the same genome. This provides users with the capability to understand the role of a BC inside a genome and to further investigate related genes functioning in the same pathway.
FIG 5 .

Biosynthetic cluster and secondary metabolite detail interfaces. (A) Interfaces to browse detailed statistics. (B) Pathway relationships. (C) Gene content. (D) Neighborhood views of bidirectional best-hit searches. (E) Secondary metabolite information for experimentally validated BCs. (F) Detailed compound annotations for SMs.
Browsing the SM database.
Similarly to BCs, the landing page to the SM Statistics section provides an overview of the database content. Through the multitabbed layout, SMs are displayed by type of chemical structure (per MeSH [25]), a tabular listing, pharmacological activity, and phylum. All compounds are integrated with structural and activity classifications available via MeSH on the PubChem website (26) and can be visualized as a clickable tree interface.
SM detail interface.
The page for each SM contains extensive information and links to external databases associated with the compound of interest. Additionally, it includes lists and links to all BCs within IMG-ABC that are connected with that compound (Fig. 5E). Besides the essential chemical descriptors (e.g., molecular weight, chemical formula, etc.), users have direct access to SMILES and InChI identifiers that can be linked to metabolites present in available genome-scale metabolic models (e.g., Streptomyces models that use flux balance to predict antibiotic production [27]), which can aid in the reconstruction of genome-scale models for SM production in less-studied microorganisms. The compound detail page also provides clickable interfaces for both structural classifications and activity hierarchical trees. Last, this page displays the two-dimensional chemical structure of the compound (Fig. 5F).
Searching biosynthetic clusters and secondary metabolites by annotation.
The IMG-ABC database contains a large number of experimentally validated and predicted BCs, with the former group connected to the SMs that they are known to produce. To enable users to efficiently narrow their search based on their specific interests, two search interfaces can be implemented (Fig. 6). Both SMs and BCs can be searched by a combination of chemical names, SM chemical classification, SM activity, number of atoms, molecular weight, and chemical formula, while the search space can be limited to specific target clades through a phylogenetic tree-based menu. Additionally, BCs are searchable by genomic features such as the number of genes, length of the BC, probability of the prediction, and type of enzymatic mechanism in the BC assigned by the AntiSMASH tool (12). Since there can be multiple combinations of BC types assigned to a cluster, we also provide users with the option to perform exact or inexact (and/or) searches for BCs with hybrid types. Another useful option is the ability to limit the results based on the presence of a single Pfam or combinations thereof, which enables “fishing” for enzymatic activities that may not be included in the AntiSMASH classification routine and therefore can aid in the discovery of new enzymatic mechanisms and/or novel chemical structures. The results are displayed in a tabular form and, in the case of BC searches, users can add selected BCs to the “Scaffold Cart” for further analysis (Fig. 6A).
FIG 6 .

Search interfaces and workflows. (A) Use of the search functions of IMG-ABC in combination with IMG’s analysis tools to investigate the distribution of BCs of specific classes. A search for a BC with the desired attributes is performed through the BC search interface (i) results and yields a list of records (ii), which can be added first to IMG’s Scaffold Cart (iii) and then to the Genome Cart (iv). From the organisms harboring BCs, matching the initial search can be grouped into different phylogenetic classes (v). These results can be exported for further analysis and visualization (vi). (B) An example of a search using a chemical structure as a query. The chemical structure descriptor string for zorbamycin was used to search IMG-ABC for secondary metabolites that matched the query with a similarity score greater than 0.3. This yielded a list of 118 compounds sorted by descending similarity score whose associated BCs could be analyzed using the Gene Cart and the Scaffold Cart.
Searching IMG-ABC based on a chemical structure.
The ability to use a chemical structure to query a genomic database is one of the new and exciting features introduced by IMG-ABC (Fig. 6B). A known SMILES descriptor (the user’s own or one retrieved through an external interface, such as NCBI PubChem’s Sketcher) is used to search compounds based on the structural similarity, computed using the Tanimoto similarity score (28, 29) to produce an interactive listing of similar SMs and related BCs, sorted by the similarity score. This search is powered through an implementation of ChemmineR functions (29) and facilitated by the precomputed atom pair descriptors for all compounds in the IMG-ABC database. SMILES strings entered by users are converted to a structure-data file (SDF) format and then to atom pair descriptors, which are used to search either all secondary metabolites or all IMG compounds, depending on the user’s input.
