

Abstract
Usher syndrome type III (USH3) is characterized by progressive loss of hearing and vision, and varying degrees of vestibular dysfunction. It is caused by mutations that affect the human clarin-1 protein (hCLRN1), a member of the tetraspanin protein family. The missense mutation CLRN1N48K, which affects a conserved N-glycosylation site in hCLRN1, is a common causative USH3 mutation among Ashkenazi Jews. The affected individuals hear at birth but lose that function over time. Here, we developed an animal model system using zebrafish transgenesis and gene targeting to provide an explanation for this phenotype. Immunolabeling demonstrated that Clrn1 localized to the hair cell bundles (hair bundles). The clrn1 mutants generated by zinc finger nucleases displayed aberrant hair bundle morphology with diminished function. Two transgenic zebrafish that express either hCLRN1 or hCLRN1N48K in hair cells were produced to examine the subcellular localization patterns of wild-type and mutant human proteins. hCLRN1 localized to the hair bundles similarly to zebrafish Clrn1; in contrast, hCLRN1N48K largely mislocalized to the cell body with a small amount reaching the hair bundle. We propose that this small amount of hCLRN1N48K in the hair bundle provides clarin-1-mediated function during the early stages of life; however, the presence of hCLRN1N48K in the hair bundle diminishes over time because of intracellular degradation of the mutant protein, leading to progressive loss of hair bundle integrity and hair cell function. These findings and genetic tools provide an understanding and path forward to identify therapies to mitigate hearing loss linked to the CLRN1 mutation.
SIGNIFICANCE STATEMENT Mutations in the clarin-1 gene affect eye and ear function in humans. Individuals with the CLRN1N48K mutation are born able to hear but lose that function over time. Here, we develop an animal model system using zebrafish transgenesis and gene targeting to provide an explanation for this phenotype. This approach illuminates the role of clarin-1 and the molecular mechanism linked to the CLRN1N48K mutation in sensory hair cells of the inner ear. Additionally, the investigation provided an in vivo model to guide future drug discovery to rescue the hCLRN1N48K in hair cells.
Keywords: clarin-1, hair cells, hearing
Introduction
Usher syndrome (USH) is a common cause of inherited deaf-blindness. Based on the severity and progression of hearing loss, the onset age of retinitis pigmentosa and the varying degree of vestibular function, USH is classified into three subtypes: USH1, USH2, and USH3, with USH3 being the least severe (Pakarinen et al., 1995; Otterstedde et al., 2001; Petit, 2001; Reisser et al., 2002). Ten genes in which mutations cause USH have been identified; of these, six cause USH1 (MYO7A, USH1C, CDH23, PCDH15, USH1G, and CIB2), three cause USH2 (USH2A, GPR98, and WHRN), and one is linked to USH3 (CLRN1; http://www.ushersyndrome.nih.gov/resources/loci.html). Nine of the proteins encoded by these 10 genes localize to the mouse inner ear mechanosensory hair cell bundle (or “hair bundle”) and are necessary for normal hair bundle development and function (Boëda et al., 2002; Adato et al., 2005; Mburu et al., 2006; McGee et al., 2006; Liu et al., 2007; Lefèvre et al., 2008; Grati and Kachar, 2011; Riazuddin et al., 2012). These data led us to hypothesize that the human clarin-1 protein (hCLRN1) has a similar expression pattern and function in the hair bundle.
Whole-mount in situ hybridization demonstrated that Clrn1 mRNA localizes to cochlear hair cells and spiral ganglion cells (Adato et al., 2002). Cochlear hair cells from the Clrn1 knock-out (Clrn1−/−) and Clrn1N48K knock-in (Clrn1N48K) mutations pointed to disrupted hair bundles as the cause of the inner ear disorder (Geng et al., 2009, 2012). However, a report based on the morpholino knockdown of clrn1 mRNA in zebrafish (Danio rerio) larvae arrived at a different set of conclusions regarding the role of clarin-1 in hair cells: knocking down clrn1 message did not affect the hair bundle morphology, but it is reported to affect localization of the synaptic ribbons in zebrafish (Ogun and Zallocchi, 2014). Therefore, it is important to examine the hair cell phenotype of zebrafish harboring a null allele of clrn1 to uncover the authentic role of clarin-1 in hair cells. To the best of our knowledge, this is the first report of successfully targeting the genomic sequence of a gene essential for hair cells in zebrafish.
The genes associated with deafness are conserved in zebrafish, including those linked to deafness in USH (Nicolson et al., 1998; Ernest et al., 2000; Whitfield, 2002; Söllner et al., 2004; Nicolson, 2005; Seiler et al., 2005; Whitfield et al., 2005; Phillips et al., 2011). Many of these genes, including clarin-1, are expressed in the hair cells of the mouse cochlea and in the ear and neuromast of zebrafish (Adato et al., 2002; Geller et al., 2009; Geng et al., 2009; Phillips et al., 2013). Based on the conservation of the gene sequence and Clrn1 mouse mutant data discussed earlier we expected loss of clrn1 expression to disrupt the hair bundle structure and function in zebrafish.
A high incidence of USH3 in the Ashkenazi Jewish population can be attributed to the CLRN1N48K mutation. For example, 40% of a cohort of 40 Ashkenazi Jews with USH were classified USH3, and all of whom carried a CLRN1N48K/N48K genotype (Ness et al., 2003). Also, 0.7% of the Ashkenazi Jews from the New York area were carriers of the N48K mutation, with a predicted USH3 frequency of 1.2 per 100, 000 (Ness et al., 2003). Individuals carrying the CLRN1N48K/N48K mutation are born hearing, but auditory function is lost over time (Ness et al., 2003; Plantinga et al., 2005). The pathogenic mechanism linking CLRN1N48K to its phenotype is not fully understood. Development of a tractable in vivo model is necessary to delineate the molecular pathology of the CLRN1N48K mutation and develop therapies to mitigate hCLRN1N48K-associated hearing loss. To address these issues, we generated a transgenic zebrafish that stably expresses hCLRN1 or hCLRN1N48K in a wild-type background. Data from these animals confirmed that hCLRN1 is a hair bundle protein and shed new light on the mechanism of hearing loss linked to hCLRN1N48K.
Materials and Methods
Zebrafish.
All experiments were conducted using the Tübingen (Tü) strain of zebrafish of either sex. The animal protocols used in this report were approved by the Case Western Reserve University Institutional Animal Care and Use Committee (Protocol Approval Number: 2009-0167). cdh23tj264a zebrafish (Söllner et al., 2004) were obtained from Teresa Nicolson (Oregon Health & Science University, Oregon). All zebrafish were maintained and bred using standard procedures (Nüsslein-Volhard and Dahm, 2002).
In situ hybridization.
To investigate the clrn1 mRNA expression pattern in zebrafish, whole-mount in situ hybridization was performed as described previously (McDermott et al., 2007). Zebrafish larvae were treated with 1-phenyl-2-thiourea to block melanization. Digoxigenin-labeled antisense and sense RNA probes recognizing clrn1 mRNA (NCBI Accession number: NM 001002671.1) and fscn2b mRNA (NCBI Accession number: EU580143.1) were synthesized from plasmids pCRII::clrn1 and pCRII::fscn2b, respectively (Chou et al., 2011). Images of whole-mount larvae were obtained using an upright bright-field microscope (Olympus BX-60) with a 40× oil-immersion objective lens and processed using MetaMorph software (Molecular Devices).
Generation and phenotype analysis of the clrn1 mutant zebrafish line.
Targeting the first coding exon of mouse Clrn1 generated a null allele (Geng et al., 2009); therefore, the corresponding exon, exon 0, in zebrafish clrn1 was targeted using ZFN (CompoZr Custom Zinc Finger Nucleases; Sigma-Aldrich; Doyon et al., 2008). The exon 0-specific DNA-binding ZFN recognition elements (left/right: ZFN1/ZFN2) were separated by a 5 bp spacer region: c.205–209; PZFN1/PZFN2: TACGGACTTTTTCACGGAcatggGATGAAACAATG (lower case letters denote the spacer site sequence and upper case letters denote the zinc finger protein-binding site). Heterodimeric ZFN pair (ZFN1/ZFN2) selection was based on the yeast MEL-1 reporter assay (Doyon et al., 2008). In the selected ZFN pair, Mel-1 activity was 134.7% of the positive control; it was the only ZFN pair to show cleavage activity in the uninduced state.
The mRNA encoding the ZFN pair was injected into single-cell stage zebrafish embryos (F0 generation) according to standard protocol (Xu, 1999). Three independent batches of embryos were injected with low and high mRNA concentrations (0.4 ng/μl, n = 1892 embryos; 1.0 ng/μl, n = 133 embryos) to efficiently induce ZFN-targeted cleavage. A higher ZFN concentration was associated with a higher percentage of embryos with nonspecific developmental abnormalities and a higher death rate within 24 h post fertilization (Table 1). Genotyping (see below) was performed to identify F0 zebrafish carrying a clrn1 mutant allele. DNA sequencing confirmed mutation at the target site. ZFN-induced clrn1 mutant alleles with variable lengths of deleted sequence in the first coding exon (exon 0) are represented by the prefix “CWR” (for Case Western Reserve University): CWR1, deletion (Δ) at nucleotide position c.205 to c.211; CWR2, Δ c.205–c.214; CWR3, Δ c.170–c.212; and CWR4, Δ c.202–c.208. Mutant alleles are referred to as clrn1CWR1, clrn1CWR2, clrn1CWR3, and clrn1CWR4, respectively, in the text.
Table 1.
Generation of ZFN-targeted zebrafish clrn1 mutants
| Dosage of ZFN RNA pair for clrn1 (ng) | Total number of embryos injecteda | Control (vehicle only) | Live embryos at 24 h |
6 dpf |
15 dpf |
>90 dpf ZFN | Carrying clrn1 mutant allele in germline (mosaic pattern)b | |||
|---|---|---|---|---|---|---|---|---|---|---|
| ZFN | Control | ZFN | Control | ZFN | Control | |||||
| 0.4 | (Batch 1) 178 | 152 | 133 (75%) | 80 (53%) | 49 (28%) | 78 (51%) | 14 (8%) | 11 (7%) | 13 (7%) | 2F |
| (Batch 2) 559 | 428 | 389 (70%) | 329 (77%) | 241 (43%) | 284 (66%) | 36 (6%) | 100 (23%) | 18 (3%) | 2F; 5M | |
| (Batch 3) 1155 | 260 | 461 (40%) | 243 (94%) | 435 (38%) | 237 (91%) | 32 (3%) | 72 (28%) | 20 (2%) | 3F; 2M | |
| 1 | (Batch 4) 133 | 47 | 109 (82%) | 45 (96%) | 98 (74%) | 45 (96%) | 52 (39%) | 32 (68%) | 5 (4%) | 1F; 1M |
a Tü embryos injected with ZFN RNA pair at single-cell stage. The ZFN RNA pair was prepared in vehicle made of 0.1 m KCl and 0.5% phenol red dye.
b No phenotype observed. F, female; M, male.
In all, 56 ZFN-injected embryos (F0) reached the adult stage, and 15 of those adults transmitted the clrn1 mutant allele through the germline to the F1 generation (Table 1). None of the F0 larvae displayed an observable phenotype. A stable F1 generation clrn1 mutant line was generated for each allele by backcrossing F0 with wild-type (clrn1+/+) zebrafish to eliminate the inheritance of any nonspecific effects of the ZFN. The genotype of the F1 individuals was either heterozygous for the clrn1 mutation (clrn1+/−) or wild-type (clrn1+/+). F1 adults carrying the clrn1 mutant allele were inbred to generate F2 larvae carrying two mutant alleles of clrn1 (clrn1−/−).
