

Abstract
Lysophosphatidic acid (LPA) is an extracellular lipid mediator involved in many physiological functions that signals through six known G-protein-coupled receptors (LPA1–LPA6). A wide range of LPA effects have been identified in the CNS, including neural progenitor cell physiology, astrocyte and microglia activation, neuronal cell death, axonal retraction, and development of neuropathic pain. However, little is known about the involvement of LPA in CNS pathologies. Herein, we demonstrate for the first time that LPA signaling via LPA1 contributes to secondary damage after spinal cord injury. LPA levels increase in the contused spinal cord parenchyma during the first 14 d. To model this potential contribution of LPA in the spinal cord, we injected LPA into the normal spinal cord, revealing that LPA induces microglia/macrophage activation and demyelination. Use of a selective LPA1 antagonist or mice lacking LPA1 linked receptor-mediated signaling to demyelination, which was in part mediated by microglia. Finally, we demonstrate that selective blockade of LPA1 after spinal cord injury results in reduced demyelination and improvement in locomotor recovery. Overall, these results support LPA–LPA1 signaling as a novel pathway that contributes to secondary damage after spinal cord contusion in mice and suggest that LPA1 antagonism might be useful for the treatment of acute spinal cord injury.
SIGNIFICANCE STATEMENT This study reveals that LPA signaling via LPA receptor type 1 activation causes demyelination and functional deficits after spinal cord injury.
Keywords: demyelination, lysophosphatidic acid, microglia, neuroprotection, oligodendrocytes, spinal cord injury
Introduction
Spinal cord injury (SCI) causes functional deficits produced by the loss of axons and neurons and contributes to the death of oligodendrocytes and the limited ability of the CNS to regenerate axons (Rowland et al., 2008; David et al., 2012a). The pathophysiology of SCI involves two stages of tissue degeneration known as primary and secondary injury. Primary injury results from the direct mechanical trauma to the spinal cord, which is followed by secondary injury, consisting of a wave of tissue degeneration that occurs over a period of several weeks, and is associated with inflammation and other mechanisms triggered by injury (David et al., 2012a,b; Popovich, 2014). Secondary injury is thought to contribute importantly to the functional deficits seen after SCI and may be preventable. Repairing the damaged spinal cord is still a clinical challenge. Neuroprotective strategies aimed at preventing damage arising from secondary injury processes provide some hope for tissue sparing and improved functional outcome. However, there is currently no effective clinical treatment for SCI. Hence, there is a need to identify factors that are involved in triggering secondary damage in SCI and to develop drug interventions to limit damage and improve functional deficits.
Lysophosphatidic acid (LPA; 1-acyl-sn-glycerol 3-phosphate) is a bioactive lipid that has a wide variety of biological activities, including cellular proliferation, prevention of apoptosis, cell migration, cytokine and chemokine secretion, platelet aggregation, smooth muscle contraction, and neurite retraction (Bot et al., 2010; Choi et al., 2010; Choi and Chun, 2013; Yung et al., 2014). LPA exerts this wide variety of effects by binding to specific G-protein-coupled receptors, such as LPA receptors (LPARs) LPA1–LPA3, which belong to the endothelial differentiation gene family, and the genetically more distant LPA4–LPA6, which belong to the P2Y purinergic receptor family (Choi et al., 2010; Choi and Chun, 2013; Kihara et al., 2014). These receptors differ in their ability to alter downstream signaling pathways, including intracellular Rho levels, mobilize Ca2+, contribute to phosphoinositol turnover, and to activate intracellular kinases, such as MAPK and ERK (Choi et al., 2010; Choi and Chun, 2013).
In vivo studies demonstrate that LPA is involved in the etiology of fetal hydrocephalus (Yung et al., 2011), fetal hypoxic brain damage (Herr et al., 2011), and the development of neuropathic pain after sciatic nerve injury and cerebral ischemia (Inoue et al., 2004; Frisca et al., 2012; Lin et al., 2012). Because of the wide variety of LPAR subtypes, it is likely that LPA may exert helpful or harmful effects in the CNS depending on the receptors it signals through. Thus, there is a need to know which LPARs contribute to neurodegeneration and/or neuroprotection. Here we show that LPA levels increase in the spinal cord parenchyma after contusion injury. We provide evidence suggesting that LPA leads to demyelination via activation of microglia LPA1. Moreover, we demonstrate that selective blockade of LPA1 after SCI reduces functional deficits and demyelination, altogether revealing important contributions of LPA–LPA1 signaling in secondary damage after SCI.
Materials and Methods
Surgical procedure
All surgical procedures were approved by the Autonomous University of Barcelona Animal Care Committee and followed the guidelines of the European Commission on Animal Care. Adult (8–10 weeks old) female C57BL/6 mice (Charles River), LPA1-deficient mice (maLPA1-null; Estivill-Torrús et al., 2008), or wild-type littermates (C57BL/6J × 129X1/SvJ background) were anesthetized with ketamine (90 mg/kg, i.m.) and xylazine (10 mg/kg, i.m.). After performing a laminectomy at the 11th thoracic vertebrae, the exposed spinal cord was either injected intraspinally or contused.
Intraspinal injections were performed using a glass needle (30 μm internal diameter; Eppendorf) coupled to a 10 ml Hamilton syringe (catalog #701; Hamilton). One microliter of saline, saline containing 5 nmol of LPA (18:1 LPA; Sigma-Aldrich), or 5 nmol of LPA plus the selective LPA1 antagonist AM095 sodium, {4'-[3-methyl-4-((R)-1-phenyl-ethoxycarbonylamino)-isoxazol-5-yl]-biphenyl-4-yl}-acetate 0.22 nmol; kindly provided by Bristol-Myers Squibb; Swaney et al., 2011) was injected into the dorsal column of the uninjured spinal cord. Injections were made at a perfusion speed of 2 μl/min controlled by an automatic injector (KDS 310 Plus; KD Scientific), and the tip of the needle was maintained inside the cord tissue 3 min after each injection to avoid liquid reflux.
SCIs were performed using the Infinite Horizon Impactor device (Precision Scientific), using a force of 50 kdyn and tissue displacement ranging from 400 to 600 μm (Klopstein et al., 2012). When indicated, AM095 or saline was administered orally at a dose of 30 mg/kg. Treatment was initiated 1 h after SCI and then given every 12 h for 1 week.
Functional assessment
Locomotor recovery was evaluated at 1, 3, 5, 7, 10, 14, 21, and 28 d post-injury (dpi) in an open-field test using the nine-point Basso Mouse Scale (BMS; Basso et al., 2006), which was developed specifically for locomotor testing after contusion injuries in mice. The BMS analysis of hindlimb movements and coordination was performed by two independent assessors who were blinded to the experimental groups and the consensus score taken. In addition, at the end of the follow-up (28 dpi), the highest locomotion speed of the mice was evaluated on a belt of a motorized treadmill. Briefly, each mouse was allowed to explore the treadmill compartment, with the motor speed set to zero, for 5 min. Then speed was gradually increased from 0 up to 35 cm/s, and the maximum speed at which each mouse was to perform for at least 5 s was recorded.
Motor-evoked potentials
At day 28, electrophysiological tests were used to evaluate spared motor central pathways after SCI. Motor-evoked potentials (MEPs) were recorded from the gastrocnemius muscle (GM) with microneedle electrodes, in response to transcranial electrical stimulation of the motor cortex by single rectangular pulses of 0.1 ms duration. Pulses were delivered through needle electrodes inserted subcutaneously, with the cathode over the skull overlying the sensorimotor cortex and the anode at the nose (García-Alías et al., 2003). Compound muscle action potential from GM muscle was recorded for internal control of normal peripheral conduction. In this case, the sciatic nerve was stimulated percutaneously by means of single pulses of 0.02 ms duration (Grass S88; Grass Instruments) delivered through a pair of needle electrodes placed at the sciatic notch. All potentials were amplified and displayed on a digital oscilloscope (Tektronix 450S).
Cell cultures
Mouse oligodendrocyte cultures.
Primary oligodendrocyte progenitor cells (OPCs) were isolated from mouse cerebral cortex at postnatal day 2 (P2) to P4 as described previously (O'Meara et al., 2011). Briefly, after removal of the meninges, cortical tissue was minced using a P1000 pipette tip and dissociated by incubating in a solution of 1.54 mg/ml papain (Worthington Biochemical), 400 μg/ml l-cysteine (Sigma), and 1 mg/ml DNase I (Roche) in MEM (Gibco) for 20 min at 37°C. Mixed glial culture media [MGCM; 10% inactivated and filtered fetal bovine serum (FBS; Sigma), 0.33% penicillin–streptomycin (P/S; Sigma), and 1% Glutamax 100× (Gibco) in DMEM (Gibco)] was added to stop papain and DNase I activity. Cells were plated into T25 tissue culture flasks coated with 10 μg/ml poly-d-lysine for 1 h at 37°C and cultured at 37°C in an humidified incubator with 5% CO2 supplementation. Three hours after plating, the floating cells were discarded by replacing the medium. Two thirds of the MGCM was replaced every 3 d with new MGCM supplemented with 5 μg/ml insulin (Seralab). Nine days later, OPCs were harvested by shaking at 37°C at 220 rpm overnight. The collected cells were plated into 10 μg/ml poly-d-lysine-coated coverslips with DMEM (Gibco) supplemented with 1% Glutamax 100×, 2% B27 (Gibco), 0.5% FBS, 50 pg/ml recombinant mouse ciliary neurotrophic factor (BioTrend), and 1% OL supplement (10 μl/ml N-2 supplement 100× (Gibco), 10 mg/ml bovine serum albumin (Sigma), and 40 μg/ml 3,3′,5-triiodo-l-thyronine (Sigma). OPC maturation in oligodendrocytes was achieved after 7 d in vitro.