Case study: analysis of the distribution of putative phenazine-encoding BCs.
A case study illustrating the power of IMG-ABC in exploration and discovery of novel BCs operating on big biological data is presented below. A global survey of BCs containing at least six of the seven core genes essential for phenazine biosynthesis (30) was conducted against the full complement of 25,000+ isolate genomes within IMG-ABC. The resulting set of nearly 1,000 hits was filtered to retain putative complete BCs present on longer scaffolds, which were subsequently narrowed to 26 phenazine and phenazine-hybrid BCs with unique pathway architectures (Fig. 7A). In addition, to establish gene clusters for phenazine biosynthesis, this analysis identified several unique and potentially novel phenazine pathway architectures in Gammaproteobacteria, Betaproteobacteria, and Actinobacteria, opening doors for the discovery of novel enzymatic activities and/or phenazine derivatives. Additionally, pathways for phenazine biosynthesis were detected for the first time in two alphaproteobacterial genomes: the root-nodulating Rhizobium leguminosarum bv. trifolii and the photosynthetic Phaeospirillum molischianum.
FIG 7 .

Diversity of architectures of phenazine biosynthetic gene clusters. (A) Architecturally unique BCs containing all core genes of phenazine biosynthesis were sorted based on similarity to the Rhizobium leguminosarum phenazine gene cluster (blue box), which were calculated by using a modification of the average nucleotide identity approach (ANI) (38). Amino acid sequences of proteins encoded in the BC were used to identify bidirectional best hits (BBH) using USEARCH (39). Additionally, amino acid similarity was evaluated instead of identity. For a set of BBH proteins, the average amino acid similarity was calculated in addition to the alignment fraction (AF). The similarity score was calculated as the product of these two values for the R. leguminosarum BC (AAS*AF). Phylum classifications are abbreviated as follow: ACT, Actinobacteria; AP, Alphaproteobacteria; BP, Betaproteobacteria; GP, Gammaproteobacteria. Phenazine clusters that have been previously studied experimentally are underlined. (B) Evidence for the horizontal transfer of the Rhizobium leguminosarum phenazine BC (box). A BLASTp search was performed with each protein in and around the BC as the query against all proteins in the IMG database. Each gene is colored (see legend) based on the phylogenetic class of its top hit (see Table S1 in the supplemental material). Genes marked with an asterisk are transposases.
The discovery of a phenazine cluster in a Rhizobium species is particularly exciting. The presence of three genes of unknown function and the novel architecture of the cluster (Fig. 7A) suggest that novel enzymatic activities may be involved, and therefore the product may be a phenazine derivative with a novel chemical structure.
Additionally, it appears that this cluster was acquired through horizontal gene transfer. First, the cluster is flanked by transposases (Fig. 7B; see also Table S1 in the supplemental material). A BLASTp search was performed with the proteins in this locus (in the cluster and flanking) against all genomes in IMG. Although all of the flanking proteins (12 upstream and 7 downstream) had their best hit against other alphaproteobacterial proteins, 8 out of the 11 proteins in the cluster were most similar to proteins from the Actinobacteria phylum, while 2 were most similar to gammaproteobacterial proteins (Fig. 7B; see also Table S1). The 11th protein was a small hypothetical protein with no hits against the IMG database. The fact that there are 32 genomes (with “permanent draft” or “finished” status) of closely related Rhizobium leguminosarum strains in the IMG database and none of them appeared as a top hit in the BLASTp search indicate that the acquisition of these genes must have been recent.
Most importantly, rhizobia are excellent root colonizers and as such they are great candidates for use in biocontrol applications. To this end, investigators have attempted to engineer a phenazine cluster from Pseudomonas into Rhizobium etli USDA9032 (31). Although the engineered strain produced the recombinant phenazine, it was unable to produce root nodules. Our discovery of a potential phenazine-producing cluster native in a Rhizobium strain isolated from clover root nodules, however, suggests the possibility of combining biocontrol and root-nodulating properties in one bacterial strain. Experiments are under way to investigate the interplay between biocontrol and root nodulation in this R. leguminosarum strain, to elucidate the structure of the SM produced by this BC, and to investigate the SM’s function, if any, in this host-symbiont relationship.