Tapping-induced vibrational startle reflex (Nicolson et al., 1998) was used to screen F2 and F3 generation larvae at 6 d post fertilization (dpf). Larvae carrying two mutant alleles of clrn1 were expected to display an attenuated startle response compared with their clrn1+/+ or clrn1+/− clutch mates. Briefly, larvae at 6 dpf were placed in a Petri dish containing E3 medium, or “fish water” (5 mm NaCl, 0.17 mm KCl, 0.33 mm CaCl2, and 0.33 mm MgSO4), and were left undisturbed for 5 min. Their swimming behavior and orientation were noted, and then the rim of the Petri dish was gently tapped with a metal probe to induce a startle response. Larvae with subtle or strong phenotypes were sorted into separate Petri dishes and observed again. Subsequently, the vibrational startle reflex (Nicolson et al., 1998) was used to screen the progenies at 6 dpf, and the clrn1 genotypes of responder and nonresponder groups were determined.
Genotyping.
PCR followed by restriction fragment length polymorphism was used for genotyping. Loss of the MslI recognition sequence in the spacer region between the ZFN-binding sites (c.205–209) was used to identify ZFN-targeted alleles. Larvae from the cdh23tj264a zebrafish (Söllner et al., 2004) were used as the reference mutant strain. Loss of the MboI restriction site in the cdh23 PCR amplicon was used to identify larvae carrying the cdh23tj264a allele. Genomic DNA from embryos and adult tail fins was isolated as described previously (Foley et al., 2009). The primers used to amplify clrn1 were KA1088 5′-AGATTGTCAGTGCGCGTCTTAG-3′ and KA1089 5′-AATATGATGAAGTTACTCACATGAAAA-3′, and those used to amplify cdh23 were KA1133 5′-ACTCCAGTTGGGACGTCAGT-3′ and KA 1134 5′-TTGTATTGCTTGATTGGTATGG-3′. PCR cycle parameters used were 94°C for 2 min, followed by 40 cycles of 94°C for 30 s, 55°C for 30 s, and 72°C for 30 s, with a final extension of 72°C for 5 min. PCR products of clrn1 (amplicon size 343 bp) and cdh23 (amplicon size 578 bp) were digested at 37°C for 2 h with MslI and MboI restriction enzymes, respectively, and resolved by 3% agarose gel electrophoresis. Two bands were observed for the wild-type clrn1 allele (269 and 74 bp) and one band (343 bp) for each of the four (CWR1–4) mutant alleles. Three bands (46, 284, and 248 bp) were observed for the wild-type cdh23 allele and two bands (330 and 248 bp) for the mutant cdh23tj264a allele. DNA sequencing was performed for samples with a clrn1 deletion/insertion at the ZFN target site.
RT-PCR.
Total RNA was isolated from the zebrafish larvae or from the adult zebrafish eye or ear using a TRIzol reagent and manufacturer's protocol (Invitrogen). RT-PCR was performed as described previously (Geng et al., 2009). Primers designed to amplify clrn1 mRNA were KA911 5′-TTTCCACTGATGAGGCTGTG-3′ and KA912 5′-GGTGGATGTCGTGTGCAG-3′. PCR cycle parameters were 94°C for 2 min, 30 cycles of 94°C for 30 s, 55°C for 30 s, and 72°C for 30 s, followed by 72°C for 5 min. PCR products were resolved by 3% agarose gel electrophoresis. The expected size of the amplicon was 790 bp. RT-PCR products were sequenced to determine changes to the coding sequence of exon 0.
Visualizing the hair cell bundle.
Phalloidin labeling of the hair cell bundle in clrn1+/+, clrn1−/−, and cdh23tj264a larvae at 5 dpf was performed as described previously (Söllner et al., 2004). F-actin-rich hair bundles were labeled with phalloidin conjugated to Alexa Fluor 488 (referred to as “fluorescent phalloidin”; 1:100 dilution; Invitrogen). Stained larvae were mounted onto a specially designed confocal stage using fine pins, and confocal images were obtained using a 63× oil-immersion objective lens on a Leica SP8 confocal microscope.
Immunohistochemistry.
To determine Clrn1 localization in zebrafish inner ear hair cells, whole-mount immunostaining of 5 dpf stage zebrafish larvae was conducted using primary rabbit anti-Clrn1 polyclonal antibody (Novus Biologicals; 1:500 dilution), secondary goat anti-rabbit IgG Alexa 594 (1:1000 dilution; Invitrogen), and fluorescent phalloidin (1:100 dilution; Invitrogen). The primary antibody is referred to as “DrClrn1” in this report. clrn1+/+ and clrn1−/− larvae were fixed with CytoSkelfix (Cytoskeleton) at −20°C for 8 min, followed by two PBS washes of 5 min each. Nonspecific binding sites were then blocked with 3% BSA in PBS for 1 h at room temperature, and the specimen was then incubated with a primary antibody at 4°C overnight. After washing with a blocking solution (4 × 30 min at room temperature), the tissue sample was incubated with a secondary antibody for 2 h at room temperature. The tissue was then counterstained with fluorescent phalloidin as described previously. Confocal images were captured using a Leica SP8 confocal microscope with a 40× oil-immersion objective lens.
Clrn1 protein sensitivity to a traditional PFA-based fixative was tested in the clrn1+/+, clrn1−/−, and transgenic hCLRN1-YFP larvae. Larvae expressing GFP in hair cells were used as controls (McDermott et al., 2010). PFA fixation and immunolabeling were performed as described previously (López-Schier et al., 2004). Various concentrations of PFA, incubation times, and temperature were tested to optimize the protocol. Briefly, for whole-mount labeling of clrn1+/+ and clrn1−/− larvae with the DrClrn1 antibody, larvae were fixed in 1–4% PFA for 15 min, 30 min, 45 min, or 1 h at room temperature, or overnight at 4°C. Fixed larvae were sequentially incubated with the primary DrClrn1 antibody (1:500 dilution) and the secondary goat anti-rabbit IgG Alexa 594 (1:1000 dilution; Invitrogen). They were then counterstained with a fluorescent phalloidin (1:100; Invitrogen). Confocal images were captured using a Leica SP8 confocal microscope with a 63× oil-immersion objective lens.
Before immunodetection of hCLRN1-YFP or GFP, hCLRN1-YFP or GFP expression was observed in live larvae using a Leica SP8 confocal microscope with a 40× oil-immersion objective lens. Larvae were then fixed in PFA, and confocal images were obtained to determine the effect of PFA on hCLRN1-YFP and GFP detection in hair cells (McDermott et al., 2010). The same larvae were immunolabeled with a primary rabbit polyclonal anti-GFP antibody (Novus Biologicals) raised against a full-length GFP that also recognizes YFP (Novus Biologicals) because of the 97% identity in amino acid sequence between the two proteins. The goat anti-rabbit IgG Alexa 594 (1:1000; Invitrogen) was used as the secondary antibody for GFP. Confocal images were captured using a Leica SP8 confocal microscope with a 40× oil-immersion objective lens.
Recording microphonic potentials in clrn1 mutant zebrafish.
Zebrafish larvae at 6 dpf were anesthetized using MS-222 (Sigma-Aldrich) dissolved in a standard bath solution containing 120 mm NaCl, 2 mm KCl, 10 mm HEPES, 2 mm CaCl2, and 0.7 mm NaH2PO4 adjusted to pH ∼7.2. The larvae were then secured in a recording chamber using dental floss tie downs (Ricci and Fettiplace, 1997). Their blood flow and heart rate were visually observed to assess viability using an upright Olympus BX51WI microscope. Recordings were made with a PC-505B amplifier (Warner Instruments) and a PCI-6221 digitizer (National Instruments). Images were taken with a Grasshopper3 CMOS camera (Point Grey) and processed with software provided by the manufacturer. To record the activity of hair cells from the lateral line neuromasts, the cupula of neuromasts were deflected with a fluid jet placed ∼50 μm from the neuromast (Trapani and Nicolson, 2010) and controlled by an HSPC-1 (ALA Scientific Instruments) that delivered sinusoidal stimuli of 50 Hz. Microphonic potentials were recorded at room temperature (22°C). For this, borosilicate glass electrodes with a resistance of 5–6 MΩ were filled with standard bath solution and placed near the apical edges of the lateral line neuromasts. Microphonic potentials were recorded using a jClamp for Windows (SciSoft, license: Joseph Santos-Sacchi, Yale University, New Haven, CT) in current-clamp mode, amplified by 20 (SIM983; Stanford Research), and low-pass filtered at 200 Hz. The data represent an average of at least 500 responses. Statistical significance was set at p < 0.05.
Evaluation of presynaptic and postsynaptic marker in the clrn1−/− larvae.
Immunolabeling for ribbon synapse markers was performed as described earlier (Mo and Nicolson, 2011). Briefly, larvae at 6 dpf were fixed in 4% PFA in PBS, 4% sucrose, and 0.01% Tween 20 at 4°C for 4 h. Permeabilization followed, with 100% acetone at −20°C for 6 min. The larvae were labeled with the antibodies against the presynaptic marker, the C-terminal-binding protein 2 (CtBP2), a component of RIBEYE and a postsynaptic marker, the membrane-associated guanylate kinase (MAGUK), respectively. The primary antibodies used are a rabbit polyclonal CtBP2 antibody (Novus Biologicals) and mouse pan-MAGUK (NeuroMab Antibodies). The secondary antibodies used against the primary antibodies are goat anti-rabbit Alexa 488 (Molecular probes) and donkey anti-mouse Alexa 594 (Molecular Probes). Z-stack images of the neuromast from clrn1+/+ and clrn1−/− larvae were captured using an SP8 Leica confocal microscope with a 63× oil-immersion objective lens. The CtBP2 puncta, MAGUK puncta, and colocalization were manually counted and entered in an Excel sheet (Microsoft). The student's t test and bar graph were done using GraphPad Software. The colocalization of presynaptic and postsynaptic puncta of CtBP2 and MAGUK at the synaptic zone of the hair cells represents the maturation of the synapse.
ERG.
ERG was performed on isolated larval eyes at 7 dpf as previously described (Sirisi et al., 2014). Briefly, after a 30 min dark-adaptation period, a single larva was anesthetized in an E3 medium containing 0.75 mm 3-aminobenzoic acid methyl ester (Sigma-Aldrich) and 4.6 mm NaHCO3. It was then transferred to the recording chamber filled with 1% agarose and moistened with Ringer's solution (111 mm NaCl, 2.5 mm KCl, 1 mm CaCl2, 1.6 mm MgCl2, 10 μm EDTA, 10 mm glucose, and 3 mm HEPES buffer, adjusted to pH 7.7–7.8 with NaOH). An eye was then removed by pressing a looped tungsten wire to cut the optical nerve and positioned to face a light source (Zeiss XBO 75 W). This provided a maximum light intensity of 5910 lux (attenuated using neutral density filters) for 100 ms periods with intervals of 10 s. Signals were collected with a recording electrode (GC100F-10; Harvard Apparatus) and amplified using a preamplifier (P55 A.C. Preamplifier; Astro-Med Grass Technology) with a bandpass between 0.1 and 100 Hz. They were then digitized by DAQ board NI PCI-6035E (National Instruments) via NI BNC-2090 accessories and displayed using a self-developed NI LabView program (Rinner et al., 2005). Statistical analysis was performed using SPSS Statistics 22 (IBM), and graphs were generated in Excel (Microsoft). Statistical significance was set at p < 0.05. All experiments were performed at room temperature (22°C), and prerecording steps were performed under dim red illumination to minimize visual pigment bleaching.
Generation of wild-type zebrafish lines stably expressing Tg hCLRN1-YFP or Tg hCLRN1N48K-YFP.