Mouse microglia cultures.
Primary microglial cells were isolated from P4 mouse cerebral cortex as described previously (Saura et al., 2003). Tissue dissociation and cell isolation was performed as described in OPC culture. Mixed glial cultures were prepared into T25 tissue culture flasks without any coating. Cells were seeded at a density of 300,000 cells/ml in DMEM/F-12 (Gibco) with 10% FBS and 5% P/S and cultured at 37°C in a humidified incubator with 5% CO2 supplementation. Medium was replaced every 4–5 d, and confluence was achieved after 10–12 d. At this point, mixed cultures were incubated with 0.25% trypsin–EDTA (Gibco) diluted 1:4 in DMEM/F-12 for 30 min at 37°C. This trypsinization resulted in the detachment of the upper layer of cells in one piece, and the remaining adherent microglial cells were cultured for 24 h before stimulation. Microglial cells were stimulated with LPA (1 μm) or LPA plus AM095 (12.5 μm) or 0.025% DMSO (vehicle for AM095) as control for 24 h. Afterward, cells were washed to remove the LPA from the medium, and microglia were then incubated with oligodendrocyte conditioned media for 24 h and then used for cytotoxicity assays in oligodendrocyte cell cultures.
Assessment of oligodendrocyte cell death
Oligodendrocytes were stimulated with LPA (0.01, 0.1, or 1 μm), LPA plus AM095 (12.5 μm), and DMSO (0.025%) as control, or with conditioned media of microglia harvested from C57BL/6 mice, maLPA1 null mice, or wild-type littermates, stimulated with DMSO, LPA, (1 μm), or LPA (1 μm) plus AM095 (12.5 μm) for 24 h. Afterward, coverslips were fixed in 4% paraformaldehyde for 20 min, washed in PBS, and incubated overnight with rat anti-MBP (1:300; Abcam), mouse anti-A2B5–Alexa Fluor 488 (1:100; Millipore), and rabbit anti-NG2 (1:100; Millipore) in 0.3% Triton X-100/PBS with 5% fetal calf serum at 4°C. After several washes in PBS, coverslips were incubated for 1 h at room temperature (RT) with anti-rat or anti-rabbit Alexa Fluor 594-conjugated antibody (1:500; Invitrogen) and DAPI (Sigma), and the total number of oligodendrocytes (MBP+ cells) or oligodendrocytes precursor cells (A2B5 and NG2) were counted under fluorescence microscope (Olympus BX51). Four independent replicates were done for each of the experiments.
Histology
At 4 d after intraspinal injection and 28 d after spinal cord contusion, mice were perfused with 4% paraformaldehyde in 0.1 m phosphate buffer (PB). A 10 mm length of spinal cord containing the injection or the contusion site centered was harvested, postfixed with 4% paraformaldehyde in 0.1 m PB for 1 h, and cryoprotected with 30% sucrose in 0.1 m PB at 4°C for a minimum of 48 h. The samples were fast-frozen at −60°C in cryoembedding compound (Tissue-Tek OCT; Sakura) and cut on a cryostat (Leica). Ten series of 10-μm-thick transversal sections were picked up on glass slides, so adjacent sections on the same slide were 100 μm apart. For demyelination and myelin sparing analyses, sections were stained with Luxol fast blue (LFB; Sigma) or fluoromyelin (Invitrogen). After graded dehydration, sections were placed in a 1 mg/ml LFB solution in 95% ethanol and 0.05% acetic acid overnight at 37°C. Sections were then washed in 95% ethanol and distilled water before being placed in a solution of 0.5 mg/ml Li2CO3 in distilled water for 1 min at RT. After washes in distilled water, sections were dehydrated and mounted in DPX mounting media (Sigma). For immunofluorescence staining, sections were rehydrated in PBS and blocked with 5% FBS in PBS with Tween 20 (PBST) for 1 h at RT. The sections were then incubated overnight at 4°C with primary antibodies against NeuN (for neurons; 1:200; Millipore), neurofilament-200 (NF-200; for axons; 1:1000; Millipore), GFAP (for astrocytes; 1:500; Invitrogen), ionized calcium-binding adapter molecule 1 (Iba1; for macrophage/microglia; 1:400; Abcam), and MBP (for myelin; 1:100; Abcam). After several washes in PBST, sections were incubated for 1 h at RT with the appropriate Alexa Fluor 594 or 488-conjugated secondary antibodies (1:200; Invitrogen) and then coverslipped in Mowiol mounting media containing DAPI (1 μg/ml; Sigma). Tissue sections were viewed with an Olympus BX51 microscope, and images were captured with an Olympus DP50 digital camera attached to it and using the Cell Image acquisition software.
The epicenter of the injection or contusion injury impact was determined for each mouse spinal cord by localizing the tissue section with the greatest demyelination. The NIH ImageJ software was used to quantify the histological parameters. For experiments injecting LPA, demyelination was measured by delineating LFB nonstained white matter in the dorsal column, whereas microglia activation was assessed by calculating the percentage of Iba1+ immunoreactivity area within a 0.025 mm2 box placed within the dorsal column. For SCI experiments, myelin sparing after SCI was calculated by delineating the spared fluoromyelin-stained tissue, whereas neuronal survival was assessed by counting the number of NeuN+ cells in the ventral horns. Moreover, axonal and demyelination in the dorsal column was assessed by counting the NF+ structures as well as fibers double stained for NF and MBP at the lesion epicenter.
Electron microscopy
Four and 21 d after intraspinal injection of LPA or saline, mice were perfused with 0.5% paraformaldehyde and 2.5% glutaraldehyde in 0.1 m PB, pH 7.4. Five-millimeter-thick cross-sections of the spinal cord containing the injection site were left in the same fixative for several days, followed by postfixation in 2% osmium tetroxide for 2 h at RT and then processed for embedding in Epon. One-millimeter-thick cross-sections of the spinal cord were stained with 1% toluidine blue for light microscopy. Then, sections were cut at 90 nm, stained with lead citrate, and viewed with a Philips CM 10 electron microscope.
Flow cytometry
Immune cells from the injured spinal cord were analyzed by flow cytometry at 7 dpi, as described previously (Klopstein et al., 2012). Briefly, spinal cords were cut in little pieces and passed through a cell strainer of 70 μm (Falcon; BD Bioscience Discovery Labware), and the cell suspension was centrifuged twice at 300 × g for 10 min at 4°C. Samples were divided, and cells alone and isotype-matched control samples were generated to control for nonspecific binding of antibodies and for autofluorescence. Isotype control antibodies were purchased from eBioscience and included phycoerythrin–cyanine 7 (PE–Cy7)-labeled rat IgG2b, adenomatous polyposis coli (APC)-labeled rat IgG2b, peridinin chlorophyll-a protein cyanine 5.5 (PerCP–Cy5.5)-conjugated rat IgG2b, and fluorescein isothiocyanate-conjugated rat IgG2b and IgG2M. The following antibodies were also purchased from eBioscience: CD45–PerCP–Cy5.5, CD11b–PE–Cy7, and F4/80–APC. After 30 min of incubation with combinations of antibodies at 4°C, the samples were washed and fixed in 1% paraformaldehyde. Microglial cells were identified as CD45low and CD11b+ cells, whereas myeloid cells were identified as CD45high and CD11b+ cells (Stirling and Yong, 2008). Macrophages were identified from myeloid cells based on F4/80 expression (CD45high, CD11b+, F4/80+). At least 3000 CD45+ cells were analyzed per spinal cord sample using FlowJo software on a FACSCanto flow cytometer (BD Biosciences). Four contused mice treated with saline or AM095 were used in this experiment.
Isolation of microglia from CNS tissue
Briefly, spinal cord and brain from adult C57BL/6 mice (8–10 weeks old) were removed and digested enzymatically with 0.2% collagenase B (Roche Diagnostics) and 0.2% trypsin–EDTA at 37°C for 30 min and then passed through a cell strainer of 40 μm (Falcon; BD Bioscience Discovery Labware). Cell suspension was centrifuged twice at 300 × g for 10 min at 4°C, and microglial cells were first isolated by magnetic sorting using a CD11b antibody (Miltenyi Biotec) and then stained with PerCP–Cy5.5-conjugated CD45 and PE–Cy7-conjugated CD11b antibodies for additional purification on a cell sorter (FACSARIA III; BD Bioscience). Microglia cells were assessed on a flow cytometer (FACSCalibur; BD Biosciences), and only populations presenting >90% purity were used for gene expression analysis.