DISCUSSION
In the last few years, there has been a resurgence of interest in the discovery of natural products, and this resurgence has been fueled by the explosion in the availability of microbial genomic sequences and the expansion of sampling to previously unstudied habitats and environments. However, a very large gap exists between the throughput of sequencing and the rate of discovery of novel pathways involved in secondary metabolism, predominantly because of the absence of tools that facilitate this intellectually and computationally difficult task. Additionally, no public resources exist that allow for the global analysis and comparison of putative biosynthetic gene clusters. These analyses have been accessible only to laboratories staffed with computational biologists and with access to high-performance computing resources.
For the first time, IMG-ABC links information regarding genomic pathways for the biosynthesis of secondary metabolites with chemical structure information on a scale of several thousand data sets. With careful efforts for quality control, it combines the predictive power of state-of-the-art computational tools, such as ClusterFinder and AntiSMASH, with the exhaustive analysis framework offered by the IMG family of systems. This combination delivers a powerful punch, predicting both familiar and novel biosynthetic gene pathways in thousands of cultured isolates, single cells, and metagenomes.
Future work: filling four existing knowledge gaps. (i) Pairwise similarities.
By computing pairwise similarities between biosynthetic pathways, we will annotate new BCs demonstrating very high similarity scores (indicating equivalence) with validated biosynthetic pathways. This process will also shed light on previously unknown enzymatic mechanisms.
(ii) Computationally linking SMs with BCs.
Traditionally, secondary metabolites were extracted from cultures of isolate organisms and tested for activity, often without any effort to identify the biosynthetic gene cluster responsible for the compound’s synthesis. However, in many cases, the genome of the producing organism has since been sequenced and, therefore, it may be feasible to computationally link SMs with BCs. We will explore new approaches to use IMG-ABC’s extensive collection of predicted BCs and whole-genome sequences to discover the relevant, but unknown, biosynthetic pathways for organisms known to produce specific compounds.
(iii) A searchable database of predicted SM backbone structures.
We will create a searchable database of predicted SM backbone structures for the more than 30,000 BCs that contain either NRPS or type I PKS enzymes (12, 32, 33). This function will be extremely useful for the identification of interesting chemical scaffolds that may be useful starting points for combinatorial chemistry.
(iv) Compatibility and synchronization with the MIBiG centralized database.
A major obstacle in collecting data describing experimentally verified BCs and SMs is the absence of a standardized way of reporting these data in public databases. Recently, a consortium of scientists have undertaken the Minimum Information about a Biosynthetic Gene cluster (MIBiG) initiative to provide structured data and high-quality annotations of BCs and the SMs they produce. In the future, we plan to adopt the database structure of MIBiG within IMG-ABC and perform regular synchronizations with the MIBiG centralized database to ensure that the content of experimentally verified data sets in IMG-ABC will continue to grow and reflect the most current and accurate public information.
Conclusion.
Computation of biosynthetic gene clusters in newly loaded genomes and metagenomes and their maintenance are now part of the IMG data-loading and annotation pipeline. The growing number of predicted BCs, in conjunction with continuous development of the analysis and search functions available through the system, will ensure that IMG-ABC will always have the latest and most complete publicly available information for the study of secondary metabolism in microbial genomes and metagenomes.
MATERIALS AND METHODS
Collection of gene clusters known to be associated with secondary metabolite biosynthesis.
The nucleotide database of NCBI (34) was searched by keywords related to biosynthetic gene clusters. GenBank records with sequences longer than 3,000 bp were retained and manually curated for their encoded SM name, whenever possible. BCs were also retrieved from ClusterMine360 (17) and DoBiscuit (16) irrespective of their length and reconciled with those retrieved from the NCBI database in order to produce a nonredundant list of BCs. The resulting BCs were mapped to isolate genomes and metagenomes in IMG/M based on BLASTN analysis (35) of their nucleotide sequences. Unmapped records were imported into IMG as “genome fragments” (19). Through this process, we acquired 1,332 experimentally verified clusters associated with a GenBank record, most of them imported as “genome fragments.” AntiSMASH (version 2.2) was used to assign a type of enzymatic mechanism (BC type) to each gene cluster, when possible (12). Whenever a BC was assigned hybrid or multiple types, the list of types was sorted and concatenated into one text string by using a semicolon separator.
Collection of secondary metabolites and determination of their associations with specific gene clusters.