The protocol described previously (Balciunas et al., 2006) was used to generate somatic cells that stably express the hCLRN1-YFP or hCLRN1N48K-YFP transgene in wild-type zebrafish. First, transgene constructs containing the desired promoter and cDNA were generated. cDNA encoding hCLRN1-YFP or hCLRN1N48K-YFP (Geng et al., 2012) was cloned downstream of the parvalbumin 3b promoter (pvalb3b; West and McDermott, 2011). The pvalb3b promoter, a zebrafish hair cell-specific promoter, was amplified from the pMT/SV/PV/mCh vector (West and McDermott, 2011) and subcloned into CMV:hCLRN1-YFP and CMV:hCLRN1N48K-YFP plasmid vectors (Geng et al., 2012). The CMV promoter was replaced with the pvalb3b promoter using the In-Fusion cloning kit (Clontech). The pvalb3b:hCLRN1-YFP or pvalb3b:hCLRN1N48K-YFP plasmids, along with Tol2 RNA, were injected into wild-type zebrafish embryos at the single-cell stage. Transgene expression was analyzed in live larvae at 5 dpf using an inverted Leica SP8 confocal microscope equipped with a 40× oil lens. All larvae were anesthetized with tricaine (Sigma-Aldrich) before screening for YFP expression. Larvae expressing YFP in their hair cells were identified and grown to adulthood. YFP-positive zebrafish were mated, and those able to pass on the transgene to their offspring (i.e., with germline transmission) were maintained for further use. After a few iterations of this process, the transgenic zebrafish lines that stably expressed Tg(pvalb3b:hCLRN1-YFP) or Tg(pvalb3b:hCLRN1N48K-YFP) were identified.
Results
Conservation of the sequence and expression pattern of zebrafish clarin-1
The amino acid sequence of the zebrafish clarin-1 protein (Clrn1) was compared with human and mouse ortholog sequences using NCBI BLASTp. This revealed a high degree of conservation of the transmembrane domains (TMDs), the glycosylation site (N48), and the C-terminal PDZ class I-binding motif (X-S/T-X-L motif) across species. Overall, the amino acid sequence of Clrn1 is ∼60% identical to the amino acid sequence of mouse and human CLRN1 (Fig. 1A). Whole-mount in situ hybridization showed that clrn1 mRNA expression in the hair cells of the inner ear (Fig. 1B,C) resemble that of the hair cell marker fscn2b (Fig. 1D,E; Chou et al., 2011). These results confirmed that the protein sequence and expression pattern generated by zebrafish clrn1 are conserved relative to the mouse and human orthologs. Furthermore, Ensembl and GenBank database searches revealed no clrn1 gene duplication in the zebrafish genome. The database also revealed clrn1 paralogs from the clarin family, clrn2 and clrn3. There could be functional redundancy with clrn2 or clrn3, but this is less likely based on the fact that mice also harbor Clrn2 and Clrn3 but show a clear phenotype when Clrn1 is knocked out (Geller et al., 2009; Geng et al., 2009,2012). These results led us to the hypothesis that loss of clrn1 would disrupt hair bundle morphology and hair cell function in the zebrafish. To test this hypothesis, we targeted clrn1 to generate null mutations and characterized the phenotype of mutant zebrafish larvae.
Figure 1.
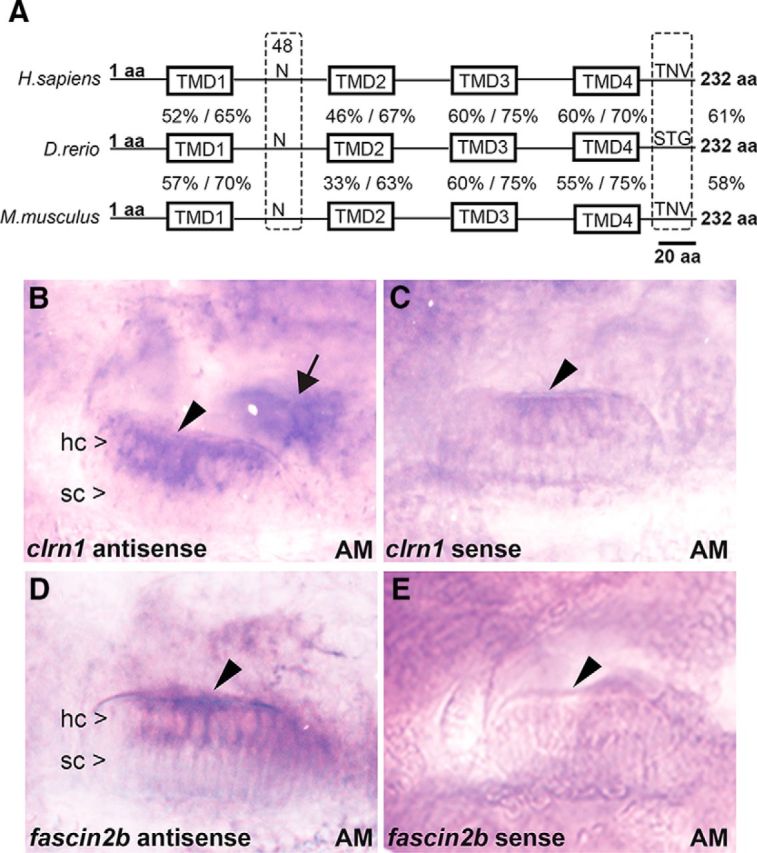
An amino acid sequence comparison and mRNA expression analysis of zebrafish clarin-1. A, The zebrafish (D. rerio) amino acid sequence is 61% identical to the human (Homo sapiens) and 58% identical to the mouse (Mus musculus) clarin-1 sequences. The TMDs (TMD1–4), asparagine “N” at position 48 (N-glycosylation site), and putative PDZ class I-binding domain (TNV, STG) at the C terminus are indicated. The percentage of identity/similarity of the amino acid sequence within each TMD in zebrafish Clrn1 is compared with the corresponding domain in human (above) and mouse (below) in the line diagram. B–E, In situ hybridization (5 dpf) shows clrn1 mRNA expression in hair cells (hc) but not in supporting cells (sc) of the anterior macula (AM; arrowhead), similar to that of the control, fscn2b. clrn1 expression is also detected in hair cells of the posterior macula (arrow). Magnification, 40×.
Successful targeting of zebrafish clrn1
To generate clrn1 knock-out zebrafish lines, targeted mutation of the sequence of exon 0 (the first coding exon of clrn1) was accomplished using ZFN (Fig. 2A). To evaluate the efficiency of ZFN in inducing clrn1 target site-specific cleavage in zebrafish in vivo, 10 embryos from each of the four injected batches (Table 1) were genotyped. The efficiency of ZFN in generating the clrn1 mutant allele ranged from 75 to 90%. DNA sequencing confirmed the targeting of clrn1 exon 0 with a deletion size ranging from 7 to 43 bp (Fig. 2B). Sequence analysis of four individual mutant zebrafish revealed disruption of the reading frame and the introduction of a stop codon in exon 0 within clrn1. The deletion size was different in each individual: the four alleles were designated CWR1, 2, 3, and 4 (Fig. 2B) and are referred to as “clrn1CWR1,” etc. In all four mutant clrn1 alleles, deletion of the “catgg” sequence at the target site resulted in loss of an MslI restriction enzyme recognition sequence. This change was used to distinguish wild-type and single or double mutant alleles of clrn1 by PCR and restriction digest (Fig. 2C). Larvae displaying the mutant phenotype (described later) always carried two mutant alleles of clrn1. The genotype/phenotype ratio of the offspring (F2) in clutches from different F1 clrn1 heterozygous crosses mirrored the Mendelian ratio and was consistent with a recessive inheritance pattern (Table 2). These data demonstrate that the phenotype is linked to a mutation in clrn1 and not to an off-target mutation.
Figure 2.
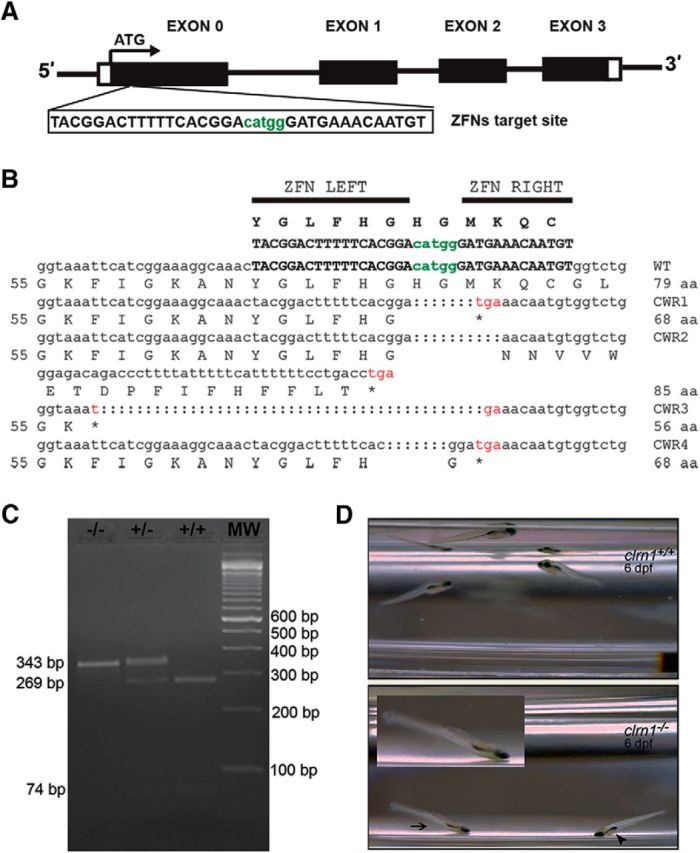
Generation of clrn1 null alleles and balance phenotype in clrn1−/− zebrafish. A, ZFN target (upper case letters) and spacer (lower case, green letters) sequences in exon 0 of clrn1. B, clrn1 mutant alleles harbor a premature stop codon in exon 0. C, For genotyping, PCR was followed by restriction digestion with MslI to distinguish wild-type, heterozygous, and homozygous mutant alleles of clrn1. D, Balance/orientation in the clrn1+/+ and clrn1−/− mutant at 6 dpf is shown. Still frames captured during swim episodes show the trajectory of clrn1+/+ larvae toward the surface or parallel to it, with a normal body orientation. clrn1−/− mutants settle to the bottom of the Petri dish in a “head down with tail tipped toward the surface” position, with their dorsal side facing the surface (arrowhead). The arrow indicates a mutant in a head-down position that is also upside down; the inset shows an enlarged image of this larva.
Table 2.
ZFN-targeted zebrafish clrn1mutants inherit the mutant alleles in a Mendelian ratio
| F1 breeding pair (parents) | F2 clutch size | Number of larvae genotypeda | Genotype |
||
|---|---|---|---|---|---|
| clrn1+/+ | clrn1+/CWR1 or clrn1+/CWR2 | clrn1CWR1/2,b | |||
| clrn1+/CWR1 × clrn1+/CWR2 | 198 | 48 | 13 (27%) | 21 (44%) | 14 (29%) |
| 94 | 50 | 13 (26%) | 26 (52%) | 11 (22%) | |
| 265 | 100 | 31 (31%) | 43 (43%) | 26 (26%) | |
clrn1 alleles segregate in a Mendelian fashion.
a Picked at random at dpf 5.
b Larvae displaying phenotype were homozygous mutants.
All clrn1 mutant experiments were conducted using 4–7 dpf larvae from the F2 or F3 generation. Zebrafish larvae carrying two mutant alleles in any combination of CWR1–4 showed the same phenotype and were indistinguishable from larvae homozygous for a specific allele. This is consistent with loss-of-function or null prediction for all four alleles (Fig. 2B). The experiments described in this report used larvae carrying two mutant copies of clrn1 in various combinations of the CWR1–4 alleles, and the larvae are represented as clrn1−/−. Larvae heterozygous for alleles CWR1–4 were indistinguishable from the wild-type larvae and are represented as clrn1+/− and clrn1+/+, respectively.