RNA isolation, reverse transcription, and real-time PCR
Mice were perfused with sterile saline, and a 5-mm-length of uninjured spinal cord was removed. Tissue was homogenized with QIAzol lysis reagent (Qiagen) and RNA extracted using the RNeasy Lipid Tissue kit (Qiagen), according to the protocol of the manufacturer. RNA was treated with DNaseI (Qiagen) to eliminate genomic DNA contamination. mRNA from cultured and in vivo sorted microglia was extracted using the RNeasy Micro kit following the guidelines of the manufacturer. One and 0.5 μg of RNA obtained from tissue or microglia, respectively, was primed with random hexamers (Promega) and reverse transcribed using the Omniscript RT kit (Qiagen). RNase inhibitor (Roche) was added (1 U/μl final concentration) to avoid RNA degradation. Primer sequences are specified in Table 1. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used as a housekeeping gene. The amount of cDNA was calculated based on the threshold cycle (CT) value and was standardized by the amount of housekeeping gene using the 2−ΔΔCT method (Livak and Schmittgen, 2001).
Table 1.
Real-time PCR primer sequences
| Gene | Forward primer (5′-3′) | Reverse primer (5′-3′) |
|---|---|---|
| LPA1 | TGTCCTGGCCTATGAGAAGTTCT | TTGTCGCGGTAGGAGTAGATGA |
| LPA2 | CTCACTGGTCAATGCAGTGGTATAT | GAAGGCGGCGGAAGGT |
| LPA3 | GGGACGTTCTTCTGCCTCTTTA | GAAAGTGGAACTTCCGGTTTGT |
| LPA4 | GATGGAGTCGCTGTTTAAGACTGA | TGTTTGATCACTAACTTCCTCTTGGATA |
| LPA5 | CCGTACATGTTCATCTGGAAGAT | CAGACTAATTTCTCTTCCCACCT |
| LPA6 | ACTGCTGCTTTGACCCTATTG | AAGGTCTGTAGGTTGTGTTGG |
| GAPDH | TCAACAGCAACTCCCACTCTTCCA | ACCCTGTTGCTGTAGCCGTATTCA |
Mass spectrometry analysis of LPA in the spinal cord
Mice were perfused with sterile saline, and a 5-mm-length of uninjured or injured spinal cord centered at the impact site was harvested at 6 h and 1, 3, 7, and 14 dpi. Samples were homogenized in 0.01% w/v butylated hydroxytoluene in PBS. After centrifugation at 15,000 × g for 20 min at 4°C, supernatants were processed for protein concentration determination using the BCA protein assay (Pierce).
The volume corresponding to 300 μg of protein was taken of every sample. Two hundred picomoles of 1-O-dodecanoyl-2-dodecanoyl phosphatidic acid was added as internal standard and water up to 100 μl. Samples were acidified with 500 μl of H2O/HCl at 19:1, and LPA species were extracted with 500 μl of water-saturated butanol twice. Organic phase was recollected, evaporated until dryness, and resuspended in 100 μl of CHCl3. Fifty microliters of this extract were injected in a high-performance liquid chromatograph equipped with a Hitachi LaChrom Elite L-2130 binary pump and a Hitachi Autosampler L-2200 (Merck) coupled to a Bruker esquire6000 ion-trap mass spectrometer (Bruker Daltonics). The column was a Supelcosil LC-Si 3 μm 150 × 3 mm column (Sigma-Aldrich) protected with a Supelguard LC-Si 20 × 3 mm guard cartridge (Sigma-Aldrich). Mobile phase was a gradient of solvent A (chloroform/methanol/32% ammonium hydroxide at 75:24.5:0.5 v/v/v) and solvent B (chloroform/methanol/water/32% ammonium hydroxide at 55:39:5.5:0.5 v/v/v/v). The gradient was started at 100% solvent A; it was decreased linearly to 50% solvent A in 2 min, maintained for 4 min, to 0% solvent A in 6 min, and maintained there for an additional 18 min. The flow rate was 0.5 ml/min in the chromatographic step and 0.2 ml/min entered into the electrospray interface of the mass spectrometer. Nebulizer gas was set to 30 lb per square inch, dry gas to 8 l/min, and dry temperature to 365°C. LPA species were detected in negative ion mode with the capillary current set at 3500 V as deprotonated molecular ions [M-H]−.
Statistical analysis
All analyses were conducted through IBM SPSS Statistics version 19. Two-tailed Student's t test was used for the single comparison between two groups. Maximal speed on a treadmill was analyzed using the Mantel–Cox test. Functional follow-up for BMS score and subscore, as well as histological analysis of myelin and neuronal sparing were analyzed using two-way repeated-measures (RM) ANOVA. The rest of the data were analyzed using one-way or two-way ANOVA depending on the appropriate design. Post hoc comparisons were performed only when a main effect showed statistical significance. p values for multiple comparisons were adjusted using Bonferroni's correction. Results are expressed as mean and SE. Differences were considered significant at p < 0.05.
Results
LPA levels are increased in the injured mouse spinal cord
We first assessed whether LPA levels increased in the spinal cord after contusion injury. Using mass spectrometry, we detected the presence of three LPA molecular variants (18:1, 18:0, and 20:1) in the uninjured spinal cord (Table 2). After contusion injury, these three LPA species were elevated significantly at all time points analyzed (one-way ANOVA; post hoc Bonferroni's test, p < 0.01; n = 4 per time point). Moreover, additional LPA variants (16:0, 20:4, 22:6, and 22:4) were also detected in the contused spinal cord (Table 2). Quantification of the different LPA species revealed that the total level of LPA increased very rapidly in the injured spinal cord (Fig. 1), being approximately fivefold higher at 6 h, rising to ∼10-fold at day 3, and the remaining approximately eightfold increased from 7 to 14 dpi compared with intact spinal cord (one-way ANOVA; post hoc Bonferroni's test, p < 0.01; n = 4 per time point). These results demonstrate that LPA levels increase in the spinal cord after contusion injury.
Table 2.
Quantification of the different LPA species in SCI
| LPA species | 16:0 | 18:1 | 18:0 | 20:4 | 20:1 | 22:6 | 22:4 | 22:1 |
|---|---|---|---|---|---|---|---|---|
| Intact | 0 | 33.5 ± 7.1 | 34.6 ± 3.2 | 0 | 7.1 ± 1.5 | 0 | 0 | 0 |
| 6 h | 7.7 ± 0.3* | 166.4 ± 9.0* | 79.7 ± 4.4* | 10.5 ± 0.9* | 77.8 ± 5.8* | 55.0 ± 4.5* | 15.1 ± 0.7* | 0.4 ± 0.1 |
| 1 d | 7.4 ± 0.9* | 167.8 ± 18.6* | 80.5 ± 8.3* | 13.1 ± 1.8* | 94.0 ± 8.4* | 60.3 ± 6.1* | 14.9 ± 1.9* | 0.5 ± 0.1 |
| 3 d | 13.4 ± 1.0* | 290.5 ± 21.2* | 143.3 ± 10.5* | 20.2 ± 1.1* | 144.0 ± 12.9* | 85.0 ± 6.2* | 25.5 ± 1.7* | 0.7 ± 0.0 |
| 7 d | 12.9 ± 0.8* | 255.4 ± 7.1* | 125.4 ± 4.1* | 21.9 ± 0.7* | 119.8 ± 6.1* | 73.4 ± 2.3* | 22.3 ± 0.5* | 0.7 ± 0.0 |
| 14 d | 0 | 307.7 ± 35.7* | 125.1 ± 8.2* | 25.7 ± 2.0* | 52.1 ± 8.2* | 82.0 ± 7.6* | 13.6 ± 2.3* | 0 |
*p < 0.01 versus intact spinal cord.
Figure 1.

Increased LPA levels in the contused spinal cord. Mass spectrometry analyses of LPA in spinal cord tissue from uninjured and contused spinal cord at 6 h and 1, 3, 7, and 14 d after SCI (n = 4 time point). *p < 0.05. Error bars indicate SEM.
Exogenous LPA exposure leads to demyelination
To assess the potential contribution of increased levels of LPA in the spinal cord parenchyma, we injected 1 μl of saline containing 5 nmol of LPA into the dorsal column of uninjured spinal cord or saline alone as a control group. The LPA variant used in these experiments was the 18:1 oleoyl LPA, because our mass spectrometry results revealed that this LPA species was the most abundant in the contused spinal cord. Microglia/macrophage activation in the spinal cord parenchyma was assessed by measuring the presence of reactive Iba1+ cells, 4 d after injection. Microglia/macrophage activation was restricted to the injection area in saline-injected mice, probably because of the mechanical injury caused by the needle insertion (Fig. 2A–C). By comparison, spinal cord sections from mice injected with LPA showed a marked activation of microglia/macrophages 4 d after injection (Fig. 2D). The presence of these inflammatory cells was not limited to the injection site but were also distributed within the whole dorsal columns (Fig. 2D). In addition, the immunoreactivity for Iba1+ cells was also increased in rostral and caudal areas from the injection site in LPA-injected compared with saline-injected mice (Fig. 2; two-way RM-ANOVA; post hoc Bonferroni's test, p < 0.05; n = 4 per group). Mild activation of astrocytes was also observed but were only restricted to areas close to the LPA injection (data not shown), indicating that LPA activates both microglia and astrocytes but has a greater preference for microglia.
Figure 2.

Intraspinal injection of LPA leads to microglia/macrophage activation. A, Quantification of microglia/macrophage immunoreactivity at 4 d after intraspinal injection of saline or LPA. B–E, Representative images of spinal cords stained against Iba1 at the epicenter (B, D) and 1800 μm rostral to the injection site (C, E) of saline-injected (B, C) and LPA-injected (D, E) mice (n = 4 per group). *p = 0.006. Error bars indicate SEM. Scale bar, 250 μm.