GenBank records of BCs were connected with chemical structures by querying the PubChem Compound database using their associated SM names (26). Exact matches preceded inexact matches. Information retrieved from the PubChem Compound records includes structural and physical descriptors, such the simplified molecular-input line-entry system (SMILES) string (36), the IUPAC international chemical identifier (InChi) (37), molecular weight, etc. Additionally, MeSH Library headings (25) related to classification and pharmaceutical action are also acquired and are represented as “SM Type” and “SM Activity” in our database, respectively. Chemical structures are rendered within IMG by using the NCI/CADD service (http://cactus.nci.nih.gov/).
Computational prediction of putative secondary metabolite biosynthetic gene clusters in genomes and metagenomes.
Putative BCs were first identified using ClusterFinder (11) for all isolate genomes and metagenomes in IMG and IMG/M, respectively. ClusterFinder predicts putative BCs based on the Pfam functional annotation of genes available in the integrated context within IMG. BCs predicted in isolate genomes were then classified by the type of biosynthetic enzymes they contain by using AntiSMASH (12). BC predictions were filtered to eliminate BCs that contained less than 6 genes, had a prediction probability of less than 0.3, and were not classified by AntiSMASH to employ a well-defined enzymatic mechanism. Additionally, BC content was analyzed to identify Pfam categories that are not known to be associated with secondary metabolism but are present as positive training features within ClusterFinder. These included Pfam categories such as prophage proteins (PF04883, PF05709, and PF06199), protein secretion systems (PF08817, PF10140, and PF10661), inorganic ion transport proteins (PF02421 and PF11604), and families representing DNA/RNA polymerases, whose presence leads to false-positive BC predictions.
SUPPLEMENTAL MATERIAL
Evidence for horizontal gene transfer of Rhizobium phenazine gene cluster.
ACKNOWLEDGMENTS
The Director, Office of Science, Office of Biological and Environmental Research, Life Sciences Division, U.S. Department of Energy under contract number DE-AC02-05CH11231; this research used resources of the National Energy Research Scientific Computing Center, which is supported by the Office of Science of the U.S. Department of Energy under contract number DE-AC02-05CH11231. Funding for open access charges was provided by the: University of California. P.C. was supported by a Howard Hughes Medical Institute Predoctoral Fellowship.
We thank Thomas Girke, Tyler Backman, and Kevin Horan at UC Riverside for their assistance and the implementation of the ChemmineR package.
Footnotes
Citation Hadjithomas M, Chen IMA, Chu K, Ratner A, Palaniappan K, Szeto E, Huang J, Reddy TBK, Cimermančič P, Fischbach MA, Ivanova NN, Markowitz VM, Kyrpides NC, Pati A. 2015. IMG-ABC: a knowledge base to fuel discovery of biosynthetic gene clusters and novel secondary metabolites. mBio 6(4):e00932-15. doi:10.1128/mBio.00932-15.
REFERENCES
- 1.Grabley S, Thiericke R. 1999. Drug discovery from nature. Springer Science & Business Media, Berlin, Germany. [Google Scholar]
- 2.Newman DJ, Cragg GM. 2012. Natural products as sources of new drugs over the 30 years from 1981 to 2010. J Nat Prod 75:311–335. doi: 10.1021/np200906s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhang F, Rodriguez S, Keasling JD. 2011. Metabolic engineering of microbial pathways for advanced biofuels production. Curr Opin Biotechnol 22:775–783. doi: 10.1016/j.copbio.2011.04.024. [DOI] [PubMed] [Google Scholar]
- 4.Piel J. 2009. Metabolites from symbiotic bacteria. Nat Prod Rep 26:338–362. doi: 10.1039/b703499g. [DOI] [PubMed] [Google Scholar]
- 5.Thomashow LS, Weller DM. 1988. Role of a phenazine antibiotic from Pseudomonas fluorescens in biological control of Gaeumannomyces graminis var. tritici. J Bacteriol 170:3499–3508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Li JW, Vederas JC. 2009. Drug discovery and natural products: end of an era or an endless frontier? Science 325:161–165. doi: 10.1126/science.1168243. [DOI] [PubMed] [Google Scholar]