Mutation in clrn1 affects hearing and balance in zebrafish
To investigate hearing and balance behavior in the clrn1−/− mutants, startle response and swimming behavior were observed at 6 dpf. Most (∼85%) of the larvae that displayed an abnormal swimming behavior or orientation were also weak or nonresponders to the startle stimulus and carried two mutant alleles of clrn1. Of the remaining larvae displaying a subtle abnormality or “normal” behavior, ∼15% carried two mutant alleles of clrn1. However, in those clrn1−/− larvae that displayed normal behavior, progressive loss of hair cell function cannot be ruled out, although it is not possible to assess this using the startle response test. Weak and nonresponders frequently sank to the bottom of the dish in a head-down position (Fig. 2D) and slowly settled on their lateral side. The clrn1+/+ larvae displayed burst swims or slow swims, and the startle stimulus induced a burst swim response. The clrn1−/− mutants showed shorter periods of spontaneous swimming. Following the stimulus, mutants swam in short spurts away from the bottom of the dish toward the surface and then quickly tumbled back to the bottom. Mutants were unable to maintain their body axis parallel to the surface (Fig. 2D). The phenotype in clrn1−/− mutants do not correlate with lack of or defective swim bladders, suggesting the phenotype is associated with a deficit in sensory input, not buoyancy, in the mutant larvae. An example of this is shown in Figure 2D, arrowhead. These results demonstrate that clrn1 is essential for inner ear function in zebrafish, and thus, clarin-1 function in the ear is conserved across species.
We compared startle response in clrn1−/− mutants with a zebrafish model representing ear disorders in USH1, cdh23tj264a (Söllner et al., 2004). Homozygous cdh23tj264a mutants examined (n ≥ 200) showed abnormal responses to startle stimuli: ∼85% failed to show evasive swimming response, and the rest swam in circles. Other USH1 zebrafish mutants reported earlier—sputnik (cdh23), mariner (myo7a), and orbiter (pcdh15a)—show little or no response, although some of the alleles of sputnik and orbiter did show some response to acoustic/vibrational startle stimuli (Nicolson et al., 1998; Ernest et al., 2000; Seiler et al., 2005). In the ush1cfh293 zebrafish line, all homozygous mutants showed abnormal response to startle stimuli: 83% failed to respond and the rest swam in circles (Phillips et al., 2011). These phenotypes in the USH1 mutants indicate a significant loss of auditory and vestibular function. In contrast, the phenotype in clrn1−/− mutants reported here is slightly milder, indicating modest preservation of auditory and vestibular function at 6 dpf in clrn1−/− mutants.
clrn1−/− mutants show loss of hair bundle integrity
To determine whether the phenotype of the clrn1−/− mutants was from disrupted hair bundle structure, the hair bundles in the ear of clrn1+/+ (n = 10) and clrn1−/− (n = 40) larvae at 5 dpf were observed after F-actin labeling with fluorescent phalloidin. In the clrn1+/+ larvae, the hair bundle appears as a cone-shaped structure in the cristae and macula (Fig. 3A,D). The cone-shaped structure is a cohesive unit formed by a set of stereocilia of graded heights. In the clrn1−/− mutants, the cone shape was often lost and the hair bundle had a less cohesive, splayed appearance (Fig. 3B,E), similar to the hair bundle phenotype observed in the cadherin 23 mutant (cdh23tj264a; Fig. 3C,F; Söllner et al., 2004). The length of the stereocilia in the clrn1−/− mutants varied between hair bundles in the same row and were shorter compared with the clrn1+/+ stereocilia. In comparison, although the cdh23tj264a mutant displayed a loss of bundle integrity, there was no indication of reduced length in the stereocilia (compare stereocilia length in Fig. 3A–C). It is important to note that not all hair bundles in the clrn1−/− mutant macula or cristae were disrupted or degenerated in nonresponders at 5 dpf. Approximately 75% had a splayed appearance, 10% were degenerated or missing, and 15% appeared cone shaped (Fig. 3M). However, the cone-shaped bundles of the clrn1−/− mutants appeared smaller or less robust (Fig. 3B) than those in clrn1+/+ (Fig. 3A) or the cdh23tj264a larvae (Fig. 3C) at the same magnification. Based on the results from nonresponders, we expect the clrn1−/− larvae that displayed normal behavior to display fewer splayed bundles compared with nonresponders at 5 dpf. However, we did not make that correlation in this study. The hair bundle phenotype observed in the clrn1−/− mutant may be a consequence of shortening or missing stereocilia and/or reduced actin content in each stereocilium. The clrn1−/− hair bundle phenotype shows that loss of clarin-1 affects hair bundle integrity. The observed hair bundle phenotype of the clrn1−/− mutants suggested that Clrn1 is a hair bundle protein, and loss of Clrn1 results in attenuated, but not completely lost, hair bundle function. We tested this hypothesis in the experiments described below.
Figure 3.
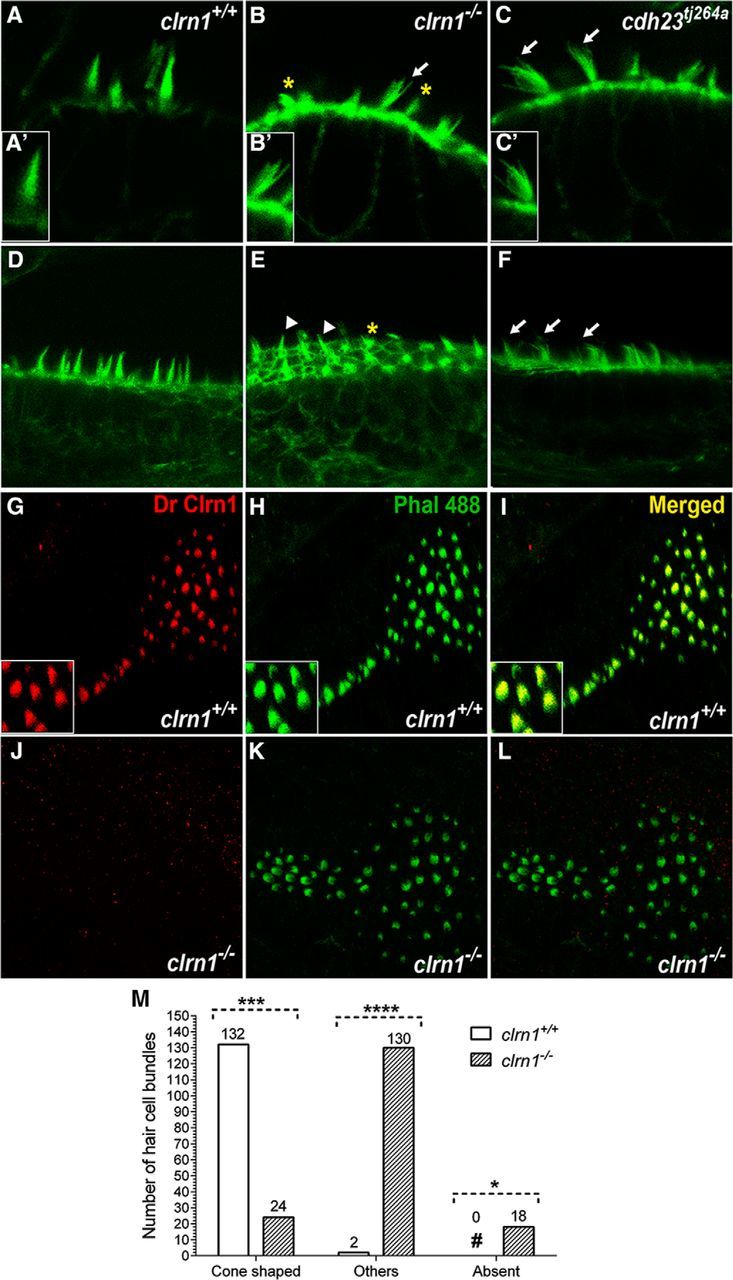
Clrn1 is an essential hair bundle protein. A–F, The inner ear hair bundle phenotype of the clrn1−/− mutant larvae at 5 dpf. F-actin-rich hair bundles were visualized using fluorescent phalloidin. The clrn1+/+ hair cells with cone-shaped bundles in the crista (A) and anterior macula (D). clrn1−/− hair cells with splayed bundles (B) similar to those of cdh23tj264a (C, arrows). Digital enlargement of a single hair bundle of A′–C′ shows more clearly the cone shape and splayed hair bundle phenotype in wild-type and mutants, respectively. Many clrn1−/− hair cells display short or fractured bundles in the crista and macula (B and E; asterisks) compared with those of cdh23tj264a bundles (C, F). Three of 19 (∼15%) clrn1−/− hair cells in this image (arrowheads, E) appear cone shaped. Magnification, 63×. Immunolabeling: Clrn1 staining of clrn1+/+ (G) or clrn1−/− (J) larvae posterior macula with a DrClrn1 antibody and F-actin with fluorescent phalloidin (H, K). Yellow bundle (I, L) in the merged image of G and H showing Clrn1 expression in the hair bundle. Insets in G–I show magnified views of a portion of the macula in each part. Magnification, 40×. M, Quantification of different hair bundle morphology types in the clrn1−/− mutants. Hair cell bundles of inner ear from the clrn1+/+ (n = 5) and clrn1−/− (n = 5) larvae at 5 dpf were classified into one of three categories based on whether they were cone shaped, splayed, or missing (degenerated). A total of 134 (132 + 2) hair cell bundles from clrn1+/+ and 172 (24 + 130 + 18) hair cell bundles from clrn1−/− larvae were evaluated. For each genotype, the number in each category is expressed as a percentage of the total number of hair cell bundles evaluated for that genotype. Approximately 2% of the wild-type hair bundles were disrupted or splayed, possibly from spontaneous defects or to the effects of mechanical force used during mounting/handling for microscopy. ****p ≤ 0.0001; ***p ≤ 0.001; *p ≤ 0.05.
Clrn1 is a hair bundle protein
To examine Clrn1 localization in the hair cell, whole-mount immunolabeling of Clrn1 in clrn1+/+ and clrn1−/− mutant larvae (5 dpf) was conducted. The anti-Clrn1 antibody (DrClrn1) recognizes the first extracellular loop of zebrafish Clrn1 (target sequence: 30–101 aa). In the clrn1+/+ larvae, Clrn1 in the hair cell was localized in the hair bundle; this was confirmed by colocalization with stereociliary F-actin (Fig. 3G–I). In contrast, no antibody signal was observed in the clrn1−/− mutant hair cells (Fig. 3J–L). Expression of Clrn1 in the hair bundle is consistent with the hair bundle defects of clrn1−/− mutants (Fig. 3A). Furthermore, the lack of Clrn1 protein in the mutant hair bundle confirmed both the presumptive null mutations in the clrn1 alleles CWR1–4 and the antibody's specificity. This is the first report to demonstrate endogenous Clrn1 localization in the hair bundle.
Disabling Clrn1 attenuates hair bundle function
To determine whether loss of Clrn1 impacts hair cell function, we recorded extracellular microphonic potentials from lateral line neuromasts in response to fluid jet mechanostimulation (Trapani and Nicolson, 2010). Microphonic potentials were recorded from the clrn1+/+ and clrn1−/− zebrafish larvae at 6 dpf (Fig. 4). The potentials represent extracellular voltages resulting from mechanotransduction current flow in hair cells of the neuromast (Trapani and Nicolson, 2010). Hair bundles of neuromasts are embedded in the gelatinous cupula. Displacement of the cupula by a fluid jet deflected the hair bundles, evoking a potential of double the frequency of the stimulus deflections exciting hair cells of opposite orientation in alternate succession (Fig. 4C). We observed a reduction in the extracellular receptor potentials in clrn1−/− mutants (Fig. 4C,D). Recordings from the clrn1+/+ larval neuromasts revealed stimulus-evoked microphonic potentials of 11.93 ± 0.98 μV (mean ± SEM; Fig. 4D); whereas neuromasts in the clrn1−/− mutants had potentials equal to 3.96 ± 0.76 μV (n = 7, p < 0.001). No significant difference was found between the number of hair cells in the neuromasts of clrn1−/− and that in clrn1+/+ larvae (Fig. 4E), indicating that the attenuated microphonic output in the mutant is from defective function of hair cells, and not from fewer hair cells. Therefore, a reduction in the microphonic potential could result from weak or degenerating hair bundles in the lateral line neuromasts, consistent with loss of integrity of the mechanosensory hair bundle in the absence of Clrn1.