We then assessed whether activation of microglia/macrophages triggered by LPA led to demyelination. At 4 dpi, LFB staining revealed the presence of a small demyelinating lesion in the dorsal column of saline-injected spinal cords (Fig. 3), which was restricted to the area of the needle insertion (Fig. 3B). We also observed a demyelinating lesion in the spinal cords injected with LPA (Fig. 3D). The area of this lesion was significantly increased in LPA-injected compared with PBS-injected mice at the injection epicenter and in adjacent areas (Fig. 3A–E; two-way RM-ANOVA; post hoc Bonferroni's test, p < 0.05; n = 4 per group). To assess whether demyelination induced by LPA was caused by axonal or myelin damage, we double stained spinal cord sections with fluoromyelin and NF. Double immunofluorescence revealed the presence of NF+ structures within the demyelinating lesion (Fig. 3F), suggesting that LPA led to demyelination. To further confirm these observations, we used electron microscopy (Fig. 3G–J). This technique confirmed the presence of fibers with degenerating myelin (Fig. 3H,I) and naked axons (Fig. 3J) within the demyelinating lesion. Therefore, these results indicate that the increased levels of LPA in the spinal cord leads to neuroinflammation and demyelination.
Figure 3.

Intraspinal injection of LPA leads to demyelination. A, Quantification of demyelination at 4 d after intraspinal injection of saline or LPA. B–E, Representative images of spinal cords stained with LFB at the epicenter (B, D) and 1800 μm rostral to the injection site (C, E) in saline-injected (B, C) and LPA-injected (D, E) mice. F, High-magnification image of spinal cord tissue section stained against NF (red) and fluoromyelin (green) showing the presence of NF+ structures within the demyelinating lesion at 4 d after intraspinal injection of LPA. G, Toluidine blue-stained section from spinal cord at 4 d after intraspinal injection of LPA. H–J, Electron microscopy images taken from the demyelinating area showing fibers with myelin breakdown (H, I) and demyelinated axons (J; see arrows; n = 4 per group). *p = 0.001. Error bars indicate SEM. Scale bar, 500 μm.
Exogenous LPA exposure mediates demyelination via LPA1
Because LPA signals through six different LPARs, we studied which LPARs were expressed constitutively in the normal spinal cord. Real-time PCR analysis revealed that all six LPARs are expressed constitutively in the uninjured spinal cord, with LPA1 being the most highly expressed (Fig. 4; n = 4 per time point). To assess whether the demyelination triggered by intraspinal injection of LPA was mediated via LPA1, we injected LPA together with AM095, a selective LPA1 antagonist (Swaney et al., 2011), into the intact spinal cord. Histological analysis revealed that 4 d after LPA administration, the demyelinating lesion was reduced substantially when LPA1 was blocked (Fig. 5A–C). Quantification of the demyelinated areas revealed that mice treated with AM095 showed reduced myelin loss at the injection site and in the adjacent sections corresponding to 600 μm rostral and caudal to the epicenter (two-way ANOVA; post hoc Bonferroni's test, p < 0.05; n = 4 per group). Spinal cord tissue sections from LPA1 null mice (maLPA1 variant) also displayed smaller demyelinating lesions (Fig. 5D–F) after intraspinal injection of LPA, further implicating LPA1 signaling in LPA-induced demyelination.
Figure 4.

Expression of LPA1–LPA5 in the spinal cord parenchyma. Note that LPA1 is the LPAR most highly expressed in the spinal cord. Data are presented as copies of receptor mRNA relative to copies of GAPDH mRNA (n = 4 per time point). Error bars indicate SEM.
Figure 5.

LPA1 signaling mediates demyelination in the spinal cord. A, Quantification of the demyelinating injury in the spinal cord of animals injected with saline, LPA, or LPA1 plus AM095. B, C, Representative images of spinal cords stained with LFB at the epicenter of the injection site in mice administered with LPA (B) or LPA plus AM095 (C). Note that demyelination was markedly reduced in the absence of LPA1 activity. D, Quantification of the demyelinating injury after intraspinal injection of LPA in wild-type or maLPA1 null mice. E, F, Representative images of spinal cords stained with LFB at the epicenter of the injection site in wild-type (E) and maLPA1 null (F) mice. Note that demyelination was reduced in the absence of LPA1 (n = 4 per group). Scale bar, 500 μm. *p < 0.05 saline versus LPA; *p < 0.05 wild-type versus maLPA1 null mice. Error bars indicate SEM.
Because LPA1 is expressed in mature oligodendrocytes (Weiner et al., 1998), we assessed whether LPA-induced demyelination was attributable to a toxic effect of LPA on oligodendrocytes. Cell culture work indicated that LPA treatment led to a mild reduction in the viability of oligodendrocytes (∼20%) at concentrations of 1 μm (Fig. 6A; n = 4 per group). However, LPA did not have an effect on oligodendrocyte precursor cell survival (Fig. 6B). Administration of AM095 failed to rescue cultured oligodendrocytes from cell death, suggesting that the LPA1 signaling does not mediate a direct cytotoxic effect in cultured oligodendrocytes (Fig. 6C; n = 4 per group), albeit with the caveat that these cells only approximate what occurs in vivo. Because LPA also triggers microglia activation when injected into the spinal cord and activated microglial cells were associated with areas of demyelination (Fig. 6D), we also assessed whether microglial LPA1 was responsible for the LPA-induced demyelination. In agreement with previous work (Möller et al., 2001; Tham et al., 2003) real-time PCR analysis identified LPA1 gene expression in cultured microglia (Fig. 6F; n = 4). Moreover, microglial cells in vivo have constitutive expression of LPA1, showing similar levels of LPA1 relative to cultured microglia (Fig. 6E,F; n = 4). Interestingly, we observed that conditioned medium of primary microglial cells stimulated with LPA led to a marked reduction (∼85%) in oligodendrocyte survival (Fig. 6G,I–F; one-way ANOVA; post hoc Bonferroni's test, p < 0.001; n = 4 per group), suggesting that microglia become cytotoxic after LPA stimulation., Oligodendrocyte cell death was reduced approximately three times when microglia LPA1 was blocked with AM095 (Fig. 6G,I–F; one-way ANOVA; post hoc Bonferroni's test, p = 0.017; n = 4 per group). Similar results were obtained from conditioned medium of LPA-stimulated microglia cells isolated from maLPA1 mice (Fig. 6H; two-way ANOVA; post hoc Bonferroni's test, p < 0.05; n = 4 per group). These data indicate that the activation of LPA1 in microglial cells is responsible, in part, for the cytotoxic effects mediated by LPA-stimulated microglia. Overall, our results suggest that demyelination triggered by intraspinal injection of LPA is mediated, in part, by the activation of microglial LPA1, with unclear participation of oligodendrocyte LPA signaling in vivo.
Figure 6.
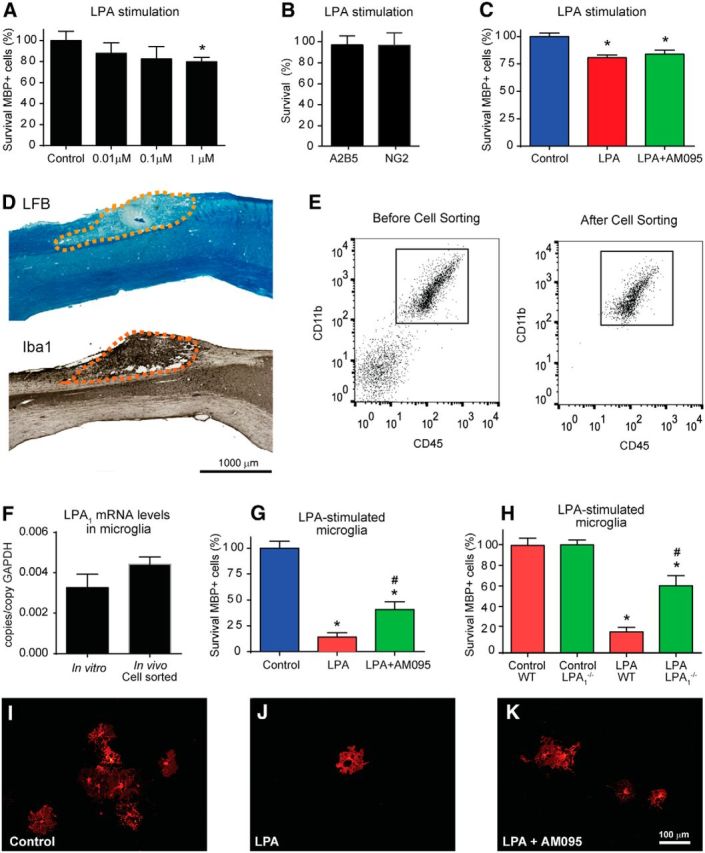
Microglia LPA1 mediates oligodendrocyte cell death. A, Effects of several concentrations of LPA on oligodendrocyte survival. B, Effects of LPA (1 μm) on OPC survival. C, Quantification of oligodendrocyte cell death after stimulation with LPA or LPA plus AM095. D, Representative spinal cord sections from mice that received intraspinal injection of LPA showing that the demyelinating lesion is associated with areas enriched in microglia. E, Representative FACS analysis of microglia immunopanned with the CD11b antibody from adult CNS before and after being cell sorted. F, Quantification of LPA1 expression in cultured and cell-sorted microglial cells. Note the microglia show similar levels of LPA1 in both conditions. G, Assessment of conditioned medium of unstimulated (control) microglia or conditioned medium of microglia stimulated with LPA or LPA plus AM095 on oligodendrocyte survival. H, Assessment of conditioned medium of unstimulated (DMSO; control) or LPA-stimulated microglial cells harvested from wild-type or LPA1 null mice. I, K, Representative images of oligodendrocytes (MBP+) from cultures treated with conditioned medium from untreated microglia (DMSO; control) or microglia treated with LPA or LPA plus AM095 stimulated microglia. Note that gene deletion of pharmacological blockade of microglia LPA1 significantly reduces oligodendrocyte cell death. Scale bar, 100 μm. *p < 0.05 versus control; #p < 0.05 versus LPA or LPA wild type). Error bars indicate SEM (n = 4 per group).