- 7.Koehn FE, Carter GT. 2005. The evolving role of natural products in drug discovery. Nat Rev Drug Discov 4:206–220. doi: 10.1038/nrd1657. [DOI] [PubMed] [Google Scholar]
- 8.McDonald JG, Smith DD, Stiles AR, Russell DW. 2012. A comprehensive method for extraction and quantitative analysis of sterols and secosteroids from human plasma. J Lipid Res 53:1399–1409. doi: 10.1194/jlr.D022285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Silva GL, Lee I, Kinghorn AD. 1998. Special problems with the extraction of plants, p 343–363. In Cannell RJP (ed), Natural products isolation. Humana Press, Totowa, NJ. [Google Scholar]
- 10.Markowitz VM, Chen IM, Chu K, Szeto E, Palaniappan K, Pillay M, Ratner A, Huang J, Pagani I, Tringe S, Huntemann M, Billis K, Varghese N, Tennessen K, Mavromatis K, Pati A, Ivanova NN, Kyrpides NC. 2014. IMG/M 4 version of the integrated metagenome comparative analysis system. Nucleic Acids Res 42:D568–D573. doi: 10.1093/nar/gkt919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cimermancic P, Medema MH, Claesen J, Kurita K, Wieland Brown LC, Mavrommatis K, Pati A, Godfrey PA, Koehrsen M, Clardy J, Birren BW, Takano E, Sali A, Linington RG, Fischbach MA. 2014. Insights into secondary metabolism from a global analysis of prokaryotic biosynthetic gene clusters. Cell 158:412–421. doi: 10.1016/j.cell.2014.06.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Blin K, Medema MH, Kazempour D, Fischbach MA, Breitling R, Takano E, Weber T. 2013. antiSMASH 2.0: a versatile platform for genome mining of secondary metabolite producers. Nucleic Acids Res 41:W204–W212. doi: 10.1093/nar/gkt449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Caboche S. 2014. Biosynthesis: bioinformatics bolster a renaissance. Nat Chem Biol 10:798–800. doi: 10.1038/nchembio.1634. [DOI] [PubMed] [Google Scholar]
- 14.Rinke C, Schwientek P, Sczyrba A, Ivanova NN, Anderson IJ, Cheng J-F, Darling A, Malfatti S, Swan BK, Gies EA, Dodsworth JA, Hedlund BP, Tsiamis G, Sievert SM, Liu W-T, Eisen JA, Hallam SJ, Kyrpides NC, Stepanauskas R, Rubin EM, Hugenholtz P, Woyke T. 2013. Insights into the phylogeny and coding potential of microbial dark matter. Nature 499:431–437. doi: 10.1038/nature12352. [DOI] [PubMed] [Google Scholar]
- 15.Lucas X, Senger C, Erxleben A, Grüning BA, Döring K, Mosch J, Flemming S, Günther S. 2013. StreptomeDB: a resource for natural compounds isolated from streptomyces species. Nucleic Acids Res 41:D1130–D1136. doi: 10.1093/nar/gks1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ichikawa N, Sasagawa M, Yamamoto M, Komaki H, Yoshida Y, Yamazaki S, Fujita N. 2013. DoBISCUIT: a database of secondary metabolite biosynthetic gene clusters. Nucleic Acids Res 41:D408–D414. doi: 10.1093/nar/gks1177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Conway KR, Boddy CN. 2013. ClusterMine360: a database of microbial PKS/NRPS biosynthesis. Nucleic Acids Res 41:D402–D407. doi: 10.1093/nar/gks993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Diminic J, Zucko J, Ruzic IT, Gacesa R, Hranueli D, Long PF, Cullum J, Starcevic A. 2013. Databases of the thiotemplate modular systems (CSDB) and their in silico recombinants (r-CSDB). J Ind Microbiol Biotechnol 40:653–659. doi: 10.1007/s10295-013-1252-z. [DOI] [PubMed] [Google Scholar]
- 19.Markowitz VM, Chen IM, Palaniappan K, Chu K, Szeto E, Pillay M, Ratner A, Huang J, Woyke T, Huntemann M, Anderson I, Billis K, Varghese N, Mavromatis K, Pati A, Ivanova NN, Kyrpides NC. 2014. IMG 4 version of the integrated microbial genomes comparative analysis system. Nucleic Acids Res 42:D560–D567. doi: 10.1093/nar/gkt963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Reddy TB, Thomas AD, Stamatis D, Bertsch J, Isbandi M, Jansson J, Mallajosyula J, Pagani I, Lobos EA, Kyrpides NC. 2015. The Genomes Online Database (GOLD) v.5: a metadata management system based on a four level (meta)genome project classification. Nucleic Acids Res 43:D1099–D1106. doi: 10.1093/nar/gku950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Challis GL, Naismith JH. 2004. Structural aspects of non-ribosomal peptide biosynthesis. Curr Opin Struct Biol 14:748–756. doi: 10.1016/j.sbi.2004.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Staunton J, Weissman KJ. 2001. Polyketide biosynthesis: a millennium review. Nat Prod Rep 18:380–416. doi: 10.1039/a909079g. [DOI] [PubMed] [Google Scholar]