Figure 4.

Microphonic potentials are reduced in the neuromasts of the clrn1−/− larvae. Oblique illumination images of the neuromasts in clrn1+/+ (A) and clrn1−/− (B) zebrafish larvae at 6 dpf. Arrows indicate single hair cell bundles. C, Stimulus (top trace) and microphonic potentials in neuromasts of clrn1+/+ (middle trace) and clrn1−/− (bottom trace) larvae. The top trace shows pressure applied to the stimulating puff pipette. D, Summary of microphonic potential peak-to-peak amplitudes at twice the stimulus frequency. Average values of microphonic potentials obtained from lateral line neuromasts of clrn1+/+ and clrn1−/− larvae. E, Number of hair cells per neuromast from clrn1+/+ and clrn1−/− larvae. Data represent the mean ± SEM. Analysis of the same larvae (n = 7) is shown in D and E. Asterisks indicate statistical significance, Student's t test, p < 0.001.
Loss of Clrn1 expression does not affect localization or the number of ribbon synapses
Previous studies suggest a possible role for the clarin-1 protein across species in the sensory synapses based on limited sequence homology to stargazin, a cerebellar synapse protein (Adato et al., 2002). Evaluation of ribbon synapses in hair cells in the neuromast of the clrn1−/− and clrn1+/+ larvae at 6 dpf, using antibodies to presynaptic and postsynaptic markers, showed no significant difference in localization or number of ribbon synapse (Fig. 5). This result shows Clrn1 is not essential for hair cell ribbon synapse development in zebrafish.
Figure 5.
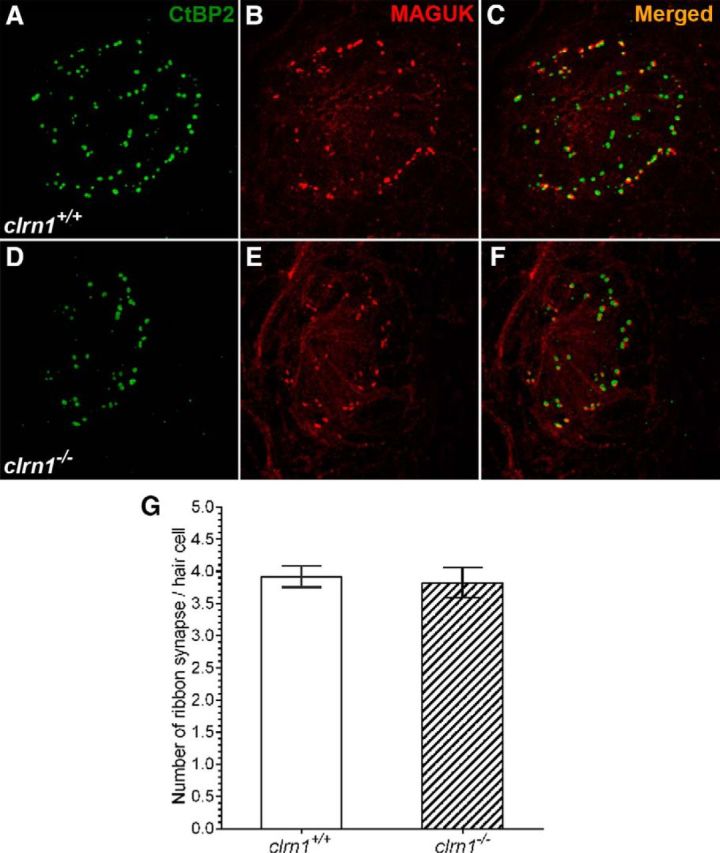
Evaluation of ribbon synapse in hair cells. A–F, Show ribbon synapse staining in the neuromast from clrn1+/+ (A–C) and clrn1−/− (B–D) zebrafish larvae at 6 dpf. A, D, Presynaptic (CtBP2). B, E, Postsynaptic (MAGUK) marker. C, F, Merged images. A–F, Maximum intensity projection of z-stack images (each section: 0.5 μm). Magnification, 63×. G, Quantification of ribbon synapse in the hair cells of clrn1+/+ and clrn1−/− zebrafish larvae at 6 dpf. The number and location of the ribbon synapse were evaluated from clrn1+/+ hair cells (n = 64) from the neuromast of three larvae and from clrn1−/− hair cells (n = 44) from the neuromast of three larvae. Data represent the mean ± SEM. The difference in ribbon synapse count between clrn1+/+ and clrn1−/− larvae is statistically not significant. Student's t test, p > 0.5.
Loss of clrn1 does not affect the visual ability of zebrafish larvae
ERG is a diagnostic test that measures the electrical responses of various cell types in the retina, including rods and cones (photoreceptor cells). Normal ERG responses were observed in 7 dpf clrn1−/− mutants. Figure 6A shows the typical ERG responses to light stimuli of increasing intensities recorded from a clrn1−/− larval eye. The negative a-wave representing photoreceptor hyperpolarization was masked by the larger positive b-wave representing depolarization of ON-bipolar cells. No significant differences were found in the averaged b-wave amplitudes at each light intensity between mutants and siblings (repeated measurement ANOVA, p > 0.1; Fig. 6B). ERG data suggest normal photoreceptor structure and function in the clrn1−/− mutant larvae at 7 dpf. However, we cannot rule out the possibility that the clrn1−/− mutants develop an eye phenotype with age. This could not be assessed because the mutants failed to survive beyond 10–15 dpf, which we attribute to morbidity associated with poor swimming and bottom-dwelling behavior (inadequate access to food is a probable cause). We have observed a similar fate in other zebrafish mutants with defective hair cells. For example, zebrafish (cdh23tj264a) carrying a mutation in the hair bundle (tip-link) protein Cdh23 (Söllner et al., 2004) typically do not survive beyond 10–15 dpf. Generation of a mutant homozygous for hypomorphic allele of clrn1 to permit survival beyond 15 dpf may allow us to test this hypothesis. Alternatively, the lack of an eye phenotype may reflect functional redundancy in the zebrafish retina that is not present in the human retina. For example, although there has been no duplication of the clrn1 gene in zebrafish, function of the clarin gene family members, clrn2 and/or clrn3, may overlap with or replace clrn1 function in the retina. Further research is needed to investigate these possibilities.
Figure 6.

Seven dpf clrn1−/− mutants show no defects in the ERG response. A, A typical ERG response recorded from a clrn1−/− mutant. The flash duration was 100 ms and the interval was 10 s. Flashes were delivered 50 ms after the start of recording. The stimulation intensity increased from 0.01 to 100% of the maximum value. Each curve represents the average of two responses. B, Mean b-wave amplitudes were plotted against the corresponding light intensities. Data represent the mean ± SEM.
Human CLRN1 localizes to the hair cell bundle in vivo
The clrn1+/+ zebrafish that stably express YFP-tagged hCLRN1 (hCLRN1-YFP) or hCLRN1N48K (hCLRN1N48K-YFP) under the control of the hair cell-specific parvalbumin 3b (pvalb3b) promoter were generated and examined. hCLRN1-YFP predominantly localized to the hair bundle (Fig. 7A–C, asterisk), with weak expression in the hair cell membrane (Fig. 7A,B, arrow) of the inner ear and neuromast. In contrast, hCLRN1N48K-YFP predominantly localized to the cell body (Fig. 7D–F). These results confirm that hCLRN1 is a hair bundle protein in vivo and that the N48K alteration leads to protein mislocalization in the cell body, thus reducing accumulation of expressed hCLRN1N48K in the hair bundle. However, while most hCLRN1N48K remained in the cell body, a small amount was translocated to the hair bundle (Fig. 7G,G′). Images from two additional hCLRN1N48K-YFP-expressing larvae are shown here to demonstrate that dual localization of hCLRN1N48K-YFP in hair cells is reproducible, with a small amount of protein observed in the bundle in all cases (Fig. 7H,H′,I,I′). We have examined >200 larvae (n > 200) from the hCLRN1N48K-YFP transgenic line, and we consistently see this dual localization in all. We propose that the small amount of hCLRN1N48K that reaches the hair bundle provides clarin-1-mediated function in the ear during the early stages of life, but is insufficient to sustain the hair bundle structure over time, leading to a progressive loss of hair bundle structure and function. This is a plausible explanation for progressive hearing loss associated with USH3 CLRN1N48K individuals. Further work is necessary to test this hypothesis.
Figure 7.
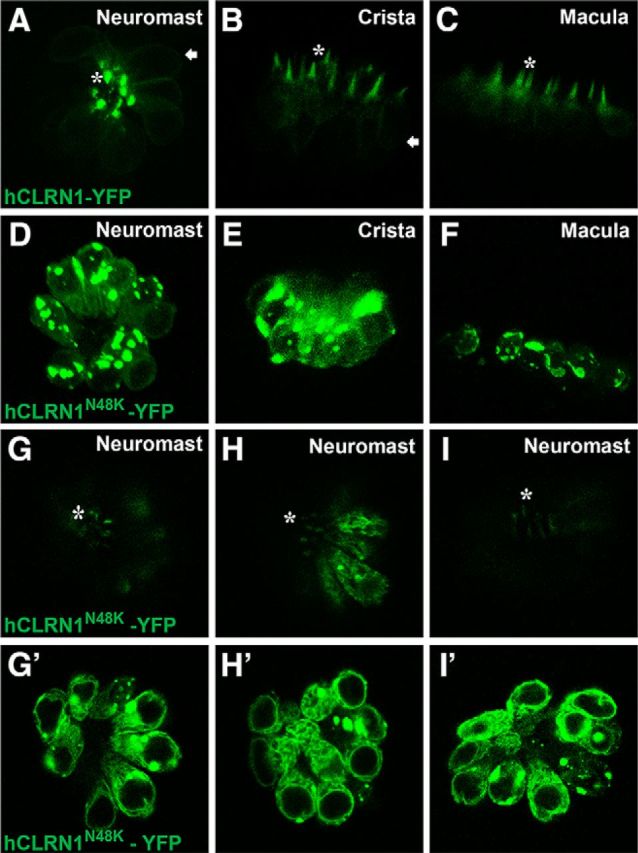
A–C, Human CLRN1 is a hair bundle protein. Live confocal images of transgenic zebrafish (5 dpf) stably expressing hCLRN1-YFP or hCLRN1N48K-YFP in hair cells show that hCLRN1-YFP localizes predominantly to the hair bundle (asterisk), with a small amount at the plasma membrane (arrows). The pathogenic mutant, hCLRN1N48K-YFP, is predominantly retained within the cell body (D–F), although a small amount of protein reaches the hair bundle (G–I, asterisks). A–F, Maximum intensity projections that retain the brightest pixel value along each projection ray. In D–F, it is hard to visualize the small amount of hCLRN1N48K-YFP localized to the hair bundle. G, G′, Images showing top and middle (optical) sections of the same neuromast; these reveal that a small amount of hCLRN1N48K protein is translocated to the hair bundle (G, asterisk), and the remaining is retained in the cell body (G′); similar confocal images from two additional hCLRN1N48K-YFP-expressing larvae are shown to demonstrate that dual localization of hCLRN1N48K-YFP in hair cells is reproducible (H, H′, I, I′). Magnification, 40×.