LPA1 contributes to SCI pathophysiology
LPA–LPA1 signaling was assessed for its role in secondary damage and functional impairment after SCI in mice. Because maLPA1 null mice showed locomotor deficits (data not shown), probably attributable to its developmental brain defects (Santin et al., 2009), we used AM095 to assess the role of LPA1 in SCI. Oral administration of AM095 significantly reduced locomotor impairments based on the BMS assessment (Fig. 7A; p < 0.001, two-way RM-ANOVA; n = 8 per group). At the end of the follow up (28 dpi), the BMS score was improved by 1.5 points after AM095 treatment (Fig. 7A). Mice treated with saline showed occasional plantar stepping and no coordination, whereas AM095 showed frequent/consistent plantar stepping and most of them (87.5%) had coordination. In addition, the BMS subscore, which assess fine aspects of locomotion, was markedly improved after LPA1 blockade (Fig. 7B; p < 0.001, two-way RM-ANOVA; n = 8 per group), whereas mice treated with AM095 were able to run at significantly higher speeds on a treadmill (Fig. 7C; p = 0.011, Mantel–Cox test; n = 8 per group).
Figure 7.
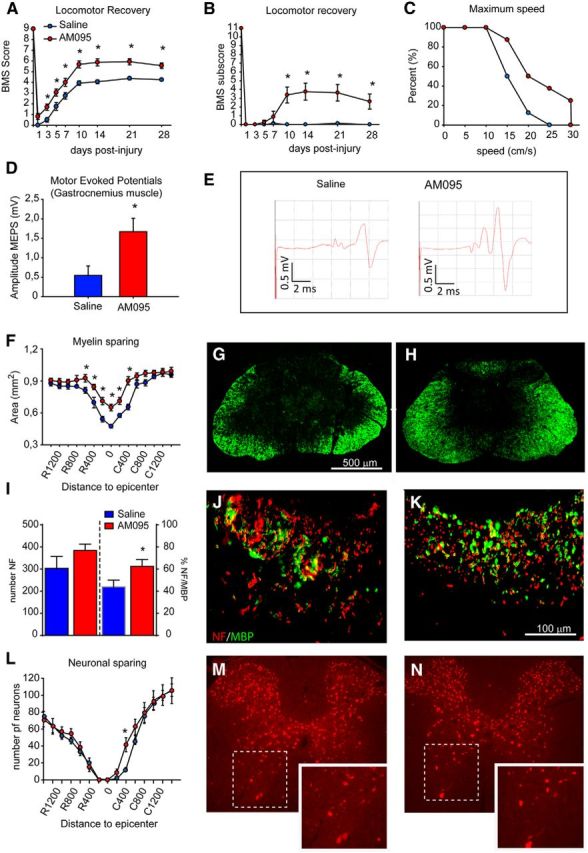
LPA1 blockade enhances functional outcomes and reduces demyelination after SCI. A, B, Animals treated with AM095 show significant improvement in locomotor performance compared with saline-treated mice using the nine-point BMS (A) and the 11-point BMS subscore (B), as well as faster locomotion on a treadmill (C). D, E, Mice treated with AM095 show greater preservation of MEPs. D, Quantification of MEP recordings from GM at 28 d after SCI. E, Representative MEP recordings from mice treated with saline or AM095. F, Quantification of myelin sparing at various distances rostral and caudal to the injury epicenter revels significant reduction in tissue loss in mice treated with AM095 at the epicenter of the injury and in adjacent sections. G, H, Representative micrographs showing myelin sparing at the injury epicenter in section stained against fluoromyelin from mice treated with saline (G) or AM095 (H; n = 8 per group). Scale bar, 500 μm. *p < 0.05. I, Quantification of axon and myelin sparing in the dorsal column at the epicenter of the injury. Treatment with AM095 resulted in greater myelin but not axonal preservation. J, K, Representative micrographs of the dorsal columns stained against NF and MBP from mice treated with saline (J) and AM095 (K). L, Quantification of ventral horn neuron survival at various distances rostral and caudal to the injury epicenter reveals significantly greater neuronal survival in mice treated with AM095. M, N, Representative micrographs showing sparing of ventral horn neurons in mice administered with saline (M) and AM095 (N) in sections stained against NeuN at 600 μm rostral to the injury epicenter. Error bars indicate SEM. n = 8 per group.
Electrophysiological tests designed to assess the preservation of descending pathways of the spinal cord based on MEP analysis were used on 28 dpi mice treated with AM095: treated animals showed significantly greater MEP amplitudes for the GM compared with controls (p = 0.018, t test; n = 8 per group), suggesting an increase in spared functional descending tracts in the spinal cord after the lesion (Fig. 7D,E). Mice administered AM095 were further assessed for amelioration of secondary tissue damage. Histological sections of the spinal cord stained with fluoromyelin revealed that animals treated with the LPA1 antagonist showed less demyelination at the injury epicenter and in adjacent regions (Fig. 7F–H; p < 0.001, two-way ANOVA; n = 8 per group). We also assessed whether the greater myelin sparing observed after AM095 treatment was attributable to reduced axonal loss or reduced demyelination. Spinal cord tissue section at the injury epicenter, the most damaged area of the spinal cord, revealed that AM095 did not enhance the number of NF+ fibers but increased the percentage of axons labeled with MBP (Fig. 7I–K; p = 0.034, t test; n = 8 per group). However, AM095 treatment led to only minor improvement in neuronal sparing, seen only at 400 μm caudal to the injury epicenter (Fig. 7L–N). These results suggest that the increased levels of LPA in the injured spinal cord contributes to demyelination and loss of function by signaling via LPA1.
We finally assessed whether AM095 attenuated the inflammatory response after SCI. FACS analysis of injured spinal cord harvested at 7 dpi, when microglial cell and macrophages peak in numbers after spinal cord contusion, revealed that AM095 did not reduce microglia counts (Fig. 8). Similarly, AM095 did not attenuate the infiltration of macrophages into the injured spinal cord (Fig. 8). Overall, our data suggest that AM095 confers protection against demyelination by reducing the cytotoxic actions of microglia LPA1 signaling but not its density.
Figure 8.

AM095 does not attenuate inflammatory response. A, B, Representative density plots of FACS analysis showing microglia and myeloid cells in the injured spinal cord of saline-treated (A) or AM095-treated (B) mice at 7 dpi. Myeloid cell were further discriminated in macrophages based on F4/80 expression. C, Graph showing microglia and macrophage counts in the injured spinal cord after LPA1 blockade. Note that treatment with AM095 did not reduce the numbers of microglia and macrophages. Error bars indicate SEM. n = 4 per group.
Discussion
The results of the present study provide clear evidence that the LPA–LPA1 pathway contributes to secondary damage after SCI. LPA levels increase in the spinal cord after contusion injury, contributing to activation of microglia/macrophages and demyelination that are, in part, mediated by LPA1 signaling. Moreover, our in vitro experiments suggest that detrimental actions of LPA1 involve signaling in microglial cells. Finally, we demonstrate that selective inhibition of LPA1 after SCI confers protection from demyelination and enhances locomotor skills. Overall, this work demonstrates for the first time that the LPA–LPA1 pathway contributes to secondary damage and is detrimental to functional recovery after SCI.
Lysophospholipids, which include LPA and sphingosine 1-phosphate (S1P), have emerged as key modulators of inflammation, including in nervous system disorders (Chun and Brinkmann, 2011; David et al., 2012c; Choi and Chun, 2013). The contribution of lysophospholipids to CNS pathology includes experiments identifying lysolecithin (also known as lysophosphatidylcholine) as a potent demyelinating and inflammation-inducing agent in the CNS (Ousman and David, 2000, 2001). S1P has been demonstrated to play a key role in multiple sclerosis (Brinkmann et al., 2010) through the actions of fingolimod (FTY720; Chun and Brinkmann, 2011), a nonselective S1P receptor modulator, that was the first worldwide oral treatment for relapsing forms of multiple sclerosis (Brinkmann et al., 2010).