- 23.Webb EC. 1992. Enzyme nomenclature 1992. Recommendations of the Nomenclature Committee of the International Union of Biochemistry and Molecular Biology on the nomenclature and classification of enzymes, p 863 Academic Press, San Diego, CA. [Google Scholar]
- 24.Finn RD, Bateman A, Clements J, Coggill P, Eberhardt RY, Eddy SR, Heger A, Hetherington K, Holm L, Mistry J, Sonnhammer ELL, Tate J, Punta M. 2013. Pfam: the protein families database. Nucleic Acids Res 42(Databsase issue):D222–D230. doi: 10.1093/nar/gkt1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rogers FB. 1963. Medical subject headings. Bull Med Libr Assoc 51:114–116. [PMC free article] [PubMed] [Google Scholar]
- 26.Wang Y, Xiao J, Suzek TO, Zhang J, Wang J, Bryant SH. 2009. PubChem: a public information system for analyzing bioactivities of small molecules. Nucleic Acids Res 37:W623–W633. doi: 10.1093/nar/gkp456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Borodina I, Krabben P, Nielsen J. 2005. Genome-scale analysis of Streptomyces coelicolor A3(2) metabolism. Genome Res 15:820–829. doi: 10.1101/gr.3364705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Holliday JD, Salim N, Whittle M, Willett P. 2003. Analysis and display of the size dependence of chemical similarity coefficients. J Chem Inf Comput Sci 43:819–828. doi: 10.1021/ci034001x. [DOI] [PubMed] [Google Scholar]
- 29.Wang Y, Backman TWH, Horan K, Girke T. 2013. fmcsR: mismatch tolerant maximum common substructure searching in R. Bioinformatics 29:2792–2794. doi: 10.1093/bioinformatics/btt475. [DOI] [PubMed] [Google Scholar]
- 30.Mavrodi DV, Ksenzenko VN, Bonsall RF, Cook RJ, Boronin AM, Thomashow LS. 1998. A seven-gene locus for synthesis of phenazine-1-carboxylic acid by Pseudomonas fluorescens 2-79. J Bacteriol 180:2541–2548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Krishnan HB, Kang BR, Hari Krishnan A, Kim KY, Kim YC. 2007. Rhizobium etli USDA9032 Engineered to produce a phenazine antibiotic inhibits the growth of fungal pathogens but is impaired in symbiotic performance. Appl Environ Microbiol 73:327–330. doi: 10.1128/AEM.02027-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Minowa Y, Araki M, Kanehisa M. 2007. Comprehensive analysis of distinctive polyketide and nonribosomal peptide structural motifs encoded in microbial genomes. J Mol Biol 368:1500–1517. doi: 10.1016/j.jmb.2007.02.099. [DOI] [PubMed] [Google Scholar]
- 33.Röttig M, Medema MH, Blin K, Weber T, Rausch C, Kohlbacher O. 2011. NRPSpredictor2: a web server for predicting NRPS adenylation domain specificity. Nucleic Acids Res 39:W362–W367. doi: 10.1093/nar/gkr323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Benson DA, Clark K, Karsch-Mizrachi I, Lipman DJ, Ostell J, Sayers EW. 2014. GenBank. Nucleic Acids Res 42:D32–D37. doi: 10.1093/nar/gkt1030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. 1990. Basic local alignment search tool. J Mol Biol 215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 36.Weininger D. 1988. SMILES, a chemical language and information system. 1. Introduction to methodology and encoding rules. J Chem Inf Comput Sci 28:31–36. doi: 10.1021/ci00057a005. [DOI] [Google Scholar]
- 37.Heller S, McNaught A, Stein S, Tchekhovskoi D, Pletnev I. 2013. InChI: the worldwide chemical structure identifier standard. J Cheminform 5:7. doi: 10.1186/1758-2946-5-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Konstantinidis KT, Tiedje JM. 2005. Genomic insights that advance the species definition for prokaryotes. Proc Natl Acad Sci U S A 102:2567–2572. doi: 10.1073/pnas.0409727102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Edgar RC. 2010. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26:2460–2461. doi: 10.1093/bioinformatics/btq461. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Evidence for horizontal gene transfer of Rhizobium phenazine gene cluster.