Clarin-1 protein from zebrafish and humans is sensitive to PFA fixation
Our initial attempts to immunolabel Clrn1 with the DrClrn1 antibody in the clrn1+/+ and clrn1−/− larvae fixed in PFA resulted in nonspecific signal artifacts. Because a PFA-based fixative is commonly used to process tissue for immunolabeling, our initial attempts to immunolocalize Clrn1 was performed in the PFA-fixed larvae. We repeated the standard fixation protocol (4% PFA at 4°C overnight) several times (n > 5) and, in parallel, executed several modified versions of the standard fixation protocol (see Materials and Methods). Results for both the standard and modified PFA fixation protocols were similar; only results from the standard fixation protocol are shown here (Fig. 8). The fluorescent signal was observed throughout the tissue, including hair cells, supporting cells, and other cells surrounding the hair cells (Fig. 8A—A″,C–C″). We believe that these signals were not linked to the Clrn1 epitope, for two reasons. First, they were observed in cell types in which clrn1 mRNA is not expressed. clrn1 mRNA is not expressed in the supporting cell of the clrn1+/+ larvae, for example (Fig. 1B). Second, an identical, nonspecific staining pattern was observed in the ear tissue of the clrn1−/− mutants (Fig. 8B,D). We hypothesize that Clrn1 is sensitive to PFA-based fixatives, resulting in loss of either Clrn1 or the Clrn1 epitope in hair cells. Further experiments demonstrated that Clrn1 can be detected in the live zebrafish larvae but not in PFA-fixed specimens.
Figure 8.
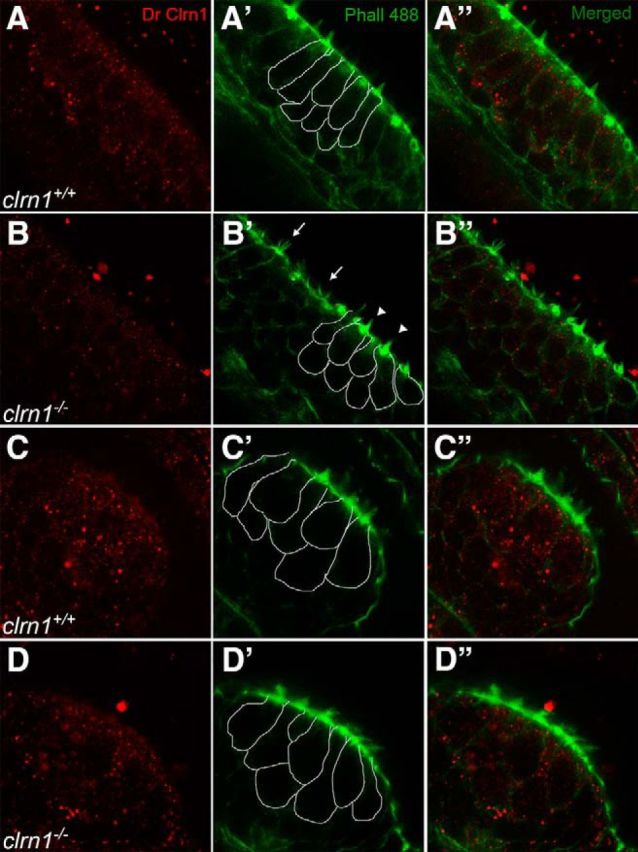
Clrn1 is sensitive to PFA. A–D, Confocal images of the DrClrn1 antibody labeling the anterior macula (A, B) and cristae (C, D) of clrn1+/+ (A, C) and clrn1−/− (B, D) zebrafish larvae at 5 dpf. Splayed (arrow) and cone-shaped (arrowhead) hair bundles within the macula (B′) confirm the genotype of larvae; crista from the same larva was imaged (D′). To distinguish hair cells from supporting cells, borders of some of the hair cells are outlined in the sections of the middle column (A′–D′). Similar scattered fluorescent dots across the tissue (hair cells, supporting cells, and beyond) are seen in both wild-type and knock-out tissue. Magnification, 63×.
The transgenic zebrafish expressing hCLRN1-YFP provided a unique opportunity to further test the hypothesis that clarin-1 protein is sensitive to PFA fixation. To test the effects of fixation on hCLRN1-YFP, protein localization was compared in live larvae and in the same larvae after fixation and immunolabeling. In parallel, a control experiment was conducted to demonstrate that the fluorescent reporter protein is not sensitive to PFA. The transgenic zebrafish larvae that stably express GFP in the hair cell body were compared with hCLRN1-YFP-expressing larvae. The amino acid sequences of GFP and YFP are 97% identical, and the anti-GFP antibody recognizes YFP. Therefore, the response of GFP to PFA serves as a proxy for the response of YFP to PFA. After live imaging, Tg(pvalb3b:hCLRN1-YFP) and Tg(pvalb3b:GFP) transgenic larvae were fixed with 1–4% PFA for four different periods (15 min, 30 min, 45 min, or 1 h) at room temperature or overnight at 4°C. After images of the fixed larvae were captured, they were processed for immunolabeling using the anti-GFP antibody. Identical results were observed for all test conditions. Images of the larvae fixed in 4% PFA for 1 h at room temperature are shown (Fig. 9). As expected, live images of the neuromasts from the stable transgenic line showed GFP expression in the cell body (Fig. 9A) and hCLRN1-YFP in the hair bundle (Fig. 9B). Images captured after PFA fixation and immunolabeling demonstrated that the GFP fluorescence was not quenched by PFA treatment, and that the GFP protein remained in the hair cell body during processing (Fig. 9A′). This result was confirmed by immunolabeling with an anti-GFP antibody (Fig. 9D). In contrast, the hCLRN1-YFP signal disappeared from the hair bundle after PFA fixation (Fig. 9B′); consistent with this observation, immunolabeling with the anti-GFP antibody failed to detect the hCLRN1-YFP protein in the hair bundle or cell body (Fig. 9G). The control data showed that loss of the fluorescent YFP signal in hair cells expressing hCLRN1-YFP was due, not to YFP sensitivity to PFA, but to hCLRN1 sensitivity to PFA. These results demonstrate that hCLRN1, similar to the endogenous zebrafish Clrn1 (Fig. 8), is sensitive to PFA fixation, and that the absence of immunolabeling is from loss of the entire protein, and not simply loss of the epitope. Thus, the clarin-1 protein is sensitive to PFA across species, and the use of PFA as a fixative can lead to erroneous conclusions about clarin-1 localization patterns in the ear.
Figure 9.
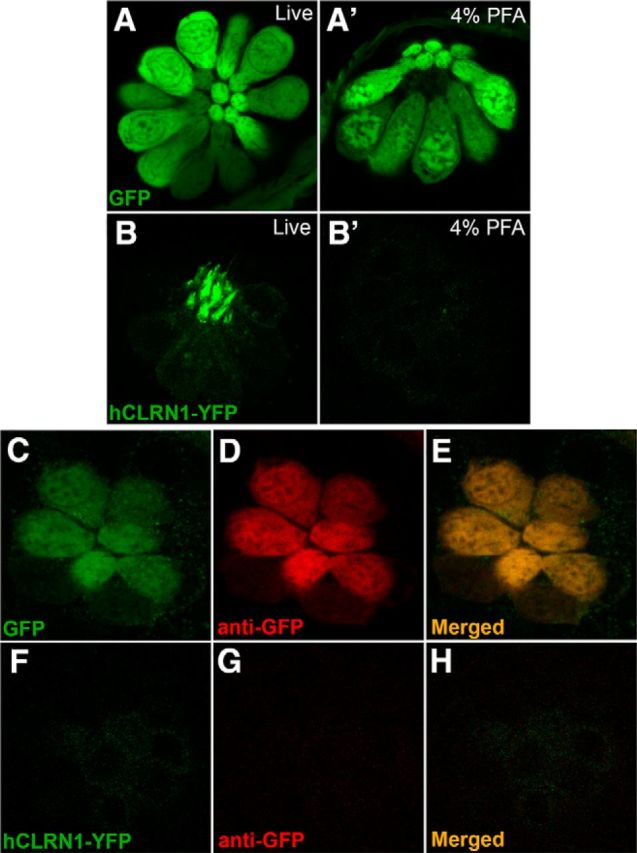
Human CLRN1 is sensitive to PFA. Confocal images of hair cells in neuromasts expressing GFP in the cell body (A) or hCLRN1-YFP in the hair bundle (B). After fixation, GFP (A′) but not hCLRN1 (B′) was detected in hair cells. In PFA-fixed larvae, anti-GFP antibodies detected GFP (C–E) but not hCLRN1-YFP (F–H) in hair cells. Magnification, 40×.
Discussion
The zebrafish clrn1−/− mutants reported here show that Clrn1 is an essential hair bundle protein. Consistent with that conclusion, we find endogenous Clrn1 localized to the hair bundle. Loss of hearing and balance associated with diminished hair bundle integrity are observed in clrn1−/− zebrafish between 4 and 6 dpf. In addition to the splayed appearance, clrn1−/− mutant hair bundles appear to be diminished compared with the wild-type hair bundles. Nevertheless, ∼15% of hair bundles in the clrn1−/− mutant ear displayed a mature (cone) shape, even though it was weakened or collapsed relative to those of the clrn1+/+ ear. This was supported by quantitative evidence of a reduction in microphonic potentials from the neuromasts of clrn1−/− mutants. The hair bundle phenotype of the clrn1−/− mutants suggest that Clrn1 is not essential for the early stages of hair cell development but is required to maintain the integrity of the hair bundles. This is consistent with data from Clrn1−/− mice and observation of individuals harboring mutations in CLRN1.
USH3 individuals are born hearing but fail to maintain that function (Joensuu et al., 2001; Ness et al., 2003; Plantinga et al., 2005), suggesting that individuals develop functional hair cells but fail to maintain them. Unfortunately, no inner ear histopathology data are available for USH3 to verify this idea. Clrn1 knock-out (Clrn1−/−) and Clrn1 knock-in (Clrn1N48K) mice display reduced hair cell transduction currents by P6, reduced cochlear microphonic potentials by P18, and profound hearing loss by P25 (Geller et al., 2009; Geng et al., 2009, 2012). Cochlear hair bundles from Clrn1−/− mice at P2 display planar polarization, staircase arrangement of stereocilia, and tip links; time point analysis from P2 to P15 revealed deterioration of hair bundle morphology over time, loss of some stereocilia, and gaps in the v-shaped bundle (Geng et al., 2012). These data suggest that loss of clairn-1 function does not affect early stages of hair cell development and hair cells in Clrn1−/− mice acquire form and function but fail to maintain hair bundle integrity. Interestingly, Clrn1−/− mice (Geller et al., 2009; Geng et al., 2009) and clrn1−/− zebrafish fail to display an eye phenotype and progressive ear disorders characteristic of USH3. The reason for lack of a progressive phenotype in animal models is unknown. However, these limitations of the clrn1−/− zebrafish do not affect the goals of this study to investigate the impact of clrn1-null mutation on hair cells and subcellular localization of Clrn1, as well as hCLRN1 and hCLRN1N48K, in hair cells.
The hair bundle phenotype in clrn1−/− mutant suggests that maintenance of the actin-rich core of the stereocilia may be dependent on clarin-1. In support of this idea, it was previously shown that clarin-1 forms membranous microdomain, and possible function of clarin-1-enriched microdomains is the local activation of actin polymerization (Tian et al., 2009). Further, transfection of hCLRN1, but not of hCLRN1N48K, leads to reorganization of actin filament structures and the induction of cellular protrusions in HEK293 cells (Tian et al., 2009). Subsequently, it was demonstrated that hCLRN1, but not hCLRN1N48K, localized to the cell membrane in HEK293 cells (Geng et al., 2012). We propose that maintenance of the actin-rich core of the stereocilia is dependent on membrane localized clarin-1.