LPA is a lipid signaling molecule capable of evoking multiple physiological responses in a wide variety of cells (Choi et al., 2010; Choi and Chun, 2013). LPA is present in low concentrations in mammalian cells and tissues, but its concentrations in the blood can range from 0.1 μm in plasma to >10 μm in serum. LPA is synthesized in vivo from membrane phospholipids by multiple pathways (Aoki et al., 2002; Choi et al., 2010; Choi and Chun, 2013). Here, we observed that LPA levels rise rapidly in the spinal cord parenchyma after injury. This is likely attributable to a combination of its de novo production in the contused spinal cord, including through phospholipase A2 enzymes that are upregulated in SCI (Titsworth et al., 2009; López-Vales et al., 2011; David et al., 2012c). However, plasma leakage and blood vessel disruption, as well as serum production during the clotting process, likely contribute to increases in LPA levels in SCI, as has been modeled for post-hemorrhagic hydrocephalus wherein LPA is an active factor in hemorrhagic fluids (Yung et al., 2011). Indeed, the presence of the 20:4 LPA in the injured spinal cord, which is a major form of LPA in plasma (Scherer et al., 2009) but that was not found in the intact cord, supports hemorrhagic blood fractions as a source of LPA in SCI.
Exogenous LPA can initiate a large range of neural sequelae within the nervous system that includes effects on most cell types and activities in disease models. Cell types include developing neural cells (Dubin et al., 1999, 2010; Kingsbury et al., 2003), astrocytes (Shano et al., 2008; Spohr et al., 2008), microglia (Möller et al., 2001; Ma et al., 2013), oligodendrocytes and Schwann cells (Weiner et al., 1998, 2001), and neurons (Fukushima et al., 2002; Trimbuch et al., 2009; Lin et al., 2012). Disease models include pain (Inoue et al., 2004; Lin et al., 2012; Ma et al., 2013), hypoxia and ischemia (Herr et al., 2011; Halder et al., 2013), demyelination (Inoue et al., 2004, 2008a,b; Nagai et al., 2010), and behavioral disorders (Contos et al., 2000, 2002; Santin et al., 2009; Castilla-Ortega et al., 2012; Pedraza et al., 2014). These physiological responses linked to LPA activation are triggered by a variety of downstream pathways, including Ca2+ mobilization, adenylyl cyclase inhibition, activation of Rho, mitogen-activated protein kinase, phospholipase C, and Akt (Yung et al., 2014, 2015; Sheng et al., 2015). The findings here identify LPA as a new trigger of secondary damage in SCI. The mechanism involves LPA1, which is the mostly highly expressed LPAR in the spinal cord, suggesting that the detrimental actions of LPA are likely to be mediated by binding to LPA1. The maLPA1 knock-out mice and mice treated with AM095, a selective LPA1 inhibitor (Swaney et al., 2011), showed reduced myelin loss when LPA is injected into the intact spinal cord, revealing that LPA1 signaling is involved in the LPA-demyelinating lesion, consistent with previous studies within the peripheral nervous system (Inoue et al., 2004, 2008a,b; Xie et al., 2008; Nagai et al., 2010; Halder et al., 2013). SCI effects appear to involve microglia/macrophages. Although we also noticed mild astrogliosis in the spinal cord after LPA injection, the effects of the LPA were more pronounced in microglia/macrophages, suggesting that they are more susceptible to activation by LPA. Importantly, we also observed that intraspinal injection of LPA led to the development of a demyelinating lesion in the spinal cord. This is of crucial importance because myelin loss contributes to functional impairments after SCI and in other neural conditions, such as multiple sclerosis.
LPA1 is found on oligodendrocytes (Weiner et al., 1998), and its expression appears shortly before maturation/myelination (Garcia-Diaz et al., 2014). Although LPA1 signaling plays a key role in the regulation of oligodendrocyte differentiation and myelination in the CNS (Garcia-Diaz et al., 2014), little is known about the effects of LPA on mature oligodendrocytes. Our data indicate that LPA exerts mild toxicity in oligodendrocytes at doses ≥1 μm but not in oligodendrocyte precursor cells. The mechanism underlying LPA toxicity is unknown, but this effect is not mediated via LPA1 because AM095 failed to rescue cell death, at least in cell culture. Hence, LPA-mediated demyelination is likely to be mediated by the activation of LPA1 in other cell types rather than to direct activation of LPA1 in oligodendrocytes, consistent with our data showing that intraspinal injection of LPA leads to activation of microglia/macrophages that express LPA1 in vivo and in vitro, as we and other authors have demonstrated (Tham et al., 2003). Conditioned medium of microglia stimulated with LPA mediates marked oligodendrocyte cell death, indicating that microglial become cytotoxic during LPA stimulation of LPA1 signaling. Interestingly, selective blockade or gene deletion of LPA1 in LPA-stimulated microglial cells appears to increase oligodendrocyte cell survival. This result suggests that microglia LPA1 is involved in the development of the demyelinating injury triggered by LPA injection into the spinal cord. However, LPA1 inhibition did not completely suppress the harmful effects of LPA-stimulated microglia, suggesting that other microglial LPARs may contribute to oligodendrocyte cell death. Similarly, myelin loss was not completely abrogated in the maLPA1 null mice or in mice administered AM095, indicating that other LPARs or other non-LPA-mediated mechanisms also contribute to demyelination. However, LPA1 blockade does not reduce microglia or macrophage accumulation in SCI, but it likely attenuates the harmful effects linked to LPA1 activation. The soluble factors released by microglial cells during LPA1 activation that mediate oligodendrocyte cell death are unidentified yet, but it can be attributed to cytokine and free radical production because LPA regulates the production of these factor in several conditions (Cummings et al., 2004; Fang et al., 2004; Sevastou et al., 2013).
Activation of Rho, which can be induced by LPA, is one of the most important intracellular pathways that inhibit axonal regeneration. Previous works revealed that LPA triggers axonal collapse and retraction (Tigyi et al., 1996; Birgbauer and Chun, 2006), in part by activating Rho (Fincher et al., 2014). However, the inhibitory effects of LPA on axonal growth are not triggered by LPA1, at least in vitro (Birgbauer and Chun, 2010).
Reports that a “B3” antibody that can bind to LPA and other lysophospholipids and mediates protection after spinal cord hemisection and traumatic brain injury (Goldshmit et al., 2012; Crack et al., 2014) are consistent with increased LPA in CNS injuries. However, mechanistic validation, including demonstration that B3 specifically lowers pathogenic LPA availability to account for its effects, remain to be established. Nevertheless, possible therapies targeting LPA, its biosynthetic enzymes, and/or its receptors may represent viable strategies for improving clinical outcomes for SCI in the future. In particular, our results suggest that LPA1 could be a novel therapeutic target for the treatment of acute SCI, as well as other CNS conditions in which demyelination contributes to the pathology.
Footnotes
This work was supported by National Institutes of Health Grant NS084398 (J.C.), Wings for Life Foundation, Marie-Curie International Reintegration Program Grant MC IRG 249274, Spanish Ministry of Economy and Competitiveness Grant SAF2013-48431-R, and the Health Research Fund of Spain [Cell Therapy Network and Center for Biomedical Research in Neurodegenerative Diseases Network (CIBERNED)] (R.L-V.) E.S.-N. is a recipient from a FPU fellowship. We thank Dr. Samuel David for critical review of this manuscript. We also acknowledge Marta Morell and Jessica Jaramillo for excellence technical assistance and Margaret Attiwell for help in electron microscopy. We thank Bristol-Myers Squibb for the gift of AM095.
References
- Aoki J, Taira A, Takanezawa Y, Kishi Y, Hama K, Kishimoto T, Mizuno K, Saku K, Taguchi R, Arai H. Serum lysophosphatidic acid is produced through diverse phospholipase pathways. J Biol Chem. 2002;277:48737–48744. doi: 10.1074/jbc.M206812200. [DOI] [PubMed] [Google Scholar]
- Basso DM, Fisher LC, Anderson AJ, Jakeman LB, McTigue DM, Popovich PG. Basso Mouse Scale for locomotion detects differences in recovery after spinal cord injury in five common mouse strains. J Neurotrauma. 2006;23:635–659. doi: 10.1089/neu.2006.23.635. [DOI] [PubMed] [Google Scholar]
- Birgbauer E, Chun J. New developments in the biological functions of lysophospholipids. Cell Mol Life Sci. 2006;63:2695–2701. doi: 10.1007/s00018-006-6155-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birgbauer E, Chun J. Lysophospholipid receptors LPA1–3 are not required for the inhibitory effects of LPA on mouse retinal growth cones. Eye Brain. 2010;2:1–13. doi: 10.2147/EB.S7666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bot M, Bot I, Lopez-Vales R, van de Lest CH, Saulnier-Blache JS, Helms JB, David S, van Berkel TJ, Biessen EA. Atherosclerotic lesion progression changes lysophosphatidic acid homeostasis to favor its accumulation. Am J Pathol. 2010;176:3073–3084. doi: 10.2353/ajpath.2010.090009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brinkmann V, Billich A, Baumruker T, Heining P, Schmouder R, Francis G, Aradhye S, Burtin P. Fingolimod (FTY720): discovery and development of an oral drug to treat multiple sclerosis. Nat Rev Drug Discov. 2010;9:883–897. doi: 10.1038/nrd3248. [DOI] [PubMed] [Google Scholar]
- Castilla-Ortega E, Pedraza C, Chun J, de Fonseca FR, Estivill-Torrús G, Santín LJ. Hippocampal c-Fos activation in normal and LPA(1)-null mice after two object recognition tasks with different memory demands. Behav Brain Res. 2012;232:400–405. doi: 10.1016/j.bbr.2012.04.018. [DOI] [PubMed] [Google Scholar]
- Choi JW, Chun J. Lysophospholipids and their receptors in the central nervous system. Biochim Biophys Acta. 2013;1831:20–32. doi: 10.1016/j.bbalip.2012.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi JW, Herr DR, Noguchi K, Yung YC, Lee CW, Mutoh T, Lin ME, Teo ST, Park KE, Mosley AN, Chun J. LPA receptors: subtypes and biological actions. Annu Rev Pharmacol Toxicol. 2010;50:157–186. doi: 10.1146/annurev.pharmtox.010909.105753. [DOI] [PubMed] [Google Scholar]
- Chun J, Brinkmann V. A mechanistically novel, first oral therapy for multiple sclerosis: the development of fingolimod (FTY720, Gilenya) Discov Med. 2011;12:213–228. [PMC free article] [PubMed] [Google Scholar]
- Contos JJ, Fukushima N, Weiner JA, Kaushal D, Chun J. Requirement for the lpA1 lysophosphatidic acid receptor gene in normal suckling behavior. Proc Natl Acad Sci U S A. 2000;97:13384–13389. doi: 10.1073/pnas.97.24.13384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Contos JJ, Ishii I, Fukushima N, Kingsbury MA, Ye X, Kawamura S, Brown JH, Chun J. Characterization of lpa(2) (Edg4) and lpa(1)/lpa(2) (Edg2/Edg4) lysophosphatidic acid receptor knockout mice: signaling deficits without obvious phenotypic abnormality attributable to lpa(2) Mol Cell Biol. 2002;22:6921–6929. doi: 10.1128/MCB.22.19.6921-6929.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crack PJ, Zhang M, Morganti-Kossmann MC, Morris AJ, Wojciak JM, Fleming JK, Karve I, Wright D, Sashindranath M, Goldshmit Y, Conquest A, Daglas M, Johnston LA, Medcalf RL, Sabbadini RA, Pebay A. Anti-lysophosphatidic acid antibodies improve traumatic brain injury outcomes. J Neuroinflammation. 2014;11:37. doi: 10.1186/1742-2094-11-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cummings R, Zhao Y, Jacoby D, Spannhake EW, Ohba M, Garcia JG, Watkins T, He D, Saatian B, Natarajan V. Protein kinase Cdelta mediates lysophosphatidic acid-induced NF-kappaB activation and interleukin-8 secretion in human bronchial epithelial cells. J Biol Chem. 2004;279:41085–41094. doi: 10.1074/jbc.M404045200. [DOI] [PubMed] [Google Scholar]
- David S, López-Vales R, Wee Yong V. Harmful and beneficial effects of inflammation after spinal cord injury: potential therapeutic implications. Handb Clin Neurol. 2012a;109:485–502. doi: 10.1016/B978-0-444-52137-8.00030-9. [DOI] [PubMed] [Google Scholar]
- David S, Zarruk JG, Ghasemlou N. Inflammatory pathways in spinal cord injury. Int Rev Neurobiol. 2012b;106:127–152. doi: 10.1016/B978-0-12-407178-0.00006-5. [DOI] [PubMed] [Google Scholar]
- David S, Greenhalgh AD, López-Vales R. Role of phospholipase A2s and lipid mediators in secondary damage after spinal cord injury. Cell Tissue Res. 2012c;349:249–267. doi: 10.1007/s00441-012-1430-8. [DOI] [PubMed] [Google Scholar]
- Dubin AE, Bahnson T, Weiner JA, Fukushima N, Chun J. Lysophosphatidic acid stimulates neurotransmitter-like conductance changes that precede GABA and l-glutamate in early, presumptive cortical neuroblasts. J Neurosci. 1999;19:1371–1381. doi: 10.1523/JNEUROSCI.19-04-01371.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubin AE, Herr DR, Chun J. Diversity of lysophosphatidic acid receptor-mediated intracellular calcium signaling in early cortical neurogenesis. J Neurosci. 2010;30:7300–7309. doi: 10.1523/JNEUROSCI.6151-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Estivill-Torrús G, Llebrez-Zayas P, Matas-Rico E, Santín L, Pedraza C, De Diego I, Del Arco I, Fernández-Llebrez P, Chun J, De Fonseca FR. Absence of LPA1 signaling results in defective cortical development. Cereb Cortex. 2008;18:938–950. doi: 10.1093/cercor/bhm132. [DOI] [PubMed] [Google Scholar]
- Fang X, Yu S, Bast RC, Liu S, Xu HJ, Hu SX, LaPushin R, Claret FX, Aggarwal BB, Lu Y, Mills GB. Mechanisms for lysophosphatidic acid-induced cytokine production in ovarian cancer cells. J Biol Chem. 2004;279:9653–9661. doi: 10.1074/jbc.M306662200. [DOI] [PubMed] [Google Scholar]
- Fincher J, Whiteneck C, Birgbauer E. G-protein-coupled receptor cell signaling pathways mediating embryonic chick retinal growth cone collapse induced by lysophosphatidic acid and sphingosine-1-phosphate. Dev Neurosci. 2014;36:443–453. doi: 10.1159/000364858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frisca F, Sabbadini RA, Goldshmit Y, Pébay A. Biological effects of lysophosphatidic acid in the nervous system. Int Rev Cell Mol Biol. 2012;296:273–322. doi: 10.1016/B978-0-12-394307-1.00005-9. [DOI] [PubMed] [Google Scholar]
- Fukushima N, Weiner JA, Kaushal D, Contos JJ, Rehen SK, Kingsbury MA, Kim KY, Chun J. Lysophosphatidic acid influences the morphology and motility of young, postmitotic cortical neurons. Mol Cell Neurosci. 2002;20:271–282. doi: 10.1006/mcne.2002.1123. [DOI] [PubMed] [Google Scholar]
- García-Alías G, Verdú E, Forés J, López-Vales R, Navarro X. Functional and electrophysiological characterization of photochemical graded spinal cord injury in the rat. J Neurotrauma. 2003;20:501–510. doi: 10.1089/089771503765355568. [DOI] [PubMed] [Google Scholar]
- Garcia-Diaz B, Riquelme R, Varela-Nieto I, Jimenez AJ, de Diego I, Gomez-Conde AL, Matas-Rico E, Aguirre JA, Chun J, Pedraza C, Santin LJ, Fernandez O, Rodriguez de Fonseca F, Estivill-Torrus G. Loss of lysophosphatidic acid receptor LPA alters oligodendrocyte differentiation and myelination in the mouse cerebral cortex. Brain Struct Funct. 2014 doi: 10.1007/s00429-014-0885-7. Advance online publication. Retrieved June 14, 2015. [DOI] [PubMed] [Google Scholar]
- Goldshmit Y, Matteo R, Sztal T, Ellett F, Frisca F, Moreno K, Crombie D, Lieschke GJ, Currie PD, Sabbadini RA, Pébay A. Blockage of lysophosphatidic acid signaling improves spinal cord injury outcomes. Am J Pathol. 2012;181:978–992. doi: 10.1016/j.ajpath.2012.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halder SK, Yano R, Chun J, Ueda H. Involvement of LPA1 receptor signaling in cerebral ischemia-induced neuropathic pain. Neuroscience. 2013;235:10–15. doi: 10.1016/j.neuroscience.2013.01.005. [DOI] [PubMed] [Google Scholar]
- Herr KJ, Herr DR, Lee CW, Noguchi K, Chun J. Stereotyped fetal brain disorganization is induced by hypoxia and requires lysophosphatidic acid receptor 1 (LPA1) signaling. Proc Natl Acad Sci U S A. 2011;108:15444–15449. doi: 10.1073/pnas.1106129108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue M, Rashid MH, Fujita R, Contos JJ, Chun J, Ueda H. Initiation of neuropathic pain requires lysophosphatidic acid receptor signaling. Nat Med. 2004;10:712–718. doi: 10.1038/nm1060. [DOI] [PubMed] [Google Scholar]
- Inoue M, Ma L, Aoki J, Chun J, Ueda H. Autotaxin, a synthetic enzyme of lysophosphatidic acid (LPA), mediates the induction of nerve-injured neuropathic pain. Mol Pain. 2008a;4:6. doi: 10.1186/1744-8069-4-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue M, Xie W, Matsushita Y, Chun J, Aoki J, Ueda H. Lysophosphatidylcholine induces neuropathic pain through an action of autotaxin to generate lysophosphatidic acid. Neuroscience. 2008b;152:296–298. doi: 10.1016/j.neuroscience.2007.12.041. [DOI] [PubMed] [Google Scholar]