Ogun and Zallocchi (2014) used morpholinos to knock down clrn1 mRNA in zebrafish embryos and studied the phenotype of the morphants at 3 dpf to deduce the role of Clrn1 in hair cells. Reduced FM1–43 uptake, shortening of the kinocilia, mislocalization of ribeye b clusters, and an absence of Pcdh15a in the hair bundle were reported in the morphants. It was also reported that Pcdh15a interacted with Clrn1, and Pcdh15a localization to the hair bundle was dependent on Clrn1. Perplexingly, hair bundle morphology was normal, but mechanotransduction was attenuated in the morphants (as reflected by reduced FM1–43 uptake). The morphant data are not consistent with the clrn1 zebrafish mutant phenotype reported here or with those reported in the literature for Clrn1 mutant mice or pcdh15a mutant zebrafish (Geller et al., 2009; Geng et al., 2009, 2012; Seiler et al., 2005). One possibility for the discrepancy between the morphant phenotype and the clrn1−/− ZFN mutant phenotype is off-target effects with morpholinos (Kok et al., 2015). The other explanations for this discrepancy are differences in the time points analyzed or variances in the maternal contribution of wild-type Clrn1. Testing those morpholinos in the clrn1−/− true null background could potentially resolve the issue of off-target effects.
Based on the auditory phenotype in USH3 described earlier, we posit that maintenance of the stereocilia structure shadows its formation and continues through the life of the organism. Therefore, a null mutation in a gene involved in the maintenance of stereocilia is likely to affect bundle integrity and function starting at an earlier time point relative to an allele carrying a missense mutation. If CLRN1 was necessary for hair cell development, severe/congenital hearing loss (like USH1) would be expected in individuals carrying stop mutation CLRN1Y176X. However, audiometric analysis shows a wide spectrum of nonlinear progressive hearing impairment among USH3 individuals (Plantinga et al., 2005). Nevertheless, CLRN1Y176X individuals typically develop more severe hearing loss earlier in life compared with CLRN1N48K subjects (Ness et al., 2003; Plantinga et al., 2005). Consistent with USH3 auditory phenotype, Clrn1N48K mice show better preservation of hair bundle structure at P2 and hearing at P18 compared with age-matched Clrn1−/− mice (Geng et al., 2012). Poor startle response and abnormal swimming behavior in the clrn1−/− mutant zebrafish, along with disrupted hair bundle morphology at a very early stage (5–6 dpf) in the clrn1−/− mutant larvae, are consistent with the Clrn1−/− mouse phenotype.
We propose two models to explain the CLRN1N48K phenotype. In model 1 diminished intrinsic function, not availability, of CLRN1N48K in the hair bundle is the cause of the phenotype. In this model, CLRN1N48K expression would be comparable to levels of CLRN1 in the hair bundle, but clarin-1-mediated function is attenuated, not sufficient to maintain hair bundle integrity over time. However, available data do not support model 1. In contrast to CLRN1, most of the hCLRN1N48K expressed in hair cells in vitro (in mouse cochlear explants; Geng et al., 2012) and in vivo remain intracellular, with only a small amount observed in the bundle (Fig. 7). In model 2 the availability, not the intrinsic function, of CLRN1N48K in the hair bundle diminishes over time because of intracellular degradation of the mutant protein. Recent literature combined with data from the transgenic zebrafish reported here provides support for the second model and illuminate the possible mechanism underlying a pathogenic mutation hCLRN1N48K. In zebrafish that express hCLRN1N48K in hair cells, some of this protein reaches the bundle but most is retained intracellularly. Whether the amount of hCLRN1N48K in the bundle diminishes over time from intracellular degradation remains to be verified. Previous work showed that hCLRN1N48K expressed in HEK293 cells accumulated in the ER in contrast to membrane localization of hCLRN1 (Geng et al., 2012). Accumulation of misfolded protein in the ER triggers ER-associated degradation (ERAD) of the mutant protein as well as ER stress signals (Wiseman et al., 2010). It was recently reported that mutation in other Usher proteins (Cdh23, Harmonin, and Myo7aa) trigger ER stress in zebrafish hair cells (Blanco-Sanchez et al., 2014). ER stress signals activate Golgi-independent trafficking of membrane proteins (Schröder and Kaufman, 2005; Claessen et al., 2012). We hypothesize that a small proportion of ER-retained hCLRN1N48K in developing hair cells “escapes” ERAD and reaches the plasma membrane via Golgi-independent or unconventional trafficking pathway. However, localization of hCLRN1N48K in the stereocilia may diminish over time because of intracellular degradation, resulting in progressive loss of clarin-1-mediated function and hair bundle integrity. There are examples of unconventional trafficking of the mutant protein involved in monogenic disorders. For example, the Golgi-independent trafficking pathway has been implicated in the weak surface expression of a ΔF508–CFTR on airway epithelial cells and restoration of chloride channel function to target cells in vitro and in vivo; however, ΔF508–CFTR individuals fail to retain CFTR-mediated function over time (Luo et al., 2009; Gee et al., 2011). In the second model, hair cells expressing hCLRN1N48K would retain hair bundle structure and function for a longer duration than hair cells harboring a stop mutation CLRN1Y176X. Consistent with that idea, CLRN1Y176X individuals typically develop more severe hearing loss earlier in life compared with CLRN1N48K subjects (Ness et al., 2003; Plantinga et al., 2005). Our findings reported here, along with zebrafish models we developed, provide a path forward to test these speculation/ideas.
In conclusion, we report the first bona fide null mutant zebrafish model for hair cell disorder in USH3, establish Clrn1 as an essential hair bundle protein, and shed light on possible molecular mechanisms linked to hCLRN1N48K in hair cells in vivo.
Footnotes
We acknowledge the use of the microscopy module of the Visual Sciences Research Core supported by P30-EY11373 from the National Eye Institute and the Leica SP8 confocal in the Genetics Department Imaging Facility at Case Western Reserve University, made available through the Office of Research Infrastructure Programs (ORIP) [National Institutes of Health (NIH)—ORIP] Shared Instrumentation Grant S10 OD016164. This work was supported in part by funds from the Anthony J. Maniglia Endowed Chair, University Hospitals Case Medical Center and NIH (R01-DC010816), to K.N.A., and by NIH Grant R01-DC009437 to B.M.M. We thank Carol Fernando in B.M.M.'s laboratory and Kara Dannenhauer in S.C.F.N.'s laboratory for technical support.
The authors declare no competing financial interests.
References
- Adato A, Vreugde S, Joensuu T, Avidan N, Hamalainen R, Belenkiy O, Olender T, Bonne-Tamir B, Ben-Asher E, Espinos C, Millán JM, Lehesjoki AE, Flannery JG, Avraham KB, Pietrokovski S, Sankila EM, Beckmann JS, Lancet D. USH3A transcripts encode clarin-1, a four-transmembrane-domain protein with a possible role in sensory synapses. Eur J Hum Genet. 2002;10:339–350. doi: 10.1038/sj.ejhg.5200831. [DOI] [PubMed] [Google Scholar]
- Adato A, Lefèvre G, Delprat B, Michel V, Michalski N, Chardenoux S, Weil D, El-Amraoui A, Petit C. Usherin, the defective protein in Usher syndrome type IIA, is likely to be a component of interstereocilia ankle links in the inner ear sensory cells. Hum Mol Genet. 2005;14:3921–3932. doi: 10.1093/hmg/ddi416. [DOI] [PubMed] [Google Scholar]
- Balciunas D, Wangensteen KJ, Wilber A, Bell J, Geurts A, Sivasubbu S, Wang X, Hackett PB, Largaespada DA, McIvor RS, Ekker SC. Harnessing a high cargo-capacity transposon for genetic applications in vertebrates. PLoS Genet. 2006;2:e169. doi: 10.1371/journal.pgen.0020169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanco-Sanchez B, Clément A, Fierro J, Jr, Washbourne P, Westerfield M. Complexes of Usher proteins preassemble at the endoplasmic reticulum and are required for trafficking and ER homeostasis. Dis Model Mech. 2014;7:547–559. doi: 10.1242/dmm.014068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boëda B, El-Amraoui A, Bahloul A, Goodyear R, Daviet L, Blanchard S, Perfettini I, Fath KR, Shorte S, Reiners J, Houdusse A, Legrain P, Wolfrum U, Richardson G, Petit C. Myosin VIIa, harmonin and cadherin 23, three Usher I gene products that cooperate to shape the sensory hair cell bundle. EMBO J. 2002;21:6689–6699. doi: 10.1093/emboj/cdf689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou SW, Hwang P, Gomez G, Fernando CA, West MC, Pollock LM, Lin-Jones J, Burnside B, McDermott BM., Jr Fascin 2b is a component of stereocilia that lengthens actin-based protrusions. PLoS One. 2011;6:e14807. doi: 10.1371/journal.pone.0014807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Claessen JH, Kundrat L, Ploegh HL. Protein quality control in the ER: balancing the ubiquitin checkbook. Trends Cell Biol. 2012;22:22–32. doi: 10.1016/j.tcb.2011.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doyon Y, McCammon JM, Miller JC, Faraji F, Ngo C, Katibah GE, Amora R, Hocking TD, Zhang L, Rebar EJ, Gregory PD, Urnov FD, Amacher SL. Heritable targeted gene disruption in zebrafish using designed zinc-finger nucleases. Nat Biotechnol. 2008;26:702–708. doi: 10.1038/nbt1409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ernest S, Rauch GJ, Haffter P, Geisler R, Petit C, Nicolson T. Mariner is defective in myosin VIIA: a zebrafish model for human hereditary deafness. Hum Mol Genet. 2000;9:2189–2196. doi: 10.1093/hmg/9.14.2189. [DOI] [PubMed] [Google Scholar]
- Foley JE, Maeder ML, Pearlberg J, Joung JK, Peterson RT, Yeh JR. Targeted mutagenesis in zebrafish using customized zinc-finger nucleases. Nat Protoc. 2009;4:1855–1867. doi: 10.1038/nprot.2009.209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gee HY, Noh SH, Tang BL, Kim KH, Lee MG. Rescue of delta F508-CFTR trafficking via a GRASP-Dependent Unconventional Secretion Pathway. Cell. 2011;146:746–760. doi: 10.1016/j.cell.2011.07.021. [DOI] [PubMed] [Google Scholar]
- Geller SF, Guerin KI, Visel M, Pham A, Lee ES, Dror AA, Avraham KB, Hayashi T, Ray CA, Reh TA, Bermingham-McDonogh O, Triffo WJ, Bao S, Isosomppi J, Västinsalo H, Sankila EM, Flannery JG. CLRN1 is nonessential in the mouse retina but is required for cochlear hair cell development. PLoS Genet. 2009;5:e1000607. doi: 10.1371/journal.pgen.1000607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geng R, Geller SF, Hayashi T, Ray CA, Reh TA, Bermingham-McDonogh O, Jones SM, Wright CG, Melki S, Imanishi Y, Palczewski K, Alagramam KN, Flannery JG. Usher syndrome IIIA gene clarin-1 is essential for hair cell function and associated neural activation. Hum Mol Genet. 2009;18:2748–2760. doi: 10.1093/hmg/ddp210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geng R, Melki S, Chen DH, Tian G, Furness DN, Oshima-Takago T, Neef J, Moser T, Askew C, Horwitz G, Holt JR, Imanishi Y, Alagramam KN. The mechanosensory structure of the hair cell requires clarin-1, a protein encoded by Usher syndrome III causative gene. J Neurosci. 2012;32:9485–9498. doi: 10.1523/JNEUROSCI.0311-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grati M, Kachar B. Myosin VIIa and sans localization at stereocilia upper tip-link density implicates these Usher syndrome proteins in mechanotransduction. Proc Natl Acad Sci U S A. 2011;108:11476–11481. doi: 10.1073/pnas.1104161108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joensuu T, Hämäläinen R, Yuan B, Johnson C, Tegelberg S, Gasparini P, Zelante L, Pirvola U, Pakarinen L, Lehesjoki AE, de la Chapelle A, Sankila EM. Mutations in a novel gene with transmembrane domains underlie Usher syndrome type 3. Am J Hum Genet. 2001;69:673–684. doi: 10.1086/323610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kok FO, Shin M, Ni CW, Gupta A, Grosse AS, van Impel A, Kirchmaier BC, Peterson-Maduro J, Kourkoulis G, Male I, DeSantis DF, Sheppard-Tindell S, Ebarasi L, Betsholtz C, Schulte-Merker S, Wolfe SA, Lawson ND. Reverse genetic screening reveals poor correlation between morpholino-induced and mutant phenotypes in zebrafish. Dev Cell. 2015;32:97–108. doi: 10.1016/j.devcel.2014.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lefèvre G, Michel V, Weil D, Lepelletier L, Bizard E, Wolfrum U, Hardelin JP, Petit C. A core cochlear phenotype in USH1 mouse mutants implicates fibrous links of the hair bundle in its cohesion, orientation and differential growth. Development. 2008;135:1427–1437. doi: 10.1242/dev.012922. [DOI] [PubMed] [Google Scholar]
- Liu X, Bulgakov OV, Darrow KN, Pawlyk B, Adamian M, Liberman MC, Li T. Usherin is required for maintenance of retinal photoreceptors and normal development of cochlear hair cells. Proc Natl Acad Sci U S A. 2007;104:4413–4418. doi: 10.1073/pnas.0610950104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- López-Schier H, Starr CJ, Kappler JA, Kollmar R, Hudspeth AJ. Directional cell migration establishes the axes of planar polarity in the posterior lateral-line organ of the zebrafish. Dev Cell. 2004;7:401–412. doi: 10.1016/j.devcel.2004.07.018. [DOI] [PubMed] [Google Scholar]
- Luo Y, McDonald K, Hanrahan JW. Trafficking of immature DeltaF508-CFTR to the plasma membrane and its detection by biotinylation. Biochem J. 2009;419:211–219. doi: 10.1042/BJ20081869. 212 p following 219. [DOI] [PubMed] [Google Scholar]
- Mburu P, Kikkawa Y, Townsend S, Romero R, Yonekawa H, Brown SD. Whirlin complexes with p55 at the stereocilia tip during hair cell development. Proc Natl Acad Sci U S A. 2006;103:10973–10978. doi: 10.1073/pnas.0600923103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDermott BM, Jr, Baucom JM, Hudspeth AJ. Analysis and functional evaluation of the hair-cell transcriptome. Proc Natl Acad Sci U S A. 2007;104:11820–11825. doi: 10.1073/pnas.0704476104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDermott BM, Jr, Asai Y, Baucom JM, Jani SD, Castellanos Y, Gomez G, McClintock JM, Starr CJ, Hudspeth AJ. Transgenic labeling of hair cells in the zebrafish acousticolateralis system. Gene Expr Patterns. 2010;10:113–118. doi: 10.1016/j.gep.2010.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGee J, Goodyear RJ, McMillan DR, Stauffer EA, Holt JR, Locke KG, Birch DG, Legan PK, White PC, Walsh EJ, Richardson GP. The very large G-protein-coupled receptor VLGR1: a component of the ankle link complex required for the normal development of auditory hair bundles. J Neurosci. 2006;26:6543–6553. doi: 10.1523/JNEUROSCI.0693-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mo W, Nicolson T. Both pre- and postsynaptic activity of Nsf prevents degeneration of hair-cell synapses. PLoS One. 2011;6:e27146. doi: 10.1371/journal.pone.0027146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ness SL, Ben-Yosef T, Bar-Lev A, Madeo AC, Brewer CC, Avraham KB, Kornreich R, Desnick RJ, Willner JP, Friedman TB, Griffith AJ. Genetic homogeneity and phenotypic variability among Ashkenazi Jews with Usher syndrome type III. J Med Genet. 2003;40:767–772. doi: 10.1136/jmg.40.10.767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicolson T. The genetics of hearing and balance in zebrafish. Annu Rev Genet. 2005;39:9–22. doi: 10.1146/annurev.genet.39.073003.105049. [DOI] [PubMed] [Google Scholar]
- Nicolson T, Rüsch A, Friedrich RW, Granato M, Ruppersberg JP, Nüsslein-Volhard C. Genetic analysis of vertebrate sensory hair cell mechanosensation: the zebrafish circler mutants. Neuron. 1998;20:271–283. doi: 10.1016/S0896-6273(00)80455-9. [DOI] [PubMed] [Google Scholar]
- Nüsslein-Volhard C, Dahm R. Zebrafish: a practical approach. New York: Oxford UP; 2002. p. 303. [Google Scholar]
- Ogun O, Zallocchi M. Clarin-1 acts as a modulator of mechanotransduction activity and presynaptic ribbon assembly. J Cell Biol. 2014;207:375–391. doi: 10.1083/jcb.201404016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otterstedde CR, Spandau U, Blankenagel A, Kimberling WJ, Reisser C. A new clinical classification for Usher's syndrome based on a new subtype of Usher's syndrome type I. Laryngoscope. 2001;111:84–86. doi: 10.1097/00005537-200101000-00014. [DOI] [PubMed] [Google Scholar]
- Pakarinen L, Karjalainen S, Simola KO, Laippala P, Kaitalo H. Usher's syndrome type 3 in Finland. Laryngoscope. 1995;105:613–617. doi: 10.1288/00005537-199506000-00010. [DOI] [PubMed] [Google Scholar]
- Petit C. Usher syndrome: from genetics to pathogenesis. Annu Rev Genomics Hum Genet. 2001;2:271–297. doi: 10.1146/annurev.genom.2.1.271. [DOI] [PubMed] [Google Scholar]
- Phillips JB, Blanco-Sanchez B, Lentz JJ, Tallafuss A, Khanobdee K, Sampath S, Jacobs ZG, Han PF, Mishra M, Titus TA, Williams DS, Keats BJ, Washbourne P, Westerfield M. Harmonin (Ush1c) is required in zebrafish Muller glial cells for photoreceptor synaptic development and function. Dis Model Mech. 2011;4:786–800. doi: 10.1242/dmm.006429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips JB, Västinsalo H, Wegner J, Clément A, Sankila EM, Westerfield M. The cone-dominant retina and the inner ear of zebrafish express the ortholog of CLRN1, the causative gene of human Usher syndrome type 3A. Gene Expr Patterns. 2013;13:473–481. doi: 10.1016/j.gep.2013.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plantinga RF, Kleemola L, Huygen PL, Joensuu T, Sankila EM, Pennings RJ, Cremers CW. Serial audiometry and speech recognition findings in Finnish Usher syndrome type III patients. Audiol Neurootol. 2005;10:79–89. doi: 10.1159/000083363. [DOI] [PubMed] [Google Scholar]
- Reisser CF, Kimberling WJ, Otterstedde CR. Hearing loss in Usher syndrome type II is nonprogressive. Ann Otol Rhinol Laryngol. 2002;111:1108–1111. doi: 10.1177/000348940211101208. [DOI] [PubMed] [Google Scholar]
- Riazuddin S, Belyantseva IA, Giese AP, Lee K, Indzhykulian AA, Nandamuri SP, Yousaf R, Sinha GP, Lee S, Terrell D, Hegde RS, Ali RA, Anwar S, Andrade-Elizondo PB, Sirmaci A, Parise LV, Basit S, Wali A, Ayub M, Ansar M, et al. Alterations of the CIB2 calcium- and integrin-binding protein cause Usher syndrome type 1J and nonsyndromic deafness DFNB48. Nat Genet. 2012;44:1265–1271. doi: 10.1038/ng.2426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ricci AJ, Fettiplace R. The effects of calcium buffering and cyclic AMP on mechano-electrical transduction in turtle auditory hair cells. J Physiol. 1997;501:111–124. doi: 10.1111/j.1469-7793.1997.111bo.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rinner O, Makhankov YV, Biehlmaier O, Neuhauss SC. Knockdown of cone-specific kinase GRK7 in larval zebrafish leads to impaired cone response recovery and delayed dark adaptation. Neuron. 2005;47:231–242. doi: 10.1016/j.neuron.2005.06.010. [DOI] [PubMed] [Google Scholar]
- Schröder M, Kaufman RJ. The mammalian unfolded protein response. Annu Rev Biochem. 2005;74:739–789. doi: 10.1146/annurev.biochem.73.011303.074134. [DOI] [PubMed] [Google Scholar]
- Seiler C, Finger-Baier KC, Rinner O, Makhankov YV, Schwarz H, Neuhauss SC, Nicolson T. Duplicated genes with split functions: independent roles of protocadherin15 orthologues in zebrafish hearing and vision. Development. 2005;132:615–623. doi: 10.1242/dev.01591. [DOI] [PubMed] [Google Scholar]
- Sirisi S, Folgueira M, López-Hernández T, Minieri L, Pérez-Rius C, Gaitán-Peñas H, Zang J, Martínez A, Capdevila-Nortes X, De La Villa P, Roy U, Alia A, Neuhauss S, Ferroni S, Nunes V, Estévez R, Barrallo-Gimeno A. Megalencephalic leukoencephalopathy with subcortical cysts protein 1 regulates glial surface localization of GLIALCAM from fish to humans. Hum Mol Genet. 2014;23:5069–5086. doi: 10.1093/hmg/ddu231. [DOI] [PubMed] [Google Scholar]
- Söllner C, Rauch GJ, Siemens J, Geisler R, Schuster SC, Müller U, Nicolson T. Mutations in cadherin 23 affect tip links in zebrafish sensory hair cells. Nature. 2004;428:955–959. doi: 10.1038/nature02484. [DOI] [PubMed] [Google Scholar]
- Tian G, Zhou Y, Hajkova D, Miyagi M, Dinculescu A, Hauswirth WW, Palczewski K, Geng R, Alagramam KN, Isosomppi J, Sankila EM, Flannery JG, Imanishi Y. Clarin-1, encoded by the Usher Syndrome III causative gene, forms a membranous microdomain: possible role of clarin-1 in organizing the actin cytoskeleton. J Biol Chem. 2009;284:18980–18993. doi: 10.1074/jbc.M109.003160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapani JG, Nicolson T. Physiological recordings from zebrafish lateral-line hair cells and afferent neurons. Methods Cell Biol. 2010;100:219–231. doi: 10.1016/B978-0-12-384892-5.00008-6. [DOI] [PubMed] [Google Scholar]
- West MC, McDermott BM., Jr Ribeye a-mCherry fusion protein: a novel tool for labeling synaptic ribbons of the hair cell. J Neurosci Methods. 2011;197:274–278. doi: 10.1016/j.jneumeth.2010.11.011. [DOI] [PubMed] [Google Scholar]
- Whitfield TT. Zebrafish as a model for hearing and deafness. J Neurobiol. 2002;53:157–171. doi: 10.1002/neu.10123. [DOI] [PubMed] [Google Scholar]
- Whitfield TT, Mburu P, Hardisty-Hughes RE, Brown SDM. Models of congenital deafness: mouse and zebrafish. Drug Discov Today. 2005;2:85–91. [Google Scholar]
- Wiseman RL, Haynes CM, Ron D. SnapShot: the unfolded protein response. Cell. 2010;140:590–590.e2. doi: 10.1016/j.cell.2010.02.006. [DOI] [PubMed] [Google Scholar]
- Xu Q. Microinjection into zebrafish embryos. Methods Mol Biol. 1999;127:125–132. doi: 10.1385/1-59259-678-9:125. [DOI] [PubMed] [Google Scholar]