- Kihara Y, Maceyka M, Spiegel S, Chun J. Lysophospholipid receptor nomenclature review: IUPHAR Review 8. Br J Pharmacol. 2014;171:3575–3594. doi: 10.1111/bph.12678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kingsbury MA, Rehen SK, Contos JJ, Higgins CM, Chun J. Non-proliferative effects of lysophosphatidic acid enhance cortical growth and folding. Nat Neurosci. 2003;6:1292–1299. doi: 10.1038/nn1157. [DOI] [PubMed] [Google Scholar]
- Klopstein A, Santos-Nogueira E, Francos-Quijorna I, Redensek A, David S, Navarro X, López-Vales R. Beneficial effects of alphaB-crystallin in spinal cord contusion injury. J Neurosci. 2012;32:14478–14488. doi: 10.1523/JNEUROSCI.0923-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin ME, Rivera RR, Chun J. Targeted deletion of LPA5 identifies novel roles for lysophosphatidic acid signaling in development of neuropathic pain. J Biol Chem. 2012;287:17608–17617. doi: 10.1074/jbc.M111.330183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- López-Vales R, Ghasemlou N, Redensek A, Kerr BJ, Barbayianni E, Antonopoulou G, Baskakis C, Rathore KI, Constantinou-Kokotou V, Stephens D, Shimizu T, Dennis EA, Kokotos G, David S. Phospholipase A2 superfamily members play divergent roles after spinal cord injury. FASEB J. 2011;25:4240–4252. doi: 10.1096/fj.11-183186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma L, Nagai J, Chun J, Ueda H. An LPA species (18:1 LPA) plays key roles in the self-amplification of spinal LPA production in the peripheral neuropathic pain model. Mol Pain. 2013;9:29. doi: 10.1186/1744-8069-9-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Möller T, Contos JJ, Musante DB, Chun J, Ransom BR. Expression and function of lysophosphatidic acid receptors in cultured rodent microglial cells. J Biol Chem. 2001;276:25946–25952. doi: 10.1074/jbc.M102691200. [DOI] [PubMed] [Google Scholar]
- Nagai J, Uchida H, Matsushita Y, Yano R, Ueda M, Niwa M, Aoki J, Chun J, Ueda H. Autotaxin and lysophosphatidic acid1 receptor-mediated demyelination of dorsal root fibers by sciatic nerve injury and intrathecal lysophosphatidylcholine. Mol Pain. 2010;6:78. doi: 10.1186/1744-8069-6-78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Meara RW, Ryan SD, Colognato H, Kothary R. Derivation of enriched oligodendrocyte cultures and oligodendrocyte/neuron myelinating co-cultures from post-natal murine tissues. J Vis Exp. 2011;pii:3324. doi: 10.3791/3324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ousman SS, David S. Lysophosphatidylcholine induces rapid recruitment and activation of macrophages in the adult mouse spinal cord. Glia. 2000;30:92–104. doi: 10.1002/(SICI)1098-1136(200003)30:1<92::AID-GLIA10>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- Ousman SS, David S. MIP-1alpha, MCP-1, GM-CSF, and TNF-alpha control the immune cell response that mediates rapid phagocytosis of myelin from the adult mouse spinal cord. J Neurosci. 2001;21:4649–4656. doi: 10.1523/JNEUROSCI.21-13-04649.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedraza C, Sánchez-López J, Castilla-Ortega E, Rosell-Valle C, Zambrana-Infantes E, García-Fernández M, Rodriguez de Fonseca F, Chun J, Santín LJ, Estivill-Torrús G. Fear extinction and acute stress reactivity reveal a role of LPA(1) receptor in regulating emotional-like behaviors. Brain Struct Funct. 2014;219:1659–1672. doi: 10.1007/s00429-013-0592-9. [DOI] [PubMed] [Google Scholar]
- Popovich PG. Neuroimmunology of traumatic spinal cord injury: a brief history and overview. Exp Neurol. 2014;258:1–4. doi: 10.1016/j.expneurol.2014.05.001. [DOI] [PubMed] [Google Scholar]
- Rowland JW, Hawryluk GW, Kwon B, Fehlings MG. Current status of acute spinal cord injury pathophysiology and emerging therapies: promise on the horizon. Neurosurg Focus. 2008;25:E2. doi: 10.3171/FOC.2008.25.11.E2. [DOI] [PubMed] [Google Scholar]
- Santin LJ, Bilbao A, Pedraza C, Matas-Rico E, López-Barroso D, Castilla-Ortega E, Sánchez-López J, Riquelme R, Varela-Nieto I, de la Villa P, Suardíaz M, Chun J, De Fonseca FR, Estivill-Torrús G. Behavioral phenotype of maLPA1-null mice: increased anxiety-like behavior and spatial memory deficits. Genes Brain Behav. 2009;8:772–784. doi: 10.1111/j.1601-183X.2009.00524.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saura J, Tusell JM, Serratosa J. High-yield isolation of murine microglia by mild trypsinization. Glia. 2003;44:183–189. doi: 10.1002/glia.10274. [DOI] [PubMed] [Google Scholar]
- Scherer M, Schmitz G, Liebisch G. High-throughput analysis of sphingosine 1-phosphate, sphinganine 1-phosphate, and lysophosphatidic acid in plasma samples by liquid chromatography-tandem mass spectrometry. Clin Chem. 2009;55:1218–1222. doi: 10.1373/clinchem.2008.113779. [DOI] [PubMed] [Google Scholar]
- Sevastou I, Kaffe E, Mouratis MA, Aidinis V. Lysoglycerophospholipids in chronic inflammatory disorders: the PLA(2)/LPC and ATX/LPA axes. Biochim Biophys Acta. 2013;1831:42–60. doi: 10.1016/j.bbalip.2012.07.019. [DOI] [PubMed] [Google Scholar]
- Shano S, Moriyama R, Chun J, Fukushima N. Lysophosphatidic acid stimulates astrocyte proliferation through LPA1. Neurochem Int. 2008;52:216–220. doi: 10.1016/j.neuint.2007.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheng X, Yung YC, Chen A, Chun J. Lysophosphatidic acid signalling in development. Development. 2015;142:1390–1395. doi: 10.1242/dev.121723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spohr TC, Choi JW, Gardell SE, Herr DR, Rehen SK, Gomes FC, Chun J. Lysophosphatidic acid receptor-dependent secondary effects via astrocytes promote neuronal differentiation. J Biol Chem. 2008;283:7470–7479. doi: 10.1074/jbc.M707758200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stirling DP, Yong VW. Dynamics of the inflammatory response after murine spinal cord injury revealed by flow cytometry. J Neurosci Res. 2008;86:1944–1958. doi: 10.1002/jnr.21659. [DOI] [PubMed] [Google Scholar]
- Swaney JS, Chapman C, Correa LD, Stebbins KJ, Broadhead AR, Bain G, Santini AM, Darlington J, King CD, Baccei CS, Lee C, Parr TA, Roppe JR, Seiders TJ, Ziff J, Prasit P, Hutchinson JH, Evans JF, Lorrain DS. Pharmacokinetic and pharmacodynamic characterization of an oral lysophosphatidic acid type 1 receptor-selective antagonist. J Pharmacol Exp Ther. 2011;336:693–700. doi: 10.1124/jpet.110.175901. [DOI] [PubMed] [Google Scholar]
- Tham CS, Lin FF, Rao TS, Yu N, Webb M. Microglial activation state and lysophospholipid acid receptor expression. Int J Dev Neurosci. 2003;21:431–443. doi: 10.1016/j.ijdevneu.2003.09.003. [DOI] [PubMed] [Google Scholar]
- Tigyi G, Fischer DJ, Sebök A, Yang C, Dyer DL, Miledi R. Lysophosphatidic acid-induced neurite retraction in PC12 cells: control by phosphoinositide-Ca2+ signaling and Rho. J Neurochem. 1996;66:537–548. doi: 10.1046/j.1471-4159.1996.66020537.x. [DOI] [PubMed] [Google Scholar]
- Titsworth WL, Cheng X, Ke Y, Deng L, Burckardt KA, Pendleton C, Liu NK, Shao H, Cao QL, Xu XM. Differential expression of sPLA2 following spinal cord injury and a functional role for sPLA2-IIA in mediating oligodendrocyte death. Glia. 2009;57:1521–1537. doi: 10.1002/glia.20867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trimbuch T, Beed P, Vogt J, Schuchmann S, Maier N, Kintscher M, Breustedt J, Schuelke M, Streu N, Kieselmann O, Brunk I, Laube G, Strauss U, Battefeld A, Wende H, Birchmeier C, Wiese S, Sendtner M, Kawabe H, Kishimoto-Suga M, et al. Synaptic PRG-1 modulates excitatory transmission via lipid phosphate-mediated signaling. Cell. 2009;138:1222–1235. doi: 10.1016/j.cell.2009.06.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiner JA, Hecht JH, Chun J. Lysophosphatidic acid receptor gene vzg-1/lpA1/edg-2 is expressed by mature oligodendrocytes during myelination in the postnatal murine brain. J Comp Neurol. 1998;398:587–598. doi: 10.1002/(SICI)1096-9861(19980907)398:43.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- Weiner JA, Fukushima N, Contos JJ, Scherer SS, Chun J. Regulation of Schwann cell morphology and adhesion by receptor-mediated lysophosphatidic acid signaling. J Neurosci. 2001;21:7069–7078. doi: 10.1523/JNEUROSCI.21-18-07069.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie W, Matsumoto M, Chun J, Ueda H. Involvement of LPA1 receptor signaling in the reorganization of spinal input through Abeta-fibers in mice with partial sciatic nerve injury. Mol Pain. 2008;4:46. doi: 10.1186/1744-8069-4-46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yung YC, Mutoh T, Lin ME, Noguchi K, Rivera RR, Choi JW, Kingsbury MA, Chun J. Lysophosphatidic acid signaling may initiate fetal hydrocephalus. Sci Transl Med. 2011;3:99ra87. doi: 10.1126/scitranslmed.3002095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yung YC, Stoddard NC, Chun J. LPA receptor signaling: pharmacology, physiology, and pathophysiology. J Lipid Res. 2014;55:1192–1214. doi: 10.1194/jlr.R046458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yung YC, Stoddard NC, Mirendil H, Chun J. Lysophosphatidic acid signaling in the nervous system. Neuron. 2015;85:669–682. doi: 10.1016/j.neuron.2015.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]